

Abstract
Transforming Growth Factor Beta (TGFβ) potently induces lens epithelial to mesenchymal transition (EMT). The resultant mesenchymal cells resemble those found in plaques of human forms of subcapsular cataract. Smad signaling has long been implicated as the sole driving force of TGFβ-mediated activity. Rat lens epithelial explants were used to examine the role of the Smad-independent signaling, namely the MAPK/ERK1/2 signaling pathway, in the initiation and progression of TGFβ-induced EMT. Phase contrast microscopy was used to observe the morphological changes associated with TGFβ-induced EMT in this model, including cell elongation, cell membrane blebbing, cell loss as indicated by the area of bare capsule and capsular wrinkling. The levels of Smad2, Smad2/3 and ERK1/2 phosphorylation measured using western blotting confirmed that the addition of UO126 was sufficient in blocking all TGFβ-induced ERK1/2 activation, as well as reducing Smad signaling at 18 hours. Immunofluorescent labeling and further western blotting confirmed that TGFβ-induced EMT was associated with an increase in α-smooth muscle actin (α-SMA) and a reduction of E-cadherin at cell borders. Pre-treatment with UO126 was effective at blocking the TGFβ-induced EMT, as evidenced by a reduction of α-SMA expression and protein labeling, E-cadherin labeling at cell borders, and a reduction of cell loss, cell elongation and capsular wrinkling. Post-treatment with UO126 at 2 and 6 hours after TGFβ addition was also effective at blocking EMT while post-treatment with UO126 at 24 and 48 hours was not sufficient in hampering TGFβ-induced EMT. Our data implicates ERK1/2 signaling in the initiation but not the progression of TGFβ-induced EMT in rat lens epithelial cells. The tight regulation of intracellular signaling pathways such as ERK1/2 are required for the maintenance of lens epithelial cell integrity and hence tissue transparency. A greater understanding of the molecular mechanisms that drive the induction and progression of EMT in the lens will provide the basis for potential therapeutics for human cataract.
Keywords: TGFβ, ERK1/2, lens pathology, EMT
1. Introduction
Cataract is the leading cause of blindness worldwide (Khairallah et al., 2015), with aging being a leading contributing factor (Mukesh et al, 2006). One form of cataract, Anterior Subcapsular Cataract (ASC) is primarily a pathology of the lens epithelium, characterized by multilayered cell aggregates that form anterior subcapsular plaques. The removal of such fibrotic cataracts and others through surgery can lead to secondary cataract, commonly known as Posterior Capsular Opacification (PCO) (see Wormstone et al., 2009). The fibrotic morphologic and molecular features of ASC and PCO are similar and can be mimicked in vitro with the exposure of primary lens epithelial cells to Transforming Growth Factor beta (TGFβ), resulting in an Epithelial to Mesenchymal Transition (EMT; Liu et al., 1994; Hales et al., 1994; Hales et al., 1995; de Iongh et al., 2005). The characteristic features of lens cells that have undergone an EMT include elongation into a spindle-shape with tapered ends, accumulation of α-smooth muscle actin (α-SMA) into stress fibers and changes to specialized cell-cell junctions leading to the loss of cell polarity and migration. Cell death through apoptosis is also a feature associated with TGFβ-induced EMT that appears specific to lens epithelial explants (Liu et al., 1994; Lovicu et al., 2002; Maruno et al., 2002).
Smad signal transduction is considered central to propagating TGFβ signaling. Ligand binding activates the transmembrane TGFβ receptor type II (TβR-II), enabling it to bind and activate TβR-I. Activation of these receptors facilitates the activation of receptor-regulated Smad2 and Smad3 (R-Smads) through C-terminal serine/threonine kinase activity (Massague, 1998). R-Smads consist of an N-terminal MH1 (MAD-Homology 1) and a C-terminal MH2 domain, joined by a linker region containing phosphorylation sites that facilitate cross-talk with non-Smad signaling pathways, such as ERK1/2 (Shi & Massague, 2003). Once phosphorylated, the Smad2/3 complex with the co-effector, Smad4, enter the nucleus to facilitate gene transcription (Wrana & Attisano, 2000; Zhu & Burgess, 2001). One mode of negative regulation of TGFβ-signaling is the upregulation of the inhibitory Smad, Smad7, that terminates the TGFβ signal by interrupting the R-Smad/Smad4 complex, or by directly antagonizing the TβRI (see Yan et al., 2009). Smad-independent signaling pathways have also been implicated in TGFβ-induced EMT. For example, in NMuMG cells, TGFβ-mediated ZO-1 and E-cadherin dissociation has been linked to PI3K/Akt signaling (Bakin et al., 2000), and apoptosis has been shown to be p38/MAPK-dependent (Yu et al., 2002). In α-TN4 mouse lens epithelial cells, TGFβ-induced upregulation of α-SMA was shown to be through Snail via PI3K/Akt signaling, with ERK1/2 and p38 not playing a role in TGFβ-induced EMT in this system (Cho et al., 2007). Conversely in FHL124 human lens cells, TGFβ has been shown to activate Smad-independent MAPK signaling pathways such as p38, JNK and ERK1/2 (Dawes et al., 2009); however, their direct function during TGFβ-induced EMT has not been examined.
In the lens, MAPK/ERK1/2 signaling is associated with normal cellular processes of epithelial cell proliferation and differentiation (Lovicu et al., 2001; Le & Musil, 2001; Upadhya et al., 2013). The conditional deletion of ERK2 in the developing mouse lens leads to disruption of cell proliferation and an increase in apoptosis (Upadhya et al., 2013). Fibroblast growth factor (FGF) is the only known growth factor present in the ocular media that is able to induce both proliferation and differentiation of lens cells, via MAPK/ERK1/2 signaling, in a dose dependent manner (Lovicu & McAvoy, 2001; Le & Musil, 2001). Given these distinct cellular responses are tightly regulated, so must FGF-induced ERK1/2 signaling. FGF ligand binding activates FGF receptor tyrosine kinases (RTKs), that results in the presentation of intracellular docking sites, and the recruitment and interaction with the Grb2/Sos complex that activates the GTPase Ras that in turn recruits Raf to the cell membrane, where it too is activated. Once activated, Raf phosphorylates MEK1/2 that in turn phosphorylates ERK1/2 (Schlessinger, 2000). In the lens and other tissues, a number of different RTK-antagonists have been identified, including Sprouty (Spry), Sef and Spred (Kramer et al., 1999; Tefft et al., 1999; Chambers & Mason, 2000; Boros et al., 2006; Shin et al., 2012; Zhao et al., 2015). These different antagonists are reported to regulate ERK1/2-signaling at different levels (Kovalenko et al., 2003; Mason et al., 2006; Meng et al., 2012).
Earlier studies in our laboratory with transgenic mice have revealed a putative role for ERK1/2 signaling during TGFβ-induced EMT (Shin et al., 2012). The deletion of Spry in the lens disrupts normal ERK1/2 signaling leading to aberrant lens cell transdifferentiation, with a rise in Smad-activation prior to an EMT that leads to cataract. Moreover, overexpression of Spry in the lens not only suppresses ERK1/2 signaling, but also TGFβ signaling, effectively blocking EMT and the development of cataract (Shin et al., 2012). TGFβ-induced EMT in rat lens epithelial explants was also recently shown to be repressed when cells were transfected with other RTK-inhibitors, including Spred and Sef (Zhao et al., 2015). TGFβ-induced EMT can be suppressed through the inhibition of ERK1/2 signaling in different cellular contexts, such as primary human lens epithelial cells, SRA01/04 human lens epithelial cells, NMuMG epithelial cells, mouse cortical tubule (MCT) epithelial cells and HaCaT cells (Zavadil et al., 2001; Xie et al., 2004; Chen et al., 2014; Tiwari et al., 2016). Taken together, these studies strongly suggest a requirement for ERK1/2 signaling in TGFβ-induced EMT.
While TGFβ-induced EMT is a well-defined process; the precise intracellular signaling events that drive EMT are yet to be fully elucidated. A role for non-canonical TGFβ signal transduction is implicated in the EMT process and appears to be context and tissue specific. In the present study, we examine the role for ERK1/2 phosphorylation not only for the initiation of TGFβ-induced EMT, but in the progression of this pathological process.
2. Materials and Methods
Lens tissue was obtained from postnatal day 21 (P21) albino Wistar rats (Rattus norvegivus). All animal handling procedures were in accordance with the National Health and Medical Research Council (Australia), the ARVO statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Animal Ethical Review Committee of the University of Sydney, Australia.
2.1. Preparation of lens epithelial explants
Tissue collection and culture was carried out in fresh equilibrated (37°C, 5% CO2) Medium 199 with Earl’s salts (M199; Life Technologies, Waltham, MA, USA) supplemented with 50μg/ml L-glutamine, 50 IU/ml penicillin, 50 μg/ml streptomycin, 2.5 μg/ml Amphotericin B (Life Technologies) and 0.1% bovine serum albumin (BSA; Sigma-Aldrich Corp., St. Louis, MO, USA). Explanting of the lens epithelium was carried out under sterile conditions with a dissecting microscope (Wild-Leitz, Switzerland). Explants were left to equilibrate (37°C, 5% CO2) for at least 24 hours prior to treatment.
Prior to treatment, media was replaced in all dishes with 1ml of fresh medium. Human recombinant TGFβ-2 (R&D Systems, Minneapolis, MN, USA) was used at 200pg - 1ng/ml to induce EMT. The selective MEK1/2 inhibitor, UO126 (Promega, Madison, WI, USA), was used at 50μM to block ERK1/2 activation (see Lovicu & McAvoy, 2001). Explants were either pre-treated with UO126 two hours prior to the addition of TGFβ (U/TGFβ), or UO126 was added after the addition of TGFβ at 2 hours (TGFβ/U2), 6 hours (TGFβ/U6), 24 hours (TGFβ/U24) or 48 hours (TGFβ/U48). All experiments were repeated independently at least three times.
2.2. Phase Contrast Microscopy
Morphological changes were monitored daily using phase contrast microscopy (CK2, Olympus, Tokyo, Japan). Micrographs were obtained with a digital camera (Leica DFC-280, Leica, Wetzlar, Germany) and only explants that exhibited a tightly packed monolayer of cells at day 0 were included in the analysis.
In the present study the extent of visible lens capsule was used as a measure of cell loss, calculated as a percentage of the whole explant surface. At least 9 micrographs were obtained of each explant each day, and using Adobe Photoshop CS6, regions of bare lens capsule were filled to create a black and white image. The percentage of the total area of the lens capsule was analyzed using ImageJ. A two-way ANOVA with Tukey’s post-hoc analysis was used to infer statistical significance.
2.3. SDS-PAGE and Western Blotting
At the end of the respective incubation periods, explants were collected, lysed and run through a 10% SDS-PAGE gel, as described previously (Zhao et. al., 2015). The PVDF membranes (Merck Millipore, Billerica, MA, USA) were incubated overnight at 4°C with the primary antibody diluted in blocking buffer (2.5% BSA/TBST or 5% skim milk/TBST). The antibodies used were against phospho-ERK1/2 (1:1000; Cell Signaling Technology, Danvers, MA, USA), total-ERK1/2 (1:1000; Cell Signaling Technology), phospho-Smad2 (Ser245/250/255; 1:1000; Cell Signaling Technology), phospho-Smad2/3 (Ser465/467 and Ser423/425 respectively; 1:1000; Cell Signaling Technology), total-Smad2/3 (1:1000; Cell Signaling Technology), α-smooth muscle actin (1:2000; Sigma-Aldrich Corp.), E-cadherin (1:1000, BD Transduction Laboratories, San Jose, CA, USA) and GAPDH (1:5000, Sigma-Aldrich). For subsequent quantification purposes, given their distinct size differences, p-ERK1/2, p-Smad2, p-Smad2/3 or α-sma were double-labeled with GAPDH on the same blot at the same time. Following an overnight incubation, HRP-conjugated anti-rabbit (1:5000; Cell Signaling Technology) or HRP-conjugated anti-mouse (1:10000; Zymed, USA) antibodies were used in conjunction with peroxidase solution and luminol reagent for enhanced chemiluminescence (ECL; Merck Millipore) to visualize the proteins under the ChemiDoc MP imaging system (Bio-Rad Laboratories, Hercules, CA, USA). The intensity of protein bands were analyzed using Image Lab software (Bio-Rad Laboratories), and all proteins of interest were normalized relative to corresponding GAPDH levels. An ANOVA with Tukey’s or Dunnett’s post hoc test was used to infer statistical differences between band intensities and presented as S.E.M., with a p<0.05 considered statistically significant.
2.4. Immunofluorescent labeling
All immunofluorescent analysis of explants was carried out in the original culture dishes. At the conclusion of the culture period, media in dishes was replaced with 100% methanol to fix cells for 45 seconds and then rinsed with PBS five times in quick succession. Non-specific binding was blocked with 10% normal goat serum (NGS) diluted in PBS supplemented with 1% BSA (PBS-BSA) for one hour at room temperature. Explants were incubated with primary antibody, diluted in 1.5% NGS in PBS-BSA overnight at 4°C. The proteins that were visualized with immunofluorescent labeling were α-SMA (1:100; Sigma-Aldrich Corp.) and E-cadherin (1:500; BD Transduction Laboratories). The following day, an Alexa594 (1:1000; Life Technologies, USA) secondary antibody diluted in PBS/BSA was applied for 2 hours at room temperature. Cell nuclei were counterstained with 1μg/ml Hoechst 33342 (Sigma-Aldrich Corp.), prior to mounting explants with 10% PBS in glycerol. Explants were imaged using confocal microscopy (Zeiss LSM 700; Carl Zeiss AG, Jena, Germany).
2.5. Quantitative Reverse Transcriptase- Polymerase Chain Reaction (qRT-PCR)
At the end of respective culture periods, explants were collected under RNAse-free conditions as described previously (Shu et al., 2017). RNA (200ng) was reverse-transcribed with the sensiFAST cDNA synthesis kit (Bioline, Alexandria, NSW, Australia). α-SMA primers spanning the exon-exon junction were used (see Shu et al., 2017). All qRT-PCR reactions were executed with SensiFAST SYBR No-ROX (Bioline, Australia) in the LightCycler480 (Roche Diagnostics Ltd. Frorrenstrasse, Switzerland) as described previously (Shu et al., 2017). Data was analyzed with GenEX 6.0 software (MultiD Analyses AB, Goteborg, Sweden). All experiments were carried out in triplicate, and each qRT-PCR reaction (including no-template controls and minus RT controls) were carried out in duplicate. GraphPad Prism version 7.02 (Graphpad software, Inc.) was used for statistical testing. A two-way ANOVA followed by Tukey’s post hoc test was used, with p<0.05 considered statistically significant.
3. Results
3.1. Lens epithelial cells undergo TGFβ-induced EMT
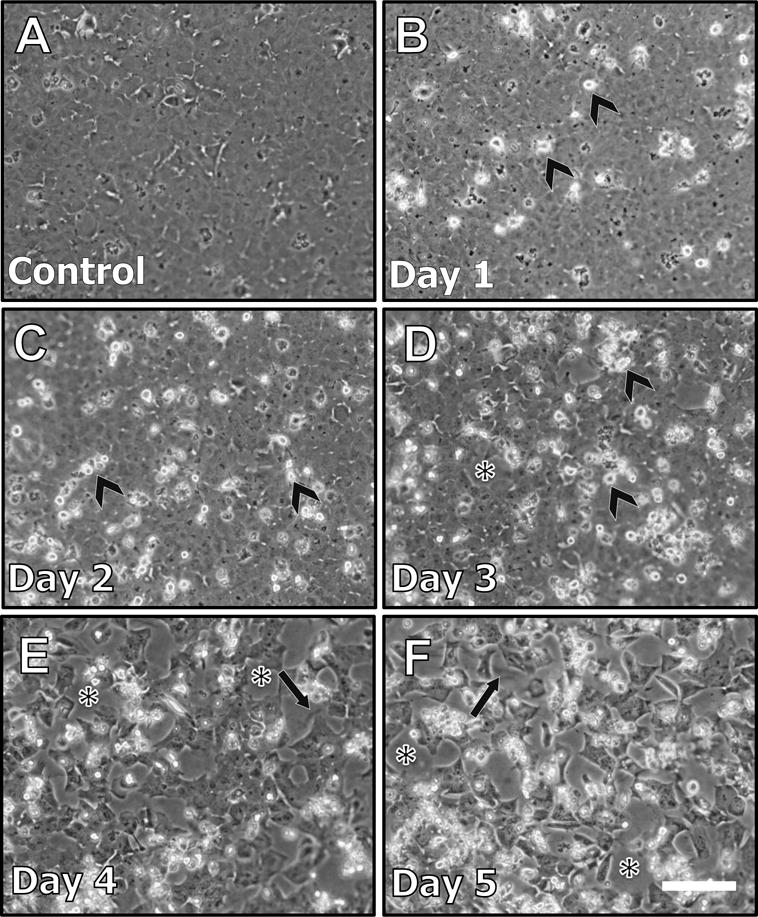
When treated with TGFβ, lens epithelial explants underwent gross morphological changes associated with EMT. Untreated control explants remained as a tightly packed epithelial monolayer throughout the 5 day culture period (see Figure 1A). After 24 hours of TGFβ treatment (day 1) some cells elongated and presented tapered ends, consistent with a myofibroblastic phenotype (Figure 1B; white arrows). This phenomenon was more pronounced on day 2 as the majority of cells present on lens explants were elongated and myofibroblastic (Figure 1C). Cellular blebbing, indicative of apoptosis was featured in explants at day 2 (Figure 1C, arrowheads) and day 3 (Figure 1D, arrowheads). Consistent with this, areas of cell loss, as revealed by bare patches of the underlying lens capsule (Figure 2D; asterisk), featured after 3 days of TGFβ treatment. There was a 62% cell loss after 3 days of TGFβ treatment (Figure 8) with clear visualization of capsular wrinkling (Figure 1D; arrows). The entire exposed lens capsule was visible at day 4 and 5 of TGFβ treatment, indicative of complete cell loss (Figure 1E, 1F).
Figure 1.
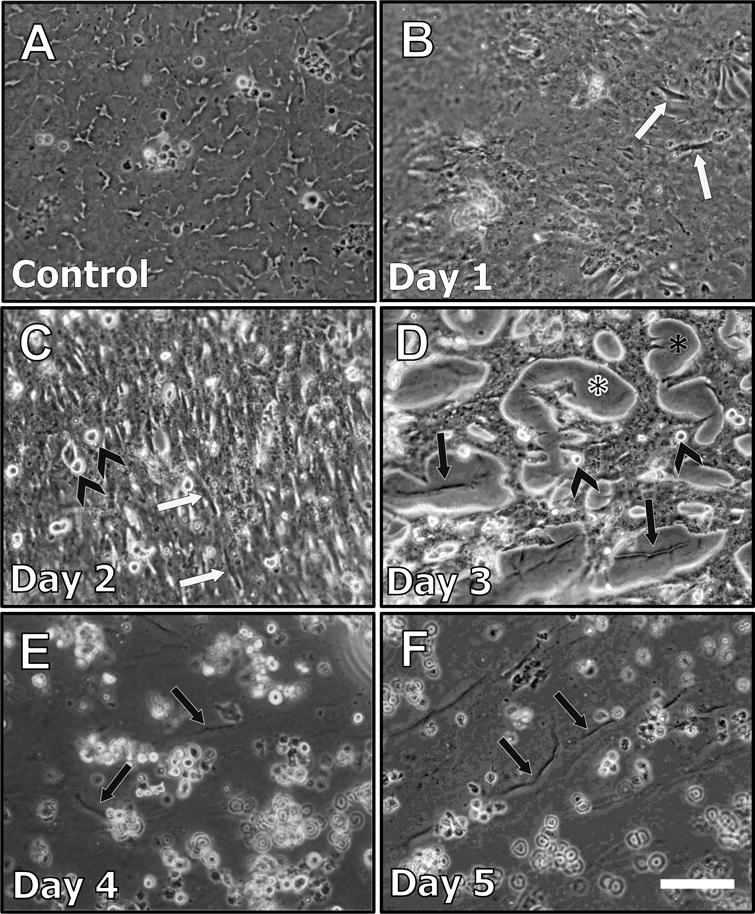
Representative phase contrast micrographs of lens epithelial explants treated with TGFβ for up to 5 days. Explants not treated with TGFβ exhibited a tightly packed monolayer (A). After 24 hours (Day 1) of TGFβ treatment (B), some cells began to elongate (white arrows). At 2 days post-treatment (C), there was more pronounced cell elongation (white arrows), and cellular blebbing was also evident (arrowheads, see also D). After 3 days of TGFβ treatment (D), cell loss led to large areas of lens capsule exposed (asterisk), revealing capsular wrinkling (black arrows). At 4 (E) and 5 days (F), viable cells are mostly absent, with pronounced capsular wrinkling more visible (black arrows) on the bare capsule (Scale bar = 100μm).
Figure 2.
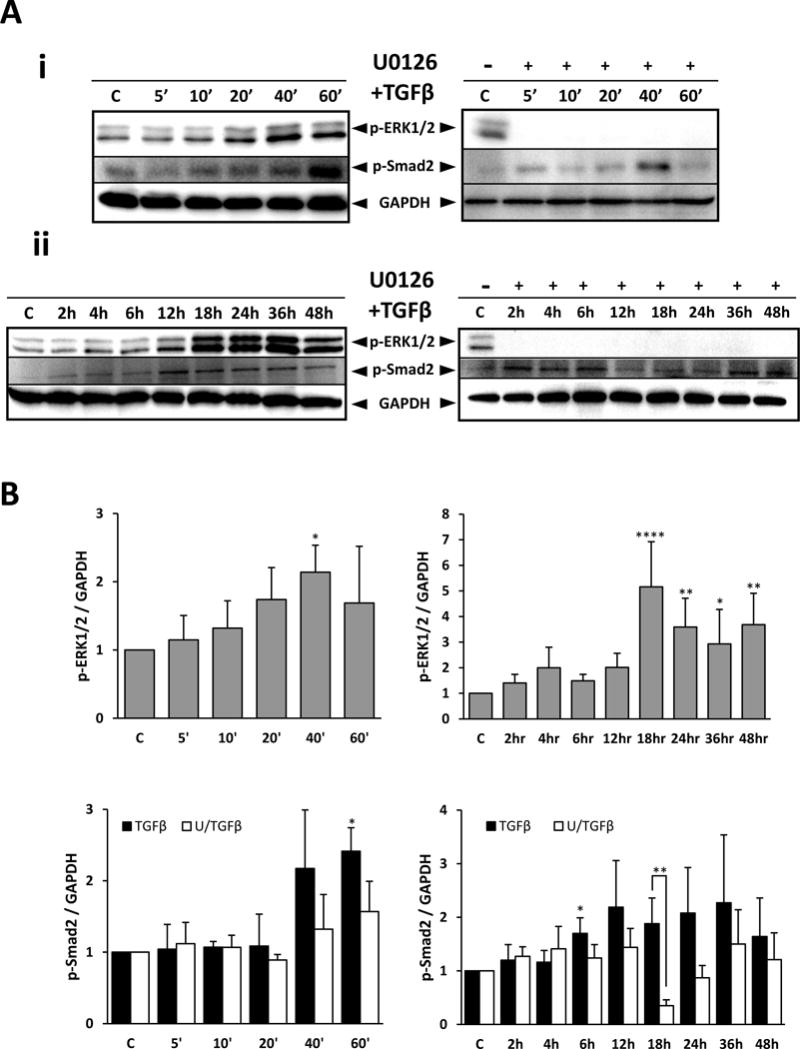
Western blot demonstrating TGFβ-induced ERK1/2 phosphorylation and Smad2 phosphorylation up to 60 minutes (Ai) and up to 48 hours (Aii), with or without UO126 pre-treatment. Levels of phosphorylated ERK1/2 and Smad2 were quantified and normalized against GAPDH (B). UO126 pre-treatment abolished all TGFβ-induced ERK1/2 signaling and modulated TGFβ-induced Smad2 signaling. ANOVA with Dunnett’s post-hoc test was used quantify ERK1/2 phosphorylation relative to untreated controls, whereas ANOVA with Tukey’s post-hoc test was used to analyze Smad2 phosphorylation. Error bars represent S.E.M., *p<0.05; **p<0.01; ****p<0.0001.
Figure 8.
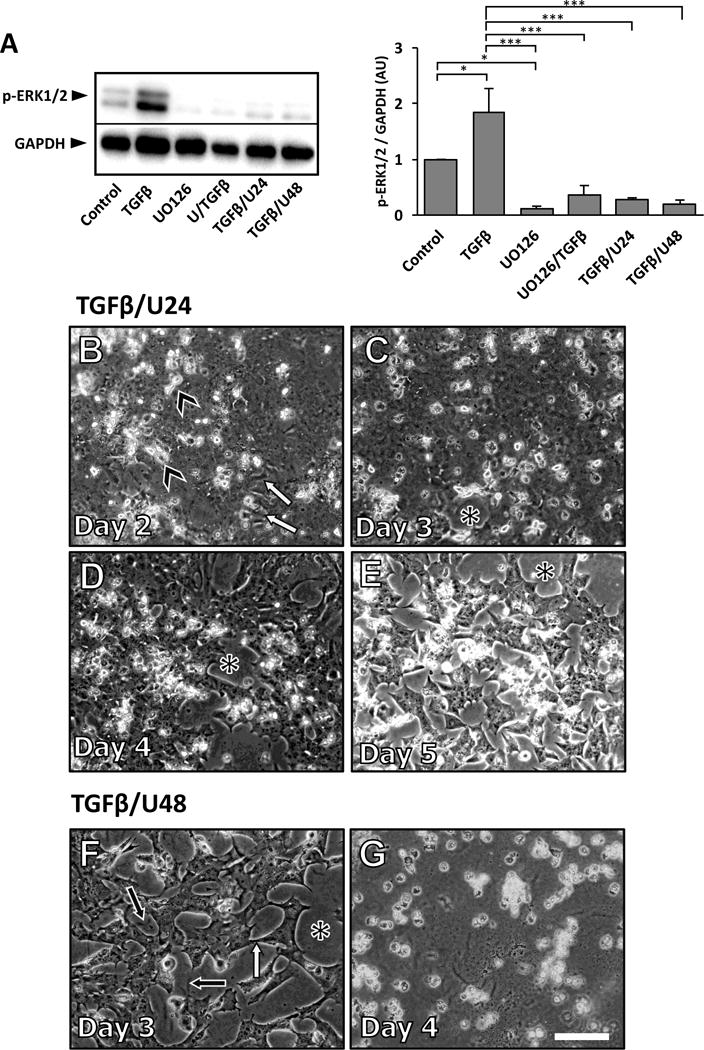
Representative western blot of TGFβ-induced ERK1/2 activation in lens epithelial explants cultured for 3 days with UO126 added prior to, or after 24 or 48 hours of TGFβ treatment (A; pERK1/2 was normalized against GAPDH; error bars represent S.E.M.; ANOVA with Tukey’s post-hoc test was used to analyze data; *p<0.05; ***p<0.001). The addition of UO126, 24 hours after TGFβ, inhibited the progression of EMT (B-E). After 2 days of culture (B), some cell elongation was present (white arrows), as was as cell blebbing (arrowheads). By day 3 some cell loss is evident (D, asterisk), and is more pronounced at day 4 (D) and 5 (E), exposing more of the underlying lens capsule (asterisk). When explants were treated with TGFβ, and UO126 was added at 48 hours, cell elongation (white arrow) was still evident at day 3 (F), as was cell loss that exposed the lens capsule (asterisk), revealing the underlying lens capsular wrinkling (black arrows). By day 4 of culture there was complete cell loss (G).
3.2. TGFβ phosphorylation of ERK1/2
Phosphorylation of ERK1/2 was induced by TGFβ in lens epithelial explants. Western blot analysis showed that the first significant amplification of ERK1/2 activation was at 40 minutes following TGFβ treatment. A 5-fold increase in ERK1/2 phosphorylation was then reached by 18 hours of TGFβ treatment (Figure 2). Following this peak at 18 hours, the level of ERK1/2 phosphorylation remained elevated; however, it did not rise beyond the level seen at 18 hours. A notable finding is that although the level of ERK1 and ERK2 phosphorylation varied throughout the culture period, TGFβ treated explants consistently showed higher levels of ERK1/2 activation compared to untreated control explants.
Pre-treatment of lens epithelial explants with UO126 for 2 hours prior to the addition of TGFβ abolished all TGFβ-induced ERK1/2 phosphorylation (Figure 2). To ensure that UO126 effectively blocked ERK1/2 signaling we examined its phosphorylation levels in all treatment groups after 3 days of culture. All groups that were treated with UO126, both pre- and post-TGFβ-treatment displayed negligible levels of ERK1/2 activation after 3 days of culture (see Figure 8A).
3.3. TGFβ-induced ERK1/2 can modulate Smad2 and Smad3 phosphorylation
A significant increase in the phosphorylation of the linker region of Smad2 was seen after 60 minutes treatment with TGFβ (Figure 2). Pre-treatment with UO126 prior to TGFβ did not significantly alter the level of phosphorylation of the Smad2 linker at this time; however, at 18 hours with UO126 pretreatment, there was a significant reduction in TGFβ-induced Smad2 phosphorylation (Figure 2). When we examined the activation of the C-terminal domain of Smad2 and Smad3 at this time point, TGFβ-treatment significantly increased the activation of Smad2/3 and UO126 pre-treatment was able to suppress this activation (Figure 3). The reduction in Smad2/3 phosphorylation was not a reflection of total Smad2/3 protein reduction, given there was no significant change in total Smad2/3 levels between treatments. Likewise, total ERK1/2 protein levels also remained constant between different treatments (Figure 3B).
Figure 3.
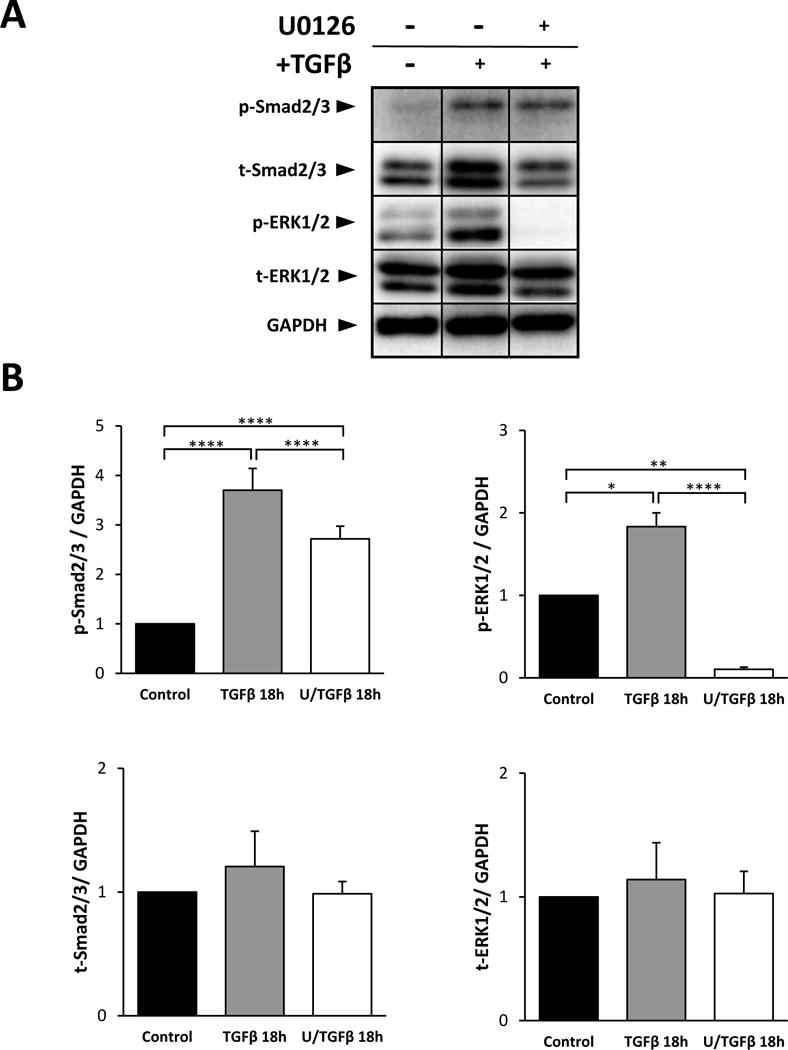
Representative western blots of explants treated for 18 hours with TGFβ with or without UO126 pre-treatment prior to TGFβ. Phosphorylated-Smad2/3, total-Smad2/3, total-ERK1/2 and GAPDH are labeled (A). p-Smad2/3, t-Smad2/3 and t-ERK1/2 were normalized against GAPDH (B; error bars represent S.E.M.). ANOVA with Sidak’s post-hoc test was used to analyze data (*p<0.05; **p<0.01; ****p<0.0001).
3.4. ERK1/2 signaling is required for the initiation of TGFβ-induced EMT
Pre-treatment with UO126 inhibited the progression of TGFβ-induced EMT in lens epithelial explants. In the presence of UO126, an epithelial monolayer was retained after 24 and 48 hours post-TGFβ treatment (Figure 4B, 4C); these explants did not display cell elongation typical of an EMT response. Blebbing of the cells, indicative of apoptosis, was still evident after 24 hours of TGFβ treatment (Figure 4B; arrowhead) and remained present throughout the culture period up to 5 days. UO126 pre-treatment was sufficient in significantly reducing TGFβ-induced cell loss (see Figure 10). After 3 days of treatment with TGFβ and UO126 (U/TGFβ) only 28% cell loss was evident; however, most notably 45–48% cells remained at day 4 and 5 of treatment, compared to no cell loss evident in control explants, and a complete cell loss in TGFβ-only-treated explants. While some evidence of capsular wrinkling was still observed in U/TGFβ explants (Figure 4E, 4F), it was noted that the overall lens capsule displayed less wrinkling than that seen in explants treated with TGFβ alone.
Figure 4.
Representative phase contrast micrographs of lens epithelial explants pre-treated with UO126 prior to TGFβ (U/TGFβ). Lens epithelial explants treated with UO126 remained as a monolayer of cuboidal cells (A). After 24 hours (Day 1) of TGFβ treatment, explants begin to exhibit some cellular blebbing (arrowheads; B) but cells do not elongate. At 2- (C) and 3-days (D) the cellular blebbing became more pronounced (arrowheads), with most cells remaining cuboidal. After 3 days of TGFβ treatment (D) regions of lens capsule were evident (asterisk), indicative of cell loss. After 4 (E) and 5 days (F), more regions of bare lens capsule became evident (asterisk), exposing the underlying capsular wrinkling (black arrows; Scale bar = 100μm).
Figure 10.
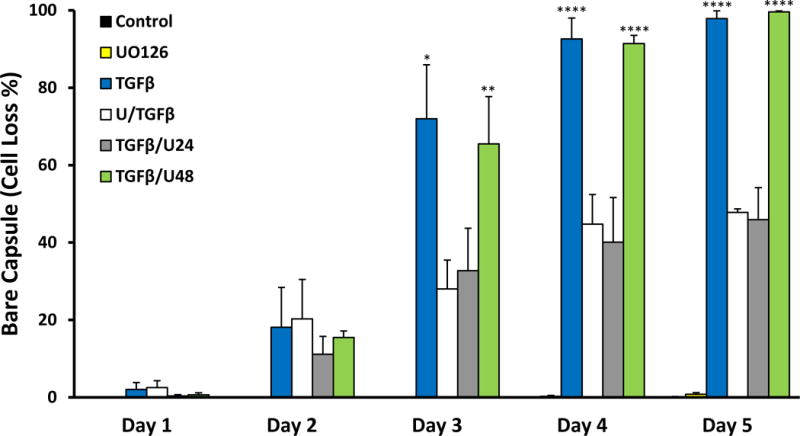
Summary of cell loss in explants treated without TGFβ (control), with UO126, TGFβ, U/TGFβ, TGFβ/U24 and TGFβ/U48 over 5 days. Trends revealed that TGFβ and TGFβ/U48 had similar rates in cell loss. U/TGFβ and TGFβ/U24 exhibited a similar progression of cell loss (error bars represent SEM; ANOVA with Tukey’s post-hoc test was used to analyze data; *p<0.05, **p<0.01, ****p<0.0001; when compared to control group). Cell loss was expressed as a percentage of bare capsule appearance (i.e. 100% bare capsule corresponds to 100% cell loss).
To determine the impact of UO126 on TGFβ-induced EMT, we next examined for molecular changes in these cells. Some cells in untreated explants showed cytoplasmic α-SMA after 3 days of culture (Figure 5A); however, when treated with TGFβ, all cells in explants displayed pronounced α-SMA incorporation into stress fibers (Figure 5C). We found that UO126 alone completely abolished α-SMA expression (Figure 5B), and that pre-treatment with UO126 reduced TGFβ-induced α-SMA expression levels, similar to that of control explants (Figure 5D). Western blot and subsequent densitometry analysis corroborated our immunofluorescent findings (Figure 5E, 5F).
Figure 5.
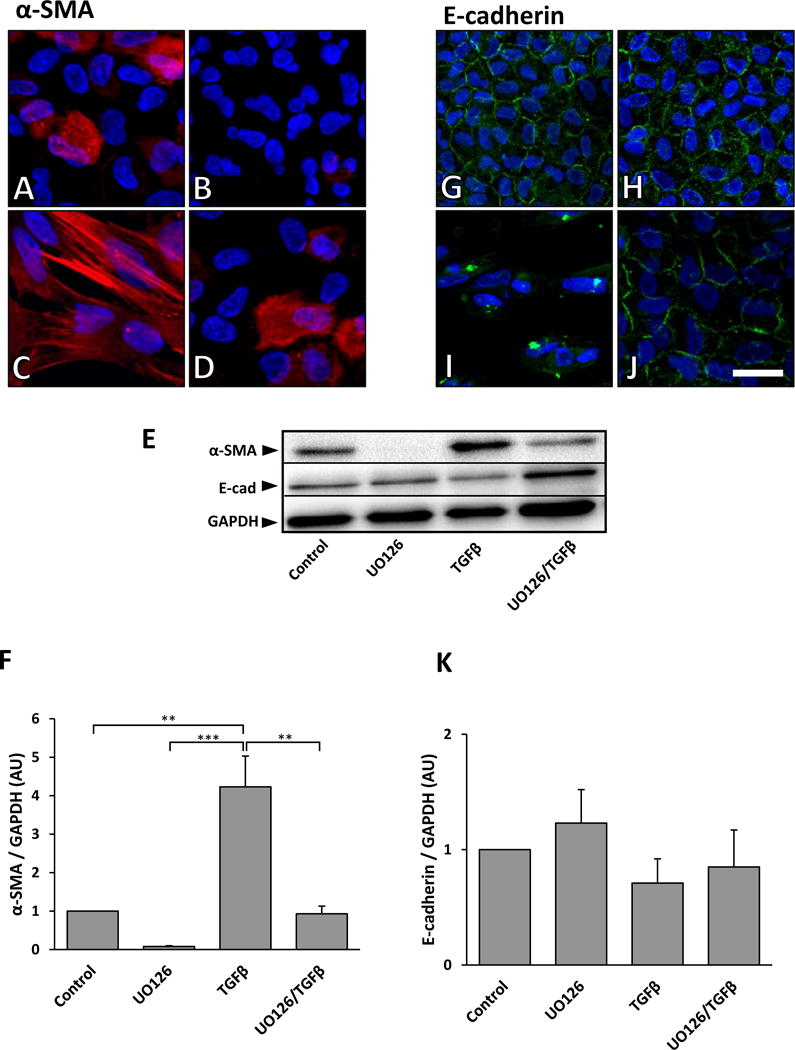
Representative immunofluorescent images of α-SMA and E-cadherin labeling of lens epithelial explants after 3 days culture with TGFβ, with or without UO126 pre-treatment. Control explants showed some cytoplasmic labeling of α-SMA (red) after 3 days (A). UO126 treatment alone completely abolished any α-SMA labeling (B). TGFβ treatment for 3 days led to prominent α-SMA expression associated with stress fibers (C). Pre-treatment with UO126 prior to TGFβ demonstrated labeling for α-SMA similar to that seen in control explants (D). Representative western blot analysis verified the immunolabeling results (E; top panel), with reduced α-SMA labeling in the presence of UO126. Densitometry analysis of western blots demonstrated a significant decrease in α-SMA expression in UO126 pre-treated explants when compared to those treated with TGFβ alone (F; α-SMA was normalized against GAPDH; error bars represent S.E.M.). E-cadherin was visualized at the cell borders of control explants and those treated with UO126 (G, H). Treatment with TGFβ led to a loss of this membrane labeling of E-cadherin (I). Pre-treatment with UO126 retained E-cadherin at the cell borders following TGFβ treatment (J; Scale bar = 20μm). Corresponding western blot analysis of E-cadherin (E, middle panel) showed no significant changes in levels of protein (K; E-cadherin was normalized against GAPDH; error bars represent S.E.M.). ANOVA with Tukey’s post-hoc test was used to analyze data (**p<0.01; ***p<0.001).
Immunofluorescent labeling of E-cadherin localized to cell borders, in control and UO126-treated explants (Figure 5G, 5H); however, after 3 days treatment with only TGFβ, E-cadherin dissociated from the cell borders and was more evient in the cytoplasm (Figure 5I). When explants were pretreated with UO126 prior to TGFβ, E-cadherin was retained and visualized at the borders of cells (Figure 5J). While the epithelial membrane marker, E-cadherin, was found to be reduced in lens explants treated with TGFβ when compared to control explants using western blotting, these differences were not statistically significant (Figure 5E, 5K).
Given we observed that TGFβ induced significantly higher ERK1/2 phosphorylation at 40 minutes and this peaked at 18 hours (Figure 2), we hypothesized that ERK1/2 activation up to 18 hours was required for TGFβ-induced EMT. To test this, explants received UO126, at select earlier time points after addition of TGFβ, at 2 hours (TGFβ/U2) and at 6 hours (TGFβ/U6). The addition of UO126 at 2 and 6 hours after TGFβ treatment resulted in a complete suppression of ERK1/2 signaling by 3 days of culture (Figure 6A, 6D). This was sufficient to block α-SMA accumulation into stress fibers shown after 3 days of culture (Figure 7G, 7J), as well as overall protein levels demonstrated using western blots (Figure 6A, 6C). The level of E-cadherin protein appeared reduced in TGFβ-treated explants (TGFβ, TGFβ/U2 and TGFβ/U6) when compared to controls; however, these differences were not significant (Figure 6A, 6B), with E-cadherin visualized at the borders of cells with immunofluorescent labeling (Figure 7H, 7K). The morphology of cells in lens explants in TGFβ/U2 and TGFβ/U6 groups after 3 days of culture was consistent with that of cells in U/TGFβ explants (Figure 7I, 7L).
Figure 6.
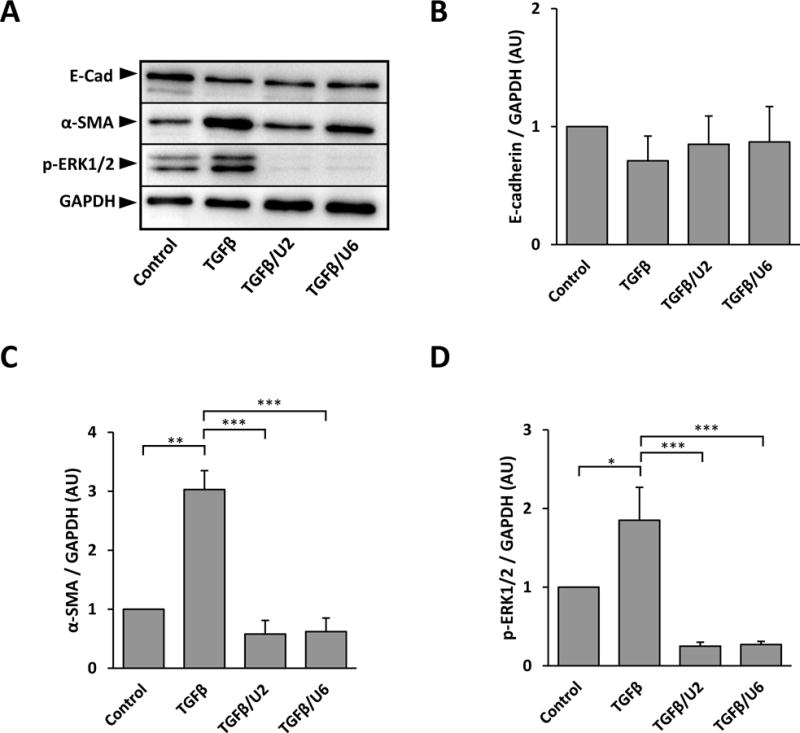
Representative western blots of E-cadherin, α-SMA and phosphorylated ERK1/2 after 3 days of culture of explants treated with UO126, added 2 (TGFβ/U2) or 6 hours (TGFβ/U6) after TGFβ addition (A). No significant differences in E-cadherin levels were found between treatment groups (B; E-cadherin was normalized against GAPDH; error bars represent S.E.M.). Analysis of α-SMA levels showed a significant decrease in both TGFβ/U2 and TGFβ/U6 explants when compared to those cultured with TGFβ alone (C; α-SMA was normalized against GAPDH; error bars represent S.E.M.). The addition of UO126 at 2 and 6 hours post-TGFβ-treatment significantly reduced the expression of phosphorylated ERK1/2 up to 3 days when compared to the untreated control (D; p-ERK1/2 was normalized against GAPDH; error bars represent S.E.M.). ANOVA with Tukey’s post-hoc test was used to analyze data (*p<0.05; **p<0.01; ***p<0.001).
Figure 7.
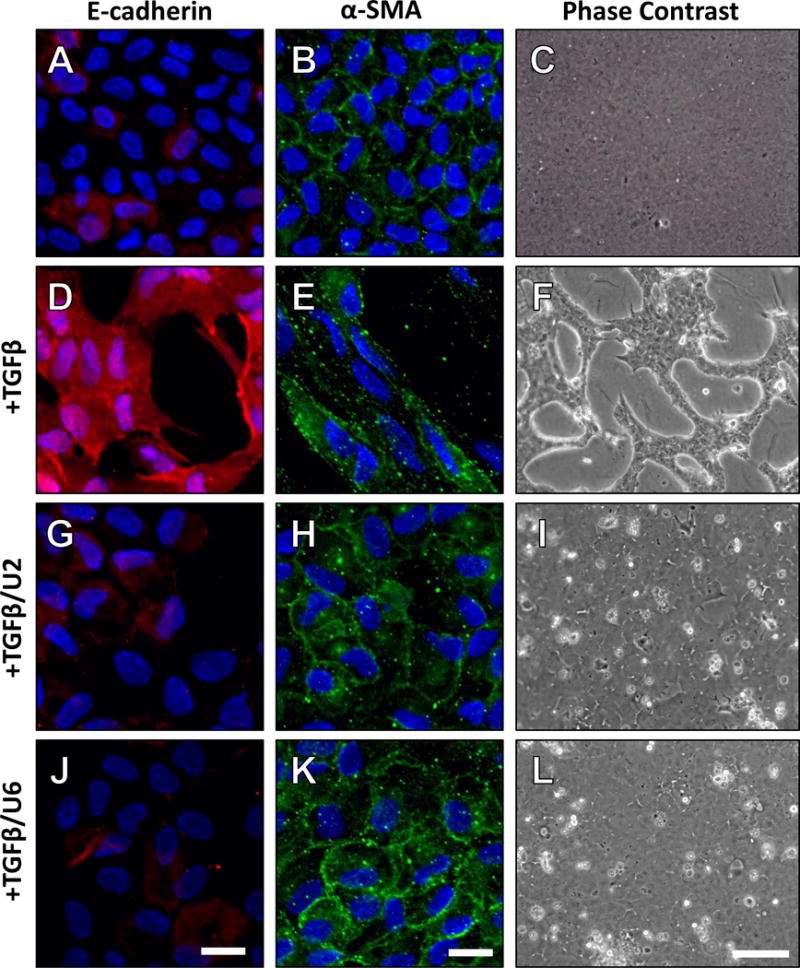
Representative micrographs of explants treated with TGFβ and post-treated with UO126 at 2 (TGFβ/U2) and 6 hours (TGFβ/U6), after 3 days. Control explants demonstrated a low level of α -SMA, not associated with stress fibers (A) and E-cadherin localized to cell borders (B), consistent with an epithelial phenotype (C). After 3 days treatment with TGFβ, α-SMA was localized to stress fibers (D) and E-cadherin was dissociated from the cell borders (E), consistent with lens epithelial cells undergoing EMT (F). UO126 addition after 2 hours of TGFβ treatment was sufficient in maintaining a low level of α-SMA, not associated with stress fibers (G), and retaining E-cadherin at the cell borders (H), consistent with an epithelial phenotype (I). Non-stress fiber α-SMA (J), and E-cadherin localization at the cell membrane (K) was also featured in explants treated with UO126, 6 hours after the addition of TGFβ (L). Scale bars are 20μm (α-SMA and E-cadherin panels) and 100μm (phase contrast).
3.5. ERK1/2 signaling is not required for the progression of EMT
To test whether ERK1/2 signaling after 18 hours is required for the progression of TGFβ-induced EMT, we added UO126 to lens epithelial explants, either at 24 hours or 48 hours after the addition of TGFβ. After 24 hours of TGFβ treatment some cells began to show elongation, typical of the TGFβ-induced EMT response (not shown). With application of UO126 at this time (TGFβ/U24), these explants retained some elongated cells 24 hours later, at day 2 of culture (Figure 8B; arrowheads), and cellular blebbing was also evident at this time (Figure 8B; white arrows). After 3 days of culture, no elongated cells were present; however, TGFβ/U24 explants showed small patches of bare capsule, indicative of cell loss (Figure 8C; asterisk). Most notably, at days 4 and 5 of treatment these explants exhibited a reduction (40–46%) in cell loss (Figure 8D, 8E) similar to explants that were pre-treated with UO126. Explants that were treated with TGFβ alone had already undergone complete cell loss by day 4 of culture (see Figure 10). Evidence of capsular wrinkling was observed; however, this was again less apparent to that seen in TGFβ-treated explants.
After 48 hours, the process of TGFβ-induced EMT is well established in lens explants. When treated with UO126 at this time (TGFβ/U48) it is not sufficient in stopping the morphological changes associated with EMT (Figure 8F, 8G). At day 3 (24 hours after the addition of UO126), explants exhibited 65% cell loss and 91% at day 4 (Figure 10), exposing the majority of the underlying wrinkled lens capsule, consistent with a TGFβ-induced EMT.
Blocking ERK1/2 signaling at 24 and 48 hours after the addition of TGFβ did not block α-SMA incorporation into stress fibers (Figure 9C, 9D). Western blot and subsequent densitometry analysis supported these immunofluorescent findings (Figure 9E, 9F). In these treatment groups, evidence of membrane labeling of E-cadherin is seen at some of the cell borders (Figure 9I, 9J). When the level of E-cadherin was assessed with western blotting, while it appeared higher in explants that received UO126 at 24 and 48 hours than those treated with TGFβ alone, these differences were not statistically significant (Figure 9E, 9K).
Figure 9.
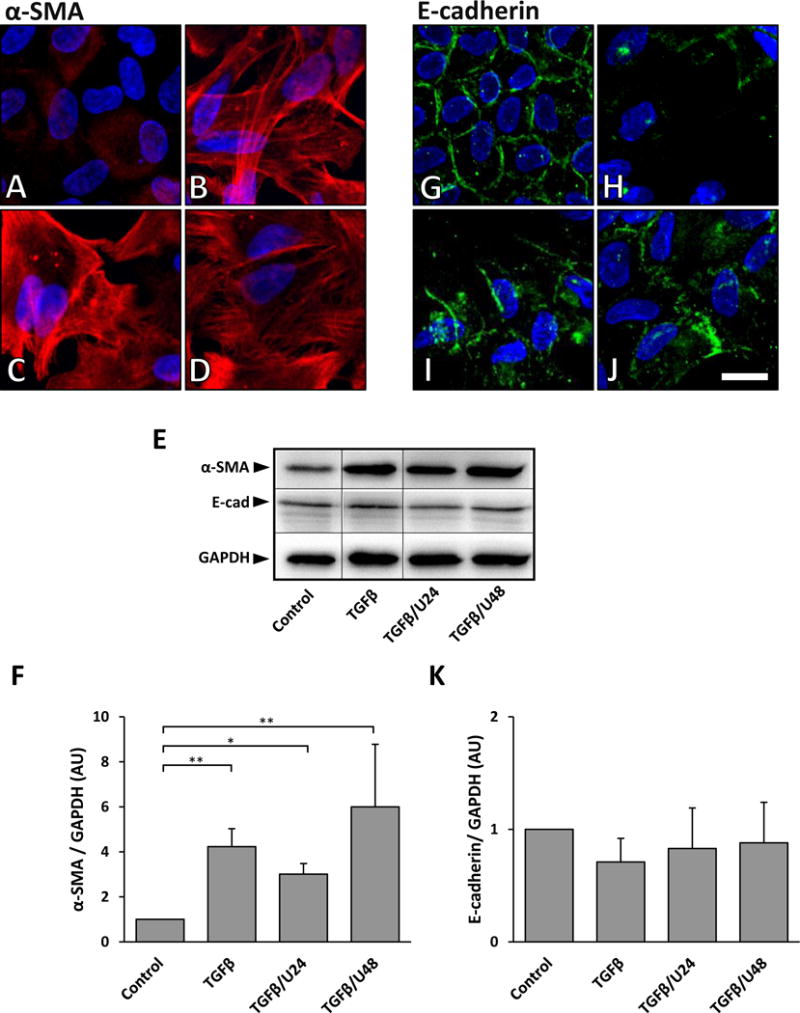
Representative images of α-SMA and E-cadherin labeling when UO126 is added to lens epithelial explants 24 or 48 hours after TGFβ. Control explants exhibited low levels of α-SMA expression after 3 days of culture (A). When treated with TGFβ, α-SMA fiber accumulation is prominent in stress fibers (B). Treatment with UO126 at 24 hours (C, TGFβ/U24) and 48 hours (D, TGFβ/U48) was not sufficient in blocking this TGFβ-induced α-SMA labeling. Representative western blots showed that after 3 days of TGFβ treatment, irrespective of UO126 post-treatment, α-SMA protein levels remained elevated relative to the untreated control (E, upper panel). Densitometry analysis demonstrated a significant increase in α-SMA expression in explants treated with TGFβ alone and TGFβ/U24 and TGFβ/U48 explants, when compared to the control (F; α-SMA was normalized against GAPDH, error bars represent SEM; ANOVA with Tukey’s post-hoc test was used to analyze data; *p<0.05; **p<0.01). E-cadherin was visualized at cell borders after 3 days of culture in control explants (G). After 3 days of culture with TGFβ alone, E-cadherin was no longer present at cell borders (H). When UO126 was added 24 hours post TGFβ treatment (I), E-cadherin labeling was present at some cell borders, and this was also seen in explants that received UO126 48 hours post TGFβ treatment (J; Scale bar = 20μm). Representative western blots of E-cadherin levels (E, middle panel) revealed no significant difference between treatment groups (K; E-cadherin was normalized against GAPDH; error bars represent S.E.M.; ANOVA with Tukey’s post-hoc test was used to analyze data).
After 5 days of treatment, explants cultured in control media (no TGFβ) remained as a tightly packed monolayer (Figure 11A). Likewise, explants cultured with UO126 over 5 days did not undergo any morphological changes (Figure 11B) and the addition of UO126 alone to media at 2, 6, 24 or 48 hours into the culture period also did not lead to any morphological changes over the 5-day-culture period (see Figure 11C–F).
Figure 11.
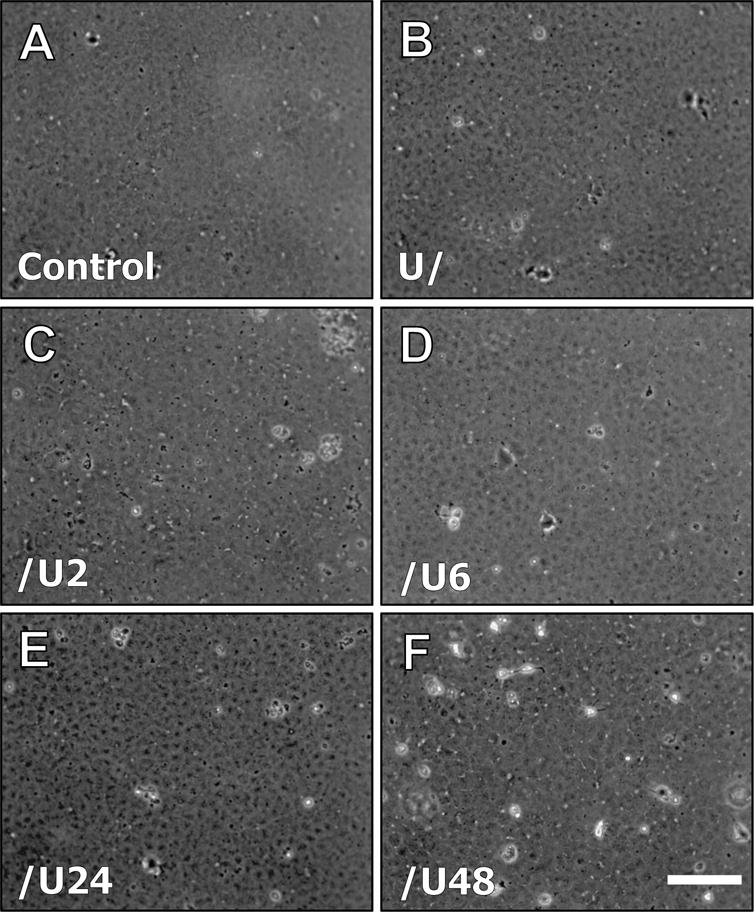
Representative phase contrast micrographs of explants at day 5, cultured with media only (A; control), UO126 pre-treatment (B), control media with U0126 added after 2hr (C), control media with U0126 added after 6hr (D), control media with U0126 added after 24hr (E) and control media with U0126 added after 48hr (F). Scale bar = 100μm.
3.6. α-SMA gene expression
To better understand why blocking ERK1/2 signaling before 24 hours of TGFβ addition was more sufficient in blocking α-SMA stress fiber association, qRT-PCR was performed to examine changes in α-SMA gene expression. It was found that the highest level of α-SMA expression was seen after 24 hours of TGFβ-treatment. This peak (5-fold increase compared to control cells) in α-SMA expression in cells treated with TGFβ reduced after this time (Figure 12). Pre-treatment with UO126 alone abolished all α-SMA expression, consistent with earlier western blot and immunofluorescent labeling (see Figure 5). Pre-treatment with UO126 reduced TGFβ-induced α-SMA expression. The addition of UO126 at 24 hours post-TGFβ treatment reduced TGFβ-induced α-SMA expression at days 2 and 3; however, post-treatment with UO126 at 48 hours was not sufficient in reducing α-SMA expression at day 3.
Figure 12.
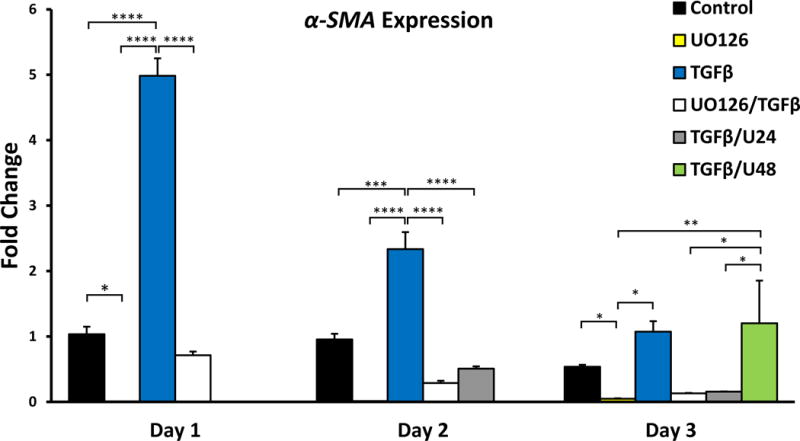
Mean fold changes in expression of α-SMA over 3 days, normalized against GAPDH expression (A; error bars represent S.E.M). UO126 reduced α-SMA expression. ANOVA followed by Tukey’s post-hoc test was used to infer statistical significance (B; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001).
4. Discussion
In the present study, we have shown that TGFβ is able to activate ERK1/2 signaling in lens epithelial explants, and that this is MEK1/2 dependent, as pharmacological inhibition with UO126 was able to abolish all ERK1/2 phosphorylation. ERK1/2 is a multifunctional signaling molecule, and in the lens it has been shown to be correlated with cell proliferation and differentiation, dependent on its duration of activity and intensity (Lovicu et al., 2001; Le & Musil, 2001). The lens epithelium is exposed to a multitude of aqueous-derived growth factors in situ such as FGF, IGF, PDGF and EGF, that when applied to lens epithelial explants in vitro are able to induce different ERK1/2 signaling profiles that can all lead to cell proliferation (Iyengar et al., 2007; Iyengar et al., 2009). RTK-antagonists, Spry, Spred and Sef, regulate ERK1/2 signaling and have been identified in the lens (Boros et al., 2006; Shin et al., 2012; Zhao et al., 2015). Given that transfection of lens epithelial explants with Spry, Spred or Sef constructs are able to inhibit TGFβ-induced EMT (Zhao et al., 2015), and the overexpression of Spry in the lens can rescue the cataractous phenotype present in TGFβ-overexpressing mice (Shin et al., 2012), it is clear that ERK1/2 signaling may also contribute to the process of TGFβ-mediated EMT. This is the first study to directly examine the temporal role of ERK1/2 signaling during TGFβ-induced lens EMT.
We have shown that TGFβ significantly increased the phosphorylation of ERK1/2 from 40 minutes, indicative of an indirect effect. Interestingly, phosphorylation of the Smad2 linker region, known to interact with ERK1/2 (Shi and Massague, 2003), occurred after the increase in ERK1/2 signaling at 60 minutes. TGFβ has been shown previously to activate ERK1/2 signaling in lens cell lines such as primary human lens cells (Tiwari et al., 2016), FHL124 (Dawes et al., 2009) and SRA01/04 (Chen et al., 2014). In SRA01/04 cells, TGFβ-induced ERK1/2 signaling was shown to be upstream of Smad2/3 (Chen et al., 2014). Similarly, in other cellular contexts, such as NIH/3T3 fibroblast cells TGFβ-induced ERK activity has been shown to be necessary for Smad activation (Li et al., 2009), whereas in Swiss3T3 and Balb3T3 cells, TGFβ failed to activate ERK signaling despite stimulating growth in these quiescent cells (Chatani et al., 1995). In rat mesangial cells, TGFβ-induced collagen IV synthesis is synergistically produced by the interaction between ERK1/2 and Smad2/3 (Jiang et al., 2010). The mechanism of crosstalk between the ERK1/2 and Smad2/3 signaling pathways appears to be cell context dependent. In the current study we found that in lens epithelial explants, Smad2 linker activation occurs after TGFβ-induced ERK1/2 phosphorylation; however, UO126 pre-treatment did not appear to modulate TGFβ-induced phosphorylation of this linker at the earlier time point of 40 mins. Given that UO126 pre-treatment significantly reduced TGFβ-induced Smad2 linker phosphorylation only at 18 hours (when TGFβ-induced ERK1/2 phosphorylation was highest), we further examined the activation of the C-terminal domain of Smad2 and Smad3 at this same time. TGFβ-induced Smad2/3 phosphorylation was significantly reduced with UO126 pre-treatment indicating that ERK1/2 may indeed facilitate the phosphorylation of Smad2/3. There is a significant reduction of TGFβ-induced phosphorylation of the Smad 2 linker and C-terminal regions of Smad2/3 in the presence of UO126. Given that it is not a complete blockage of TGFβ-signaling, indicates that Smad signaling in lens epithelial explants occurs in parallel through both ERK1/2-dependent and - independent mechanisms.
TGFβ-induced ERK1/2 signaling has been implicated in α-SMA stress fiber labeling in SRA01/04 lens cells (Chen et al., 2014) and LLC-PK1/AT1 porcine proximal tubular epithelial cells (Sebe et al., 2008). Likewise, in human lens cells, pharmacological inhibition of ERK1/2 signaling was sufficient in reducing TGFβ-induced α-SMA expression (Tiwari et al., 2016). In FHL124 lens cells, TGFβ-induced upregulation of α-SMA was shown to be independent of Smad4 (Dawes et al., 2009). These findings coincide with the current study where α-SMA protein expression was reduced by pretreatment with UO126; however, this is the first study to show a clear role for ERK1/2 in the initiation of α-SMA stress fiber association. We found that early inhibition of ERK1/2 signaling, either prior to or 2 and 6 hours after TGFβ treatment, blocked the upregulation of α-SMA and its subsequent association with stress fibers during TGFβ-induced EMT. This coincided with the peak of α-SMA expression found 24 hours after TGFβ treatment. Blocking ERK1/2 signaling 24 or 48 hours into the TGFβ-induced EMT process was not sufficient in preventing α-SMA accumulation into stress fibers. As it has been recently shown that tropomyosin facilitates the incorporation of α-SMA into stress fibers in human myofibroblasts (Prunotto et al, 2015), it would be of interest to examine how ERK1/2 influences tropomyosin in lens EMT. Silencing of α-SMA expression in FHL124 cells led to an increased contractility in response to TGFβ (Dawes et al., 2008; Dawes et al., 2009). The uncoupling of α-SMA and cell contractility was also evident in the present study, as capsular wrinkling was less evident in U/TGFβ and TGFβ/U24 explants despite strong α-SMA labeling at day 3 in TGFβ/U24 and not in U/TGFβ explants.
The expression of E-cadherin has been shown to be negatively regulated by, and dependent on the transcription factor Snail (Cano et al., 2000). In a rat whole lens model, TGFβ was found to increase the expression of Snail mRNA in subcapsular plaques (Nathu et al., 2009). In transgenic mice, TGFβ-overexpression, or deletion of the RTK/ERK1/2 antagonist Spry in the lens led to an increase in Snai1 and Snai2 labeling in cell nuclei. This was not present in mice co-overexpressing Spry and TGFβ, implicating ERK1/2 signaling in TGFβ-mediated Snai1 activation and subsequent E-cadherin down regulation (Shin et al., 2012). The loss of cell-cell contact is thought to facilitate the process of EMT; as has been highlighted in colorectal cancer biopsies where there is a negative correlation between E-cadherin loss and α-SMA expression (Valcz et al., 2012). In contrast we found that post-treatment with UO126 at 24 and 48 hours after TGFβ treatment was not able to suppress α-SMA fiber formation; however, it permitted the retention of some E-cadherin at cell borders. In the context of lens epithelial cells, it appears that α-SMA expression and cell membrane localization of E-cadherin are not mutually exclusive. Although α-SMA expression is dependent on early TGFβ-mediated ERK1/2 signaling, it appears that cell-cell adhesion may not be heavily dependent on ERK1/2 signaling. It is possible that ERK1/2 may interact with other signaling pathways to contribute to the loss of cell-cell contact, for example, ZO-1 and E-cadherin dissociation has been linked to TGFβ-induced P13K/Akt signaling in NMuMG cells (Bakin et al., 2000).
Cell loss, highlighted by changes in the area of exposed lens capsule in this study, was significantly reduced and comparable in U/TGFβ and TGFβ/U24 explants, indicating that TGFβ-induced ERK1/2 signaling may facilitate cell death in the earlier stages of culture. Blocking ERK1/2 signaling 48 hours after TGFβ treatment was not sufficient in reducing TGFβ-mediated cell loss, indicating that ERK1/2 is not necessary for the progression of cell loss after 48 hours. While UO126 was able to reduce cell loss in U/TGFβ and TGFβ/U24 explants, it did not stop the process of cell death completely, indicating that ERK1/2 may potentially interact with other signaling pathways to contribute to cell loss. For example, in NMuMG cells, TGFβ-mediated apoptosis has been shown to be dependent on p38 signaling (Yu et al., 2002). TGFβ also promotes the upregulation of Smad7 (Yan et al., 2009) that in mesangial cells, has been attributed to activation of caspase-3 during TGFβ-mediated apoptosis. This apoptosis was dependent on Smad7 and not Smad2 or Smad3 (Okado et al., 2002).
Overall, here we show that ERK1/2 signaling is required for the initiation of TGFβ-induced EMT in the lens, but not its progression. We show that earlier suppression of ERK1/2 signaling is more effective at inhibiting TGFβ-induced EMT, and as EMT progresses, it becomes less dependent on ERK1/2. This highlights the important function of normally expressed antagonists in the lens such as Spry, Spred and Sef that are known to negatively regulate ERK1/2 signaling. Their maintenance in the lens throughout life, not only to tightly regulate ERK1/2 signaling, may protect the lens from aberrant TGFβ signaling that leads to age-related cataract. This temporal relationship of TGFβ-induced ERK1/2 and EMT progression may have novel implications for the better understanding and potentially the treatment of cataract.
Research Highlights.
-
*
TGFß induces the phosphorylation of ERK1/2 in lens epithelial cells
-
*
Phosphorylation of ERK1/2 is required for the initiation of TGFβ-induced lens EMT
-
*
ERK1/2 activity is not required for the later progression of TGFβ-induced lens EMT
Acknowledgments
The authors would like to acknowledge our funding sources, including the Rebecca L. Cooper Foundation, the National Health & Medical Research Council (NHMRC), Australia, and National Institutes of Health (R01 EY0-3177; McAvoy and Lovicu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-Kinase Function Is Required for Transforming Growth Factor β-mediated Epithelial to Mesenchymal Transition and Cell Migration. The Journal of Biological Chemistry. 2000;275(47):36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Boros J, Newitt P, Wang Q, McAvoy JW, Lovicu FJ. Sef and Sprouty expression in the developing ocular lens: implications for regulating lens cell proliferation and differentiation. Seminars in cell & developmental biology. 2006;17(6):741–752. doi: 10.1016/j.semcdb.2006.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature Cell Biology. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chatani Y, Tanimura S, Miyoshi K, Hattori A, Sato M, Kohno M. Cell Type-specific Modulation of Cell Growth by Transforming Growth Factor β1 Does Not Correlate with Mitogen-activated Protein Kinase Activation. The Journal of Biological Chemistry. 1995;270(51):30686–30692. doi: 10.1074/jbc.270.51.30686. [DOI] [PubMed] [Google Scholar]
- Chen X, Ye S, Xiao W, Wang W, Luo L, Liu Y. ERK1/2 pathway mediates epithelial-mesenchymal transition by cross-interacting with TGFβ/Smad and Jagged/Noth signaling pathways in lens epithelial cells. International Journal of Molecular Medicine. 2014;33:1664–1670. doi: 10.3892/ijmm.2014.1723. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Baek KE, Saika S, Jeong MJ, Yoo J. Snail is required for transforming growth factor-β-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochemical and Biophysical Research Communications. 2007;353(2):337–343. doi: 10.1016/j.bbrc.2006.12.035. [DOI] [PubMed] [Google Scholar]
- Dawes LJ, Eldred JA, Anderson IK, Sleeman M, Reddan JR, Duncan G, Wormstone IM. TGFβ-Induced Contraction Is Not Promoted by Fibronectin-Fibronectin Receptor Interaction, of αSMA expression. Investigative Ophthalmology & Visual Science. 2008;49(2):650–661. doi: 10.1167/iovs.07-0586. [DOI] [PubMed] [Google Scholar]
- Dawes LJ, Sleeman M, Anderson IK, Reddan JR, Wormstone IM. TGFβ/Smad4-Dependent and -Independent Regulation of Human Lens Epithelial Cells. Investigative Ophthalmology & Visual Science. 2009;50(11):5318–5327. doi: 10.1167/iovs.08-3223. [DOI] [PubMed] [Google Scholar]
- de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-ß induced epithelial-mesenchymal transition in the lens: A model for cataract formation. Cells Tissues Organs. 2005;179:43–55. doi: 10.1159/000084508. [DOI] [PubMed] [Google Scholar]
- Hales AM, Chamberlain CG, McAvoy JW. Cataract induction in lenses cultured with Transforming Growth Factor-ß. Investigative Ophthalmology & Visual Science. 1995;36(8):1709–1713. [PubMed] [Google Scholar]
- Hales AM, Schulz MW, Chamberlain CG, McAvoy JW. TGF-beta 1 induces lens cells to accumulate alpha-smooth muscle actin, a marker for subcapsular cataracts. Current Eye Research. 1994;13(12):885–890. doi: 10.3109/02713689409015091. [DOI] [PubMed] [Google Scholar]
- Iyengar L, Patkunanathan B, McAvoy JW, Lovicu FJ. Growth factors involved in aqueous-induced lens cell proliferation. Growth Factors. 2009;27(1):50–62. doi: 10.1080/08977190802610916. [DOI] [PubMed] [Google Scholar]
- Iyengar L, Wang Q, Rasko JE, McAvoy JW, Lovicu FJ. Duration of ERK1/2 phosphorylation induced by FGF or ocular media determines lens cell fate. Differentiation. 2007;75:662–668. doi: 10.1111/j.1432-0436.2007.00167.x. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhang Y, Wu H, Zhang X, Gan H, Sun J, Chen Q, Guo M, Zhang Z. Role of cross-talk between the Smad2 and MAPK pathways in TGF-β1-induced collagen IV expression in mesangial cells. International Journal of Molecular Medicine. 2010;26:571–576. doi: 10.3892/ijmm_00000501. [DOI] [PubMed] [Google Scholar]
- Khairallah M, Kahloun R, Bourne R, Limburg H, Flaxman SR, Jonas JB, Keeffe J, Leasher J, Naidoo K, Pesudovs K, Price H, White RA, Wong TY, Resinkoff S, Taylor HR. Number of People Blind or Visually Impaired by Cataract Worldwide and in Word Regions, 1990 to 2010. Investigative Ophthalmology & Visual Science. 2015;56:6762–6769. doi: 10.1167/iovs.15-17201. [DOI] [PubMed] [Google Scholar]
- Kovalenko D, Yang X, Nadeau RJ, Harkins LK, Friesel R. Sef Inhibits Fibroblast Growth Factor Signaling by Inhibiting FGFR1 Tryosine Phosphorylation and Subsequent ERK Activation. The Journal of Biological Chemistry. 2003;278(16):14087–14091. doi: 10.1074/jbc.C200606200. [DOI] [PubMed] [Google Scholar]
- Kramer S, Okabe M, Hacohen N, Krasnow MA, Hiromi Y. Sprouty: a common antagonist of FGF and EGF signaling pathways in Drosophila. Development. 1999;126:2515–2525. doi: 10.1242/dev.126.11.2515. [DOI] [PubMed] [Google Scholar]
- Le AN, Musil LS. FGF Signaling in Chick Lens Development. Developmental Biology. 2001;233(2):394–411. doi: 10.1006/dbio.2001.0194. [DOI] [PubMed] [Google Scholar]
- Li L, Chen SF, Liu Y. MAP kinase phosphotase-1, a critical negative regulator of the innate immune response. International Journal of Clinical and Experimental Medicine. 2009;2:48–67. [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hales AM, Chamberlain CG, McAvoy JW. Induction of cataract-like changes in rat lens epithelial explants by Transforming Growth Factor- β. Investigative Ophthalmology & Visual Science. 1994;35(2):388–401. [PubMed] [Google Scholar]
- Lovicu FJ, McAvoy JW. FGF-induced lens cell proliferation and differentiation is dependent on MAPK (ERK1/2) signalling. Development. 2001;128:5075–5084. doi: 10.1242/dev.128.24.5075. [DOI] [PubMed] [Google Scholar]
- Lovicu FJ, Schulz MW, Hales AM, Vincent LN, Overbeek PA, Chamberlain CG, McAvoy JW. TGFβ induces morphological and molecular changes similar to human anterior subcapsular cataract. British Journal of Ophthalmology. 2002;86:220–226. doi: 10.1136/bjo.86.2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruno KA, Lovicu FJ, Chamberlain CG, McAvoy JW. Apoptosis is a feature of TGF beta-induced cataract. Clinical and Experimental Optometry. 2002;85(2):76–82. doi: 10.1111/j.1444-0938.2002.tb03012.x. [DOI] [PubMed] [Google Scholar]
- Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends in Cell Biology. 2006;16(1):45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-β Signal Transduction. Annual Review of Biochemistry. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Meng S, Zhang M, Pan W, Li Z, Anderson DH, Zhang S, Ge B, Wang C. Tyrosines 303/343/353 within the Sprouty-related domain of Spred2 are essential for its interaction with p85 and inhibitory effect on Ras/ERK activation. The International Journal of Biochemistry & Cell Biology. 2012;44:748–758. doi: 10.1016/j.biocel.2012.01.014. [DOI] [PubMed] [Google Scholar]
- Nathu Z, Dwivedi DJ, Reddan JR, Sheardown H, Margetts PJ, West-Mays JA. Temporal changes in MMP mRNa expression in the lens epithelium during anterior subcapsular catarat formation. Experimental Eye Research. 2009;88:323–330. doi: 10.1016/j.exer.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okado T, Terada Y, Tanaka H, Inoshita S, Nakao A, Sasaki S. Smad7 mediates transforming growth factor-β-induced apoptosis in mesangial cells. Kidney International. 2002;62:1178–1186. doi: 10.1111/j.1523-1755.2002.kid583.x. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Sebe A, Leivonen S, Fintha A, Masszi A, Rosivall L, Kahari V, Musci I. Transforming growth factor-β-induced alpha-smooth muscle actin expression in renal proximal tubular cells is regulated by p38β mitogen-activated protein kinase, extracellular signal-regulated protein kinase1,2 and the Smad signalling during epithelial-myofibroblast transdifferentiation. Nephrology Dialysis Transplantation. 2008;23:1537–1545. doi: 10.1093/ndt/gfm789. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shin EHH, Basson MA, Robinson ML, McAvoy JW, Lovicu FJ. Sprouty is a negative regulator of transforming growth factor β-induced epithelial-to-mesencymal transition and cataract. Molecular Medicine. 2012;18:861–873. doi: 10.2119/molmed.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu DY, Wojciechowski MC, Lovicu FJ. Bone Morphogenetic Protein-7 suppresses TGFβ2-induced epithelial-mesenchymal transition in the lens: implications for cataract prevention. Investigative Ophthalmology & Visual Science. 2017;58:781–796. doi: 10.1167/iovs.16-20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefft JD, Lee M, Smith S, Leinwand M, Zhao J, Bringas P, Jr, Crowe DL, Warburton D. Conserved function of mSpry-2, a murine homolog of Drosophila sprouty, which negatively modulates respiratory organogenesis. Cuurent Biology. 1999;9(4):219–222. doi: 10.1016/s0960-9822(99)80094-3. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Kumar R, Ram J, Sharma M, Luthra-Guptasarma M. Control of fibrotic changes through the synergistic effects of anti-fibronectin antibody and an RGDS-tagged form of the same antibody. Scientific Reports. 2016;6:30872. doi: 10.1038/srep30872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya D, Ogata M, Reneker LW. MAPK1 is required for establishing the pattern of cell proliferation and for cell survival during lens development. Development. 2013;140:1573–1582. doi: 10.1242/dev.081042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcz G, Spios F, Krenacs T, Molnar J, Patai AV, Leiszter K, Toth K, Wichmann B, Molnar B, Tulassay Z. Increase of α-SMA+ and CK+ Cells as an Early Sign of Epithelial-Mesenchymal Transition during Colorectal Carcinogenesis. Pathology and Oncology Research. 2012;18:371–376. doi: 10.1007/s12253-011-9454-z. [DOI] [PubMed] [Google Scholar]
- Wormstone IM, Wang L, Liu CS. Posterior capsule opacification. Experimental Eye Research. 2009;88:257–269. doi: 10.1016/j.exer.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L. The Smad Pathway. Cytokine & Growth Factor Reviews. 2000;11:5–13. doi: 10.1016/s1359-6101(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-β1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Liu Z, Chen Y. Regulation of TGF-β signaling by Smad7. Acta Biochimica et Biophysica Sinica. 2009;41(4):263–272. doi: 10.1093/abbs/gmp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Hebert C, Zhang YE. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. The EMBO Journal. 2002;21(14):3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, Yang Y, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transformning growth factor-β. Proceedings of the National Academy of Sciences. 2001;98(12):6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Wojciechowski MC, Jee S, Boros J, McAvoy JW, Lovicu FJ. Negative regulation of TGFβ-induced lens epithelial to mesenchymal transition (EMT) by RTK antagonists. Experimental Eye Research. 2015;132:9–16. doi: 10.1016/j.exer.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Burgess AW. Regulation of Transforming Growth Factor-β Signaling. Molecular Cell Biology Research Communications. 2001;4:321–330. doi: 10.1006/mcbr.2001.0301. [DOI] [PubMed] [Google Scholar]