

SUMMARY
Genome-wide association studies have struggled to identify functional genes and variants underlying complex phenotypes. We recruited a multi-ethnic cohort of healthy volunteers (n = 91) and used their tissue to generate induced pluripotent stem cells (iPSCs) and hepatocyte-like cells (HLCs) for genome-wide mapping of expression quantitative trait loci (eQTLs) and allele-specific expression (ASE). We identified many eQTL genes (eGenes) not observed in the comparably sized Genotype-Tissue Expression project’s human liver cohort (n = 96). Focusing on blood lipid-associated loci, we performed massively parallel reporter assays to screen candidate functional variants and used genome-edited stem cells, CRISPR interference, and mouse modeling to establish rs2277862-CPNE1, rs10889356-DOCK7, rs10889356-ANGPTL3, and rs10872142-FRK as functional SNP-gene sets. We demonstrated HLC eGenes CPNE1, VKORC1, UBE2L3, and ANGPTL3 and HLC ASE gene ACAA2 to be lipid-functional genes in mouse models. These findings endorse an iPSC-based experimental framework to discover functional variants and genes contributing to complex human traits.
Graphical abstract
INTRODUCTION
Genome-wide association studies (GWAS) have emerged as a robust unbiased approach to identify single nucleotide polymorphisms (SNPs) associated with incidence of a particular phenotype or disease (Manolio, 2010). Only a small fraction of GWAS lead variants lie within coding sequence and thus directly implicate a functional gene at a locus; the vast majority of lead SNPs fall in noncoding sequence. Moreover, most of these SNPs are not themselves functional but exist in linkage disequilibrium (LD) with the true functional variants. Because many human disease-associated variants are believed to regulate gene expression, expression quantitative trait locus (eQTL) and allele-specific expression (ASE) studies may illuminate potential downstream targets of functional variants. These regulated genes then become candidates for experimental manipulation to ascertain their relevance to the phenotype of interest. However, functional studies elucidating the mechanisms of identified variants have remained a challenge due to the need for laborious experiments and the lack of suitable model systems for noncoding sequence studies.
Recently emergent technologies make it feasible to identify and interrogate the function of noncoding variants at eQTL and ASE loci in human model systems. Human pluripotent stem cells (hPSCs), especially induced pluripotent stem cells (iPSCs), make it possible to generate cohorts of person-specific, renewable, differentiated cell lines in vitro (Zhu et al., 2011). In theory, when drawn from a population with diverse genotypes of common genetic variants, these cohorts might offer the opportunity to validate known eQTL/ASE loci and discover new eQTL/ASE loci “in the dish.” Massively parallel reporter assays (MPRAs) allow investigators to generate high-complexity pools of reporter constructs where each regulatory element or variant of interest is linked to a synthetic reporter gene that carries an identifying barcode (Melnikov et al., 2012; Patwardhan et al., 2012). The reporter construct pools are introduced into cells, and the relative transcriptional activities of the individual elements or variants are measured by sequencing the transcribed reporter mRNAs and counting their specific barcodes. This approach can be used to rapidly profile the regulatory activity of thousands of variants at GWAS loci (Tewhey et al., 2016; Ulirsch et al., 2016). Finally, advances in genome-editing technologies—most notably clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated 9 (Cas9) systems—have opened up new avenues to rigorously assess the functional impact of genetic variation (Musunuru, 2013).
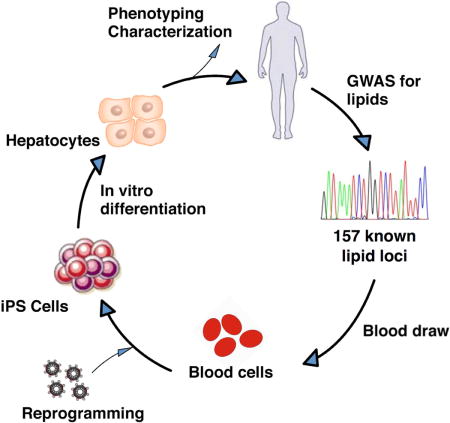
In this study, we asked two overarching questions. First, can population cohorts of iPSCs and iPSC-differentiated cells be used to perform unbiased genome-wide eQTL/ASE studies in a manner that is complementary to traditional primary tissue-based studies such as the Gene-Tissue Expression (GTEx) project? Second, can we better understand the functional role of human genetic variation in influencing quantitative phenotypic traits, particularly those related to liver metabolism such as blood lipid levels? As part of the NHLBI Next Generation Genetic Association Studies Consortium, we generated population-based cohorts of iPSCs and iPSC-differentiated hepatocyte-like cells (HLCs) to perform genome-wide mapping and characterize known and new eQTL/ASE loci. We thereafter employed gene overexpression mouse models as well as a combination of MPRAs and CRISPR-Cas9 in hPSCs, other types of cultured cells, and mouse models to screen, identify, and validate functional variants and/or genes in several blood lipid-associated eQTL/ASE loci.
RESULTS
Generation and gene expression profiles of iPSCs and HLCs
We generated iPSCs from peripheral blood mononuclear cells isolated from 91 individuals, predominantly African Americans (43%) and European Americans (55%), with more women (60%) than men (40%) (Table S1). All established iPSC lines were confirmed to be free of exogenous Sendai viral reprogramming factor expression and then tested for pluripotency by fluorescence-activated cell sorting (FACS) staining for SSEA4 and Tra-1-60 (Figure S1A, Table S1). Samples passing these criteria were differentiated into HLCs. Differentiated HLCs were morphologically similar to primary hepatocytes, and for a subset of HLC samples, expression of HNF4α was confirmed by immunofluorescence (Figure S1B). The HLCs also secreted apolipoprotein B (apoB), triglycerides, and albumin (Figure S2A), a set of properties unique to functional hepatocytes.
In addition to molecular and qualitative assessments of pluripotency and differentiation efficiencies, we used computational means to further validate the iPSCs and HLCs used for our study. We generated RNA sequencing (RNA-seq) data from iPSCs (n = 89), HLCs (n= 86), and primary human hepatocytes (n = 4). In addition, we used RNA-seq data of human whole-liver samples from GTEx (n = 96) (Aguet et al., 2016). With the use of singular value decomposition (SVD), expression levels of a panel of differentiation markers (Carcamo-Orive et al., 2016) demonstrated that our iPSCs were extremely similar to other iPSCs and human embryonic stem cells (ESCs) with previously published gene expression data (Choi et al., 2015) (Figure S3A). We characterized the global gene expression profiles of the iPSCs, HLCs, primary hepatocytes, and GTEx livers and in general found that the HLCs had been successfully differentiated from the iPSCs and approximated the gene expression profiles of the primary hepatocytes more closely than the profiles of the livers (Figure S3B). We specifically assessed the expression levels of six hepatocyte-specific genes in the HLCs (Figure S2B), using these levels as compared to levels in primary human hepatocytes as a means to assess the quality of the HLCs (scored from zero to five) (Table S1).
Differentially expressed genes are associated with liver function
Fully understanding the underlying variance between HLCs and whole livers merits further investigation, as hepatocytes constitute only one of several cell types in the liver. We quantified differential gene expression using DESeq2 (Love et al., 2014). We identified 3,709 genes more highly expressed in livers compared to iPSCs and 2,040 genes more highly expressed in HLCs compared to iPSCs at FDR < 5%. We sought to assess the liver-specific functions associated with 1,349 genes that were common to both sets (differentially expressed in both HLCs and livers compared to iPSCs) by estimating the enrichment of these genes for gene ontology (GO) terms. We identified 126 common GO terms including functions specific to hepatic regulation and lipid metabolism (Table S2).
Cis-regulatory variation in HLCs
With a goal of identifying hepatic cis-regulatory variation, we mapped genome-wide cis-eQTLs in the iPSCs, HLCs, and GTEx livers using linear models as implemented in Matrix eQTL (Shabalin, 2012) (Figure 1A). We identified a total of 3,587 eGenes in the iPSCs, 2,392 eGenes in the HLCs, and 1,511 eGenes in the GTEx livers at a false discovery rate (FDR) < 5% (Table S3). The HLCs and livers shared a total of 477 eGenes, 176 of which were not associated in the iPSCs at FDR < 5% (Figure 1B). The eQTLs were primarily associated with protein coding genes (83%), and the majority of eGenes were nominally associated (P < 0.05) in more than one cell type.
Figure 1. Genome-wide Mapping of Expression Quantitative Trait Loci (eQTLs).
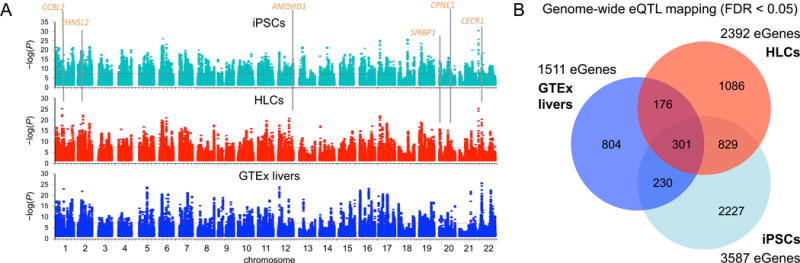
(A) Overview of the single-tissue cis-eQTL mapping results. Representative HLC eGenes are indicated.
(B) Venn diagram of mapped eGenes with a stringent cutoff of FDR < 5%.
See also Figures S1, S2, and S3.
To complement the eQTL analyses, and to assess the effects of cis-regulatory variants at lower frequencies that eQTL analyses are underpowered to detect, we quantified allele-specific expression (ASE) in iPSCs and HLCs (individual-level data not provided in the manuscript due to privacy concerns). Genome-wide, we identified 1,721 independent SNPs in 494 genes that exhibited significant ASE in iPSCs (FDR < 5%) and 2,137 independent SNPs in 631 genes that exhibited significant ASE in HLCs. Furthermore, 876 independent SNPs in 541 genes exhibited differential ASE (FDR < 5%) between iPSCs and HLCs (Table S4), indicating cell-type-specific regulatory effects. Genes exhibiting differential ASE were significantly enriched for cell-type-specific eGenes (odds ratio = 3.3, P < 1.8 × 10−13), independently confirming the cell-type-specific regulatory effects identified with the two methodologies.
Tissue specificity of cis-regulatory variation in iPSCs and HLCs
To further refine cell-type-specific effects of cis-eQTLs between iPSCs and HLCs, we conducted a mixed model meta-analysis. Considering our paired sample design we used METASOFT (Han and Eskin, 2012) and Meta-Tissue (Sul et al., 2013) to estimate effect sizes of each variant-gene pair accounting for covariates used in single-tissue analyses as well as tissue sharing among individuals. All SNPs within 100 kb of 20,488 genes expressed in either iPSCs or HLCs were used for meta-analysis. The probability of tissue specificity was estimated using m-values (Han and Eskin, 2012). Briefly, m-values indicate the posterior probability that the effect exists in each study or cell type. We considered m-values ≥ 0.9 as indicating a particular variant had a molecular effect in each cell type. The most significant HLC eGenes with respect to meta-analysis P-values were AMDHD1, FA2H, and ACAA2 (Table S5).
We assessed tissue specificity of HLC and iPSC eGenes by selecting SNP-eGene pairs (the most significant single-tissue SNP per gene) (Table S5). Among 2,392 HLC eGenes, we found 267 HLC-specific eGenes (HLC m-value ≥ 0.9; iPSC m-value < 0.1) and 1,051 eGenes with evidence of tissue specificity in both HLCs and iPSCs (HLC m-value ≥ 0.9; iPSC m-value ≥ 0.9). Among 3,587 iPSC eGenes, we found 474 iPSC-specific eGenes (HLC m-value < 0.1; iPSC m-value ≥ 0.9) and 1,279 eGenes with evidence of effects in both HLCs and iPSCs (HLC m-value ≥ 0.9; iPSC m-value ≥ 0.9). We also indirectly assessed potential HLC and iPSC specificity of GTEx liver eGenes by comparing GTEx liver eGenes to m-values estimated from our data. We found 24 GTEx liver eGenes (HLC m-value ≥ 0.9; iPSC m-value < 0.1) and 10 GTEx liver eGenes (HLC m-value < 0.1; iPSC m-value ≥ 0.9) that potentially share HLC or iPSC tissue specificity, respectively.
Our identified 267 HLC-specific eGenes were significantly enriched for genes with known functions in decreased cholesterol level (MP:0003983; Benjamini-Hochberg FDR = 0.03), decreased sterol level (MP:0012225; FDR = 0.03), and decreased circulating cholesterol level (MP:0005179; FDR = 0.04) (Table S6). Perhaps not surprisingly, these HLC-specific eGenes were also enriched for liver selective genes as defined by MSigDB (M13283; FDR = 0.04).
Identifying blood lipid-associated eQTL and ASE loci
In order to assess whether the eQTL/ASE analyses could identify new functional variants and genes at GWAS loci, we chose to focus on blood lipid traits [total cholesterol (TC), LDL-C, HDL-C, and triglyceride (TG) levels] because of the major influence of the liver on these traits, as well as the ability to interrogate the functional effects of candidate genes on blood lipid levels in mice. To identify potential functional variants and genes in the HLC, iPSC, and GTEx liver cohorts, we interrogated 7,240 SNPs across 157 loci associated with one or more of the four lipid traits (P < 5 × 10−8) from the Global Lipids Genetics Consortium (GLGC) (Global Lipids Genetics Consortium, 2013) for association with the expression of any gene within 1 Mb (eQTL FDR < 5%). Across the 157 loci, there were 32 eGenes in the HLCs, 43 eGenes in the iPSCs, and 30 eGenes in the GTEx livers (Figure 2A, Table S7). In the HLCs, 205 genes at the GLGC loci exhibited significant ASE (FDR < 5%) in at least one individual, potentially identifying additional candidate functional genes that were missed by the eQTL analyses.
Figure 2. HLC eGenes and Candidate Functional Variants from MPRAs.

(A) Venn diagram of mapped eGenes at GLGC loci with a stringent cutoff of FDR < 5% (also see Table S7). In bold and underlined are eGenes for which functional studies were performed.
(B) Top-ranked GLGC HLC eGenes. The displayed SNP in the third column is the GWAS SNP (or one of a set of GWAS SNPs in perfect LD in the cohort) in the locus that displayed the strongest association with the eGene. The displayed SNP in the seventh column is the GWAS SNP in the locus that displayed the strongest association in a sensitivity analysis with 63 higher-quality HLC samples. In bold are eGenes for which functional studies were performed.
(C) MPRAs identified rs2277862, rs10889356, and rs10872142 as the SNPs with the greatest allele-specific regulatory activity in the CPNE1, ANGPTL3, and FRK loci, respectively (also see Table S9). Each candidate SNP was represented on a 145-bp tile that was either left-shifted, centered, or right-shifted relative to the SNP, in order to increase the probability of capturing the correct regulatory context for that SNP. For each tile, the individual signals for the two alleles are shown for two independent experiments (where signal refers to the log of median barcode counts for the given tile divided by median barcode counts for all tiles). In the final two columns, a log-ratio of the signals of the two alleles is calculated, along with a P-value (Mann-Whitney U test) for the null hypothesis that the two alleles generate equal signals.
We were particularly interested in the HLC eGenes, irrespective of whether they were found only in HLCs or were shared by iPSCs (i.e., not exclusive to HLCs), because of their potential to modulate hepatocyte lipid metabolism. We rank-ordered the identified GLGC HLC eGenes by strength of association, judged by eQTL P-value (Figure 2B). Among the top GLGC HLC eGenes, we deferred studying SYPL2 and IGF2R; for each of these genes, the best eQTL SNP is in weak LD with an extremely strongly lipid-associated SNP in the SORT1 locus (r2 = 0.01 between rs10857787 and rs12740374 in Europeans) and the LPA locus (r2 = 0.035 between rs3777404 and rs1564348 in Europeans), respectively—so strongly that the SYPL2 and IGF2R SNPs meet the statistical threshold of P < 5 × 10−8 for lipid association even though they are located in different loci than the SORT1 and LPA lead SNPs, simply by being in weak LD with them. We also deferred studying FUT2, since its best eQTL SNP is a coding variant in the gene. Among the other top GLGC HLC eGenes, we sought to identify functional regulatory variants in the loci of CPNE1, ANGPTL3, and FRK, as described below. We note that in eQTL analyses performed only on higher-quality HLCs (the 63 HLC samples with scores of at least three in Table S1, out of the total 86 HLC samples), all of the top GLGC HLC eGenes remained strongly associated with SNPs in their loci (Figure 2B).
Colocalization of SNP-gene associations
To assess the hypothesis that lipid and eQTL associations are driven by the same variant, we assessed colocalization of each of the top eight GLGC HLC eGenes and the four relevant GLGC traits (Giambartolomei et al., 2014). Fifteen trait-eGene pairs exhibited moderate to strong evidence of colocalization (PP.H4.ABF) > 0.4; Table S8). In particular, VKORC1 showed strong evidence of colocalization in both HLCs (TG, PP.H4.ABF = 0.97; LDL-C, PP.H4.ABF = 0.52) and GTEx livers (TG, PP.H4.ABF = 0.98; LDL-C, PP.H4.ABF = 0.65) but not in iPSCs (TG, PP.H4.ABF = 0.07; LDL-C, PP.H4.ABF = 0.006). ANGPTL3 colocalized in HLCs with all four GLGC traits (LDL-C, PP.H4.ABF = 0.77; TG, PP.H4.ABF = 0.76; TC, PP.H4.ABF = 0.49; HDL-C, PP.H4.ABF = 0.48). ANGPTL3 did not show evidence of colocalization in iPSCs or GTEx livers (Table S8).
Functional analysis of rs2277862 at the CPNE1/ERGIC3 locus
We focused first on the locus of CPNE1 (Figure 3A), an HLC eGene that did not independently qualify as an eGene in the GTEx liver cohort (i.e., did not have any associated SNPs with FDR < 5%), although it qualified as an eGene in a cohort of ~1000 liver samples (Teslovich et al., 2010). To identify candidate functional variants among all the SNPs in LD within the CPNE1 locus in an unbiased fashion, we performed MPRA experiments to rapidly profile the regulatory activity of 239 variants linked (r2 ≥ 0.5) to the lead SNP at the locus. Duplicate MPRAs were performed, and we rank-ordered SNPs by magnitude of allele-specific regulatory activity as measured by reporter expression (Table S9). The top MPRA SNP in the locus was rs2277862 (Figure 2C). Of note, rs2277862 was also the lead SNP for TC in the locus in the GLGC study, with each alternate allele copy associated with a 1.19 mg/dL change in TC (Teslovich et al., 2010).
Figure 3. Evidence for rs2277862-CPNE1 as a Functional SNP-Gene Set.
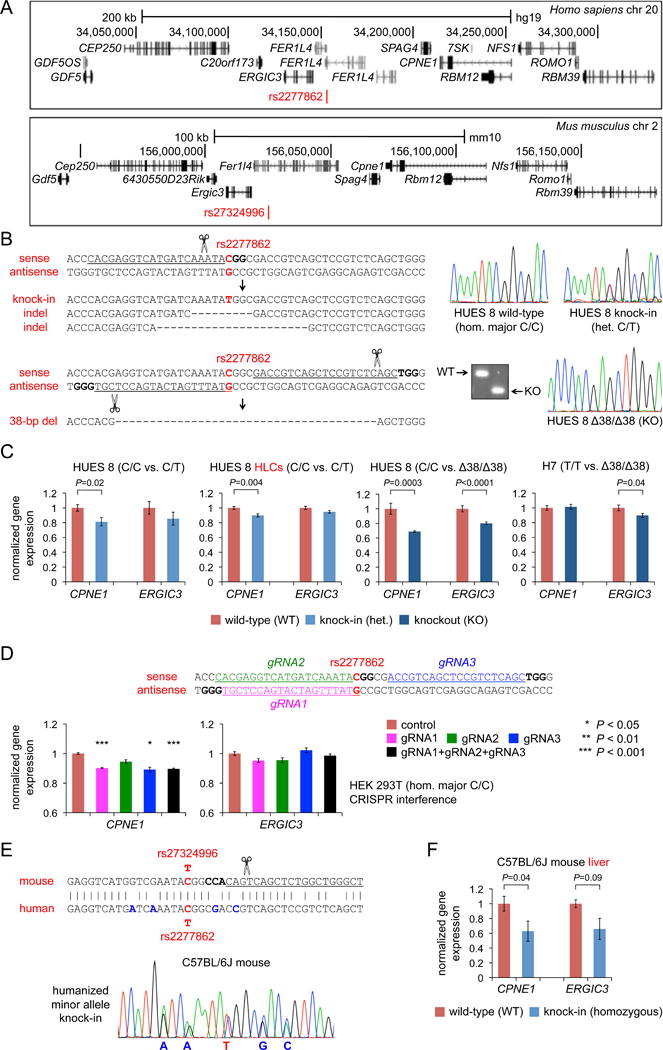
(A) Schematics of human chromosome 20q11 locus showing the relative positions of rs2277862, CPNE1, and ERGIC3 and mouse chromosome 2qH1 locus showing the relative positions of rs27324996, Cpne1, and Ergic3.
(B) Top panels: heterozygous knock-in of rs2277862 minor allele with a single-strand DNA oligonucleotide. Representative indels in non-knock-in clones are also shown. Bottom panels: homozygous 38-bp deletions (“knockout” or Δ38/Δ38) encompassing rs2277862 using a dual gRNA approach. A representative agarose gel of PCR amplicons is shown. The protospacers are underlined, the PAMs are bolded, and the SNP position is indicated in red.
(C) Left panels: gene expression in undifferentiated HUES 8 cells (n = 2 wild-type clones and 1 knock-in clone; 6 wells per clone) and differentiated HUES 8 HLCs (n = 2 wild-type clones and 1 knock-in clone; 6 wells per clone) normalized to mean expression level in wild-type clones. Right panels: gene expression in undifferentiated HUES 8 cells (n = 10 wild-type and 10 knockout clones, 3 wells per clone) and undifferentiated H7 cells (n = 8 wild-type and 6 knockout clones, 3 wells per clone) normalized to mean expression level in wild-type clones.
(D) CRISPR interference at the rs2277862 site. The three gRNA protospacers are underlined, the PAMs are bolded, and the SNP position is indicated in red. The graphs show gene expression, normalized to mean expression level in control cells, in HEK 293T cells transfected with catalytically dead Cas9 (dCas9) with the gRNAs (either singly or in combination, 3 wells per condition). Control cells were transfected with dCas9 without an accompanying gRNA.
(E) The noncoding rs2277862 site is well conserved in mouse, including allelic variants of the SNP itself, with the murine equivalent being rs27324996. The SNP position is indicated in red, non-conserved nucleotides are indicated in blue, the gRNA protospacer used to generate the knock-in mouse is underlined, and the PAM is bolded. The electropherogram is from a mouse in which the minor allele of rs2277862/rs27324996 (T) has been knocked into one chromosome, along with four non-conserved nucleotides to “humanize” the site.
(F) Gene expression in liver from littermates of the C57BL/6J background (n = 18 wild-type mice and 10 homozygous knock-in mice), normalized to mean expression level in wild-type mice.
Data are displayed as means and s.e.m. P-values were calculated with two-tailed Welch’s t-tests.
The locus harbors eGenes in the HLC cohort (CPNE1), the iPSC cohort (CPNE1), and the GTEx liver cohort (ERGIC3); CPNE1 exhibited ASE in HLCs and iPSCs (lowest CPNE1 HLC P = 3 × 10−8; lowest CPNE1 iPSC P = 6 × 10−9). The functions of these genes are poorly understood. We reasoned that rs2277862 might modulate transcription of CPNE1 in both HLCs and undifferentiated human pluripotent stem cells (hPSCs). To test if rs2277862 regulates CPNE1 and/or ERGIC3 expression, we performed four types of experiments, two based in hPSCs. First, in an hPSC line, HUES 8 (C/C, major/major at rs2277862), we used CRISPR-Cas9 to knock in one minor allele (Figure 3B). Using a single-strand DNA oligonucleotide as a repair template, we obtained only a single recombinant heterozygous (C/T) clone at a frequency of 0.15% (1 out of 672 clones screened), highlighting the inefficiency of the procedure. We observed significantly decreased CPNE1 expression in both undifferentiated knock-in hPSCs (down 19%) and differentiated knock-in HLCs (down 10%), with non-significant effects on ERGIC3 (Figure 3C).
Second, in two hPSC lines, HUES 8 and H7 (T/T, minor/minor at rs2277862), we used multiplexed CRISPR-Cas9 to cleanly and efficiently delete ~38 bp around the SNP (168/285 clones) (Figure 3B), which was far more efficient than knock-in. Homozygous deletion of 38 bp in undifferentiated HUES 8 cells decreased expression of CPNE1 (down 31%) and ERGIC3 (down 20%), whereas homozygous deletion in undifferentiated H7 cells did not affect CPNE1 and decreased expression of ERGIC3 to a lesser degree (down 10%) (Figure 3C). These data are concordant with the data from the heterozygous knock-in clone.
Third, we used catalytically dead Cas9 (dCas9) with three different guide RNAs (gRNAs) at the site of the SNP to attempt CRISPR interference, or CRISPRi (Qi et al., 2013) in HEK 293T cells (C/C, major/major at rs2277862). The rationale was that if the variant were truly functional and lay within a transcriptional regulatory element, then the presence of the bulky Cas9 protein at the site should sterically hinder recruitment of transcription factors to the regulatory site and thus interfere with transcriptional regulation. We found that the gRNAs generally resulted in decreased expression of CPNE1 but not ERGIC3 (Figure 3D).
Fourth, we noted that, atypically, the noncoding region encompassing rs2277862 is well conserved in mouse (Figure 3A and 3E), and the orthologous nucleotide in mouse also displays naturally occurring variation and has been previously cataloged as rs27324996, with the same two alleles (C and T) as in humans. We used CRISPR-Cas9 in single-cell mouse embryos of the C57BL/6J strain (C/C at rs27324996) to generate a knock-in mouse with a minor allele (T) as well as four additional nucleotide changes both to prevent CRISPR-Cas9 re-cleavage of the knock-in allele and to “humanize” the sequence (i.e., make a perfect match to the orthologous human sequence) (Figure 3E). There was decreased expression of Cpne1 (down 37%) and Ergic3 (down 34%, albeit not with P < 0.05) in the liver in homozygous knock-in (T/T) mice compared to wild-type (C/C) littermates (Figure 3F); cholesterol levels were unchanged, as expected from the small effect in humans (1.19 mg/dL change in cholesterol per allele). These data are highly concordant with the data from the genome-edited hPSC clones with respect to both the direction and magnitude of effects. Taken together, all of these findings support rs2277862-CPNE1 as a functional SNP-gene set.
Functional analysis of rs10889356 at the ANGPTL3/DOCK7 locus
We next focused on the locus of ANGPTL3 (Figure 4A), another HLC eGene that did not independently qualify as an eGene in the GTEx liver cohort, although it qualified as an eGene in a cohort of ~1000 liver samples (Teslovich et al., 2010). In duplicate MPRA experiments, we profiled the regulatory activity of 210 variants linked (r2 ≥ 0.5) to the lead SNP at the locus (Table S9). The top MPRA SNP in the locus was rs10889356 (Figure 2C). In Europeans, rs10889356 is tightly linked to rs2131925 (r2 = 0.90), the lead SNP in the locus in the GLGC study, with each alternate allele copy of rs2131925 associated with a 4.9 mg/dL change in TG (Teslovich et al., 2010). ANGPTL3, which is generally believed to be the functional gene at this locus, encodes a liver-specific secreted protein that increases blood cholesterol and triglyceride levels in mice (Koishi et al., 2002) and in humans (Musunuru et al., 2010a). ANGPTL3 lies within an intron of DOCK7, a poorly understood non-liver-specific gene. As with the CPNE1/ERGIC3 locus, we used several complementary approaches to interrogate the relationship of rs10889356 with ANGPTL3 and DOCK7.
Figure 4. Evidence for rs10889356-DOCK7 and rs10889356-ANGPTL3 as Functional SNP-Gene Sets.
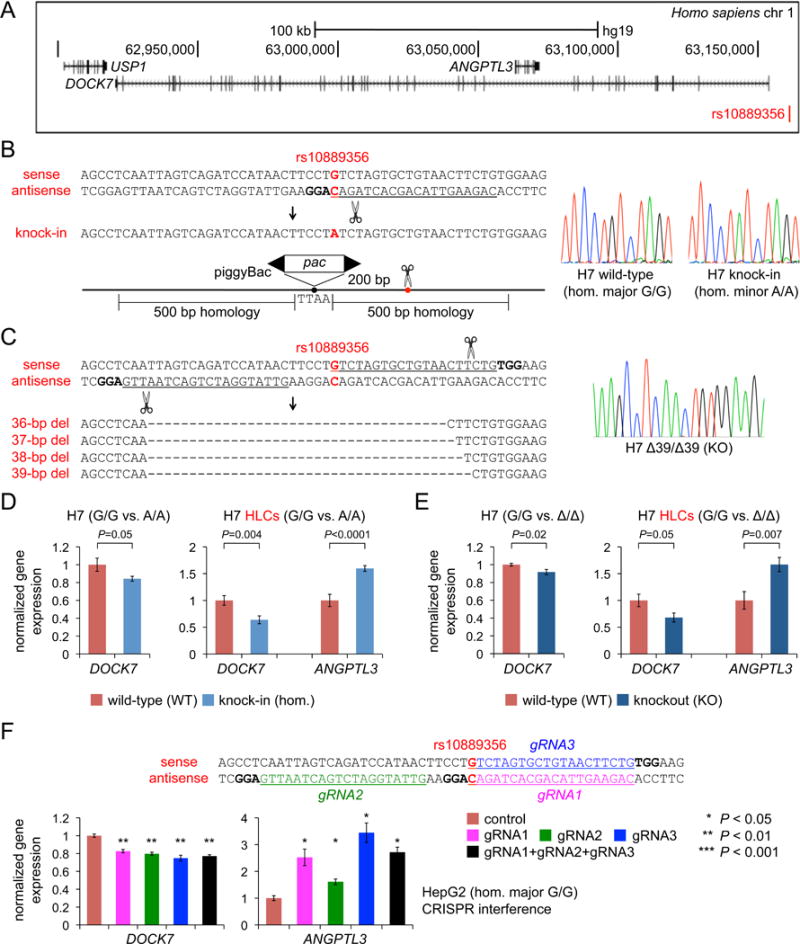
(A) Schematic of human chromosome 1p31 locus showing the relative positions of rs10889356, DOCK7, and ANGPTL3.
(B) Homozygous knock-in of rs10889356 minor allele using a targeting vector with puromycin resistance encoded within a scarless-excision piggyBac transposon. The protospacer is underlined, the PAM is bolded, and the SNP position is indicated in red.
(C) Homozygous 36- to 39-bp deletions (“knockout” or Δ/Δ) encompassing rs10889356 using a dual gRNA approach. The protospacer is underlined, the PAM is bolded, and the SNP position is indicated in red.
(D) Gene expression in undifferentiated H7 cells (n = 12 wild-type and 10 homozygous knock-in clones, 6 wells per clone) and differentiated H7 HLCs (n = 6 wild-type and 4 homozygous knock-in, 3 wells per clone), normalized to mean expression level in wild-type clones.
(E) Gene expression in undifferentiated H7 cells (n = 12 wild-type and 8 knockout clones, 3 wells per clone) and differentiated H7 HLCs (n = 4 wild-type and 4 knockout clones, 2 wells per clone).
(F) CRISPR interference at the rs10889356 site. The three gRNA protospacers are underlined, the PAMs are bolded, and the SNP position is indicated in red. The graphs show gene expression, normalized to mean expression level in control cells, in HepG2 cells transfected with dCas9 with the gRNAs (either singly or in combination, 3 wells per condition).
Data are displayed as means and s.e.m. P-values were calculated with two-tailed Welch’s t-tests.
First, in the H7 hPSC line (G/G, major/major at rs10889356), we used CRISPR-Cas9 to knock in the minor allele (Figure 4B). We attempted to use a single-strand DNA oligonucleotide as a repair template but were unsuccessful in obtaining any heterozygous or homozygous knock-in clones after screening a large number of clones. We thereafter used a targeting vector with 500-bp homology arms and a puromycin resistance cassette on a transposon that could undergo scarless removal from a TTAA site with piggyBac (Figure 4B). The closest endogenous TTAA site to rs10889356 was 200 bp away. We obtained several clones in which puromycin selection facilitated knock-in of the rs10889356 minor allele into both chromosomes (i.e., homozygous knock-in). We pooled these clones and introduced piggyBac, which yielded a large number of clones in which scarless excision was achieved. Homozygous knock-in (A/A) clones had decreased expression of DOCK7 compared to wild-type clones (down 16%) (Figure 4D). When differentiated into HLCs, homozygous knock-in cells had decreased expression of DOCK7 (down 36%) and substantially increased ANGPTL3 expression (up 60%) (Figure 4D).
Second, in the same H7 hPSC background (G/G, major/major), we used multiplexed CRISPR-Cas9 to delete 36–39 bp around the SNP (Figure 4C). Homozygous deletion of 36–39 bp in undifferentiated H7 cells decreased expression of DOCK7 (down 8%) (Figure 4E). When differentiated into HLCs, homozygous deleted H7 cells had decreased expression of DOCK7 (down 32%) and increased ANGPTL3 expression (up 67%) (Figure 4E). These data are extremely concordant with the data from the homozygous knock-in clones.
Third, CRISPRi with three gRNAs in HepG2 cells (G/G, major/major at rs10889356; used because being of hepatic origin they express ANGPTL3) both decreased DOCK7 expression and increased ANGPTL3 expression (Figure 4F). These data are concordant with the data from the genome-edited hPSC clones with respect to the direction of effects for both genes. Taken together, all of these findings support rs10889356-DOCK7 and rs10889356-ANGPTL3 as functional SNP-gene sets.
Functional analysis of rs10872142 at the FRK locus
We investigated a third locus with a highly ranked HLC eGene that did not independently qualify as an eGene in the GTEx liver cohort, the FRK locus, although FRK qualified as an eGene in a cohort of ~1000 liver samples (Teslovich et al., 2010) (Figure 5A). In duplicate MPRA experiments, we profiled the regulatory activity of 76 variants linked (r2 ≥ 0.5) to the lead SNP at the locus (Table S9). The top MPRA SNP in the locus was rs10872142 (Figure 2C). In Europeans, rs10872142 is tightly linked to rs11153594 (r2 = 0.965), the lead SNP for LDL-C in the locus in the GLGC study, with each alternate allele copy of rs2131925 associated with a 0.9 mg/dL change in LDL-C (Teslovich et al., 2010). rs10872142 lies within intron 1 of FRK, whose protein product belongs to the TYR family of protein kinases. FRK was the only eGene at this locus in both the iPSC and HLC cohorts.
Figure 5. Evidence for rs10872142-FRK as a Functional SNP-Gene Set.
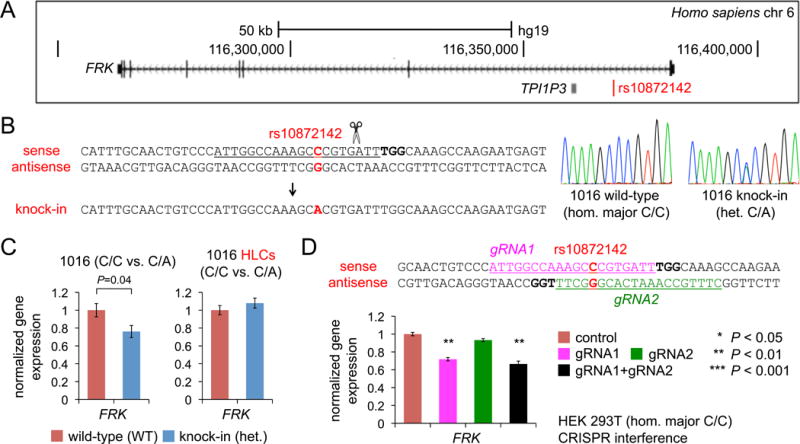
(A) Schematic of human chromosome 6q22 locus showing the relative positions of rs10872142 and FRK.
(B) Heterozygous knock-in of rs10872142 minor allele with a single-strand DNA oligonucleotide. The protospacer is underlined, the PAM is bolded, and the SNP position is indicated in red.
(C) Gene expression in undifferentiated 1016 cells (n = 3 wild-type and 3 heterozygous clones, 3 wells per clone) and differentiated 1016 HLCs (n = 3 wild-type and 3 heterozygous clones, 4 wells per clone), normalized to mean expression level in wild-type clones.
(D) CRISPR interference at the rs10872142 site. The two gRNA protospacers are underlined, the PAMs are bolded, and the SNP position is indicated in red. The graphs show gene expression, normalized to mean expression level in control cells, in HEK 293T cells transfected with dCas9 with the gRNAs (either singly or in combination, 6 wells per condition).
Data are displayed as means and s.e.m. P-values were calculated with two-tailed Welch’s t-tests.
In the DiPS 1016 SevA iPSC line (called 1016 for short; C/C, major/major at rs10872142), we used CRISPR-Cas9 to knock in the minor allele (Figure 5B). Using a single-strand DNA oligonucleotide as a repair template, we obtained several heterozygous (C/A) clones (out of 140 clones screened). Heterozygous knock-in clones had decreased expression of FRK compared to wild-type clones (down 24%); when differentiated into HLCs, no difference in expression was observed (Figure 5C). CRISPRi with one of two gRNAs and the combination of the two gRNAs in HEK 293T cells (C/C, major/major at rs10872142) decreased FRK expression (Figure 5D), concordant with the data from the genome-edited iPSC clones. These findings support rs10872142-FRK as a functional SNP-gene set in iPSCs but not in HLCs.
Identifying functional genes associated with lipid metabolism via the HLC cohort
We next sought to assess whether HLC eGenes and ASE genes are functional for lipid metabolism in mouse models. We used adeno-associated virus (AAV) serotype 8, which effectively transduces mouse hepatocytes in vivo, to heterologously express candidate genes from the liver-specific TBG promoter. We first assessed the candidate genes in the CPNE1/ERGIC3 locus, for which CPNE1 had stronger evidence of being a hepatocyte eGene than ERGIC3 (see above). We found that CPNE1 but not ERGIC3 reproducibly decreased blood HDL-C levels when overexpressed in mouse liver with AAV (Figure 6A), establishing CPNE1 as a lipid-functional gene. In the ANGPTL3/DOCK7 locus, we confirmed ANGPTL3 to be a lipid-functional gene by using CRISPR-Cas9 to delete the entire ANGPTL3 gene in mice and observing substantial decreases in both TG and cholesterol levels (Figure 6B).
Figure 6. Interrogation of candidate genes for effects on blood lipid levels in mice.
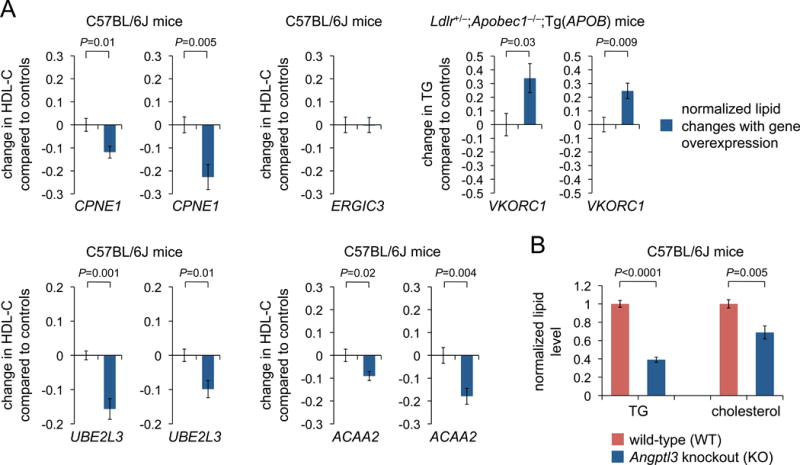
(A) Proportional changes in blood TG or HDL-C levels before versus after AAV8-mediated gene overexpression in liver, normalized to mice that received control AAV8 vectors (n = 7 mice per group). Two independent experiments are shown for each gene except ERGIC3.
(B) Blood triglyceride and cholesterol levels in wild-type versus Angptl3 knockout mice (n = 17 wild-type and 6 knockout mice), normalized to mean expression levels in wild-type mice.
Data are displayed as means and s.e.m. P-values were calculated with two-tailed Welch’s t-tests.
We interrogated two other GLGC HLC eGenes, VKORC1 (locus associated with TG) and UBE2L3 (locus associated with HDL-C). Whereas VKORC1 independently qualified as a GTEx liver eGene (FDR < 5%), UBE2L3 did not. For VKORC1, we used a lipid-humanized mouse model to better reflect human triglyceride physiology (Burkhardt et al., 2010). Each of the two genes reproducibly altered the associated lipid trait (Figure 6A).
Finally, to assess whether the HLC cohort could be used to discover lipid-functional genes that were not directly nominated by the GLGC HLC eGene analysis, we interrogated ACAA2. ACAA2 was notable in that it was one of the top-ranked HLC-specific eGenes in the genome-wide iPSC/HLC meta-analysis (P = 4 × 10−13; Table S6) and exhibited HLC-specific differential ASE relative to iPSCs in the genome-wide ASE analysis (P = 2 × 10−4; Table S4). We found that expression of ACAA2 reproducibly altered blood HDL-C levels in mice (Figure 6A). Taken together, all of these findings establish that cohorts of iPSC-differentiated cells can discover new functional genes underlying complex traits.
DISCUSSION
One of the principal challenges of the post-GWAS era has been identifying functional variants and genes underlying complex disease susceptibility. In this work, we describe a methodological framework for functional pathway discovery that involves characterizing eQTL and ASE loci in population cohorts of iPSCs and iPSC-differentiated cells, high-throughput identification of candidate functional variants underlying eQTLs and ASE with MPRAs, and validation of prioritized variants in genome-edited cellular and mouse models. We focused our experimental efforts on interrogating lipid-associated eQTLs in HLCs and defining functional SNP-gene sets in three different eQTL loci.
Efforts such as the GTEx project (The GTEx Consortium, 2015; Aguet et al., 2016) seek to identify functional eGenes, but these studies rely on scarce primary tissues from postmortem donors or from surgical patients, and the tissues typically represent mixes of different cell types, potentially obscuring eGenes with functional significance in one of the cell types. The distinction between postmortem whole-liver samples and primary human hepatocytes was quite apparent in the global gene expression profiling (Figure S2B). In principle, iPSCs offer an alternative, more expedient approach to eQTL/ASE studies because they can be obtained from healthy living donors non-invasively, are expandable in vitro, and can be differentiated to a single desired cell type. However, a serious shortcoming of in vitro iPSC-differentiated hepatocytes—and, generally, all in vitro iPSC-differentiated cells—is that the resulting cells are immature, heterogeneous, and lack some characteristics of authentic cells in their natural environment in vivo (Schwartz et al., 2014). As with the liver samples, the distinction between HLCs and primary hepatocytes was evident with respect to global gene expression profiling (Figure S3) as well as the expression of individual genes and secretion of proteins (Figure S2). Thus, our analyses confirm that neither liver samples nor HLCs faithfully reflect the cellular state of primary hepatocytes, an important limitation of eQTL/ASE studies performed with either sample type. In light of these limitations, we suggest that whole-liver eQTL studies and HLC eQTL studies should be pursued in a complementary fashion, and that functional studies should be prioritized for eGenes that are independently observed in the two types of studies, as these genes are more likely to reflect bona fide hepatocyte biology.
Our HLC cohort did successfully identify a large number of eGenes shared with the GTEx liver cohort, as well as a large number of eGenes that did not independently qualify as eGenes in the GTEx liver cohort, including genes expressed only in hepatocytes (such as ANGPTL3). We were able to use these findings to discover and validate a number of functional genes—HLC eGenes CPNE1, VKORC1, and UBE2L3 and HLC-specific differential ASE gene ACAA2 modulated blood lipid levels in mouse models—as well as functional variants. One possible reason why many HLC eGenes were not independently observed in the GTEx liver cohort (at FDR < 5%) is a limitation in power offered by the currently available GTEx cohort. Indeed, observations from much larger primary human liver eQTL studies (as large as ~1,000 samples) provide evidence for CPNE1, ANGPTL3, and FRK being liver eGenes (Teslovich et al., 2010; Innocenti et al., 2011). Thus, we expect that comparisons of much larger, better-powered HLC cohorts and liver cohorts would reveal substantially more overlap of eGenes than we observed in the present study. The establishment of iPSC biobanks by efforts such as the NHLBI Next Generation Genetic Association Studies Consortium (http://www.wicell.org/home/stem-cell-lines/collections/collections.cmsx) should make it feasible for investigators to perform future iPSC-based eQTL/ASE studies with thousands of samples.
We note that the availability of many other tissue types besides liver in GTEx offers the opportunity to assess for eQTLs in loci of interest. Even if the power of the GTEx liver cohort is limited, other GTEx tissue cohorts might reveal eQTLs of relevance in hepatocytes. This strategy would be more informative for eQTLs that are shared widely among tissues than for eQTLs that are specific to hepatocytes. Even so, non-liver-tissue eQTL findings should be interpreted with caution. For example, whereas there is a strong eQTL for VKORC1 in the GTEx liver cohort, there is also a strong eQTL for VKORC1 in the GTEx adrenal gland cohort—but with the opposite directionality (as assessed at the GTEx portal at the time of this writing, http://www.gtexportal.org/), suggesting different regulatory mechanisms in the two tissue types.
One salient advantage of iPSC-based regulatory variation studies such as eQTL/ASE studies is that candidate functional genes and variants can be rigorously tested and validated within the same system (i.e., pluripotent stem cells and differentiated cells), one that is readily amenable to CRISPR-Cas9 genome editing for the purpose of studying gene expression (Soldner et al., 2016). This cannot easily be done with findings from human tissues such as whole liver, except in unusual cases where there is a high degree of orthology between the corresponding loci in human and an appropriate animal model (such as mouse) that allows for genome editing of a conserved candidate noncoding variant in the animal model in vivo (as was the case with rs2277862 and rs27324996 in the CPNE1/Cpne1 loci). We were able to rigorously assess for functionality of the MPRA-nominated rs2277862, rs10889356, and rs10872142 through the generation of isogenic wild-type and variant hPSCs via genome editing. Consistent with prior studies, we found the generation of knock-in clones via HDR to be very inefficient compared to the generation of clones with defined deletions via multiplexed NHEJ. Nonetheless, we were able to generate knock-in clones for all three SNPs, and the gene expression changes observed in knock-in clones were concordant with those observed from SNP-deleted clones.
In addition, we employed the alternative approach of CRISPR interference, a term that is typically used to describe the repression of transcription via the positioning of dCas9 to a gene promoter or coding sequence to block transcriptional initiation or elongation via steric interference (Qi et al., 2013); alternatively, dCas9 can be attached to a repressor domain such as Krüppel associated box (KRAB) to effect transcriptional silencing via epigenetic chromatin modification (Gilbert et al., 2013). We opted to use the unadorned dCas9 protein rather than attaching an extra domain, since the regulatory elements in the vicinity of the three SNPs were undefined and it was unclear whether each SNP allele acted as an enhancer or repressor or was neutral. Instead, we relied on steric interference to reverse any effect of the SNP allele. Within each locus the various guide RNAs did not show equal effects on gene expression, reflecting that CRISPR-Cas9 can display considerable site-to-site variability in its activity, in some cases showing no activity. Nonetheless, for all three SNPs, CRISPRi yielded results that were concordant with the effects observed in the genome-edited cells—decreased CPNE1 expression vis-à-vis the major allele of rs2277862, decreased DOCK7 and increased ANGPTL3 expression vis-à-vis the major allele of rs10889356, and decreased FRK expression vis-à-vis the major allele of rs10872142. It should be noted that while our overall approach was successful in identifying and validating functional variants in the three eQTL loci, it might not have identified all functional variants in the loci, as multiple variants in a locus can contribute to a phenotype.
In summary, we generated a resource of iPSC lines from a multi-ethnic cohort of healthy individuals and used them to generate differentiated cells with which to perform informative genome-wide eQTL and ASE mapping in a manner analogous to more traditional primary tissue-based studies such as GTEx. Our studies highlight the utility of population cohorts of iPSCs and iPSC-differentiated cells and functional genomics approaches to elucidate functional variants and genes underlying complex human phenotypes. Furthermore, they set the stage for the use of HLCs for a variety of –omics studies, studies of responses to drugs and environmental perturbations, and other types of studies that can be more easily performed “in the dish” than in living persons. Thus, we anticipate an iPSC-based framework of discovery and validation being broadly applied to gain new insights into a variety of human diseases.
STAR*METHODS
KEY RESOURCES TABLE
See attached file.
CONTACT FOR REAGENT AND RESOURCE SHARING
The iPSC lines generated in this study are publicly available through the WiCell Research Institute depository for the NHLBI Next Generation Genetic Association Studies Consortium collection (Table S1 includes the designations): http://www.wicell.org/home/stem-cell-lines/catalog-of-stem-cell-lines/collections/nhlbi-next-gen-rader.cmsx. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kiran Musunuru (kiranmusunuru@gmail.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
The human study included 91 subjects who were free of cardiovascular disease and in generally good health. The individuals were specifically recruited for this study [Phenotyping Lipid traits in iPS derived hepatocytes Study (PhLiPS Study)]. The University of Pennsylvania Human Subjects Research Institutional Review Board approved the study protocol, and all subjects gave written informed consent for study participation. The study was wholly performed at the Perelman School of Medicine at the University of Pennsylvania. Peripheral blood samples obtained from the subjects were used for blood lipid measurements as well as the generation of iPSC lines and genome-wide genotyping. See Table S1 for demographic data and blood lipid data.
Animals
All animal procedures performed in this study and described below were reviewed and approved by the pertinent Institutional Animal Care and Use Committees at the University of Pennsylvania and Harvard University and were consistent with local, state and federal regulations as applicable. Euthanasia in all instances was via terminal inhalation of carbon dioxide, consistent with the 2013 AVMA Guidelines on Euthanasia. C57BL/6J animals were obtained from the Jackson Laboratory (stock number 000664) and were bred in-house. Ldlr+/−;Apobec1−/−;Tg(APOB) mice, which were used to better model TG and LDL-C metabolism for genes in loci associated with one of those traits (Burkhardt et al., 2010), were bred in-house. rs27324996 knock-in mice and Angptl3 knockout mice were generated on the C57BL/6J background with CRISPR-Cas9 as described below and were bred in-house. All mice were fed a standard chow diet and maintained on a 12 hour light/12 hour dark cycle. Assignments to groups (experimental vs. control, comparisons between genotypes) were performed to achieve age-matched groups of littermates/colonymates with shared housing and were otherwise random. All mice used for experiments were healthy, free of drug exposure, and had not undergone any previous procedures. Details on the ages and sexes of mice used for experiments are provided in the relevant sections below.
Cell Lines
All cultured cell lines were maintained in a humidified 37°C incubator with 5% CO2. HEK 293T, HepG2, and NIH 3T3 cells were all obtained directly from American Type Culture Collection and were cultured in high-glucose DMEM (Thermo Fisher Scientific) supplemented with 10% Fetal Bovine Serum (FBS; Thermo Fisher Scientific) and 1% penicillin/streptomycin (P/S). HUES 8 cells (directly obtained from the Harvard Stem Cell Institute iPS Core Facility), H7 cells (directly obtained from the WiCell Research Institute), DiPS 1016 SevA cells (directly obtained from the Harvard Stem Cell Institute iPS Core Facility), and all of the iPSC lines generated for this study (see below) were grown under feeder-free conditions on Geltrex (Thermo Fisher Scientific)-coated plates in chemically defined mTeSR1 medium (STEMCELL Technologies) supplemented with 1% P/S and 5 μg/mL Plasmocin (InvivoGen). Medium was changed every 24 hours. The cell lines were routinely tested and confirmed to be negative for mycoplasma contamination, as described below.
METHOD DETAILS
Generation of iPSC lines
Blood was collected from donors, and iPSC lines were derived from the blood cells as previously described in detail (Yang et al., 2012). Briefly, peripheral blood mononuclear cells (PBMCs) were isolated from blood and cultured in media supplemented with cytokines for expansion of erythroblasts. Erythroblast-enriched cultures were transduced with Sendai viral (SeV) vectors (CytoTune; Thermo Fisher Scientific) expressing the four Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc). Transduced cells were then transferred onto irradiated mouse embryonic fibroblast (MEF) feeder cells (Global Stem) and cultured at 37 °C at 5% oxygen/5% CO2 in hESC medium consisting of DMEM/F12 supplemented with 20% knockout serum replacement (KOSR), 2 mM L-glutamine, MEM nonessential amino acids (NEAA), 1% penicillin/streptomycin (P/S), and 10 ng/mL basic fibroblast growth factor (bFGF). All ingredients for hESC medium were obtained from Thermo Fisher Scientific. iPSC colonies were picked and further cultured in hESC medium to at least cell passage 12 to establish iPSC lines. Two to four iPSC lines were established from each blood sample. Established iPSC lines were transitioned to feeder-free culture conditions for at least 5 passages in mTeSR1 medium and on Geltrex-coated tissue culture plates before they were used for HLC differentiation.
Quality control and characterization of iPSC lines
Established iPSC lines were cultured in hESC medium without P/S for 3 days without refreshing medium. Spent medium was subjected to the MycoAlert Mycoplasma Detection Kit (Lonza) for the presence of mycoplasma species. For flow cytometric analysis of cell surface pluripotency markers SSEA4 and Tra-1-60, iPSCs were dissociated into single cells with Accutase (Innovative Cell Technologies) and incubated with Alexa Fluor 647 anti-human SSEA-4 (BioLegend) and PE anti-human Tra-1-60 (BD Biosciences) antibodies. Samples were analyzed using the BD FACSCalibur system (BD Biosciences). Cells were plotted according to forward scatter and side scatter profiles and gated to exclude cell doublets and debris. Data were analyzed with FlowJo software (FlowJo). Table S1 includes the proportion of double-positive cells for each iPSC line. For confirmation of loss of SeV reprogramming vector expression, total RNA was isolated from iPSC lines cultured under feeder-free conditions using the miRNeasy Mini Kit (QIAGEN) and quantified with the RNA 6000 Nano Kit (Agilent) on the Agilent Bioanalyzer system. 1 μg of RNA was used for cDNA synthesis using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) for pluripotency gene expression was performed using TaqMan reagents and SeV-specific probe-primer sets (Mr04269880_mr; Thermo Fisher Scientific). A subset of iPSC samples were cultured in T25 tissue culture flasks on MEF feeder cells and sent to Cell Line Genetics for cytogenetic analysis with G-banded karyotyping. An average of 20 cells per cell line were analyzed for chromosome integrity.
Differentiation of hPSCs into HLCs
Following the protocol of Cai et al. (2012), we used feeder-free differentiation conditions entailing the addition of a variety of growth factors and chemicals to the growth media. We incubated cells in (1) RPMI-B27 (RPMI-1640 from Thermo Fisher Scientific; 2% B-27 Supplement Minus Insulin from Thermo Fisher Scientific) medium supplemented with recombinant human/mouse/rat activin A (100 ng/mL; R&D Systems), recombinant human BMP-4 (10 ng/mL; Peprotech), and recombinant human FGF basic (20 ng/mL; R&D Systems) for 2 days, followed by 3 days of incubation in RPMI-B27 (minus insulin) supplemented with activin A alone, in ambient oxygen/5% CO2, yielding definitive endoderm; (2) RPMI-B27 (with insulin, i.e., made with 2% B-27 Supplement) supplemented with BMP-4 (20 ng/mL; PeproTech) and FGF basic (10 ng/mL) for 5 days in 5% oxygen/5% CO2, yielding hepatic progenitor cells; (3) RPMI-B-27 (with insulin) supplemented with recombinant human HGF (20 ng/mL; PeproTech) for 5 days in 5% oxygen/5% CO2, yielding immature HLCs; and (4) HCM Hepatocyte Culture Medium (Lonza) without EGF and supplemented with recombinant human oncostatin M (20 ng/mL; R&D Systems) for 5 days in ambient oxygen/5% CO2, yielding mature HLCs. A subset of HLC samples were assessed with DAPI and immunofluorescence with an HNF-4α antibody (Santa Cruz Biotechnology). RNA-seq data (see below) confirmed the expression of hepatocyte-specific genes in the HLC samples. ApoB, triglyceride, and albumin secreted into the media by HLCs were measured with the Human apoB ELISAPRO kit (Mabtech), Infinity Triglycerides Reagent (Thermo Fisher Scientific), and Human Albumin ELISA Quantitation Set (Bethyl Laboratories) according to the manufacturers’ instructions.
Primary human hepatocytes
Single vials containing 5 to 15 million viable primary human hepatocytes each from lots derived from individuals were obtained from Thermo Fisher Scientific (Human Plateable Hepatocytes, Metabolism Qualified). All were rated to survive in culture for at least three days after plating. The cells from each vial were thawed into Cryopreserved Hepatocyte Recovery Medium (Thermo Fisher Scientific), gently spun down, and then resuspended in William’s Medium E with added Hepatocyte Plating Supplement (fetal bovine serum, dexamethasone, and a cocktail solution of penicillin-streptomycin, bovine insulin, GlutaMAX, and HEPES) (Thermo Fisher Scientific) and plated at 400,000 cells/well in collagen-coated 24-well plates, according to the manufacturer’s instructions. After 6 hours, each well was refed with 500 μL maintenance medium [William’s Medium E with added Hepatocyte Maintenance Supplement (dexamethasone and a cocktail solution of penicillin-streptomycin, insulin, transferrin, selenium complex, BSA, linoleic acid, GlutaMAX and HEPES)] (Thermo Fisher Scientific). Each well was refed with 500 μL maintenance medium daily for the next 2 days. ApoB, triglyceride, and albumin secreted into the media by HLCs were measured as described above. For RNA-seq analyses, primary human hepatocytes from each vial were thawed and spun down as described above and then immediately processed for RNA isolation as described below.
Genotyping and quality assessments
Buffy coat DNA from donors was extracted using the QIAsymphony SP system (QIAGEN) and submitted for genotyping on the Infinium Human CoreExome-24 BeadChip (Illumina) at the Center for Applied Genomics of the Children’s Hospital of Philadelphia. After filtering for missing genotypes (>5%) and deviation from Hardy-Weinberg equilibrium, haplotypes were estimated by statistical phasing using SHAPEIT2 (Delaneau et al., 2011). Missing genotypes were imputed with IMPUTE2 (Howie et al., 2009) using the 1000 Genomes Project multi-ethnic panel (Phase 3). Post-imputation SNPs with a missingness rate >5% or deviation from the Hardy-Weinberg equilibrium with P < 1 × 10−6 were removed. SNPs that had minor allele frequency ≥ 5% remained in eQTL analyses, resulting in a total of 5,313,398 autosomal SNPs. We estimated the population substructure among individuals used in our study by principal component analysis (PCA) of genotypes using smartpca as implemented in the EIGENSOFT suite (Price et al., 2006). A combined dataset containing all 90 individuals with one or both cell types represented in our study was used to estimate PCs. Resulting genotype PC estimates were also used as covariates for RNA-seq analysis.
RNA-seq data generation and quality assessments
RNA was extracted from iPSCs, HLCs, and primary human hepatocytes using the miRNeasy Mini Kit (QIAGEN), and RNA integrity was assayed using the RNA 6000 Nano Kit (Agilent) on the Agilent Bioanalyzer system. All samples had an RNA integrity number (RIN) greater than 7.5. Libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina). Sequencing was performed at the Next Generation Sequencing Center of the University of Pennsylvania on Illumina HiSeq 2000/2500 systems with paired-end, 100-bp/125-bp read lengths with a target read depth of 50 million reads per sample. We generated and used whole transcriptome data (RNA-seq) for a total of 89 iPSC samples and 86 HLC samples. All individuals included had both iPSCs and HLCs generated for the study except for five individuals (one iPSC-only and four HLC-only).
Raw fastq files were assessed for quality measures using FastQC (Andrews, 2010). Adaptor trimming for Illumina paired-end libraries was applied using TrimGalore! (Krueger, 2012). We then aligned all remaining reads to the hg19/GRCh37 reference using the STAR aligner on the 2-pass mode (Dobin et al., 2013). We quantified the level of gene-level expression variation (transcripts per million; TPMs) using RSEM (Li and Dewey, 2011). We downloaded raw fastq files from GTEx (dbGaP Accession phs000424.v6.p1) and processed whole-liver sample data in an identical pipeline to iPSCs and HLCs. For additional tissue specificity assessments, we downloaded gene expression quantification files from the GTEx Portal (www.gtexportal.org). As data processing pipelines differed greatly between our samples and GTEx (Aguet et al., 2016), our use of the GTEx processed data was limited to quality control steps. To maximize compatibility and reduce batch effects between the two cohorts, we calculated TPMs from RPKMs provided by the GTEx Consortium with the following formula:
where m is all transcripts per given individual. Subsequently, TPMs across all individuals and all genes were scaled to normal distribution.
Assessment of pluripotency for generated iPSCs
We quantified pluripotency (or lack thereof, indicating successful differentiation) of iPSCs using differentiation marker expression levels as previously described (Carcamo-Orive et al., 2016). Of the total 24 differentiation markers, we used 16 markers with minimum estimated three log2 fold change (FC) across samples. Previously published human iPSC and ESC gene expression quantifications (Choi et al., 2013) were included as controls. We downloaded relevant TPM quantification matrices from the Gene Expression Omnibus (GEO) database (GEO Accession: GSE 73211). TPM quantifications from our cohorts and those of Choi et al. were quantile normalized per tissue, log2 transformed to estimate FC, and then normalized with respect to each other as a merged dataset. Eigenvalues were estimated using singular value decomposition (SVD) as implemented in the base R function svd() for the centered dataset. Variance contributed by each component was estimated as D2/ΣD2.
Gene expression profiling of iPSCs, HLCs, and GTEx livers
We analyzed the genome-wide gene expression profiles of the iPSCs, HLCs, and GTEx livers using SVD as described above for all genes expressed at minimum three log2 transformed fold changes across all samples included in our study. All gene expression quantifications were log2 transformed for fold change estimates then filtered to exclude genes with minimal FC < 3. The resulting gene expression matrices were then quantile normalized, wherein expression estimates of all genes per individual were ranked by magnitude and the ranks were transformed into normal distribution. Gene expression levels were scaled across all samples after merging the three datasets for analyses.
Differential gene expression among iPSCs, HLCs, and GTEx livers
For differential gene expression analyses, gene expression estimates (TPM) were log-transformed after the addition of a constant (1) to each expression estimate. The top 58% (27,446 genes) most highly expressed genes across cell types (mean expression in iPSCs, HLCs, primary hepatocytes, and whole livers) were used for genome-wide PCA using the pcaMethods R package (Stacklies et al., 2007).
Single-tissue eQTL analyses
The linear regression coefficients for genotypes associated with gene expression variation were estimated using Matrix eQTL (Shabalin, 2012). PEER factors were estimated from transcriptome-wide gene expression matrices for each cell types using the PEER R package (Stegle et al., 2010). We used four genotype-based PCs and 20 PEER factors as covariates, and all SNPs within 100 kb of the transcriptional start site (TSS) for each gene were considered. Standard errors (SE) for the linear regression coefficients of cis-eQTL effects were calculated as the proportion of the regression coefficient, β, and the test statistic. A local false discovery rate as implemented in the qvalue R package (Bass et al., 2015) was used to estimate multiple testing-corrected P-values. All cis-eQTLs for each sample type with minimum P < 0.05 were then compared across sample types. An eQTL SNP was considered genome-wide significant if it was associated with at least one gene in one sample type at a false discovery rate (FDR) < 5%. Any gene with at least one genome-wide significant cis-eQTL SNP was considered an eGene in this study. Additionally, we further investigated a total of 7,240 genome-wide significant SNPs that were previously reported to be associated with one or more blood lipid traits (Global Lipids Genetics Consortium, 2013); the results in GRCh37/hg19 annotations were downloaded from the GLGC website. In this study, these loci were referred to as the GLGC loci. We included all SNPs and genes with TSSs within 1 Mb of each sentinel GLGC SNP and used four genotype-based PCs as covariates in the linear model implemented in Matrix eQTL (Shabalin, 2012) as described above to estimate the effects of these SNPs.
Pathway analysis of HLC-specific eGenes
We interrogated all HLC- and iPSC-specific eGenes as determined by meta-analysis for enrichment of functional pathways or gene sets using ToppGene (Chen et al., 2009). Default parameters were used at a multi-testing correction threshold of Benjamini-Hochberg FDR < 5% and maximum of 2000 genes per set. We used 267 HLC-specific eGenes and 473 iPSC-specific eGenes for pathway analyses with the ToppFun tool implemented in the ToppGene Suite. A total of 19 HLC-specific eGenes and 52 iPSC-specific eGenes were excluded due to lack of matching IDs in the ToppFun database, leaving 248 HLC-specific and 421 iPSC-specific eGenes.
Allele-specific expression analyses of iPSCs and HLCs
Genotyped and imputed variants as described above were phased using SHAPEIT2 (Delaneau et al., 2011) for allele-specific expression (ASE) analyses. Haplotype phasing was performed based on reference haplotypes in the 1000 Genomes Project multi-ethnic panel (Phase 3) as described above for the genotype imputation. All variants without high phasing probability or missing 1000 Genomes references were excluded from ASE analyses. For allele-specific RNA-seq quantification, we used hg19/GRCh37 genome-based alignment of STAR for the second pass in the 2-pass mode (Dobin et al., 2013). Reference-mapping bias and potential PCR duplicates were removed with WASP (van de Geijn et al., 2015). Allele-specific read counts were quantified using SAMtools mpileup (Li et al., 2009). Single-cell-type ASE was quantified using QuASAR (Harvey et al., 2015), which quantifies ASE with a beta-binomial model, and differential ASE was quantified with Fisher’s exact test. False discovery rates were estimated using the method of Benjamini and Hochberg (1995).
Meta-analysis of iPSC and HLCs
Considering paired individuals among two datasets (iPSCs and HLCs), we further validated the tissue specificity of the iPSC and HLC cis-eQTLs by considering correlated data structure in the mixed effects model as implemented in Meta-Tissue (Sul et al., 2013). Three genotype PCs estimated from PCA of genotypes were used as covariates in mixed effect estimates calculations. As suggested by the developers, we subsequently used METASOFT (Han and Eskin, 2012) to estimate posterior probabilities (m-values) of tissue specificity incorporating correlations estimated by Meta-Tissue. In order to distinguish between iPSC and HLC eGenes identified via single tissue eQTL analyses, we used the m-value threshold of 0.9, as recommended by the developers, to determine the significance of the predicted cell-type effect. An eQTL-eGene pair was considered strongly associated with both cell types if m-value ≥ 0.9 in both cell types. Accordingly, an eQTL-eGene pair was considered HLC-specific if m-value ≥ 0.9 for HLCs and m-value < 0.1 for iPSCs. An eQTL-eGene pair was considered iPSC-specific if it had m-value ≥ 0.9 for iPSCs and m-value < 0.1 for HLCs. All other eQTL-eGene pairs with m-value < 0.9 and single-tissue FDR < 0.05 were considered eGenes for the relevant tissue without predicted tissue-specific effects. Due to potential biases in combining paired samples among iPSCs and HLCs with the independent set of GTEx liver samples, meta-analysis was not performed across the three datasets. However, we further assessed predicted tissue specificity of iPSC- and HLC-specific eQTL-eGene pairs by comparing to the list of GTEx liver eQTL-eGene pairs post hoc.
Colocalization analysis of functionally validated genes
We used the R package coloc (Giambartolomei et al., 2014) to assess statistical evidence of colocalization between each of four GLGC traits (LDL-C, HDL-C, TG, and TC) and each of eight top GLGC HLC eGenes (ANGPTL3, CPNE1, FRK, FUT2, IGF2R, SYPL2, UBE2L3, and VKORC1). We used P-values obtained from single-tissue eQTL analyses to calculate approximate Bayes factors as implemented in coloc. We estimated credible sets from these associations at 95% confidence as suggested previously (Wellcome Trust Case Control Consortium, 2012). We used minor allele frequencies estimated by the 1000 Genomes Project (Phase 3). All SNPs were compared and matched for genomic positions in GRCh37/hg19 coordinates and for reference and alternate alleles between the GWAS and eQTL summary statistics. Any mismatching SNPs were: (1) flipped to match strands; (2) allele-switched in cases where the effect allele of the GWAS SNP was not the effect allele of the corresponding eQTL SNP, with the corresponding beta estimate multiplied by −1; or (3) excluded if we could not resolve the discrepancies.
Massively parallel reporter assays
MPRA experiments were performed as previously described (Melnikov et al., 2012). Approximately 240,000 145-bp oligonucleotides representing ~11,000 distinct “tiles” with the major or minor alleles of 1,837 candidate functional variants (including 525 candidate functional variants in the CPNE1, ANGPTL3, and FRK loci) in the center, shifted by 40 nt towards the 5′ end (right-shifted), or shifted by 40 nt towards the 3′ end (left-shifted) and coupled to distinguishing barcodes were generated by microarray-based DNA synthesis (Agilent). To minimize barcode- and amplification-associated biases, each tile was coupled to ~22 distinct barcodes. Two common restriction sites separated the tiles and barcodes. The oligonucleotides were PCR amplified using universal primer sites and directionally cloned into a pMPRA1 (Addgene plasmid #49349) backbone using Gibson assembly. A minimal promoter-firefly luciferase segment from pMPRAdonor2 (Addgene plasmid #49353) was inserted between the tiles and barcodes via double digestion and directional ligation. The resulting reporter plasmid pools were co-transfected into NIH 3T3 fibroblasts using FuGENE 6 (Promega). Two independent, biological replicate MPRA experiments were performed. The relative enhancer activities of the different tiles were calculated by comparing the corresponding barcodes from the cellular mRNA and the transfected plasmid pool.
CRISPR-Cas9 plasmid construction
Guide RNAs were designed by manual inspection of the genomic sequences flanking rs2277862, rs10889356, and rs10872142 and evaluated for potential off-target activity using the CRISPR Design tool at http://crispr.mit.edu. Protospacers were cloned into the BbsI site of pGuide (Addgene plasmid #64711) via the oligonucleotide annealing method, and, if not already present, a G was added to the 5′ end to facilitate U6 polymerase transcription. Genome editing was performed using pCas9_GFP (Addgene plasmid #44719), which co-expresses a human codon-optimized Cas9 nuclease and GFP via a viral 2A sequence. To generate the targeting construct for minor allele knock-in at rs10889356, 500-bp homology arms flanking a TTAA site 200 bp upstream of rs10889356 were amplified from genomic DNA (from the H7 hPSC line)—with the 3′ arm undergoing PCR-based mutagenesis to introduce the rs10889356 minor allele—and subcloned into the PB-MV1Puro-TK vector (Transposagen), which harbors a piggyBac transposon containing a puromycin selection cassette, to make PB-rs10889356. For CRISPRi studies, pAC154-dual-dCas9VP160- sgExpression (gift from Dr. Rudolph Jaenisch, Addgene plasmid #48240), a dual expression construct that expresses dCas9-VP160 and sgRNA from separate promoters, was modified by PCR-based methods to remove the VP160 domain and include a viral 2A sequence and GFP after dCas9. Additionally, the gRNA sequence was modified to include a 5-bp hairpin extension, which improves Cas9-gRNA interaction, and a single-basepair substitution (A-U flip) that removes a putative Pol III terminator sequence, as described previously (Chen et al., 2013).
Cultured cell line transfection
HEK 293T and HepG2 cells (CRISPRi experiments) and NIH 3T3 cells (testing of gRNAs) were seeded into 6-well plates and transfected 24 hours later using Lipofectamine 3000 (Thermo Fisher Scientific) or TransIT-2020 Reagent (Mirus Bio) according to the manufacturer’s instructions.
CRISPR-Cas9 targeting of hPSCs
For electroporation, cells in a 60–70% confluent 10-cm plate were dissociated into single cells with Accutase, resuspended in PBS, and combined with 25 μg pCas9_GFP and 25 μg gRNA plasmid (or 12.5 μg of two different gRNA plasmids, for multiplexed targeting) in a 0.4 cm cuvette. For knock-in, 15 μg pCas9_GFP, 15 μg gRNA plasmid, and either 30 μg ssODN (Integrated DNA Technologies; for rs2277862, 5′-GTGGTGGCTATAGAATCGGTTTTCCAGATCAATGTGGGTCTCCCCGATGAGGTCGTCAGAACCCACGAGGTCATGATCAAATATGGCGACCGTCAGCTCCGTCTCAGCTGGGAGAGAGATGCTTAGCTCCAGGATCCTGGCAAGGAGGGAGAGGGACTGAGGTCACT-3′; for rs10872142, 5′-AGAGAACTAAGAAAGAAAACTCCAATGTGCAAGTGCTTTTTAAGTCTCCACTTTTGTCACATTTGCAACTGTCCCATTGGCCAAAGCACGTGATTTGGCAAAGCCAAGAATGAGTGTGGGAGGAGACTACCCCAAGAATGCAGATGCAAGGAAGCACAAAAAGATGGGACAGTTACTACAACTTACACACCAC-3′) or 30 μg PB-rs10889356 targeting vector were used instead. A single pulse was delivered at 250 V/500 μF (Bio-Rad Gene Pulser), and the cells were recovered and plated in mTeSR1 with 0.4 μM ROCK inhibitor (Y-27632, Cayman Chemical). In most cases, cells were dissociated with Accutase 48 hours post-electroporation, and GFP-positive cells were isolated by FACS (FACSAriaII, BD Biosciences) and replated onto 10-cm Geltrex-coated plates (15,000 cells/plate) with conditioned medium and 0.4 μM ROCK inhibitor to facilitate recovery; in the case of targeting with PB-rs10889356, cells were instead selected with 1 μg/mL puromycin (Sigma) for 7 days starting 24 hours post-electroporation. After expansion and screening for the desired recombinants, PB-rs10889356-targeted clones were pooled and electroporated with excision-only piggyBac transposase (PBx) expression vector (Transposagen) in the same way as described above.
Isolation and screening of clonal hPSC populations
Following FACS or puromycin selection, single cells were permitted to expand for 7–14 days to establish clonal populations. Colonies were manually picked and replated into individual wells of a 96-well plate. Once the wells reached 80–90% confluence, cells were dissociated with Accutase and split at a 1:3 ratio to create a frozen stock and two working stocks that were maintained in culture. For genomic DNA isolation, cells from one of the working stocks were lysed in 50 μL lysis buffer (10 mM Tris pH 7.5, 10 mM EDTA, 10 mM NaCl, 0.5% Sarcosyl) with 40 μg/mL Proteinase K (New England BioLabs) for 1–2 hours in a humidified incubator at 56°C. Genomic DNA was precipitated by addition of 100 μL 95% ethanol with 75 mM NaCl, followed by incubation at −20°C for 2 hours. Precipitated DNA was washed three times with 70% ethanol, resuspended in 30–50 μL TE buffer with 0.1 mg/mL RNase A (Thermo Fisher Scientific), and allowed to dissolve at room temperature overnight. hPSC clones were screened by PCR amplification of a small region surrounding the targeted site using BioReady rTaq DNA Polymerase (Bulldog Bio) and the following cycling conditions: 94°C for 5 min, [94°C for 30 sec, 54–56.5°C for 30 sec, 72°C for 30 sec] × 40 cycles, 72°C for 5 min. The following primer pairs were used: for rs2277862, F: 5′-TGCTGGACCCACACTTCATA-3′ and R: 5′-CTCAGTCCCTCTCCCTCCTT-3′; for rs10889356, F: 5′-CCATTAGGTCACTTGCCAGA-3′ and R: 5′-ACAGGGGGATTCTGTCTAAAA-3′; for rs10872142, F: 5′-GCTGATCTTAGCTGGGCTTG-3′ and R: 5′-TTTGTGCTTCCTTGCATCTG-3′. PCR amplicons were separated on a high-percentage agarose gel, and clones with indels were identified based on size shifts relative to the wild-type band. Clones were confirmed by Sanger sequencing of the PCR products. Multiple mutant clones were retrieved from the frozen stock, or if possible, from the second working stock and expanded for experiments. Additionally, several clones that underwent the targeting procedure but remained genetically wild-type at the intended site were expanded as controls.
Generation of viruses
To generate AAVs, plasmids containing cDNAs of the mouse Vkorc1, Ube2l3, Acaa2, Cpne1, and Ergic3 genes were commercially obtained (GE Dharmacon). The nucleotide sequences of the cDNA clones used in this study can be accessed using the following identifiers: Vkorc1 (BC031732), Ube2l3 (BC093503), Acaa2 (BC028901), Cpne1 (BC057554), Ergic3 (BC043720). Coding sequences were subcloned into the pENN.AAV.TBG.PI.rBG vector (Penn Vector Core), and AAV serotype 8 particles expressing the candidate genes under the control of the liver-specific TBG promoter were prepared by the Penn Vector Core at the University of Pennsylvania. Empty vectors were used as controls.
Mouse liver overexpression studies
Groups of 7 male mice at ~8 weeks of age were used for AAV8 expression studies. Mice received 1 × 1012 viral particles via intraperitoneal injection. Blood was collected prior to injection and at 2 weeks post-injection via retro-orbital bleed, following a 4-hour fast. Mice were sacrificed at 2 weeks post-injection and livers were collected. Gene overexpression was confirmed by qRT-PCR, as described below. Plasma triglyceride, HDL cholesterol, and ALT levels were measured on individual samples using analytical chemistry (Infinity reagents; Thermo Fisher Scientific) on a Cobas Mira Autoanalyzer (Roche Diagnostic Systems). ALT levels were checked to confirm that the lipid phenotypes were not influenced by liver damage (data not shown).
CRISPR knock-in and knockout mice
For targeting of rs27324996, candidate guide RNAs with a predicted cleavage site near rs27324996 were designed by manual inspection, and the corresponding protospacers were cloned into the pGuide plasmid as described above. Each gRNA plasmid was co-transfected with pCas9_GFP into mouse NIH 3T3 cells using TransIT-2020 Reagent (Mirus Bio) according to the manufacturer’s instructions. Two days post-transfection, GFP-positive cells were isolated by FACS, and genomic DNA was isolated using the DNeasy Blood & Tissue Kit (QIAGEN). The region flanking rs27324996 was PCR amplified (F: 5′-TGGGAATGGCTTCTTAGGGC-3′ and R: 5′-CATCCCCAAGCAACTCAACC-3′) using AccuPrime Taq DNA Polymerase (Thermo Fisher Scientific) with the following cycling conditions: 94°C for 2 min, [94°C for 30 sec, 55°C for 30 sec, 68°C for 30 sec] × 40 cycles, 68°C for 5 min. PCR products were purified using the DNA Clean & Concentrator kit (Zymo Research) and analyzed for the presence of indels using the Surveyor Mutation Detection Kit (Integrated DNA Technologies) according to the manufacturer’s instructions. CEL I nuclease-treated PCR products were resolved on a 1.5% agarose gel to detect mutagenesis activity. The gRNA sequence exhibiting the highest mutation rate was PCR amplified, and the purified PCR product was used as a template for in vitro transcription using the MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific). The transcribed RNA was purified by phenol/chloroform extraction, ethanol precipitated, and resuspended in injection buffer (5 mM Tris-HCl pH 7.6, 0.1 mM EDTA). The Genome Modification Facility at Harvard University performed one-cell embryo injections. Superovulated C57BL/6J females were mated with C57BL/6J males, and fertilized embryos were harvested from the oviducts. One-cell embryos were injected with a mixture of 100 ng/μL Cas9 mRNA (TriLink BioTechnologies), 50 ng/μL gRNA, and 100 ng/μL ssODN (5′-AGCCCACAGTTGGCTCTGTGGTGGCTATAGAATCTGTTTTCCAGGTCAATGTGGGTCTCCCCGATGAGGTCATCTGAACCCACGAGGTCATGATCAAATATGGCGACCGTCAGCTCTGGCTGGGCTGGGAGGGAGACGCTCAGCTCCAGGACCCTGGGCAGGAAGGGAAATTGACTAACCACAGCTCCATGCCCTCAGAG-3′). Injected embryos were implanted into the uteri of pseudopregnant foster mothers. DNA was prepared from tail biopsies of 3-week-old founder mice by the hot hydroxide method, and genotyping was performed with the same PCR primers and cycling conditions used for the CEL I nuclease assay. Positive founders were identified by Sanger sequencing of PCR products. The single positive founder was bred to a wild-type C57BL/6J mouse, and the resulting progeny were intercrossed for one to two generations to breed the knock-in allele to homozygosity. Genotyping of progeny was performed in the same manner. Wild-type and homozygous knock-in littermates of both sexes from several litters, ~12 weeks of age, were used for gene expression analyses.
The generation of Angptl3 knockout mice was performed essentially as described above, except that a mix of two gRNAs and no ssODNs were used for embryo injections. gRNAs with predicted cleavage sites flanking the entire Angptl3 gene (protospacers 5′-AATTACTAAGAGTGTGACTC-3′ and 5′-TAATGCCAATCCACGAGCAT-3′) were used to cleanly delete the gene (~9.1 kb). PCR amplification around the junction of the predicted cleavage sites was used to confirm the deletion (F: 5′-TGCAGCTATCCCAATGAATGAG-3′ and R: 5′-AGAGAAACGACACCCTTCAC-3′). Wild-type and homozygous knockout littermates of both sexes from several litters, ~8 weeks of age, were used for plasma lipid measurements using Infinity Triglycerides Reagent (Thermo Fisher Scientific) and Infinity Cholesterol Reagent (Thermo Fisher Scientific).
Quantitative reverse transcriptase-polymerase chain reaction
Snap-frozen livers were used for RNA preparation with either the QIAsymphony RNA Kit on the QIAsymphony SP system (QIAGEN) or homogenization in TRIzol Reagent (Thermo Fisher Scientific). Wells of cells were washed with ice-cold PBS and lysed directly in TRIzol Reagent. RNA isolation according to the manufacturers’ instructions was followed by reverse transcription to cDNA using the First-Strand cDNA Synthesis Kit (GE Healthcare), the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific), or SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific) with an equimolar mixture of random hexamers and oligo-dT. Gene expression was measured using the following TaqMan Gene Expression Assays along with TaqMan Gene Expression Master Mix (Thermo Fisher Scientific): Mm00624282_m1 for Acaa2, Hs00537765_m1 for CPNE1, Hs00211070_m1 for ERGIC3, Mm00467970_m1 for Cpne1, and Mm00499400_m1 for Ergic3, Hs00290630_m1 for DOCK7, Hs00205581_m1 for ANGPTL3, and Hs00176619_m1 for FRK. Human B2M (Assay ID 4326319E), human ACTB (Assay ID 4310881E), or mouse Actb (Assay ID 4352341E) was used as the reference gene as appropriate. For Vkorc1, the primers F: 5′-GCCCTCTCACTGTACGCACT-3′ and R: 5′-CCCACCGAGAGGAGAAGAC-3′ were used with SYBR Green (Thermo Fisher Scientific). For Ube2l3, the primers F: 5′-GGCTGATGAAGGAGCTTGAA-3′ and R: 5′-CAGGAACAATAAGCCCTTGC-3′ were used with SYBR Green. Each 10 μL qPCR reaction contained 1 μL cDNA (diluted 1:3 with water) and was performed in technical duplicate or triplicate. Reactions were carried out on the ViiA 7 Real-Time PCR system or the 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific). Relative expression levels were quantified by the 2−ΔΔCt method.
QUANTIFICATION AND STATISTICAL ANALYSIS
All details related to the statistical parameters for the reported experiments are included in the figure legends. For the hPSC experiments, CRISPRi experiments, and mouse studies, the average gene expression levels or changes in lipid levels were compared between groups using the two-tailed Welch’s t-test, due to the groups having unequal sizes in some experiments. Statistical analyses regarding these functional studies were performed using GraphPad Prism 6 for Mac OS X.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The RNA-seq and genotype data generated as a part of this study have been deposited in dbGaP (Study ID: 15979).
Supplementary Material
Table S1. Characteristics of population cohort, iPSCs, and HLCs, Related to Figure 1
Table S10. Oligonucleotides used for this study. Related to STAR Methods.
Table S2. Gene ontology enrichment analysis for differentially expressed genes in both HLCs and livers, Related to Figure 1
Table S3. Summary statistics of genome-wide significant cis-eQTLs identified in HLCs, iPSCs, and GTEx livers, Related to Figure 1
Table S4. Summary statistics of genes with both significant differential allele-specific expression variation between iPSCs and HLCs and genome-wide significant eQTLs, Related to Figure 1
Table S5. Summary statistics of genome-wide meta-analysis of cis-eQTLs in iPSCs and HLCs, Related to Figure 1
Table S6. Summary of pathway analysis for HLC-specific eGenes, Related to Figure 1
Table S7. Summary statistics of genome-wide significant cis-eQTLs identified in HLCs, iPSCs, and GTEx livers at GLGC loci, Related to Figure 2
Table S8. Colocalization of SNP-gene associations, Related to Figure 2
Table S9. Results of MPRA experiments with SNPs in CPNE1, ANGPTL3, and FRK loci, Related to Figure 2
Acknowledgments
We are grateful to the participants of the study, the staff of the University of Pennsylvania Clinical Translational Research Center, and the staff of the iPSC Core Facility of the Institute for Regenerative Medicine at the University of Pennsylvania. This work was supported in part by the Howard Hughes Medical Institute Medical Research Fellows Program (A.R.); grant R01-GM104464 from the United States National Institutes of Health (NIH) (K.M. and T.S.M.); grants R01-HL118744 and R01-DK099571 from the NIH (K.M.); the Harvard Stem Cell Institute (K.M.); grant U01-HG006398 from the NIH (D.J.R., E.E.M., and S.A.D.); RC2-HL101864 from the NIH (D.J.R.); grant UL1-TR000003 from the NIH/NCATS (D.J.R.); and R01-MH101822 from the NIH (C.D.B.). E.P. is now an employee of Pfizer, P.R. is now an employee of Neon Therapeutics, I.M.S. is now an employee of Xenon Therapeutics, and T.S.M. is now an employee of 10X Genomics. The other authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
E.E.P., Y.P., X.W., A.R., W.Y., D.A., D.T.P., J.A., M.H., N.K., W.L., Z.L., Y.L., W.L., S.L.L.-G., D.M., P.R., J.S., K.J.S., I.M.S., L.W., R.Y., X.Z., C.D.B., and K.M. carried out experimental work and/or performed data analyses. W.Y., S.K., S.A.D., T.S.M., E.E.M., D.J.R., C.D.B., and K.M. supervised various aspects of the study. D.J.R., C.D.B., and K.M. conceived and designed the study. E.E.P., Y.P., C.D.B., and K.M. wrote the manuscript.
References
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010 Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Aguet F, Brown AA, Castel S, Davis JR, Mohammadi P, Segre AV, Zappala Z, Abell NS, Fresard L, Gamazon ER, et al. Local genetic effects on gene expression across 44 human tissues. bioRxiv. 2016 doi: https://doi.org/10.1101/074450.
- Bass AJ, Storey JD, Dabney A, Robinson D. qvalue: Q-value estimation for false discovery rate control. 2015 Available online at: http://github.com/jdstorey/qvalue.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- Burkhardt R, Toh SA, Lagor WR, Birkeland A, Levin M, Li X, Robblee M, Fedorov VD, Yamamoto M, Satoh T, et al. Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J Clin Invest. 2010;120:4410–4414. doi: 10.1172/JCI44213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, DeLaForest A, Fisher J, Urick A, Wagner T, Twaroski K, Cayo M, Nagaoka M, Duncan SA. StemBook [Internet] Cambridge, Massachusetts: Harvard Stem Cell Institute; 2012. Protocol for directed differentiation of human pluripotent stem cells toward a hepatocyte fate. [PubMed] [Google Scholar]
- Carcamo-Orive I, Hoffman GE, Cundiff P, Beckmann ND, D’Souza SL, Knowles JW, Patel A, Papatsenko D, Abbasi F, Reaven GM, et al. Analysis of transcriptional variability in a large human iPSC library reveals genetic and non-genetic determinants of heterogeneity. Cell Stem Cell. 2016 doi: 10.1016/j.stem.2016.11.005. http://dx.doi.org/10.1016/j.stem.2016.11.005. [DOI] [PMC free article] [PubMed]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Lee S, Mallard W, Clement K, Tagliazucchi GM, Lim H, Choi IY, Ferrari F, Tsankov AM, Pop R, et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Eskin E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012;8:e1002555. doi: 10.1371/journal.pgen.1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CT, Moyerbrailean GA, Davis GO, Wen X, Luca F, Pique-Regi R. QuASAR: quantitative allele-specific analysis of reads. Bioinformatics. 2015;31:1235–1242. doi: 10.1093/bioinformatics/btu802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, Ramirez J, Liu W, Lin YS, Moloney C, et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet. 2011;7:e1002078. doi: 10.1371/journal.pgen.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, Horikoshi H, Furukawa H. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30:151–157. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- Krueger F. Trim Galore! 2012 Available online at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Jr, Kinney JB, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis Model Mech. 2013;6:896–904. doi: 10.1242/dmm.012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010a;363:2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010b;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal component analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature. 2016;533:95–99. doi: 10.1038/nature17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacklies W, Redestig H, Scholz M, Walther D, Selbig J. pcaMethods–a bioconductor package providing PCA methods for incomplete data. Bioinformatics. 2007;23:1164–1167. doi: 10.1093/bioinformatics/btm069. [DOI] [PubMed] [Google Scholar]
- Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol. 2010;6:e1000770. doi: 10.1371/journal.pcbi.1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul JH, Han B, Ye C, Choi T, Eskin E. Effectively identifying eQTLs from multiple tissues by combining mixed model and meta-analytic approaches. PLoS Genet. 2013;9:e1003491. doi: 10.1371/journal.pgen.1003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016;165:1519–1529. doi: 10.1016/j.cell.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell. 2016;165:1530–1545. doi: 10.1016/j.cell.2016.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B, McVicker G, Gilad Y, Pritchard JK. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet. 2012;44:1294–1301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Mills JA, Sullivan S, Liu Y, French DL, Gadue P. StemBook [Internet] Cambridge, Massachusetts: Harvard Stem Cell Institute; 2012. iPSC reprogramming from human peripheral blood using Sendai virus mediated gene transfer. [PubMed] [Google Scholar]
- Zhu H, Lensch MW, Cahan P, Daley GQ. Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet. 2011;12:266–275. doi: 10.1038/nrg2951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of population cohort, iPSCs, and HLCs, Related to Figure 1
Table S10. Oligonucleotides used for this study. Related to STAR Methods.
Table S2. Gene ontology enrichment analysis for differentially expressed genes in both HLCs and livers, Related to Figure 1
Table S3. Summary statistics of genome-wide significant cis-eQTLs identified in HLCs, iPSCs, and GTEx livers, Related to Figure 1
Table S4. Summary statistics of genes with both significant differential allele-specific expression variation between iPSCs and HLCs and genome-wide significant eQTLs, Related to Figure 1
Table S5. Summary statistics of genome-wide meta-analysis of cis-eQTLs in iPSCs and HLCs, Related to Figure 1
Table S6. Summary of pathway analysis for HLC-specific eGenes, Related to Figure 1
Table S7. Summary statistics of genome-wide significant cis-eQTLs identified in HLCs, iPSCs, and GTEx livers at GLGC loci, Related to Figure 2
Table S8. Colocalization of SNP-gene associations, Related to Figure 2
Table S9. Results of MPRA experiments with SNPs in CPNE1, ANGPTL3, and FRK loci, Related to Figure 2