

Abstract
Mutations in the HFE (hemochromatosis) gene cause hereditary hemochromatosis, an iron overload disorder that is hallmarked by excessive accumulation of iron in parenchymal organs. The HFE mutation p.Cys282Tyr is pathologically most relevant and occurs in the Caucasian population with a carrier frequency of up to 1 in 8 in specific European regions. Despite this high prevalence, the mutation causes a clinically relevant phenotype only in a minority of cases. In this review, we summarize historical facts and recent research findings about hereditary hemochromatosis, and outline the pathological consequences of the associated gene defects. In addition, we discuss potential advantages of HFE mutations in asymptomatic carriers.
Introduction
Iron plays a key role in various physiological pathways. All cells of the human body contain iron as an integral part of FeS-proteins. These are essential for oxidation-reduction reactions that occur, for example, in the mitochondrial respiratory chain where biochemical energy is generated. In red blood cells, iron binds oxygen in the hemoglobin molecule to enable its transport from respiratory organs throughout the human body. Sufficient amounts of serum iron are mainly ensured by macrophages, which recycle approximately 25 mg of iron a day from aging or damaged erythrocytes. Iron losses resulting from bleeding or skin desquamation are compensated for by dietary iron absorption, which amounts to approximately 1–2 mg/day.1 Duodenal iron absorption is a highly regulated process. If duodenal iron uptake exceeds the requirements for physiological iron consuming processes, such as erythropoiesis, excess iron accumulates in parenchymal organs, including the liver, heart, and pancreas. Iron overload causes oxidative damage in these organs and, ultimately, the iron-overload disease hereditary hemochromatosis (HH) may develop when genetic and environmental risk factors are present.2
Ever since it was first described in the 19th century, scientists and clinicians have been increasing their efforts to unravel the causes and consequences of HH. Modern research has been successful in identifying HH-causing gene mutations and unraveling molecular mechanisms responsible for elevated dietary iron uptake. HH is mainly caused by mutations in the HFE (hemochromatosis) gene. The most pathologically influential HFE mutation, p.Cys282Tyr (C282Y), is frequently inherited in a heterozygous state (overall carrier frequency approx. 1:16) in the Caucasian population, but accounts for a phenotype only in a minority of cases.3 This observation has led to the hypothesis that HFE mutations may confer a genetic advantage to asymptomatic hemochromatosis ‘patients’ and have, therefore, continued to spread.4
A history of hereditary hemochromatosis
Hemochromatosis was first described in the mid-1800s by French physicians who referred to the disease as “bronze diabetes” and “pigmented cirrhosis”.5–7 A few years later, the German pathologist Friedrich Daniel von Recklinghausen linked the syndrome to iron metabolism after he had observed an excess of iron in the tissues of patients and introduced the present-day term hemochromatosis.8 In those days, scientists and physicians believed that the syndrome was exclusively caused by environmental factors and conditions such as diabetes, alcohol abuse, metabolic disturbances, or infections. The English gerontologist Sheldon rejected this hypothesis in 1935 and postulated that hemochromatosis was a hereditary disease.9 This assumption was experimentally verified in the 1970s in a study that associated hemochromatosis with the HLA (human leukocyte antigen) gene locus and identified it as an autosomal recessive disease that mainly affects men.10
The discovery of the intracellular iron storage protein ferritin11 and the iron-binding blood plasma protein transferrin12 in the first half of the 20th century gave researchers the first molecular hints about the biological mechanisms regulating iron metabolism. Those and subsequent mechanistic studies have provided an understanding of the molecular processes involved in hemochromatosis. Twenty years ago, the genetic factor responsible for HLA-linked HH was finally identified as a mutation in the gene encoding the non-classical MHC class I-like molecule HFE.13 Subsequently, rare non-HFE-related HH subtypes were discovered, hallmarked by mutations in newly identified iron-related proteins such as hemojuvelin (HJV; hemochromatosis type IIA),14 hepcidin, the master regulator of systemic iron homeostasis, (HAMP; hemochromatosis type IIB),15 transferrin receptor 2 (TfR2; hemochromatosis type III),16 or ferroportin (FPN or SLC40A1; type IV or ferroportin disease).16,17
Large-scale population studies have revealed that most HFE mutation carriers with a mild iron overload-phenotype lack any clinically relevant disease symptoms.19 This has led to a debate about the potential benefits of HFE gene defects and has been motivating the scientific community to identify potential advantages of these mutations.
The molecular and genetic causes of hereditary hemochromatosis
The iron-regulated hormone hepcidin is of central importance to the pathogenesis of HH. It is mainly produced in hepatocytes and secreted into the blood stream. It binds to and degrades its target receptor, the iron exporter ferroportin, to inhibit iron release from duodenal enterocytes, iron-recycling macrophages, and hepatocytes. Any defect that ultimately impairs hepcidin function and inhibits the hormone’s ability to monitor and regulate serum iron levels may provoke an iron overload phenotype. In HH patients, hepcidin levels are reduced, causing excess ferroportin-mediated iron export and, as a consequence, increased dietary iron uptake and release from macrophages.20 Elevated iron levels in the blood stream subsequently lead to the saturation of the binding capacity of the iron transporter transferrin. Above a transferrin saturation of approximately 75%21 highly reactive, non-transferrin bound iron species (especially labile plasma iron, LPI) appear in the blood, which will preferentially be taken up by parenchymal cells of the liver, the pancreas and other organs,22 and ultimately provoke an HH phenotype.
Mutations in either the hepcidin gene itself, in genes affecting upstream activators of hepcidin expression (HFE, TFR2, HJV), or in ferroportin (the iron exporter which acts as the hepcidin receptor) can cause different classes and subtypes of HH20 (Figure 1). Most cases of HH are due to mutations in the HFE gene. The most prevalent disease-causing HFE mutation in the general population is the 845G polymorphism, which causes a p.Cys282Tyr amino acid substitution (C282Y) in the HFE protein. Today, approximately 0.4% of Caucasians carry a homozygous23,24 and approximately 6% a heterozygous HFE C282Y mutation.24 HFE is a non-classical major histocompatibility (MHC) protein13 located on the cell surface. The C282Y mutation disrupts the formation of a disulfide bond in the HFE protein and impairs its capability to bind β2-microglobulin.25 As a consequence, HFE is unable to reach the cell surface and aggregates intracellularly.25,26 This causes impaired signaling leading to reduced hepcidin mRNA expression, decreased plasma hepcidin levels, and excessive systemic iron accumulation in adults (aged over 40 years).20 The molecular mechanism by which HFE regulates hepcidin expression is not yet fully understood. It has been proposed that HFE plays a regulatory role in the sensing of serum TF-Fe concentrations, involving protein-protein interactions between HFE, the ubiquitous transferrin receptor TfR1 and the liver-specific transferrin receptor TfR2. High concentrations of iron-bound transferrin dissociate HFE from TfR127,28 which have been found to strengthen the interaction between HFE and Tfr2.29 Increased iron levels further stimulate the expression of BMP6 (bone morphogenetic protein 6), a member of the TGFβ superfamily, whose genetic impairment causes severe iron overload in mice30,31 and in patients.32 BMP6 binds to the GPI-anchored receptor hemojuvelin (HJV) at the cell surface,33 as well as to type 1 (ALK2 and ALK334) and type 2 (BMPR2 and ACTR2a35) serine threonine kinase receptors. As a result, SMAD1/5/8 is phosphorylated and binds to SMAD4, which translocates to the nucleus to induce hepcidin transcription.36 Stimulation of hepcidin expression by BMP6 has recently been reported to also take place in an HJV-independent manner.37 HFE has been found to prevent the ubiquitination and proteasomal degradation of the BMP6-receptor ALK3,38 as well as to engage in a tertiary complex with the BMP6-receptor Hjv and TfR2.39 These observations suggest a role of HFE in BMP/SMAD signaling and provide a first mechanistic explanation for the impairment of BMP/SMAD signaling in patients with HFE-hemochromatosis.40
Figure 1.

Hereditary hemochromatosis (HH). Three different HH classes (HFE-related HH, non-HFE-related HH, and ferroportin disease) are subcategorized into different subtypes (I, II A + B, III and IV) depending on mutations in iron-related genes (HFE, HJV, HAMP, TFR2, FPN/SLC40A1) and related pathophysiology. FD: ferroportin disease.
HFE gene sequencing approaches have identified additional HFE mutations with different pathological impact. These include the amino acid alteration S65C, which is not considered clinically meaningful,20,41,42 or H63D, which may, in rare cases, contribute to abnormal iron parameters in H63D/C282Y compound heterozygote individuals.43–45 By contrast, individuals carrying one H63D and one healthy allele are asymptomatic as long as no additional risk factors are present.41 In addition, researchers discovered a deletion (p.Y231del) in an Huh7 hepatoma cell line derived from a Japanese HH patient46 which prevents HFE cell surface expression. The identical mutation has more recently been discovered in another Japanese patient,47 showing, for the first time, that HFE-associated HH can also occur in Asians. Furthermore, a few Sardinian individuals show deletions of the entire HFE gene.48,49 Additional HFE mutations have been detected to influence iron levels when co-inherited with heterozygous C282Y mutations, such as the p.Arg226Gly (R226G) mutation50 or the nonsense mutations HFE-Brianza and HFE-Ossola, termed after the Italian provinces they were detected in.51 Although the prevalence of such mutations is low, they show that the presence of genetic factors may contribute to the clinical manifestation of hemochromatosis in C282Y heterozygotes.
Non-HFE-hemochromatosis (commonly referred to as hemochromatosis type II to IV) is much rarer than HFE-HH. In contrast to HFE-hemochromatosis, it also occurs in individuals of non-European descent, in both adult and juvenile onset forms.52 Mutations causing non-HFE-HH are detected in genes encoding hepatocytic membrane proteins [HJV (hemojuvelin), TFR2], which play a role in the monitoring of iron levels and signaling to hepcidin (see above), or HAMP itself. Mutations in HJV14 and HAMP15 cause juvenile non-HFE-HH forms (HH types II A and II B) which are characterized by very low circulating hepcidin levels. Patients usually develop a severe iron overload phenotype before the age of 30 years, including cardiovascular, liver, and endocrine complications.53 Adult onset non-HFE-HH forms (HH types III and IV) are either caused by mutations in TFR2 encoding transferrin receptor 2,16 or the hepcidin receptor ferroportin (FPN/SLC40A1), generally referred to as HH type IV.55 Gain-of-function mutations within ferroportin confer resistance to hepcidin binding and thus prevent ferroportin internalization and degradation (HH type IV, classical ferroportin disease with gain-of-function mutation). As a consequence, uncontrolled iron export from cell types expressing ferroportin, such as duodenal enterocytes or macrophages, causes high levels of iron overload.56 By contrast, loss-of-function mutations in ferroportin diminish the ability of ferroportin for iron export (HH type IV, classical ferroportin disease with loss-of-function mutations), which is characterized by iron accumulation in macrophages of the spleen and the liver, and is associated with low serum iron levels.2
Symptoms, diagnosis and treatment of HH
Elevated liver enzymes and/or iron parameters indicative of iron overload (serum ferritin and transferrin saturation levels) usually precede a symptomatic manifestation of hemochromatosis.20 Individuals with elevated body iron levels despite fully functional erythropoiesis are thus commonly diagnosed with hemochromatosis, regardless of whether or not they show disease symptoms.57 Genetic and biochemical screening of the general population for HFE mutations has been discussed but is currently not recommended because of its high costs and the low penetrance of the disease.20,58
Depending on the degree of iron accumulation, cytotoxic hydroxyl and lipid radicals will be produced causing various organ pathologies, including those of the liver, heart, and pancreas, and ultimately lead to organ failure in affected individuals who remain untreated.2 The symptoms of hemochromatosis range from chronic fatigue, hyperpigmentation, joint and bone symptoms to diabetes, and liver diseases, such as fibrosis, cirrhosis, and hepatocellular carcinoma20 (Table 1 and Figure 2). The correlation between increased ferritin levels and transferrin saturation with organ damage is poor,59,60 highlighting the need for better markers for iron-induced organ damage. Despite this, symptoms develop especially in men with homozygous p.Cys282Tyr mutations and serum ferritin concentrations greater than 1000 μg/L.61 Liver biopsies may thus be performed to test for liver damage in HH when serum ferritin levels exceed 1000 μg/L, while diverse arthropathies may also be observed in C282Y mutation carriers without severe iron overload.60 Patients diagnosed with clinical HH face a higher risk of developing liver cancer (when cirrhosis is present at diagnosis) compared to controls.62 Although highly debated, several studies have suggested a direct association of HFE mutations with the progression of various cancers including colon, breast, prostate, and epithelial ovarian cancer.63–66 Patients with iron overload are also more susceptible to some infections, such as those caused by Listeria monocytogenes, Vibrio vulnificus, or HIV.67,68 Consistently, studies in mice with iron overload resulting from a hepcidin deficiency showed decreased survival of animals infected by Vibrio vulnificus69 or the malaria-causing Plasmodium berghei.70 In addition, Hfe-deficiency in mice prevents a proper response to infection in that recruitment of pulmonary neutrophils upon an inflammatory stimulus is reduced.71
Table 1.
Symptoms of hereditary hemochromatosis.

Figure 2.

The positive and negative effects of HFE mutations. Hereditary hemochromatosis (HH)-associated mutations affect the health of their carriers in various ways. Besides provoking classical HH disease symptoms (left), these mutations can provide benefits for affected individuals (right). Some of the benefits associated with HH have remained speculative to date and need further experimental validation (marked with a question mark).
Until today, traditional phlebotomy (venesection) has remained the standard treatment for hemochromatosis. It was introduced in 1950 and has been applied ever since to improve the survival of HH patients.72 Phlebotomy removes large amounts of iron localized in red blood cells, which stimulates erythropoiesis and mobilizes iron stored in peripheral tissues. It is a relatively simple and inexpensive treatment but has limited effects on specific HH-associated pathologies, including musculoskeletal symptoms.73 Whether unaffected mutation carriers should undergo phlebotomy has been highly debated, especially because evidence for a beneficial effect of phlebotomy in these individuals has not been documented.20 More recent insights into the molecular mechanisms maintaining iron homeostasis led to the development of alternative therapies, including the use of iron chelators, (eg. the most recent available oral chelator deferasirox, which effectively removes excess iron in HFE-HH patients74), and minihepcidin,75 which might be applicable for patients with HH in the future.76
Prevalence and penetrance of HFE-HH
The HFE mutation C282Y occurs in approximately 6% of Caucasians77 and thus represents the most common genetic variant among this population. The frequency of the C282Y allele decreases from Northern to Southern Europe suggesting that it initially occurred as a ‘master’ mutation in the Neolithic Age in the Celtic population and then spread throughout the rest of Europe later on.78 Immigration from Europe to the US and Australia led to the widespread distribution of HH among Caucasian adults living in those geographic areas.79
Despite the high prevalence of the C282Y allele, numerous population screening studies provided evidence that the mutation clinically manifests in hemochromatosis in only a subgroup of carriers (Table 2). Differences in study design, inclusion criteria, and the definition of disease penetrance make it difficult to confirm an exact number for the overall penetrance of the C282Y mutation. A meta-analysis taking into account the data from 16 independent studies reported a penetrance of C282Y homozygosity of 14%.24 It was shown that homozygous men face a much higher risk of developing an iron overload phenotype than women,61 which may be explained by the recurrent physiological blood loss in women or by the frequency disparity of certain HLA haplotypes that occurs between male and female patients.80 The penetrance of the heterozygous C282Y mutation is even lower, with only approximately 3% of mutation carriers showing disease symptoms.3 Despite the fact that HFE mutations are not always pathogenic per se, but rather represent polymorphisms that predispose to iron overload, murine disease models with Hfe-deficiency81 or engineered mutations corresponding to the human C282Y mutation show an iron overload phenotype. Like in humans, the genetic background of the mouse strain strongly affects the severity of tissue iron accumulation.82 The disease manifestation of other HFE mutations, such as H63D in combination with C282Y, is negligible41 (Table 2). In rare cases, H63D mutations can lead to phenotypic expression of HH when additional risk factors such as heavy alcohol consumption or hepatitis virus infections are present.83
Table 2.
Prevalence and penetrance of HFE (hemachromatosis) gene mutations.
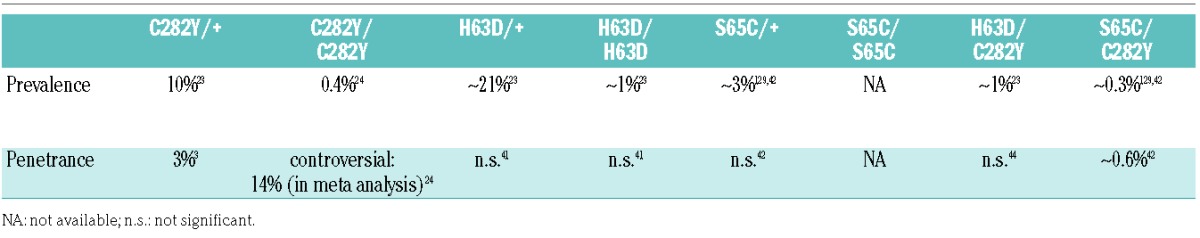
The discrepancy between the frequency of HFE mutations and their phenotypic expression indicates that additional environmental or genetic factors contribute to the manifestation of HH in affected individuals. Despite significant efforts to determine these parameters, only a few risk factors have been identified, including alcohol abuse, being heavily overweight, liver disease, or viral infections.57 The consumption of more than 60 g of alcohol per day leads to higher serum iron and ferritin concentrations, contributing to the progression of cirrhosis in C282Y homozygotes.84 Certain dietary habits, such as heme iron intake and meat consumption, are also associated with increased iron loading.85 In addition, recent genome-wide association studies have identified SNPs in so-called ‘modifier’ genes that are associated with iron-related phenotypes. These include polymorphisms in genes with known roles in iron metabolism, such as those encoding transferrin receptor 1 (TFRC) and ferroportin (FPN/SCL40A1),86 but also in novel genomic loci, eg. in transmembrane protease, serine 6 (TMPRSS6)87 and in the vicinity of the genes FADS2, NAT2, ABO and TEX14.86 SNPs in transferrin (TF),86,88 transferrin receptor 2 (TFR2),86 bone morphogenetic protein 2 (BMP2),89 aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL),20 glyceronephosphate O-acyltransferase (GNPAT),90 and cytochrome B reductase 1 (CYBRD1)91 were further found to influence iron-related parameters in C282Y homozygotes. In addition, a large cohort study reported an association between common variants in the genes coding for bone morphogenetic protein 4 (BMP4) and hemojuvelin (HJV) and increased serum ferritin levels in homozygous C282Y mutation carriers.89 Moreover, a polymorphism in the PCSK7 (proprotein convertase subtilisin/kexin type 7) gene has been linked with liver cirrhosis and advanced fibrosis in C282Y homozygotes.92 These variants are extremely rare and may thus determine disease development on an individual basis. Although carriers of heterozygous HFE mutations mostly lack a hemochromatosis phenotype, enhanced iron loading and manifestation of HH may occur in persons with heterozygous HFE mutations and additional mutations in the hepcidin (HAMP)93,94 HJV,93,95 or TFR296 genes. Additional HH modifiers, such as haptoglobin (HP)97 and ceruloplasmin (CP) have been identified in mice. Hfe knock-out mice (Hfe −/−) with a heterzogous R435X nonsense mutation in the CP gene and Hfe(+/−) mice with a homozygous R435X nonsense mutation showed lower liver iron levels compared to single mutant animals, revealing a protective effect of this specific ceruloplasmin variant in mice.98 In line with this, ceruloplasmin levels were found to be lower in HH patients when compared to control subjects.99
Do HFE mutations offer any advantage to asymptomatic patients?
The observation that C282Y HFE mutations are frequent but only cause a disease-related phenotype in a subgroup of carriers led to the hypothesis that this HFE gene variant may be of an environmental or genetic advantage to asymptomatic carriers and that this is the reason for which it has been inherited with such a high frequency. Increased iron uptake, a hallmark of HH, may have helped humankind, and especially women of reproductive age, to better cope with the iron-reduced cereal grain-based diet which replaced the paleo diet rich in red meat in Europe in the Neolithic Age, at the time when the first HFE mutation occurred.100 Over centuries, the mutation may have spread throughout the Caucasian population because it actively promoted the health of its carriers. Recent studies have provided evidence that the HFE C282Y variant as well as the H63D and S65C variants can positively influence the immune system, the general fitness and reproductive status of mutation carriers, and might even diminish the risk of developing diseases such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease, and atherosclerosis (Figure 2).
Micro-organisms heavily depend on the availability of iron for their proliferation. Iron levels in macrophages of carriers of the C282Y allele are reduced, which makes them less susceptible to those bacteria that depend on macrophage iron, such as Mycobacterium tuberculosis,101 Leishmania amazonensis,102 and Chlamydia or Legionella species.103 In line with this, proliferation of Salmonella Typhimurium in macrophages of mice with hetero- and homozygotic Hfe-deficiencies is strongly attenuated.104 In addition, in a mouse model with an HFE H63D mutation, increased survival over wild-type mice was observed following an infection with Plasmodium falciparum, a parasite that causes cerebral malaria.105 Besides being critical for micro-organismal growth, intramacrophage iron levels influence the inflammatory state of macrophages. While reduced iron levels attenuate the inflammatory response of macrophages to Salmonella infection or LPS-stimulation,106 increased iron levels trigger a phenotypic switch of macrophages towards a pro-inflammatory state.107 HFE mutations may thus interfere with the inflammatory response of macrophages by reducing iron levels in macrophages in a hepcidin-dependent manner and protect carriers from infectious diseases. This may provide an explanation of why HFE mutations were inherited with such a high frequency.
In addition to influencing the host immune system, mutations in the HFE gene have also been linked to the increased fitness of affected individuals. Intravenous and oral iron supplementation can treat fatigue in non-anemic women with low iron stores,108,109 showing that elevated iron levels positively influence fitness. A recent study demonstrated that 80% of successful French athletes carry a heterozygous HFE mutation (C282Y, H63D or S65C) suggesting that the resulting enhanced iron supply during physical activity accounts for the superior physical performance of these sportsmen.110 Large-scale studies among the Sicilian population further showed that HFE C282Y heterozygous individuals, particularly women, have a significantly increased life expectancy compared to controls.111,112 A trend towards an extended life span has also been observed in Sardinian women carrying the H63D mutation.113 Treated C282Y homozygotes with serum ferritin concentrations less than 1000 μg/L further exhibit a lower mortality related to cardiovascular events and extrahepatic cancers when compared to the general population.114 Interestingly, people diagnosed with HFE-HH and verified iron overload are also taller on average, potentially because augmented iron absorption has a beneficial effect on growth.115
Unlike the carriers of homozygous HFE C282Y mutations, who may suffer from hypogonadism, heterozygous individuals may also have a reproductive advantage. This is supported by a study that reported higher levels of the sex hormone-binding globulin in the blood of C282Y heterozygous men.116 Women of reproductive age carrying a heterozygous C282Y mutation were found to suffer less commonly from low serum ferritin concentrations compared to control subjects, which might positively affect their fertility.117
HFE C282Y mutations have been associated with the attenuation of various disease states. For example, a lower incidence rate of atherosclerosis in HFE C282Y carriers has been reported. Possible explanations include lower serum cholesterol and low-density lipoprotein cholesterol levels,118,119 or iron deficiencies of macrophages that may contribute to a diminished inflammatory response106 and a decreased tendency to form atherosclerotic plaques.120 However, other studies failed to establish a link between hemochromatosis and atherosclerosis.121–124
Screening studies and corresponding meta-analyses further associated mutations in the HFE gene with a decreased risk of developing neurodegenerative diseases. So far, experimental validations for these findings are lacking and they are still considered highly controversial. Screening among patients with AD detected an association with the C282Y allele,125 a result in line with data from a meta-analysis that revealed a correlation between AD and the C282Y but not the H63D polymorphism.126 Similarly, two different meta-analyses reported an association of the C282Y mutation and sporadic ALS127 or Parkinson’s disease,128 both excluding a role for the H63D polymorphism in disease manifestation.
Taken together, several studies suggest that HFE mutation carriers might benefit from mutation-associated increased iron levels as long as the iron overload phenotype is mild. This demonstrates that we clearly need to distinguish between hemochromatosis ‘patients’ who suffer from the pathophysiological complications associated with iron overload and individuals who carry HH-associated mutations without showing any disease symptoms. In fact, it is possible that the beneficial effects of the mutations observed in heterozygotes might also manifest in homozygote individuals during growth and early adulthood before significant organ iron overload develops. Future clinical and biochemical studies may reveal more detailed information about the impact of HFE mutations on the health of their carriers and may provide an answer to the long-standing question as to why HFE mutations have so far been inherited with such a high frequency.
Supplementary Material
Acknowledgments
The authors would like to thank Prof. Peter Nielsen, Prof. Pierre Brissot and Prof. Graça Porto for their critical reading of the manuscript. MUM acknowledges funding from the Deutsche Forschungsgemeinschaft (SFB1118 and SFB1036) and the Dietmar Hopp Stiftung. We would like to apologize to all colleagues whose work could not be cited because of space constraints.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/5/809
References
- 1.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell. 2017;168(3):344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yun S, Vincelette ND. Update on iron metabolism and molecular perspective of common genetic and acquired disorder, hemochromatosis. Crit Rev Oncol Hematol. 2015;95(1):12–25. [DOI] [PubMed] [Google Scholar]
- 3.Gallego CJ, Burt A, Sundaresan AS, et al. Penetrance of Hemochromatosis in HFE Genotypes Resulting in p.Cys282Tyr and p.[Cys282Tyr];[His63Asp] in the eMERGE Network. Am J Hum Genet. 2015;97(4):512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton JC. Hemochromatosis and iron overload: from bench to clinic. Am J Med Sci. 2013;346(5):403–412. [DOI] [PubMed] [Google Scholar]
- 5.Trousseau A. Glycosurie; diabete sucre. Clinique Med de l’Hotel de Paris. 1865;2: 663–698. [Google Scholar]
- 6.Hanot V, Chauffard A. [Cirrhose hypertrophique pigmentaire dans le diabète sucré]. Rev Med. 1882;2:385–403. [Google Scholar]
- 7.Troisier M. [Diabète sucré]. Bull Soc Anatomique Paris. 1871;44:231–235. [Google Scholar]
- 8.von Recklinghausen FD. [Uber Haemochromatose. Taggeblatt der 62 Versammlung deutscher Naturforscher and Aerzte in Heidelberg]. 1889:324–325. [Google Scholar]
- 9.Sheldon JH. Haemochromatosis London: Oxford University Press, 1935. [Google Scholar]
- 10.Simon M, Pawlotsky Y, Bourel M, Fauchet R, Genetet B. [Idiopathic hemochromatosis associated with HL-A 3 tissular antigen]. Nouv Presse Med. 1975;4(19):1432. [PubMed] [Google Scholar]
- 11.Laufberger V. [Sur la cristallisation de la ferritine]. Soc Chim Biol. 1937. [Google Scholar]
- 12.Schade AL, Caroline L. An iron-binding component in human blood plasma. Science. 1946;104(2702):340. [PubMed] [Google Scholar]
- 13.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. [DOI] [PubMed] [Google Scholar]
- 14.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004; 36(1):77–82. [DOI] [PubMed] [Google Scholar]
- 15.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–22. [DOI] [PubMed] [Google Scholar]
- 16.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25(1):14–15. [DOI] [PubMed] [Google Scholar]
- 17.Montosi G, Donovan A, Totaro A, et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001; 108(4):619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Njajou OT, Vaessen N, Joosse M, et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet. 2001;28(3):213–214. [DOI] [PubMed] [Google Scholar]
- 19.Seckington R, Powell L. HFE-Associated Hereditary Hemochromatosis. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., eds. GeneReviews(R). Seattle (WA), 1993. [Google Scholar]
- 20.Porto G, Brissot P, Swinkels DW, et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet. 2016;24(4):479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garbowski MW, Ma YM, Fucharoen S, Srichairatanakool S, Hider R, Porter JB. Clinical and methodological factors affecting non-transferrin-bound iron values using a novel fluorescent bead assay. Transl Res. 2016;177:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brissot P, Ropert M, Le Lan C, Loreal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820(3):403–410. [DOI] [PubMed] [Google Scholar]
- 23.Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352(17):1769–1778. [DOI] [PubMed] [Google Scholar]
- 24.European Association For The Study Of The L. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3–22. [DOI] [PubMed] [Google Scholar]
- 25.Feder JN, Tsuchihashi Z, Irrinki A, et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J Biol Chem. 1997;272(22):14025–14028. [DOI] [PubMed] [Google Scholar]
- 26.Waheed A, Parkkila S, Zhou XY, et al. Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with beta2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci USA. 1997;94(23):12384–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giannetti AM, Bjorkman PJ. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J Biol Chem. 2004;279(24):25866–25875. [DOI] [PubMed] [Google Scholar]
- 28.West AP, Jr, Giannetti AM, Herr AB, et al. Mutational analysis of the transferrin receptor reveals overlapping HFE and transferrin binding sites. J Mol Biol. 2001;313(2):385–397. [DOI] [PubMed] [Google Scholar]
- 29.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281(39):28494–28498. [DOI] [PubMed] [Google Scholar]
- 30.Andriopoulos B, Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41(4):482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41(4):478–481. [DOI] [PubMed] [Google Scholar]
- 32.Daher R, Kannengiesser C, Houamel D, et al. Heterozygous Mutations in BMP6 Propeptide Lead to Inappropriate Hepcidin Synthesis and Moderate Iron Overload in Humans. Gastroenterology. 2016; 150(3):672–683e4. [DOI] [PubMed] [Google Scholar]
- 33.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–539. [DOI] [PubMed] [Google Scholar]
- 34.Steinbicker AU, Bartnikas TB, Lohmeyer LK, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011;118(15):4224–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayeur C, Leyton PA, Kolodziej SA, Yu B, Bloch KD. BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood. 2014;124(13):2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2(6):399–409. [DOI] [PubMed] [Google Scholar]
- 37.Latour C, Besson-Fournier C, Meynard D, et al. Differing impact of the deletion of hemochromatosis-associated molecules HFE and transferrin receptor-2 on the iron phenotype of mice lacking bone morphogenetic protein 6 or hemojuvelin. Hepatology. 2016;63(1):126–137. [DOI] [PubMed] [Google Scholar]
- 38.Wu XG, Wang Y, Wu Q, et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood. 2014;124(8):1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol. 2012;57(5):1052–1060. [DOI] [PubMed] [Google Scholar]
- 40.Ryan JD, Ryan E, Fabre A, Lawless MW, Crowe J. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology. 2010;52(4):1266–1273. [DOI] [PubMed] [Google Scholar]
- 41.Gochee PA, Powell LW, Cullen DJ, Du Sart D, Rossi E, Olynyk JK. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. 2002;122(3):646–651. [DOI] [PubMed] [Google Scholar]
- 42.Holmstrom P, Marmur J, Eggertsen G, Gafvels M, Stal P. Mild iron overload in patients carrying the HFE S65C gene mutation: a retrospective study in patients with suspected iron overload and healthy controls. Gut. 2002;51(5):723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramakrishna R, Gupta S, Sarathy K, Bowen A. Phenotypic and clinical manifestations of compound heterozygous genetic haemochromatosis (CHGH): a non-invasive approach to clinical management. Intern Med J. 2013;43(3):254–261. [DOI] [PubMed] [Google Scholar]
- 44.Gurrin LC, Bertalli NA, Dalton GW, et al. HFE C282Y/H63D Compound Heterozygotes Are at Low Risk of Hemochromatosis-Related Morbidity. Hepatology. 2009;50(1):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walsh A, Dixon JL, Ramm GA, et al. The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis. Clin Gastroenterol Hepatol. 2006;4(11):1403–1410. [DOI] [PubMed] [Google Scholar]
- 46.Vecchi C, Montosi G, Pietrangelo A. Huh-7: a human “hemochromatotic” cell line. Hepatology. 2010;51(2):654–659. [DOI] [PubMed] [Google Scholar]
- 47.Takano A, Niimi H, Atarashi Y, et al. A novel Y231del mutation of HFE in hereditary haemochromatosis provides in vivo evidence that the Huh-7 is a human haemochromatotic cell line. Liver Int. 2011;31(10):1593–1597. [DOI] [PubMed] [Google Scholar]
- 48.Le Gac G, Gourlaouen I, Ronsin C, et al. Homozygous deletion of HFE produces a phenotype similar to the HFE p.C282Y/p.C282Y genotype. Blood. 2008;112(13):5238–5240. [DOI] [PubMed] [Google Scholar]
- 49.Pelucchi S, Mariani R, Bertola F, Arosio C, Piperno A. Homozygous deletion of HFE: the Sardinian hemochromatosis? Blood. 2009;113(16):3886. [DOI] [PubMed] [Google Scholar]
- 50.Cezard C, Rabbind Singh A, Le Gac G, Gourlaouen I, Ferec C, Rochette J. Phenotypic expression of a novel C282Y/R226G compound heterozygous state in HFE hemochromatosis: molecular dynamics and biochemical studies. Blood Cells Mol Dis. 2014;52(1):27–34. [DOI] [PubMed] [Google Scholar]
- 51.Piperno A, Arosio C, Fossati L, et al. Two novel nonsense mutations of HFE gene in five unrelated italian patients with hemochromatosis. Gastroenterology. 2000; 119(2):441–445. [DOI] [PubMed] [Google Scholar]
- 52.Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol. 2007;13(35):4690–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos PC, Dinardo CL, Cancado RD, Schettert IT, Krieger JE, Pereira AC. Non-HFE hemochromatosis. Rev Bras Hematol Hemoter. 2012;34(4):311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roetto A, Totaro A, Piperno A, et al. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood. 2001;97(9):2555–2560. [DOI] [PubMed] [Google Scholar]
- 55.Detivaud L, Island ML, Jouanolle AM, et al. Ferroportin Diseases: Functional Studies, a Link Between Genetic and Clinical Phenotype. Hum Mutat. 2013;34(11):1529–1536. [DOI] [PubMed] [Google Scholar]
- 56.Schimanski LM, Drakesmith H, Merryweather-Clarke AT, et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood. 2005;105(10):4096–4102. [DOI] [PubMed] [Google Scholar]
- 57.Pietrangelo A. Genetics, Genetic Testing, and Management of Hemochromatosis: 15 Years Since Hepcidin. Gastroenterology. 2015;149(5):1240–1251.e4. [DOI] [PubMed] [Google Scholar]
- 58.Coppin H, Bensaid M, Fruchon S, Borot N, Blanche H, Roth MP. Longevity and carrying the C282Y mutation for haemochromatosis on the HFE gene: case control study of 492 French centenarians. BMJ. 2003;327(7407):132–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Allen KJ, Bertalli NA, Osborne NJ, et al. HFE Cys282Tyr Homozygotes With Serum Ferritin Concentrations Below 1000 mu g/L Are at Low Risk of Hemochromatosis. Hepatology. 2010;52(3):925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221–230. [DOI] [PubMed] [Google Scholar]
- 62.Fracanzani AL, Conte D, Fraquelli M, et al. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33(3):647–651. [DOI] [PubMed] [Google Scholar]
- 63.Gannon PO, Medelci S, Le Page C, et al. Impact of hemochromatosis gene (HFE) mutations on epithelial ovarian cancer risk and prognosis. Int J Cancer. 2011;128(10):2326–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Osborne NJ, Gurrin LC, Allen KJ, et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology. 2010;51(4):1311–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaheen NJ, Silverman LM, Keku T, et al. Association between hemochromatosis (HFE) gene mutation carrier status and the risk of colon cancer. J Natl Cancer Inst. 2003;95(2):154–159. [DOI] [PubMed] [Google Scholar]
- 66.Syrjakoski K, Fredriksson H, Ikonen T, et al. Hemochromatosis gene mutations among Finnish male breast and prostate cancer patients. Int J Cancer. 2006;118(2):518–520. [DOI] [PubMed] [Google Scholar]
- 67.Ashrafian H. Hepcidin: the missing link between hemochromatosis and infections. Infect Immun. 2003;71(12):6693–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khan FA, Fisher MA, Khakoo RA. Association of hemochromatosis with infectious diseases: expanding spectrum. Int J Infect Dis. 2007;11(6):482–487. [DOI] [PubMed] [Google Scholar]
- 69.Arezes J, Jung G, Gabayan V, et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe. 2015;17(1):47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang HZ, He YX, Yang CJ, Zhou W, Zou CG. Hepcidin Is Regulated during Blood-Stage Malaria and Plays a Protective Role in Malaria Infection. J Immunol. 2011; 187(12):6410–6416. [DOI] [PubMed] [Google Scholar]
- 71.Benesova K, Vujic Spasic M, Schaefer SM, et al. Hfe deficiency impairs pulmonary neutrophil recruitment in response to inflammation. PloS One. 2012;7(6):e39363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanwar P, Kowdley KV. Diagnosis and treatment of hereditary hemochromatosis: an update. Expert Rev Gastroenterol Hepatol. 2013;7(6):517–530. [DOI] [PubMed] [Google Scholar]
- 73.Brissot P. Optimizing the diagnosis and the treatment of iron overload diseases. Expert Rev Gastroenterol Hepatol. 2016;10(3):359–370. [DOI] [PubMed] [Google Scholar]
- 74.Phatak P, Brissot P, Wurster M, et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52(5):1671–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramos E, Ruchala P, Goodnough JB, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120(18): 3829–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706–716. [DOI] [PubMed] [Google Scholar]
- 77.Liver EAFTSOT. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3–22. [DOI] [PubMed] [Google Scholar]
- 78.Distante S, Robson KJ, Graham-Campbell J, Arnaiz-Villena A, Brissot P, Worwood M. The origin and spread of the HFE-C282Y haemochromatosis mutation. Hum Genet. 2004;115(4):269–279. [DOI] [PubMed] [Google Scholar]
- 79.Merryweather-Clarke AT, Pointon JJ, Jouanolle AM, Rochette J, Robson KJ. Geography of HFE C282Y and H63D mutations. Genet Test. 2000;4(2):183–198. [DOI] [PubMed] [Google Scholar]
- 80.Barton JC, Wiener HW, Acton RT, Go RC. HLA haplotype A*03-B*07 in hemochromatosis probands with HFE C282Y homozygosity: frequency disparity in men and women and lack of association with severity of iron overload. Blood Cells Mol Dis. 2005;34(1):38–47. [DOI] [PubMed] [Google Scholar]
- 81.Muckenthaler M, Roy CN, Custodio AO, et al. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet. 2003;34(1):102–107. [DOI] [PubMed] [Google Scholar]
- 82.Levy JE, Montross LK, Andrews NC. Genes that modify the hemochromatosis phenotype in mice. J Clin Invest. 2000;105(9):1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piperno A, Sampietro M, Pietrangelo A, et al. Heterogeneity of hemochromatosis in Italy. Gastroenterology. 1998;114(5):996–1002. [DOI] [PubMed] [Google Scholar]
- 84.Fletcher LM, Dixon JL, Purdie DM, Powell LW, Crawford DHG. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology. 2002;122(2):281–289. [DOI] [PubMed] [Google Scholar]
- 85.Cade JE, Moreton JA, O’Hara B, et al. Diet and genetic factors associated with iron status in middle-aged women. Am J Clin Nutr. 2005;82(4):813–820. [DOI] [PubMed] [Google Scholar]
- 86.Benyamin B, Esko T, Ried JS, et al. Novel loci affecting iron homeostasis and their effects in individuals at risk for hemochromatosis. Nat Commun. 2015;6:6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benyamin B, Ferreira MAR, Willemsen G, et al. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat Genet. 2009;41(11):1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Tayrac M, Roth MP, Jouanolle AM, et al. Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis. J Hepatol. 2015;62(3):664–672. [DOI] [PubMed] [Google Scholar]
- 89.Milet J, Dehais V, Bourgain C, et al. Common variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance. Am J Hum Genet. 2007;81(4):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McLaren CE, Emond MJ, Subramaniam VN, et al. Exome sequencing in HFE C282Y homozygous men with extreme phenotypes identifies a GNPAT variant associated with severe iron overload. Hepatology. 2015;62(2):429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pelucchi S, Mariani R, Calza S, et al. CYBRD1 as a modifier gene that modulates iron phenotype in HFE p.C282Y homozygous patients. Haematol-Hematol J. 2012;97(12):1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stickel F, Buch S, Zoller H, et al. Evaluation of genome-wide loci of iron metabolism in hereditary hemochromatosis identifies PCSK7 as a host risk factor of liver cirrhosis. Hum Mol Genet. 2014;23(14):3883–3890. [DOI] [PubMed] [Google Scholar]
- 93.Barton JC, LaFreniere SA, Leiendecker-Foster C, et al. HFE, SLC40A1, HAMP, HJV, TFR2, and FTL mutations detected by denaturing high-performance liquid chromatography after iron phenotyping and HFE C282Y and H63D genotyping in 785 HEIRS Study participants. Am J Hematol. 2009;84(11):710–714. [DOI] [PubMed] [Google Scholar]
- 94.Merryweather-Clarke AT, Cadet E, Bomford A, et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Blood. 2003;102(11): 758a–758a. [DOI] [PubMed] [Google Scholar]
- 95.Pietrangelo A, Caleffi A, Henrion J, et al. Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology. 2005; 128(2):470–479. [DOI] [PubMed] [Google Scholar]
- 96.Biasiotto G, Belloli S, Ruggeri G, et al. Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload. Clin Chem. 2003;49(12):1981–1988. [DOI] [PubMed] [Google Scholar]
- 97.Tolosano E, Fagoonee S, Garuti C, et al. Haptoglobin modifies the hemochromatosis phenotype in mice. Blood. 2005;105(8): 3353–3355. [DOI] [PubMed] [Google Scholar]
- 98.Gouya L, Muzeau F, Robreau AM, et al. Genetic study of variation in normal mouse iron homeostasis reveals ceruloplasmin as an HFE-hemochromatosis modifier gene. Gastroenterology. 2007;132(2):679–686. [DOI] [PubMed] [Google Scholar]
- 99.Cairo G, Conte D, Bianchi L, Fraquelli M, Recalcati S. Reduced serum ceruloplasmin levels in hereditary haemochromatosis. Br J Haematol. 2001;114(1):226–229. [DOI] [PubMed] [Google Scholar]
- 100.Naugler C. Hemochromatosis: a Neolithic adaptation to cereal grain diets. Med Hypotheses. 2008;70(3):691–692. [DOI] [PubMed] [Google Scholar]
- 101.Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J Leukoc Biol. 2007;81(1):195–204. [DOI] [PubMed] [Google Scholar]
- 102.Ben-Othman R, Flannery AR, Miguel DC, Ward DM, Kaplan J, Andrews NW. Leishmania-Mediated Inhibition of Iron Export Promotes Parasite Replication in Macrophages. Plos Pathog. 2014;10(1): e1003901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112(3):866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nairz M, Theurl I, Ludwiczek S, et al. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007; 9(9):2126–2140. [DOI] [PubMed] [Google Scholar]
- 105.Leitner DF, Stoute JA, Landmesser M, Neely E, Connor JR. The HFE genotype and a formulated diet controlling for iron status attenuate experimental cerebral malaria in mice. Int J Parasitol. 2015;45(12):797–808. [DOI] [PubMed] [Google Scholar]
- 106.Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol. 2008;181(4):2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vinchi F, Costa da Silva M, Ingoglia G, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127(4):473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Krayenbuehl PA, Battegay E, Breymann C, Furrer J, Schulthess G. Intravenous iron for the treatment of fatigue in nonanemic, pre-menopausal women with low serum ferritin concentration. Blood. 2011;118(12):3222–3227. [DOI] [PubMed] [Google Scholar]
- 109.Vaucher P, Druais PL, Waldvogel S, Favrat B. Effect of iron supplementation on fatigue in nonanemic menstruating women with low ferritin: a randomized controlled trial. CMAJ. 2012;184(11):1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hermine O, Dine G, Genty V, et al. Eighty percent of French sport winners in Olympic, World and Europeans competitions have mutations in the hemochromatosis HFE gene. Biochimie. 2015;119:1–5. [DOI] [PubMed] [Google Scholar]
- 111.Lio D, Balistreri CR, Colonna-Romano G, et al. Association between the MHC class I gene HFE polymorphisms and longevity: a study in Sicilian population. Genes Immun. 2002;3(1):20–24. [DOI] [PubMed] [Google Scholar]
- 112.Balistreri CR, Candore G, Almasio P, et al. Analysis of hemochromatosis gene mutations in the Sicilian population: implications for survival and longevity. Arch Gerontol Geriatr Suppl. 2002;8:35–42. [DOI] [PubMed] [Google Scholar]
- 113.Carru C, Pes GM, Deiana L, et al. Association between the HFE mutations and longevity: a study in Sardinian population. Mech Ageing Dev. 2003;124(4):529–532. [DOI] [PubMed] [Google Scholar]
- 114.Bardou-Jacquet E, Morcet J, Manet G, et al. Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemochromatosis. J Hepatol. 2015;62(3):682–689. [DOI] [PubMed] [Google Scholar]
- 115.Cippa PE, Krayenbuehl PA. Increased height in HFE hemochromatosis. N Engl J Med. 2013;369(8):785–786. [DOI] [PubMed] [Google Scholar]
- 116.Yeap BB, Beilin J, Shi Z, et al. The C282Y polymorphism of the hereditary hemochromatosis gene is associated with increased sex hormone-binding globulin and normal testosterone levels in men. J Endocrinol Invest. 2010;33(8):544–548. [DOI] [PubMed] [Google Scholar]
- 117.Bulaj ZJ, Griffen LM, Jorde LB, Edwards CQ, Kushner JP. Clinical and biochemical abnormalities in people heterozygous for hemochromatosis. N Engl J Med. 1996;335(24):1799–1805. [DOI] [PubMed] [Google Scholar]
- 118.Pankow JS, Boerwinkle E, Adams PC, et al. HFE C282Y homozygotes have reduced low-density lipoprotein cholesterol: the Atherosclerosis Risk in Communities (ARIC) Study. Transl Res. 2008;152(1):3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Adams PC, Pankow J, Barton JC, et al. Hfe C282y Homozygosity Is Associated with Lower Total and Ldl Cholesterol: The Hemochromatosis and Iron Overload Screening (Heirs) Study. Hepatology. 2008; 48(4):437a–438a. [DOI] [PubMed] [Google Scholar]
- 120.Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol. 2014;5:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miller M, Hutchins GM. Hemochromatosis, multiorgan hemosiderosis, and coronary artery disease. JAMA. 1994;272(3):231–233. [PubMed] [Google Scholar]
- 122.Sullivan JL, Zacharski LR. Hereditary haemochromatosis and the hypothesis that iron depletion protects against ischemic heart disease. Eur J Clin Invest. 2001;31(5):375–377. [DOI] [PubMed] [Google Scholar]
- 123.Munoz-Bravo C, Gutierrez-Bedmar M, Gomez-Aracena J, Garcia-Rodriguez A, Navajas JF. Iron: protector or risk factor for cardiovascular disease? Still controversial. Nutrients. 2013;5(7):2384–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sullivan JL. Do Hemochromatosis Mutations Protect Against Iron-Mediated Atherogenesis? Circ-Cardiovasc Gene. 2009;2(6):652–657. [DOI] [PubMed] [Google Scholar]
- 125.Correia AP, Pinto JP, Dias V, Mascarenhas C, Almeida S, Porto G. CAT53 and HFE alleles in Alzheimer’s disease: a putative protective role of the C282Y HFE mutation. Neurosci Lett. 2009;457(3):129–132. [DOI] [PubMed] [Google Scholar]
- 126.Lin M, Zhao L, Fan J, et al. Association between HFE polymorphisms and susceptibility to Alzheimer’s disease: a meta-analysis of 22 studies including 4,365 cases and 8,652 controls. Mol Biol Rep. 2012;39(3):3089–3095. [DOI] [PubMed] [Google Scholar]
- 127.Li M, Wang L, Wang W, Qi XL, Tang ZY. Mutations in the HFE gene and sporadic amyotrophic lateral sclerosis risk: a meta-analysis of observational studies. Braz J Med Biol Res. 2014;47(3):215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xia J, Xu H, Jiang H, Xie J. The association between the C282Y and H63D polymorphisms of HFE gene and the risk of Parkinson’s disease: A meta-analysis. Neurosci Lett. 2015;595:99–103. [DOI] [PubMed] [Google Scholar]
- 129.Mura C, Raguenes O, Scotet V, Jacolot S, Mercier AY, Ferec C. A 6-year survey of HFE gene test for hemochromatosis diagnosis. Genet Med. 2005;7(1):68–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.