

Abstract
While c-Myc dysregulation is constantly associated with highly proliferating B-cell tumors, nuclear factor (NF)-κB addiction is found in indolent lymphomas as well as diffuse large B-cell lymphomas, either with an activated B-cell like phenotype or associated with the Epstein-Barr virus. We raised the question of the effect of c-Myc in B cells with NF-κB activated by three different inducers: Epstein-Barr virus-latency III program, TLR9 and CD40. Induction of c-Myc overexpression increased proliferation of Epstein-Barr virus-latency III immortalized B cells, an effect that was dependent on NF-κB. Results from transcriptomic signatures and functional studies showed that c-Myc overexpression increased Epstein-Barr virus-latency III-driven proliferation depending on NF-κB. In vitro, induction of c-Myc increased proliferation of B cells with TLR9-dependant activation of MyD88, with decreased apoptosis. In the transgenic λc-Myc mouse model with c-Myc overexpression in B cells, in vivo activation of MyD88 by TLR9 induced splenomegaly related to an increased synthesis phase (S-phase) entry of B cells. Transgenic mice with both continuous CD40 signaling in B cells and the λc-Myc transgene developed very aggressive lymphomas with characteristics of activated diffuse large B-cell lymphomas. The main characteristic gene expression profile signatures of these tumors were those of proliferation and energetic metabolism. These results suggest that c-Myc is an NF-κB co-transforming event in aggressive lymphomas with an activated phenotype, activated B-cell like diffuse large B-cell lymphomas. This would explain why NF-κB is associated with both indolent and aggressive lymphomas, and opens new perspectives on the possibility of combinatory therapies targeting both the c-Myc proliferating program and NF-κB activation pathways in diffuse large B-cell lymphomas.
Introduction
NF-κB associated aggressive B-cell lymphomas can be subdivided into at least two main categories: diffuse large B-cell lymphomas (DLBCLs) of immunocompetent patients with an activated B-cell-like (ABC) gene expression signature, and Epstein-Barr virus (EBV) associated DLBCLs, either in elderly or immunocompromised patients. ABC-DLBCLs exhibit an NF-κB addiction and have a worse prognosis when compared to DLBCLs with a germinal center B-cell-like (GCB) signature, the other main molecular DLBCL subtype.1 In ABC-DLBCLs, activation of NF-κB is due either to autoactivation of the CD40 signalosome2 or to NF-κB activating mutations, among them mutations of TNFAIP3 (A20) or MYD88 (the most frequent, concerning 39% of ABC-DLBCLs).3 Prognosis of EBV-associated DLBCLs of the elderly who have an ABC-DLBCL profile is poor.4,5 In these cases, NF-κB activation is due to the expression of the latent membrane protein 1 (LMP1), the main oncoprotein of EBV. NF-κB inhibition in these tumors induces apoptosis.5,6 Immunodeficient patients are also prone to aggressive B-cell lymphoproliferative disorders, which are often associated with EBV and inhibition of NF-κB leading to apoptosis of tumor cells.7–10
The activated B-cell phenotype of all these aggressive B-cell lymphomas is largely due to NF-κB activation. These tumors also exhibit a high proliferative index,11 likely due to the dysregulation of c-Myc activity.12 Genetic alterations of MYC, although more frequent in GCB-DLBCLs, are also found in ABC-DLBCL, and c-Myc overexpression is a negative predictor of survival in both ABC and GCB-DLBCLs.13 In EBV-positive DLBCLs, Epstein-Barr virus nuclear antigen 2 (EBNA2) expression is a poor prognosis factor.14 By subverting the Notch pathway through targeting of the RBP-Jκ nuclear factor, EBNA2 is the EBV protein responsible for the EBV-latency III program (also called proliferating program)15 and directly upregulates LMP1 gene expression, which in turns activates NF-κB.16 EBNA2 itself directly contributes to protection against apoptosis.15,17 EBNA2 is also responsible for c-Myc deregulation.18 In vitro, EBV-driven B-cell proliferation is directly related to the activity of two master transcription factors: c-Myc and NF-κB.19
c-Myc is the master transcription factor for cell proliferation and is involved in numerous hematological and solid cancers.20 Several transgenic mice models, including Eμ-Myc, λc-Myc or 3′RR-c-Myc models21–23 as well as Burkitt’s lymphomas (BLs) demonstrate that c-Myc overexpression in B cells leads to the emergence of aggressive B-cell lymphomas, but with a non-activated phenotype.
Based on these features, we raised the question concerning the effect of c-Myc overexpression in B cells with an NF-κB activated B-cell-like phenotype. We showed that c-Myc constantly promoted B-cell proliferation of NF-κB activated B cells in different models depending either on EBV, MyD88 or CD40, in vitro and in vivo. Co-regulation of c-Myc and CD40 in a mouse model led to very aggressive B-cell lymphomas with an activated phenotype.
Methods
See the Online Supplementary Materials and Methods for a description of the techniques.
Cells
EREB2.5 cells are a non-classical LCL with an estradiol inducible EBV-latency III proliferation program.24 The P493.6 cell line is an EREB2.5 derivative transfected with a Tet-Off inducible c-Myc expressing vector.25
EMSAs, Plasmid Constructs, Western Blotting and RT-PCR
Methods for nuclear extracts, electrophoretic mobility shift assays (EMSAs), Western blots and reverse transcription-polymerase chain reactions (RT-PCRs) are described elsewhere.26 Complementary DNA for IκBαS32,36A (super-repressor form of IκBα) has already been published.27
Cell Labeling, proliferation and immunohistochemistry
Red blood cell lysis buffer came from eBioscience, San Diego, CA, USA. To assess proliferation, carboxyfluorescein diacetate succinimidyl ester (CFSE) and 5-ethynyl-2′-deoxyuridine (EdU) were both obtained from Life Technologies, while 5-bromo-2′-deoxyuridine (BrdU) came from Sigma-Aldrich, Saint-Louis, MO, USA. Ki-67 labeling was also used to follow proliferation using imaging flow cytometry with the ImageStream 100 apparatus (Amnis®; Merck, Darmstadt, Germany).
Gene Expression Profiling
Amplification of ribonucleic acids (RNAs) and hybridization onto microarrays were performed on an Affymetrix GeneAtlas® System with: Affymetrix® Human Genome U219 Array Strip, and Affymetrix® Mouse Gene 2.1 ST Array Strip as previously described.26
Mouse Models
Information on λc-Myc mice and mice with the CD19-Cre conditional LMP1.CD40 fusion transgene have already been published.22,28 All procedures were conducted under an approved protocol according to European guidelines for animal experimentation (French national authorization number: 87-022 and French ethics committee registration number “CREEAL”: 09-07-2012).
Results
c-Myc increases NF-κB dependant EBV-latency III B-cell proliferation
c-Myc and NF-κB are the two master transcriptional factors of EBV-latency III proliferating B cells.19 To understand how c-Myc interferes with the EBV-latency III proliferation program, we used the EBV-infected P493.6 B-cell line, a cell line that is double conditional for c-Myc and EBNA2, allowing for growth under the c-Myc or EBV-latency III program.25 Indeed, P493.6 cells are thought to be an in vitro model of normal B cells that can be forced to adopt four different proliferation statuses: quiescent state (c-Myc−/EBNA2−), EBV-latency III proliferating program (c-Myc−/EBNA2+), BL-like c-Myc proliferating program (c-Myc+/EBNA2−), and both programs (c-Myc+/EBNA2+) (see the Online Supplementary Figure S1 and its legend for a detailed description of this model).
We first analyzed transcriptional changes induced by EBV-latency III when associated with c-Myc overexpression. Supervised analysis led to the selection of 1,648 probes with at least a two-fold signal variation in one of the four P493.6 cell conditions when compared to the median of each probe. Genes could be grouped into five clusters by hierarchical clustering. Their main relevant biological functions are shown in Figure 1 (see also Online Supplementary Table S1). Cluster 1 corresponds to genes that were strongly repressed when both EBV-latency III and c-Myc programs were induced (c-Myc+/EBNA2+). Most genes were associated with immune response and the induction of apoptosis. Cluster 2 genes (such as CD80, CFLAR/c-Flip, TRAF1, EBI3, and TNFAIP3/A20) were induced by the EBV-latency III program alone (c-Myc−/EBNA2+), these genes are known to be NF-κB targets. These genes were repressed by c-Myc (compare c-Myc+/EBNA2− and c-Myc−/EBNA2+ conditions) but were still expressed in the presence of both proliferating programs. Genes belonging to cluster 3 were likely to be targets of signaling pathways repressed by c-Myc in the presence or absence of EBV-latency III (c-Myc+/EBNA2+ and c-Myc+/EBNA2−). These signaling pathways included interferon, JAK/STAT, NF-κB, Jun, Erk, and Akt pathways. The last two clusters (clusters 4 and 5) were genes induced either by EBV-latency III (c-Myc+/EBNA2+) or c-Myc (c-Myc+/EBNA2−) programs alone for which expression was over-induced when both programs were switched on (c-Myc+/EBNA2+). The functions of these genes were nucleic acid metabolic processes, energy metabolism, and proliferation.
Figure 1.
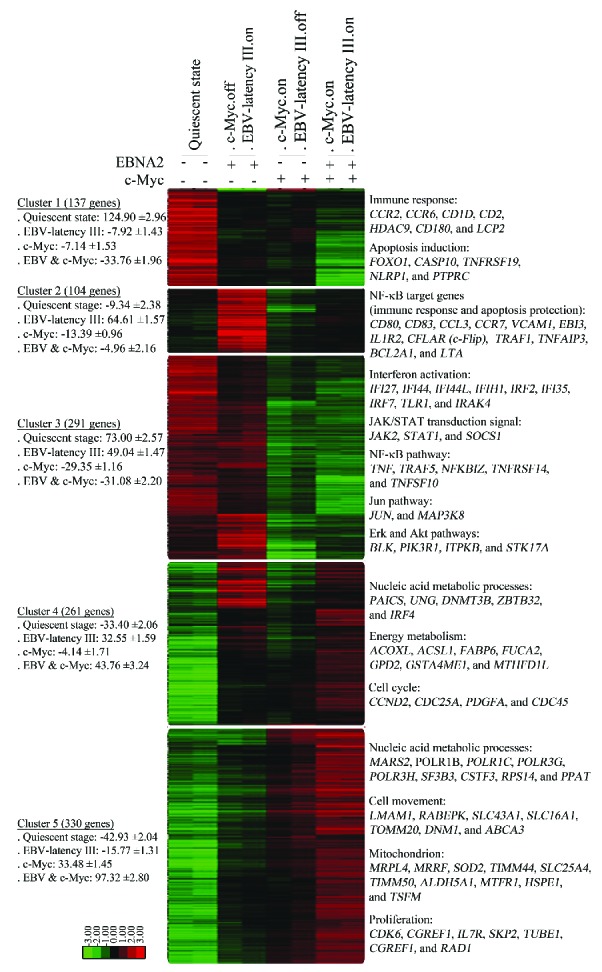
Transcriptomic changes induced by c-Myc overexpression on EBV-latency III proliferating P493.6 cells. Gene expression profiles of P493.6 cells induced (+) or not (−) for EBNA2 and/or c-Myc for 48 hours. Each cell condition was repeated once. Selection of the 1,648 probes was based on at least a two-fold variation in one of the four cell conditions when compared to the median. After ascending hierarchical clustering on median centered values, five main clusters were defined as the five ultimate ascending clusters with a global positive correlation value. For each cluster and each cell condition, centered means of gene expression values with standard errors (±SE) are indicated on the left. Most representative functions with most significant genes are mentioned on the right. Log2 value color codes are shown beside the heat map. EBNA2: Epstein-Barr virus nuclear antigen 2; EBV: Epstein-Barr virus; NF-κB: nuclear factor-κB.
We subsequently studied the functional consequences of co-activation of both EBV-latency III and c-Myc programs. Induction of c-Myc by tetracycline retrieval increased proliferation of EBV-latency III P493.6 cells in a dose-dependent manner, reaching 45% with 1 μM estradiol (c-Myc+/EBNA2+) compared to 29% and 37% with EBV-latency III or c-Myc alone, respectively (Figure 2A). Using an imaging flow cytometer, the strongest labeling of the Ki-67 proliferation marker was observed when both proliferation programs were induced (Figure 2B–D). Morphological evaluation of apoptotic cells by quantification of nuclear fragmentation29 revealed that, as expected, induction of the EBV-latency III program increased protection against apoptosis and that induction of c-Myc alone was not deleterious in this cell type, as reported by Schuhmacher et al.30 (Figure 2E). These results suggest that proliferation was increased when c-Myc overexpression was associated with the EBV-latency III program (c-Myc+/EBNA2+), maintaining protection from apoptosis. Since P493-6 cells could have been selected to resist the potential deleterious effect of c-Myc overexpression, we also transfected two classical LCLs, named PRI and JEF, with a doxycycline-inducible PRT-1 vector overexpressing c-Myc.31 Results suggest that mild induction of c-Myc increased proliferation of both classical LCLs while, as a control, proliferation of these cells was stable over luciferase induction levels (data not shown).
Figure 2.
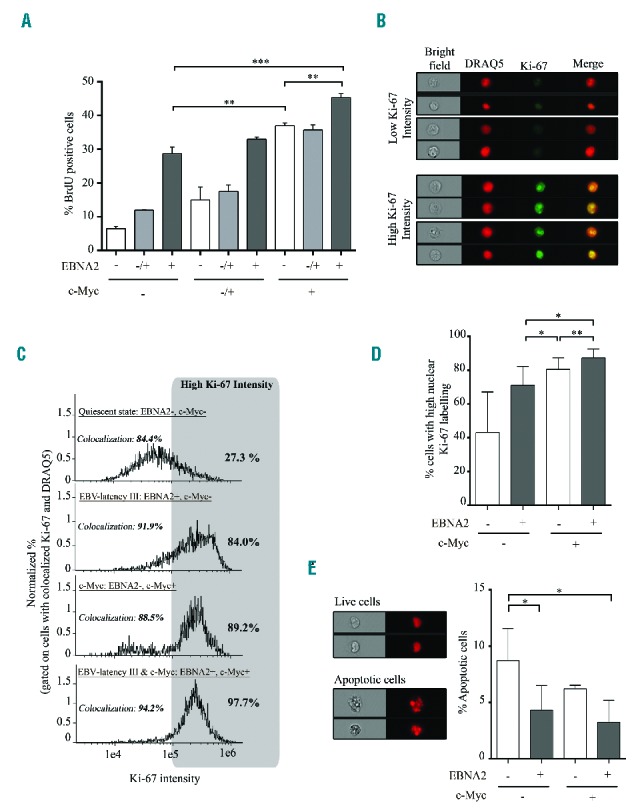
Proliferation and apoptosis of P493.6 cells after induction of c-Myc programs in EBV-latency III B cells. (A) Percentage of BrdU positive cells was assessed by flow cytometry on P493.6 cells treated with two doses of estradiol (0.1 μM: EBNA2−/+ and 1 μM: EBNA2+) or not (EBNA2−) and/or with tetracycline (100 μg/ml: c-Myc−; 3 μg/ml: c-Myc−/+; and not: c-Myc+) for 48 hours. Data are shown as mean ± standard deviation (n=3 independent experiments). (B to E) P493.6 cells were treated or not with 1 μM estradiol and/or 100 μg/ml tetracycline for 48 hours to induce either cell quiescence (EBNA2−, c-Myc−), EBV-latency III alone (EBV+, c-Myc−), c-Myc alone (EBNA2−, c-Myc+), and both c-Myc and EBV-latency III (EBNA2+, c-Myc+). (B,C and D) Percentage of cells with strong staining of the Ki-67 proliferation marker in the nucleus, assessed by imaging flow cytometry. (B) Examples of cells under the c-Myc program with low or high Ki-67 expression. Selection of cells was based on colocalization of Ki-67 and the DRAQ5® nuclear dye. (C) Representative monoparametric histograms of Ki-67 intensity gated on cells with colocalization of Ki-67 and DRAQ5®. Each cell condition is indicated at the top left of each histogram. Threshold for strong Ki-67 labeling is indicated by the gray box. Percentage of cells with high Ki-67 labeling is indicated on the top right of each histogram. (D) Total percentage of high nuclear stained Ki-67 cells in the four culture conditions of P493.6 cells. Data are shown as mean ± standard deviation (n=5 independent experiments). (E) Apoptosis of the uncolocalized Ki-67 and DRAQ5® population using imaging flow cytometry. Left panel: example of living and apoptotic cells under the c-Myc condition. Right panel: total percentage of apoptotic cells in the four culture conditions. Quantification was based on morphometric parameters.29 Data are shown as mean ± standard deviation (n=3 independent experiments). Statistical significance was determined by Student’s t-test (*** P<0.001; **P<0.01; *P<0.05). BrdU: 5-bromo-2′-deoxyuridine; EBNA2: Epstein-Barr virus nuclear antigen 2; EBV: Epstein-Barr virus; DRAQ5: deep red anthraquinone 5.
Altogether, the results presented suggest that both EBV-latency III and c-Myc programs brought complementary advantages for cell transformation, both acting concomitantly on cell proliferation and cell metabolism, with EBV-latency III acting more specifically on protection against apoptosis and c-Myc on repression of interferon response genes.
Albeit in a less intense manner than in EBV-latency III proliferating cells, NF-κB was clearly activated in c-Myc+/EBNA2+ P493.6 cells when compared to c-Myc+/EBNA2− cells (see the Online Supplementary Figure S2 for details on NF-κB activation in the P493.6 cell line). We thus tested the effect of NF-κB inhibition on these cells. NF-κB was first repressed by overexpression of the super-repressor IκBαS32,36A.27 Inhibition of NF-κB decreased cell proliferation regardless of whether c-Myc was induced or not in EBV-latency III proliferating P493.6 cells (Figure 3A). The percentage of sub-G1 cells also increased after NF-κB inhibition (data not shown). We also treated cells with PHA-408 (Figure 3B,C), an inhibitor of the IκB kinase 2 (IKK2) subunit of the IKK complex.32 PHA-408 significantly decreased NF-κB DNA binding activity when the EBV-latency III program was switched on (Online Supplementary Figure S3; Figure 3B: lanes 1, 5, 9 in the presence of c-Myc, and lanes 3, 7, 11 in the absence of c-Myc). In the presence of PHA-408, P493.6 proliferation driven by both EBV-latency III plus c-Myc programs was similar to that of cells driven by c-Myc alone. As control, the addition of PHA-408 marginally affected proliferation of P493.6 cells when c-Myc alone was turned on (Figure 3C). This suggests that when both c-Myc and the EBV-latency III program were induced, increased proliferation was mainly due to both c-Myc and NF-κB.
Figure 3.
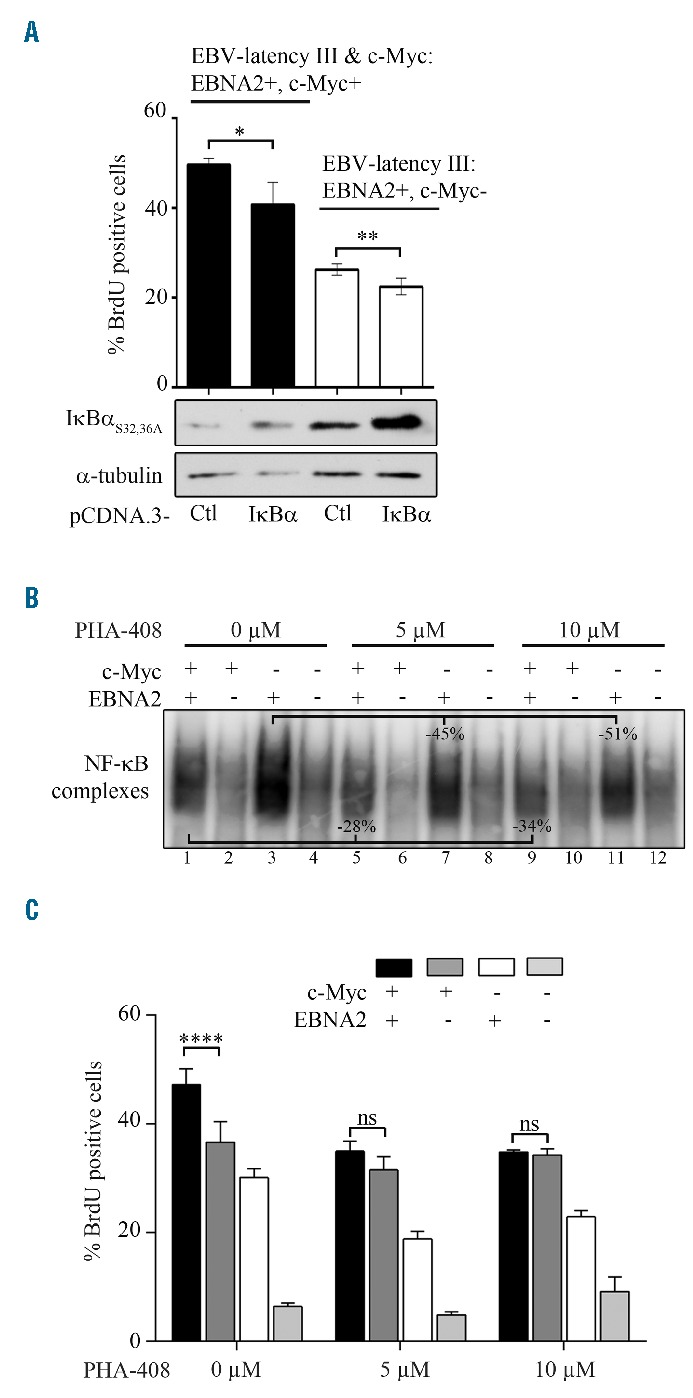
Activation of NF-κB in P493.6 cells after induction of c-Myc on EBV-latency III. P493.6 cells were treated or not with estradiol 1 μM (EBNA2− or EBNA2+) and/or with tetracycline at 100 ng/mL (c-Myc+ or c-Myc−) for 48 hours. (A) Percentage of BrdU positive cells after transient transfection with the pCDNA.3 empty vector (Ctl) or expressing the super-repressor IκBαS32,36 A for 48 hours (upper panel). Representative analysis of IκBαS32,36A, and α-tubulin expression by Western blot (lower panel). (B and C) Effect of the IKK2 inhibitor PHA-408 (5 μM and 10 μM) for 48 hours on NF-κB. Representative DNA binding by EMSA (panel B) and BrdU incorporation (panel C). In the histograms, data are shown as the mean ± standard deviation (at least 3 independent experiments). Statistical significance was determined by Student’s t-test (****P<0.001; **P<0.01; *P<0.05). ns: not significant; BrdU: 5-bromo-2′-deoxyuridine; EBNA2: Epstein-Barr virus nuclear antigen 2; EBV: Epstein-Barr virus; Ctl: control; NF-κB: nuclear factor-κB.
C-Myc increases TLR9 induced B-cell proliferation both in vitro and in vivo
Since activating mutations of MyD88 are frequently found in aggressive B-cell lymphomas with activation of NF-κB,33 we decided to assay the effect of both c-Myc and TLR9 activation in B cells. In vitro, resting P493.6 cells (c-Myc-/EBNA2-) were stimulated with or without either short control oligodeoxynucleotides (ODN-ctl) or with unmethylated CpG motifs (ODN-CpG). TLR9 activation moderately but significantly increased the S-phase entry of resting P493.6 cells (Figure 4A). c-Myc induction (c-Myc+/EBNA2−) led to a considerable acceleration of cell proliferation; an effect that was further enhanced on TLR9 activated cells (Figure 4A). As expected, NF-κB targets, such as TRAF1, were upregulated upon ODN-CpG treatment (Figure 4B). When NF-κB was inhibited after the addition of PHA-408, as shown by inhibition of TRAF1 expression, a significant decrease in TLR9 activated B-cell proliferation was seen. This shows that enhanced proliferation of TLR9 activated P493.6 cells with c-Myc overexpression was dependent on NF-κB. This proliferative effect of TLR9 stimulation was confirmed in c-Myc overexpressing BL2 and BL41 BL cell lines (data not shown).
Figure 4.
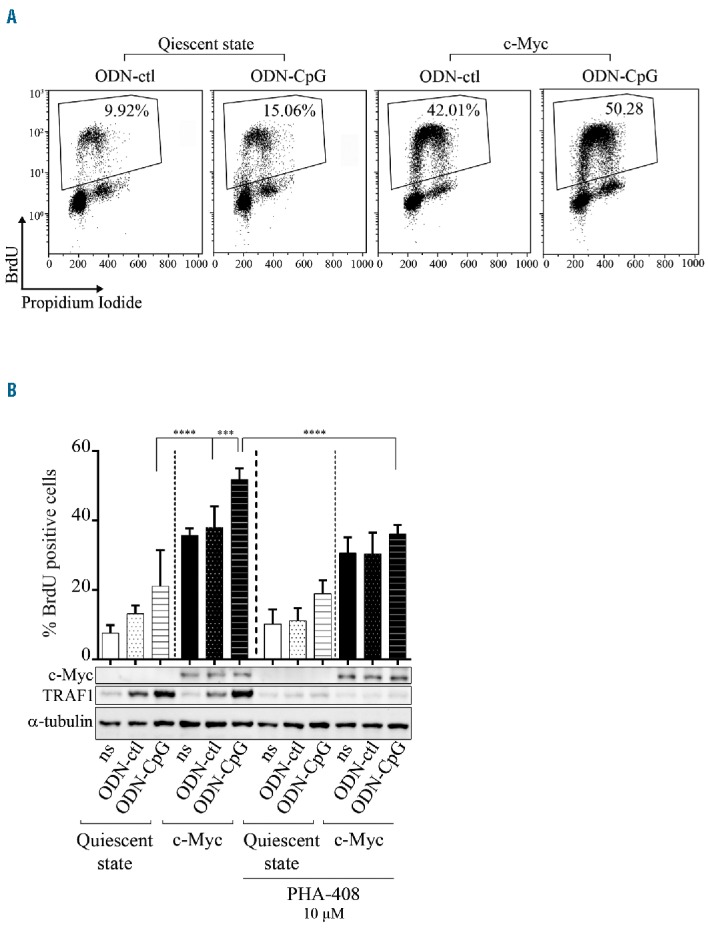
Effect of TLR9 stimulation and NF-κB implication on c-Myc overexpressing P493.6 cells. P493.6 cells were treated (quiescent state) or not (c-Myc condition) with 100 ng/mL tetracycline and stimulated with 0.5 μM oligodeoxynucleotides control (ODN-ctl) or containing CpG (ODN-CpG) for 48 hours. (A) Representative bivariate BrdU propidium iodide cytograms. Percentage of S-phase cells is indicated on each cytogram. (B) Upper panel: percentage of BrdU positive cells after ODN stimulation or not (ns) and 10 μM PHA-408 treatment. Data are shown as the mean ± standard deviation (at least 4 independent experiments). Statistical significance was determined by Student’s t-test (****P<0.0001; ***P<0.001). Lower panel: representative expression of c-Myc, TRAF1 and α-Tubulin as loading control by Western blot detection. BrdU: 5-bromo-2′-deoxyuridine.
We used the λc-Myc mice published by Kovalchuk et al.22 carrying an insertion of a translocated MYC gene cloned from the human BL60 cell line in order to investigate the cooperation of c-Myc overexpression and TLR9 signaling in vivo. As reported, all λc-Myc mice developed high grade B-cell lymphomas with similarities to BL, and median survival was 14 weeks (Online Supplementary Figure S4A). At pre-tumor stage (5–6-week old) mice had increased spleen weight, white blood cell count, and percentage of activated CD86 positive B cells, but without affecting the percentage of total B cells in the spleen (Online Supplementary Figure S4B,C). TLR9 stimulation with ODN-CpG led to strong NF-κB activation in splenocytes from both λc-Myc and wild-type (wt) mice (Online Supplementary Figure S5A). In vivo repeated ODN-CpG intraperitoneal injections induced a significant increase in spleen size and weight in both wt and λc-Myc mice (Figure 5A,B). This splenomegaly was much higher for half of the ODN-CpG injected λc-Myc mice when compared to the wt mice which were also stimulated: 361.3±49.7 mg vs. 216.9±14.5 mg (P = 0.0043). In vivo, ODN-CpG injection induced proliferation of B cells in spleens from both wt and λc-Myc mice. This increased proliferation was higher in ODN-CpG injected λc-Myc mice (Figure 5C). Similar results were obtained from lymph node B cells of the same mice (data not shown).
Figure 5.
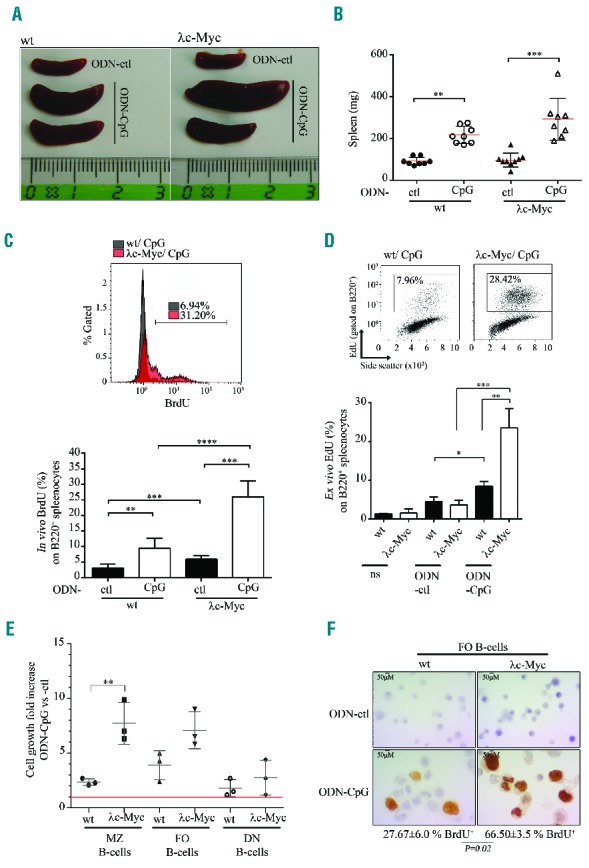
Effect of TLR9 stimulation on B cells from 4–5-week old λc-Myc (pre-tumor stage) and wild-type (wt) mice. In vivo stimulation with control (ODN-ctl) or CpG containing (ODN-CpG) oligodeoxynucleotides: 10-fold 50 μg ODN intraperitoneal injection every two days. Representative spleen size (A) and scatter-plot from spleen weight (B). B220 positive splenocyte proliferation after in vivo BrdU incorporation for about 24 hours (C): upper panel, overlay of representative BrdU labeling histograms gated on B220 positive cells from ODN-CpG injected λc-Myc and wt mice; and lower panel, mean of in vivo BrdU percentage on B220 positive splenocytes. Data are shown as mean ± standard deviation (n=6 mice in each experiment group). (D) Total splenocytes were ex vivo stimulated or not (ns) with control (ODN-ctl) or CpG containing motif (ODN-CpG) oligodeoxynucleotides. Proliferation at day 4 was assessed by EdU incorporation and labeling of B220 positive splenocytes. Upper panel: representative biparametric EdU side scatter cytograms gated on B220 positive cells. Percentages of EdU positive cells are indicated in each graph. Lower panel: mean percentages of EdU+ and B220+-cells. Data are shown as mean ± standard deviation (n=4 mice in each experiment group). (E) Cell growth of marginal zone (MZ), follicular (FO) and double negative (DN) spleen B cells sorted from wt and λc-Myc mice. Cells were counted by Trypan blue staining exclusion after 4 days stimulation. Red line indicates no cell growth fold increase in ODN-CpG condition versus ODN-ctl condition (value = 1). Data are shown as mean ± standard deviation (n=3 mice in each experiment group). (F) BrdU labeling of sorted FO B cells from wt (left panels) and λc-Myc mice (right panels) treated (CpG, lower panels) or not (ctl, upper panels) with CpG oligonuclotides for 4 days. BrdU incorporation was performed 4 hours before cells were cytocentrifuged. No BrdU incorporation was detectable in ctl conditions (upper panels). Mean and standard deviation of percentages of BrdU positive CpG treated cells are indicated (n=3 mice in each experiment group). Statistical significance was determined by Student’s t-test (****P<0.0001; ***P<0.001; **P<0.01; *P<0.05). EdU: 5-ethynyl-2′-deoxyuridine; BrdU: 5-bromo-2′-deoxyuridine.
To eliminate an indirect proliferating effect on ODN-CpG on B cells via stimulation of the microenvironment, ex vivo ODN-CpG stimulations of total splenocytes were done. TLR9 stimulation induced a significant increase in proliferation and viability of λc-Myc splenocytes when compared to wt cells (Online Supplementary Figure S5B,C). B-cell labeling after EdU incorporation showed that proliferating splenocytes belonged to the B-cell fraction with a higher proliferation when B cells were from λc-Myc spleens (Figure 5D). Cell growth assay and Ki-67 labeling on sorted B-cell subsets showed that this increased proliferation was mainly due to follicular (CD23-high, CD21-low) and marginal zone (CD23-low, CD21-high) B cells (Figure 5E,F). These data show that in vivo MYC/IG translocation with c-Myc overexpression provided an additional signal driving a stronger proliferation and survival of TLR9 activated B cells.
In vivo dysregulation of both CD40 signaling and c-Myc leads to aggressive diffuse large B-cell lymphoma with an activated phenotype
To assess the role of c-Myc in an in vivo model of NF-κB dependent B-cell lymphoma, we crossbred λc-Myc with L.CD40 transgenic mice (L.CD40/λc-Myc transgenic mice). L.CD40 mice are characterized by surface membrane expression of a chimeric LMP1/CD40 protein in CD19 positive B cells, resulting in a constitutively active CD40 signal in the B-cell lineage with constitutive activation of the NF-κB pathway as well as the mitogen-activated protein kinases Jnk and extracellular signal–regulated kinase.28 In these mice, deregulated CD40 signaling induces the development of indolent splenic B-cell lymphomas with an increase in the marginal zone B-cell compartment in more than 60% of mice older than 1 year.
Spleen size and weight were much higher in L.CD40/λc-Myc transgenic mice when compared to control mice (wt, L.CD40, and λc-Myc mice) (Figure 6A,B). L.CD40/λc-Myc mice prematurely developed enlarged lymph nodes at 10±2 weeks (vs. 12±3 weeks for λc-Myc mice). In contrast, lymph nodes were not enlarged in L.CD40 and wt mice (Figure 6B). If percentages of CD80 or CD86 activated B cells were increased in a comparable manner in both λc-Myc and L.CD40/λc-Myc mice (Figure 6C), CD80 and CD86 mean fluorescent intensities on B cells were significantly increased in L.CD40/λc-Myc mice compared to λc-Myc mice, both in spleen and lymph nodes (Figure 6D,E). The viability of splenocytes after three days culture in complete medium without stimulation indicated that protection against cell death was increased in L.CD40/λc-Myc tumor cells when compared to other conditions (Online Supplementary Figure S6). In both λc-Myc and L.CD40/λc-Myc mice, lymphoid tissue architecture was lost with a massive tumor infiltration, and the Ki-67 proliferation index was high (>90%) (Figure 6F). Tumor lymph nodes from λc-Myc histologically resembled BL with medium sized monomorphic tumor cells and the presence of numerous tingible body macrophages with apoptotic bodies giving a starry sky effect. In L.CD40/λc-Myc mice, lymphoma cells were highly atypical, being large and irregular, with abundant cytoplasm, large nuclei that possessed one to three or four prominent nucleoli usually located in the periphery of the nucleus, evoking the aspect of immunoblastic lymphomas according to the Bethesda classification of lymphoid neoplasms in mice34 (Figure 6F), for which the human counterpart would be DLBCLs with an activated phenotype.
Figure 6.
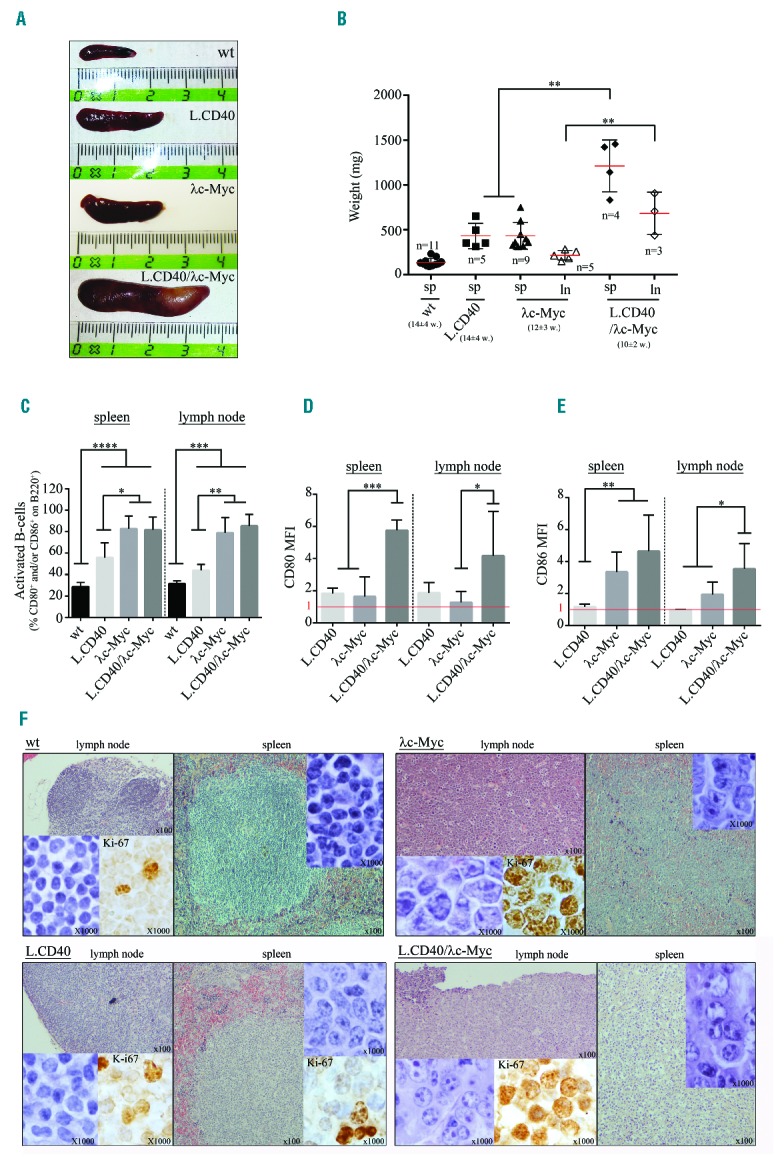
Phenotypic analysis of L.CD40/λc-Myc mice compared to wild-type (wt), L.CD40, and λc-Myc control mice. (A) Representative spleen size. (B) Scatter-plot of spleen (sp) and lymph node (ln) weights. For each group, number (n) and mean age of mice (w, week) are indicated. Data are shown as mean ± standard deviation (n=3–11 mice in each experiment group). Statistical significance was determined by Student’s t-test (**P<0.01). (C, D, and E) B220, CD80, and CD86 labeling from total splenocytes and lymph node cells. Histograms of activated B-cell percentages on B220+ cells i.e., percentage of CD80+ and/or CD86+ cells (C), mean fluorescent intensity (MFI) of CD80 (D) and CD86 (E) on B220 positive cells. MFI was normalized from wt mice (MFI=1 in red). Data are shown as mean ± standard deviation (n=3–11 mice in each experiment group). Statistical significance was determined by Student’s t-test (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001). (F) H&E and Ki-67 staining on lymph node and spleen sections.
Taking wt mice as a reference, gene expression profiles of tumors from lymph node samples suggest that deregulated genes could be separated into thirteen clusters. These clusters were grouped into six categories: upregulated genes in λc-Myc mice alone, in both λc-Myc and L.CD40/λc-Myc mice, in L.CD40 mice alone, in L.CD40/λc-Myc when compared to λc-Myc mice, downregulated genes in both λc-Myc and L.CD40/λc-Myc mice, and in L.CD40/λc-Myc mice alone (Figure 7, see also Online Supplementary Table S2). In agreement with the morphology, a specific macrophage signature was found in upregulated genes in λc-Myc mice alone. Both λc-Myc and L.CD40/λc-Myc tumors had numerous upregulated genes involved in cell cycle regulation, such as E2f7/8, Cemp, Ccnb2 and Ccne1/2. Metabolism processes were also among the highly upregulated functions in both mice models. Genes whose expression was increased in L.CD40 mice alone include those of the CD8 cytotoxic T cells and natural killer cells response, as well as those of the inflammatory and interferon response. This is in agreement with the original publication reporting an activated T-cell phenotype in these mice.28 In the meantime, expression of the immunosuppressive CD274/PDL.1 was increased in L.CD40 mice. CD274/PDL.1 was also increased in L.CD40/λc-Myc when compared to λc-Myc mice. Among over-expressed genes in L.CD40/λc-Myc compared to λc-Myc mice, the main represented biological functions were inflammation, transformation, cell activation, angiogenesis, transcription regulators, and NF-κB regulators such as Ltb, Tlr3, CD40, RelB, and Traf1. CD40 expression was increased in both L.CD40 and L.CD40/λc-Myc mice. This could be due to expression of the L.CD40 transgene or to endogenous CD40 expression, since one of the three probe sets spanning CD40 partially matched the C-terminal CD40 moiety of the L.CD40 protein while the two others did not. NF-κB target genes were induced in L.CD40/λc-Myc mice, including, for example, CD80, Fas, Tnfaip3, Bcl2, Ebi3 and Ccnd2. Repression of interferon response was significantly attenuated in these double L.CD40/λc-Myc mice when compared to λc-Myc mice, but was still present if compared to wt or L.CD40 mice. Genes coding for T-cell signature and T-cell activation markers were decreased in both λc-Myc and L.CD40/λc-Myc mice. Thus, as regards P493.6 cells in the c-Myc+/EBNA2+ condition, the main transcriptomic features of L.CD40/λc-Myc mice evoked increased proliferation, metabolism with apoptosis protection and attenuated interferon response as well as expression of the CD274/PDL1 molecule that inhibits the T-cell antitumor response. Moreover, expression of some B-cell markers such as CD79a, CD19 and CD22 was specifically decreased in L.CD40/λc-Myc. Even if lower CD19 expression could be the result of using CD19-Cre mice, its strongest decrease in L.CD40/λc-Myc mice together with the decrease of the B-cell receptor (BCR) associated CD22 molecule, of two BCR signaling molecules (CD79a and BANK1) as well as of a molecule normally expressed in germinal center B-cells (BCL11a), evokes features found in B cells with partial engagement in plasma cell differentiation.
Figure 7.
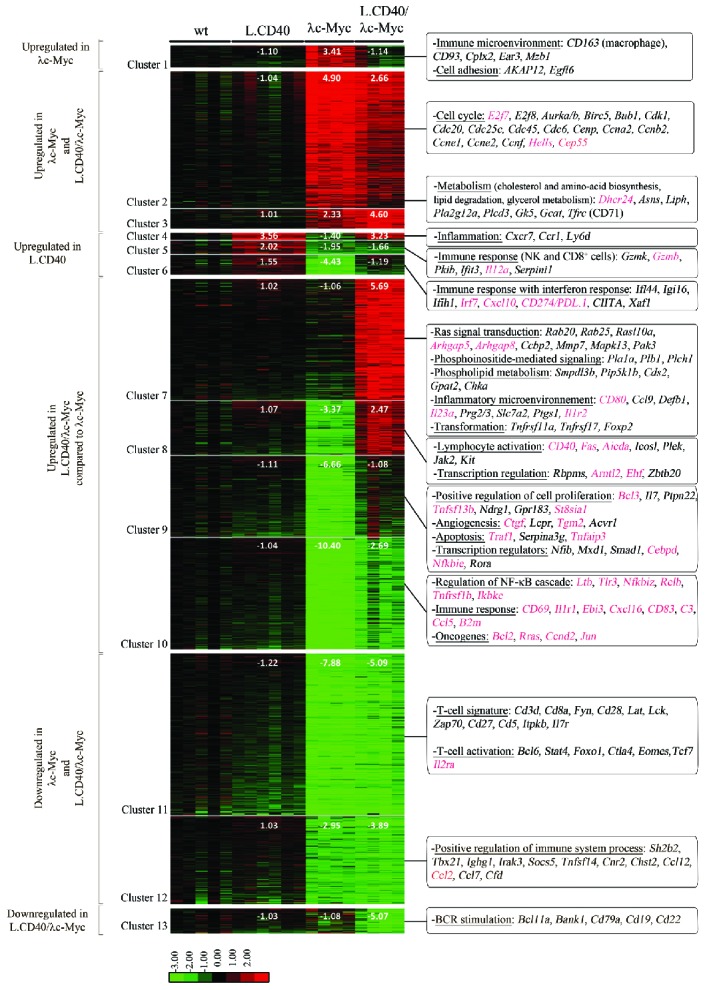
Gene expression profile of L.CD40/λc-Myc tumors compared to λc-Myc mice tumors. Supervised analysis led to the selection of 2,437 significant probes, which were initially partitioned into 30 clusters by K-means clustering so that the number of probes was above 15. The closest groups were merged 2 by 2 according to their proximity by hierarchical clustering and principal component analysis of the mean vectors. This was repeated until maximization of the absolute value of χ2.19 The final resulting number of clusters was 13 (Cluster 1 to 13). For each cluster and each sample condition, fold change value compared to wild-type (wt) samples were calculated from the mean of gene expression values and are indicated on the heat map. Most representative functions with most significant genes are mentioned on the right. NF-κB target genes are indicated in red. Log2 value color codes are shown beside the heat map. NF-κB: nuclear factor-κB; BCR: B-cell receptor.
These results suggest that c-Myc overexpression confers an aggressive DLBCL phenotype with a high proliferative index and an activated phenotype to L.CD40 activated B cells.
Discussion
EBV-positive DLBCLs of the elderly are characterized by prominent NF-κB and JAK/STAT activation,4,35 able to upregulate c-Myc expression in ABC-DLBCLs.36 B-cell lymphomas from HIV-infected patients very often have an ABC-DLBCL phenotype and are associated with MYC/IGH translocation.37 In LCLs, blocking c-Myc directly arrests cell growth and survival.19 In our EBV-infected human cell lines, the addition of c-Myc clearly favored cell growth with increased proliferation and metabolism, meanwhile, genes involved in immune surveillance and/or the inflammatory or interferon response were repressed. Enforced MyD88-L252P expression by retroviral infection of Eμ-Myc fetal liver cells dramatically shortened the time of tumor onset after transplantation in irradiated mice.38 In agreement, our results show that activation of MyD88 by TLR9 stimulation increased cell growth both in vitro in human cell lines and in vivo in the λc-Myc mouse model. c-Myc may play an important role in aggressive lymphomas with an activated phenotype related to NF-κB.
It is a paradox that, as a primary event, NF-κB activation is associated with both indolent and aggressive B-cell non-Hodgkin lymphomas (NHL). For example, mucosa-associated lymphoid tissue (MALT) indolent lymphomas frequently exhibit recurrent inactivating mutations of A20/TNFAIP3 that results in continuous activation of the classical NF-κB pathway, and these mutations are also found in ABC-DLBCLs.39,40 Waldenström macroglobulinemias are characterized by recurrent activating MyD88 mutations in 90% of cases, mainly MyD88-L265P, first described in ABC-DLBCLs,33,41,42 which again leads to the activation of NF-κB. This could suggest that NF-κB addiction in DLBCLs is associated with other genetic events that promote cell proliferation.
To our knowledge, only three mouse models for indolent lymphomas of the spleen have been published, one mimicking TRAF3 inactivation,43 the second with constitutive expression of Bcl10,44 and the last one with continuous CD40 signaling.28 The three models are characterized by an increased activation of RelB (i.e., the NF-κB alternative pathway), and B-cell lymphomas preferentially located in the spleen, with expansion of the marginal zone. B-TRAF3−/− deficient mice developed lymphomas that have been reported to be either low or high grade and to diffuse into other lymphoid organs as well as the bone marrow.43 Bcl10 transgenic mice were characterized by an expansion of marginal zone B cell due to lymphocyte accumulation, and some mice older than 8 months developed lymphoma resembling the human marginal zone lymphomas of the spleen.44 The evolution of L.CD40 transgenic mice was characterized by a progressive increase in splenomegaly and considerable expansion of the spleen B-cell area with acquisition of monoclonality, but the proliferative index remained constantly low, with no evidence for transformation into high grade lymphoma.28 Regarding the aggressive transformation of B cells in mice, Calado et al. reported that a mouse model with constitutive activation of IKK2 in B cells only developed plasma cell hyperplasia with a serum immunoglobulin peak.45 Additional ablation of the Blimp1/Prdm1 gene blocked plasma cell differentiation and led to the development of aggressive B-cell lymphomas resembling ABC-DLBCLs. Zhang et al. showed that in vivo enforced expression of NIK in germinal center B cells led to plasma cell hyperplasia, again without lymphoma. But the combination of NIK and Bcl6 enforced expression led to the development of aggressive lymphomas resembling ABC-DLBCLs.46 These results on animal models suggest that in vivo continuous NF-κB activation would favor B cells, but other events, such as Blimp1 inactivation or Bcl6 dysregulation, seem to be needed for transformation into an aggressive DLBCL.
Both BLIMP1 and BCL6 are transcriptional repressors of the MYC gene.47,48 Consequently, BLIMP1 disruption can cause increased c-Myc expression.20 In DLBCLs tumor B cells become refractory to c-Myc repression by BCL6.49 Furthermore, BCL6 expression represses that of BLIMP1.48 This suggests that BCL6 may indirectly activate c-Myc transcription through repression of BLIMP1 expression.20 Thus, c-Myc could be the good endpoint target of various secondary events to then cooperate with NF-κB to promote aggressive B-cell lymphomagenesis.
Contrasting with mice models with constitutive NF-κB activation in B cells; all mice models with c-Myc overexpression in B cells develop aggressive B-cell tumors. For example, λc-Myc mice had an early aggressive tumor onset primarily located in lymph nodes with secondary dissemination to the spleen.22 Morphology of these tumors evokes that of human BL, with a very high proliferative index. Our results show that double L.CD40/λc-Myc mice have very aggressive tumors with systemic dissemination to all lymphoid compartments of the organism. Immunoblastic morphology of these tumors evokes that of ABC-DLBCLs in humans, being completely different from that of both L.CD40 and λc-Myc mice. These features can be viewed either as the c-Myc-driven transformation of a CD40-dependent indolent lymphoma, or as a shift from a c-Myc-driven aggressive B-cell lymphoma resembling human BL to an activated phenotype due to continuous CD40 activation. Functional studies clearly indicate cooperation between L.CD40 and λc-Myc transgenes in B-cell transformation. This cooperation was also found at the gene expression level, with upregulated genes involved in transformation, cell activation, and angiogenesis, such as Tnfaip3, Rab20/25, CD40, Arntl2, Ctgf, Bcl2, Rras, Ccnd2, and Jun. Most of these genes are known to be NF-κB targets.
λc-Myc and L.CD40/λc-Myc tumors were characterized by a marked decrease in the T-cell signature. This could be either due to massive infiltration of the tumor or to lack of immune control. The growth of c-Myc-driven tumors is rather likely to be independent of their immune microenvironment.50 By contrast, escaping immune surveillance is clearly a key step for tumor emergence and/or tumor progression in DLBCLs.50 As illustrated herein by our transcriptome results, Hömig-Hölzel et al. have previously shown that L.CD40 mice exhibited activating T-cell expansion.28 However, CD274/PDL1 expression was found to be increased in L.CD40 tumors. Even if attenuated, such CD274/PDL1 expression was also increased in L.CD40/λc-Myc tumors when compared to those of λc-Myc. Thus, combining the lack of T-cell signatures, including T-cell activation markers, with CD274/PDL1 expression by the tumor (a L.CD40 effect) and decreased interferon response (a c-Myc effect) in L.CD40/c-Myc mice could in part explain the aggressiveness of tumor growth, which is under a lesser degree of immune control. Therefore, c-Myc increased the tumor potency of NF-κB activated B cells in terms of proliferation, protection against apoptosis and tumor aggressiveness.
In conclusion, we demonstrated that adding c-Myc to B cells with NF-κB activated by either LMP1, TLR9 or CD40, constantly increased the proliferation potential of cells and/or the aggressiveness of the tumor both in vitro and in animal models. Furthermore, we showed that adding c-Myc to CD40 activation in mice transformed indolent into aggressive lymphomas with features of NF-κB-activated cells. These results highlight the role of these two master transcriptional systems in B-cell lymphomagenesis, explaining why NF-κB is associated with both indolent and aggressive lymphomas and opening new perspectives on the possibility of combinatory therapies targeting both the c-Myc proliferating program and NF-κB activation pathways in DLBCLs.
Supplementary Material
Acknowledgments
We are especially grateful to Pr GW Bornkamm, Research Unit Gene Vectors, Helmholtz Center Munich, German Research Center for Environmental Health GmbH, Germany, for helping us to initiate the project and for continuous support and helpful discussions. We thank Dr A Marfak, Statistical Unit, IFCS, Rabat, Morocco, for transcriptome analysis. We thank Dr J Cook Moreau, UMR CNRS 7276, Limoges, France, for English editing.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/5/883
Funding
The group of J Feuillard is supported by grants from the Ligue National contre le Cancer (Equipe Labellisée Ligue), the Institut National contre le Cancer (INCa), the Comité Orientation Recherche Cancer (CORC), the Limousin Region and the Haute Vienne and Corrèze comitees of the Ligue Nationale contre le Cancer and by the Lyons Club of Corrèze. U Zimber Strobk was supported by the DFG grant: GZ: ZI 1382/4-1.
References
- 1.Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362(15):1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pham LV, Tamayo AT, Yoshimura LC, et al. A CD40 Signalosome anchored in lipid rafts leads to constitutive activation of NF-kappaB and autonomous cell growth in B cell lymphomas. Immunity. 2002;16(1):37–50. [DOI] [PubMed] [Google Scholar]
- 3.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11(1):12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montes-Moreno S, Odqvist L, Diaz-Perez JA, et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod Pathol Off J U S Can Acad Pathol Inc. 2012;25(7):968–982. [DOI] [PubMed] [Google Scholar]
- 5.Castillo JJ, Beltran BE, Miranda RN, Young KH, Chavez JC, Sotomayor EM. EBV-positive diffuse large B-cell lymphoma of the elderly: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91(5):529–537. [DOI] [PubMed] [Google Scholar]
- 6.Bavi P, Uddin S, Bu R, et al. The biological and clinical impact of inhibition of NF- B-initiated apoptosis in diffuse large B cell lymphoma (DLBCL). J Pathol. 2011; 224(3):355–366. [DOI] [PubMed] [Google Scholar]
- 7.Liebowitz D. Epstein-Barr virus and a cellular signaling pathway in lymphomas from immunosuppressed patients. N Engl J Med. 1998;338(20):1413–1421. [DOI] [PubMed] [Google Scholar]
- 8.Feuillard J, Schuhmacher M, Kohanna S, et al. Inducible loss of NF-kappaB activity is associated with apoptosis and Bcl-2 downregulation in Epstein-Barr virus-transformed B lymphocytes. Blood. 2000; 95(6):2068–2075. [PubMed] [Google Scholar]
- 9.Zou P, Kawada J, Pesnicak L, Cohen JI. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J Virol. 2007; 81(18):10029–10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao C, Silverberg MJ, Martínez-Maza O, et al. Epstein-Barr virus infection and expression of B-cell oncogenic markers in HIV-related diffuse large B-cell Lymphoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18(17):4702–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y-H, Xu F-P, Zhuang H-G, et al. Clinicopathologic significance of immunophenotypic profiles related to germinal center and activation B-cell differentiation in diffuse large B-cell lymphoma from Chinese patients. Hum Pathol. 2008; 39(6):875–884. [DOI] [PubMed] [Google Scholar]
- 12.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Hematol Educ Program Am Soc Hematol Am Soc Hematol Educ Program. 2013;122(24):3884–3891. [DOI] [PubMed] [Google Scholar]
- 13.Valera A, López-Guillermo A, Cardesa-Salzmann T, et al. MYC protein expression and genetic alterations have prognostic impact in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Haematologica. 2013;98(10):1554–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuhlmann-Laeisz C, Borchert A, Quintanilla-Martinez L, et al. In Europe expression of EBNA2 is associated with poor survival in EBV-positive diffuse large B-cell lymphoma of the elderly. Leuk Lymphoma. 2016;57(1):39–44. [DOI] [PubMed] [Google Scholar]
- 15.Kohlhof H, Hampel F, Hoffmann R, et al. Notch1, Notch2, and Epstein-Barr virus-encoded nuclear antigen 2 signaling differentially affects proliferation and survival of Epstein-Barr virus-infected B cells. Blood. 2009;113(22):5506–5515. [DOI] [PubMed] [Google Scholar]
- 16.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004; 4(10):757–768. [DOI] [PubMed] [Google Scholar]
- 17.Lee JM, Lee K-H, Weidner M, Osborne BA, Hayward SD. Epstein-Barr virus EBNA2 blocks Nur77- mediated apoptosis. Proc Natl Acad Sci USA. 2002;99(18):11878–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J Virol. 1999;73(5):4481–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faumont N, Durand-Panteix S, Schlee M, et al. c-Myc and Rel/NF-kappaB are the two master transcriptional systems activated in the latency III program of Epstein-Barr virus-immortalized B cells. J Virol. 2009; 83(10):5014–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wierstra I, Alves J. The c-myc promoter: still MysterY and challenge. Adv Cancer Res. 2008;99:113–333. [DOI] [PubMed] [Google Scholar]
- 21.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell. 1986. October 10;47(1):11–18. [DOI] [PubMed] [Google Scholar]
- 22.Kovalchuk AL, Qi CF, Torrey TA, et al. Burkitt lymphoma in the mouse. J Exp Med. 2000;192(8):1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truffinet V, Pinaud E, Cogné N, et al. The 3’ IgH locus control region is sufficient to deregulate a c-myc transgene and promote mature B cell malignancies with a predominant Burkitt-like phenotype. J Immunol. 2007;179(9):6033–6042. [DOI] [PubMed] [Google Scholar]
- 24.Kempkes B, Zimber-Strobl U, Eissner G, et al. Epstein-Barr virus nuclear antigen 2 (EBNA2)-oestrogen receptor fusion proteins complement the EBNA2-deficient Epstein-Barr virus strain P3HR1 in transformation of primary B cells but suppress growth of human B cell lymphoma lines. J Gen Virol. 1996;77(Pt 2):227–237. [DOI] [PubMed] [Google Scholar]
- 25.Pajic A, Spitkovsky D, Christoph B, et al. Cell cycle activation by c-myc in a burkitt lymphoma model cell line. Int J Cancer. 2000;87(6):787–793. [DOI] [PubMed] [Google Scholar]
- 26.Chanut A, Duguet F, Marfak A, et al. RelA and RelB cross-talk and function in Epstein-Barr virus transformed B cells. Leukemia. 2014;28(4):871–879. [DOI] [PubMed] [Google Scholar]
- 27.Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14(12):2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hömig-Hölzel C, Hojer C, Rastelli J, et al. Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-kappaB pathway and promotes lymphomagenesis. J Exp Med. 2008; 205(6):1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Clorennec C, Ouk T-S, Youlyouz-Marfak I, et al. Molecular basis of cytotoxicity of Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) in EBV latency III B cells: LMP1 induces type II ligand-independent autoactivation of CD95/Fas with caspase 8-mediated apoptosis. J Virol. 2008;82(13): 6721–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuhmacher M, Kohlhuber F, Hölzel M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornkamm GW, Berens C, Kuklik-Roos C, et al. Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic Acids Res. 2005;33(16):e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mbalaviele G, Sommers CD, Bonar SL, et al. A novel, highly selective, tight binding IkappaB kinase-2 (IKK-2) inhibitor: a tool to correlate IKK-2 activity to the fate and functions of the components of the nuclear factor-kappaB pathway in arthritis-relevant cells and animal models. J Pharmacol Exp Ther. 2009;329(1):1–25. [DOI] [PubMed] [Google Scholar]
- 33.Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011; 470(7332):115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morse HC, Anver MR, Fredrickson TN, et al. Bethesda proposals for classification of lymphoid neoplasms in mice. Blood. 2002; 100(1):246–258. [DOI] [PubMed] [Google Scholar]
- 35.Kato H, Karube K, Yamamoto K, et al. Gene expression profiling of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014;105(5):537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding BB, Yu JJ, Yu RY-L, et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. 2008;111(3):1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morton LM, Kim CJ, Weiss LM, et al. Molecular characteristics of diffuse large B-cell lymphoma in human immunodeficiency virus-infected and -uninfected patients in the pre-highly active antiretroviral therapy and pre-rituximab era. Leuk Lymphoma. 2014;55(3):551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knittel G, Liedgens P, Korovkina D, et al. B-cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood. 2016;127(22):2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–716. [DOI] [PubMed] [Google Scholar]
- 40.Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012; 367(9):826–833. [DOI] [PubMed] [Google Scholar]
- 42.Gachard N, Parrens M, Soubeyran I, et al. IGHV gene features and MYD88 L265P mutation separate the three marginal zone lymphoma entities and Waldenström macroglobulinemia/lymphoplasmacytic lymphomas. Leukemia. 2013;27(1):183–189. [DOI] [PubMed] [Google Scholar]
- 43.Moore CR, Liu Y, Shao C, Covey LR, Morse HC, Xie P. Specific deletion of TRAF3 in B lymphocytes leads to B-lymphoma development in mice. Leukemia. 2012;26(5):1122–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Z, Wang H, Xue L, et al. Emu-BCL10 mice exhibit constitutive activation of both canonical and noncanonical NF-kappaB pathways generating marginal zone (MZ) B-cell expansion as a precursor to splenic MZ lymphoma. Blood. 2009;114(19):4158–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calado DP, Zhang B, Srinivasan L, et al. Constitutive canonical NF- B activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18(6):580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Calado DP, Wang Z, et al. An oncogenic role for alternative NF-κB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep. 2015;11(5):715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin Y, Wong K, Calame K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science. 1997;276(5312):596–599. [DOI] [PubMed] [Google Scholar]
- 48.Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13(2):199–212. [DOI] [PubMed] [Google Scholar]
- 49.Ci W, Polo JM, Cerchietti L, et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood. 2009; 113(22):5536–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fowler NH, Cheah CY, Gascoyne RD, et al. Role of the tumor microenvironment in mature B-cell lymphoid malignancies. Haematologica. 2016;101(5):531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.