

Abstract
Chemical reactions that are named in honor of their true or at least perceived discoverers are known as “name reactions”. This review is a collection of biological representatives of named chemical reactions. Emphasis is placed on reaction types and catalytic mechanisms that showcase both the chemical diversity in natural product biosynthesis as well as the parallels with synthetic organic chemistry. An attempt has been made to describe the enzymatic mechanisms of catalysis within the context of their synthetic counterparts whenever possible and to discuss the mechanistic hypotheses for those reactions that are presently active areas of investigation. This review has been categorized by reaction type, e.g., condensation, nucleophilic addition, reduction and oxidation, substitution, carboxylation, radical-mediated, and rearrangements, which are subdivided by named reactions.
Keywords: name reaction, enzyme, mechanism, biosynthesis, catalysis
1. Introduction
The field of organic chemistry is extraordinarily rich due to the seemingly limitless number of structurally diverse compounds that can be constructed with carbon. A significant catalog of chemical reactions has been compiled in the last several decades. Many of these reactions have been developed by generation after generation of scientists, each working, often in obscurity, to make their own small addition to this vast collection of chemical knowledge. For those lucky enough to discover a new process involving a particularly useful and exciting transformation or one that simply came to represent an established paradigm, the honor of having the reaction named after them stamps their mark on the history of scientific discovery. Approximately 500 named reactions exist in the Merck index, from the Pelouze synthesis,[1] first described in 1834 to the Shi epoxidation[2] described in 1996. While the majority of these named reactions do not have a direct biological counterpart, the ongoing study of primary and secondary metabolic pathways has shown that a good number of them do.
In contrast to the tools available to the synthetic organic chemist, for whom there is considerable leeway with regard to efficiency as well as a full panoply of available solvents, temperatures and reagents, biochemical reactions are generally constrained by limited resources and restricted to the relatively narrow range of conditions that living systems can tolerate. Thus, it is not surprising that a large number of biological reactions are characterized by fairly standard Lewis acid-base chemistry. However, while a researcher will use thought and reason in the lab to discover new and possibly beneficial reactions, nature has used mutation and selection in approximately 1030 cells[3] over billions of years to expand life’s chemical processes to the current level of diversity. The Bergman cyclization in the activation of enediyne antibiotics[4] or the Baeyer-Villiger oxidation in the degradation of xenobiotic toxins[5] are just two examples of how biology uses novel chemical processes to expand its capabilities. Thus, named organic reactions are indeed well represented in biology.
As we delve deeper into the inner workings of the cell we find that just as often as chemistry can explain biology, biology can inspire chemistry. This symbiotic relationship drives both fields to new heights of understanding. In this review we highlight the parallels in the “lab” chemistry to the chemistry of the cell by describing a number of examples of named reactions that have also been identified in biology. We included the enzymatic reaction mechanism whenever it is known, but have also included examples where the reaction mechanism for the biological process has strong support, even the specific role of the enzyme remains obscure. This review is categorized according to the type of reaction being discussed: condensation, nucleophilic addition, reduction and oxidation, substitution, carboxylation, radical-mediated, and rearrangements. These sections are then subdivide into the named reactions with the examples of their biological parallels.
2. Condensation Reactions
2.1. Aldol Condensation/Aldol Reaction
The aldol condensation reaction is the nucleophilic attack by the α-carbon of an aldehydic or ketonic enolate upon the carbonyl carbon of another aldehyde or ketone to form a β-hydroxy product (Figure 1). This is followed by dehydration of the nascent aldol product to give a conjugated enone product. The aldol condensation, first reported by Charles Adolph Wurtz in 1872,[6] derives its name from a particular case in which an enolate reacts with an aldehyde carbonyl to afford an alcohol. While Aldol condensation is not a real “name reaction” by strict definition, we decide to include it here since a number of name reactions are variations of the aldol condensation, such as Claisen condensation, which is an aldol condensation between two esters to generate a β-keto ester,[7] and Dieckmann condensation which is an intramolecular version of Claisen condensation.[8] It should be noted that many biological reactions involve nucleophilic addition of an enolate to an aldehyde to yield a β-hydroxyl aldol product without subsequent dehydration. In this review, we opt to group reactions leading to the formation of aldol products under the class of aldol condensation and aldol reaction. The aldol reaction and the related Claisen reaction are the methods-of-choice for enzymes catalyzing C-C bond formation (biosynthetic mode) and C-C bond cleavage (degradative mode) reactions. In fact, aldol reaction is the underlining theme of a great number of enzymes catalyzing diverse reactions.
Figure 1.

Aldol condensation and related reactions.
2.1.1. Aldolases
Many enzymes are capable of catalyzing aldol reactions. A group of them known as aldolases are especially synthetically useful. These enzymes can be divided into two classes, type I and type II aldolases, and have distinct evolutionary origins (Figure 2).[9] The reaction mechanism of type I aldolases includes the formation of a Schiff base (2) between the donor substrate (1) and a conserved lysine residue in the active site, which after α-deprotonation adds stereospecifically to the carbonyl group of an acceptor (3). Instead of relying on employing an iminium ion to facilitate enolate formation, type II aldolases utilize a bound Zn2+ ion to stabilize the enolate intermediate generated after deprotonation (5). Type I aldolases are found primarily in animals and higher plants and the type II enzymes are found in microorganisms.
Figure 2.

Reaction mechanisms of type I and II aldolases. (a) Fructose 1,6-diphosphate aldolase (type I) and (b) fuculose 1-phosphate aldolase (type II).
The canonical aldolase enzyme, fructose 1,6-diphosphate (FDP) aldolase, catalyzes the fourth step in the glycolytic pathway – the reversible cleavage of FDP (4) to glyceraldehyde-3-phosphate (G3P, 3) and dihydroxyacetone phosphate (DHAP, 1). FDP aldolase can accept a wide range of aldehyde acceptor substrates with DHAP as the donor to generate (3S,4R) vicinal diols as the products.[9] Besides DHAP, pyruvate, phosphoenol pyruvate, or acetaldehyde can also be used by various aldolases as the donor substrate to generate the reactive enolate intermediate. Because the enzymatic aldol C-C bond formation and cleavage reactions are versatile and highly stereospecific, they are useful for synthetic applications. For a comprehensive review on aldolases see Brovetto et. al.[10]
2.1.2. Porphobilinogen synthase-catalyzed aldol condensation reaction
While it is not always apparent, many enzyme-catalyzed reactions consisting of multi-step transformations involve aldol addition as a key reaction. For example, the first common step in the biosynthesis of tetrapyrroles in heme and vitamin B12 catalyzed by porphobilinogen synthase is the condensation of two molecules of 5-aminolevulinic acid (8) to give porphobilinogen (12, Figure 3).[11] The reaction is initiated by Schiff base (9) formation between the first 5-aminolevulinic acid (at P-side) with a lysine residue in the active site. The second 5-aminolevulinic acid (at A-side) is bound to a Zn2+ ion, which facilitates the formation of the second Schiff base between the amino group from the substrate at P-side with the 4-keto group of the substrate at A side.[12] α-Deprotonation to give the enamine 10 followed by an intramolecular aldol addition leads to a 5-membered ring intermediate (11, Figure 3, route A). It is also possible that ring formation is a direct aldol addition of the C-3 enolate of the substrate at A-side to the 4-iminium group of the substrate at the P-side (Figure 3, route B). Subsequent elimination and tautomerization gives porphobilinogen (12) as the final product. It is important to note that the conversion of 10 to 11 is reminiscent to that of Stork Enamine Alkylation reaction, which was developed by Gilbert Stork in 1960s and is a widely used C-C bond coupling reaction.[13]
Figure 3.

Proposed reaction mechanism for porphobilinogen synthase.
2.1.3. Enzyme-catalyzed retro-aldol reactions
Since aldol addition reaction is reversible, a number of enzymes catalyze the retro-aldol reaction as their primary biological functions. For example, dihydroneopterin aldolase catalyzes the cleavage of 7,8-dihydroneopterin (15) to 6-hydroxymethyl-7,8-dihydropterin (18) and glycolaldehyde (17) during folic acid biosynthesis (Figure 4).[14] The pyridoxal-5′-phosphate (PLP, 21)-dependent serine hydroxymethylase catalyzes the reversible conversion of L-serine (22) to glycine (27) and a one-carbon fragment at the oxidation level of formaldehyde (24), which is then transferred to tetrahydrofolate (THF, 20) to form N5-N10-methylene-THF (28), an important methyl source in biological systems (Figure 5).[15] The transformation from 23 to 25 is a retro-aldol reaction and the stabilized glycine-coenzyme imine intermediate (26) is eventually hydrolyzed to glycine (27).
Figure 4.

The folic acid biosynthetic pathway in bacteria.
Figure 5.

Proposed mechanism of serine hydroxymethylase.
The first committed step in the non-mevalonate pathway to produce isoprenes is the conversion of 1-deoxy-D-xylulose 5-phosphate (DXP, 29), to methyl-D-erythritol 4-phosphate (MEP, 30) catalyzed by the NADPH-dependent DXP reductoisomerase (DXR, also known as MEP synthase, see Figure 6).[16] Studies of the secondary kinetic isotope effects measured in the equilibrium perturbation experiments with [3-2H]- and [4-2H]-DXP provided direct evidence supporting a retro-aldol/aldol mechanism for the DXR-catalyzed rearrangement of DXP (29) to MEP (30).[17, 18]
Figure 6.

Proposed mechanism of 1-deoxy-D-xylulose-5-phosphate reducto-isomerase.
In a recent study of the biosynthesis of baumycin A1 (31), the flavin adenine dinucleotide (FAD)-dependent nitrososynthase (DnmZ) was shown to catalyze the oxidation of TDP-L-epi-vancosamine (32) to a vicinal-hydroxyl-nitrososugar (33) which then underwent C3−C4 bond cleavage to give an acyclic aldehyde-oxime product (34, Figure 7).[19] The overall rearrangement is a retro oxime-aldol transformation and bears a resemblance to that by Type I aldolases. The polarized nitroso functional group in this case is the surrogate for the iminium group of an aldolase. This aldehyde-oxime product (34) will eventually be reduced and incorporated (in the form of 35) into baumycin A1 (31).
Figure 7.

Proposed mechanism of DnmZ.
2.2. Claisen Condensation
The Claisen condensation is named after Rainer Ludwig Claisen, who first reported this reaction in 1887.[7] Similar to the aldol condensation, the Claisen condensation is a common mode of C-C bond formation in nature. It differs from aldol reaction in which the acceptor is an ester or thioester instead of an aldehyde or ketone, and the product is a β-keto ester or thioester (Figure 1). The Claisen condensation fulfills a prominent role in biological chemistry as the means by which C-C bonds are formed during the biosynthesis of fatty acids and polyketides, which will be highlighted in the following section. Many enzyme-catalyzed C-C bond cleavage reactions are effectively the reverse Claisen reactions. Some well-known examples include citrate lyase[20] and thiolase.[21] The reaction catalyzed by PLP-dependent kynureninase also involves a retro-Claisen step to cleave L-kynurenine (36) to anthranilic acid (37) and L-alanine (38, Figure 8).[22] Interestingly, enzymes catalyzing Claisen condensation generally produce a new C-C bond with inversion of stereochemistry relative to the α-proton that is abstracted; this is in contrast to the retention of configuration observed for the enzyme-catalyzed aldol reactions.
Figure 8.

Kynureninase catalyzes a retro-Claisen reaction.
2.2.1. Claisen condensations in fatty acid biosynthesis
Fatty acid biosynthetic systems can be divided into two groups. The type II system found in plants and bacteria is composed of seven discrete enzymes,[23] whereas the type I system found in animals is a single, high molecular weight protein dimer containing all of the functionalities of the type II system in each polypeptide chain.[24] The designations of the enzymes described below is based on the type II system of Escherichia coli but is generalizable to the type I system as well. The proposed reaction mechanisms are based largely on X-ray crystal structures of the enzymes.[25]
Fatty acid carbon chains are elongated two carbons at a time in a reaction catalyzed by β-ketoacyl-acyl carrier protein (ACP) synthases (Figure 9a). The two-carbon unit is provided by malonyl-ACP (40), which is derived from coupling of malonyl-CoA (39) to ACP via a phosphopantetheinyl linkage by malonyl-CoA-ACP transacylase, known as FabD (where “Fab” stands for fatty acid biosynthesis). Fatty acid biosynthesis is initiated by β-ketoacyl-ACP synthase III (FabH), which uploads an acetate from acetyl-CoA (41) to a conserved cysteine residue in its active site to form an enzyme-linked thioester (acetyl-FabH). The second half of the FabH reaction is a decarboxylating Claisen condensation. As shown in Figure 9b, malonyl-ACP (40) undergoes decarboxylation and the resulting enolate attacks the thioester carbonyl carbon of acetyl-FabH to give acetoacetyl-ACP (42). The β-carbonyl group of acetoacetyl-ACP is reduced to a hydroxyl group and the reduced product (43) undergoes dehydration resulting in a C-C double bond, which is subsequently saturated. These steps are catalyzed by FabG, FabA or Z, and FabI, respectively, and occur after each round of condensation.
Figure 9.

(a) Type II fatty acid biosynthesis. (b) Mechanisms of Fab-H catalyzed and (c) FabB or FabF-catalyzed decarboxylating Claisen condensations.
After the first condensation reaction mediated by FabH, carbon chain elongation is carried out by further decarboxylating Claisen condensations catalyzed by β-ketoacyl-ACP synthases I and II (FabB and FabF, respectively). Both enzymes operate via the same mechanism as FabH (Figure 9c). The acyl chain is transferred from ACP to a conserved active site cysteine residue on β-ketoacyl-ACP synthase and the resulting enzyme-thioester carbonyl carbon is attacked by acetyl-ACP enolate (derived from decarboxylation of malonyl-ACP). The difference between FabB and FabF is that FabB is specifically involved in the elongation of unsaturated fatty acids and FabF controls the temperature-dependent regulation of fatty acid composition. The final fatty acid product is hydrolytically cleaved from ACP by the thioesterase (TEs).[26]
There is also a family of enzymes known as thiolases that catalyze non-decarboxylating Claisen condensations. They fall into two classes: biosynthetic and degradative thiolases. Biosynthetic thiolase catalyzes the condensation of acetyl-CoA with an acetate, which is derived from a second molecule of acetyl-CoA, to give acetoacetyl-CoA.[27] Degradative thiolase, known as FadA in E. coli, catalyzes the reverse reaction – the thiolytic cleavage of an acetate from β-ketoacyl-CoA, which is part of fatty acid breakdown commonly referred as β-oxidation.[28] The activities of both degradative and biosynthetic thiolases are reversible and their general reaction mechanism is depicted in Figure 10.
Figure 10.

Thiolase-catalyzed biosynthetic and degradative reactions.
2.2.2. Claisen condensations in polyketide biosynthesis
Similarly, β-ketoacyl synthase mechanisms are operative in the biosynthesis of polyketides, which are one of the most important classes of natural products. The formation of polyketides is catalyzed by polyketide synthases (PKSs), which are divided into type I and II PKS (Figure 11). These designations mirror those described for fatty acid biosynthesis in that a type I PKS is a single, large enzyme containing multiple functional domains, including ketosynthase (KS), acyl transferase (AT), ACP domains and possibly ketoreductase (KR), dehydratase (DH), enoyl reductase (ER), methyltransferase (MT), TE domains. In contrast, a type II PKS is a catalytic protein complex consisting of two ketosynthase units (KSα and KSβ) and an ACP. Biosynthesis of the polyketide chain is constructed via a series of decarboxylative Claisen condensations catalyzed by ketosynthases (KSs) in a manner analogous to fatty acid biosynthesis (Figure 11a).[29] PKSs can be further classified as iterative or non-iterative systems based on whether the KS domain is reused in the chain elongation or not.
Figure 11.
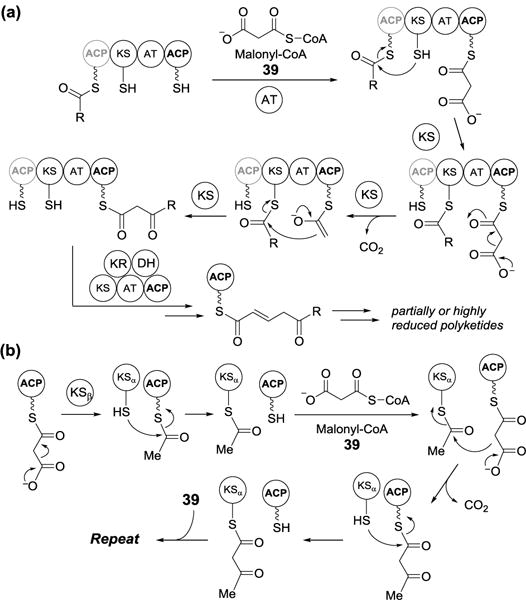
(a) Type 1 and (b) type II polyketide synthase-catalyzed reactions.
Interestingly, a third class of PKSs (type III) are singular bifunctional enzymes that catalyze both Claisen condensation and cyclization reactions.[30] The prototypical type III PKSs is chalcone synthase (CHS), which catalyzes the condensation of p-coumaric acid (from p-coumaryl-CoA, 48) to 3 molecules of acetate (from malonyl-CoA, 39). The linear ketide intermediate then undergoes cyclization and aromatization to yield chalcone (49), a starting material for a number of plant metabolites (Figure 12).[31] The cyclization portion of the CHS reaction is a Claisen-like C6→C1 condensation. This is a contrast to the C2→C7 aldol-like cyclization catalyzed by the type III PKS stilbene synthase (STS). The difference in the cyclization mechanism between these two enzymes is the result of subtle, electrostatic variation between their active sites (Figure 12).[32] Since PKSs have been extensively reviewed,[33–36] the details of their catalyses will not be discussed here.
Figure 12.
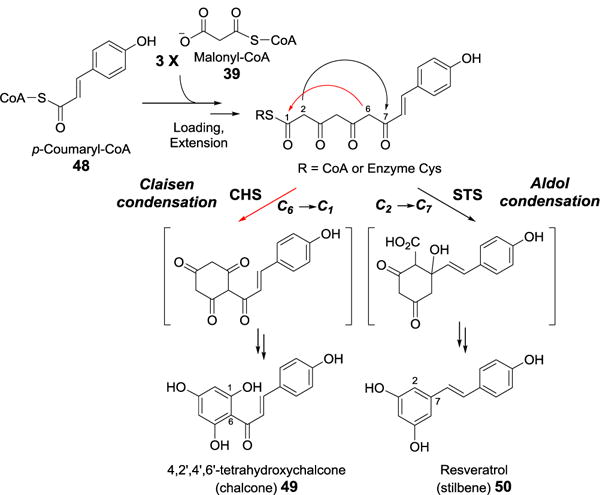
Reaction mechanisms of chalcone and stilbene synthase.
2.3. Dieckmann Condensation
The Dieckmann condensation is an intramolecular cyclization of a diester to form a β-ketoester product under basic conditions (Figure 1). It is named after the German chemist, Walter Dieckmann, who first reported this reaction in 1894.[8] This reaction has been widely used to prepare five- and six-membered cyclic β-ketoester structures. A few biological precedents of Dieckmann-like condensations have recently been identified, such as formation of tetramic acid catalyzed by several hybrid PKS-non-ribosomal peptide synthases (PKS-NRPS), the MenB-catalyzed naphthoquinone formation in the biosynthesis of vitamin K, and the BadI-mediated production of pimelyl-CoA in the catabolism of benzoate. Although the molecules involved in these examples are not exactly diesters, the intramolecular condensations between two carbonyl-containing functional groups catalyzed by these enzymes show strong resemblance to Dieckmann condensation.
2.3.1. Formation of tetramic acid catalyzed by hybrid polyketide-non-ribosomal peptide synthase
A number of hybrid polyketide-non-ribosomal peptides, or their precursors, contain pyrrolidine-2,4-dione ring systems, also known as tetramic acids (51, Figure 13). These compounds are produced by an array of terrestrial and marine organisms such as fungi, cyanobacteria, and actinobacteria and display various bioactivities.[37] Examples include equisetin,[38] cyclopiazonic acid,[39] chaetoglobosin A,[40] heat-stable antifungal factor (HSAF),[41] and ikarugamycin (52).[42]
Figure 13.
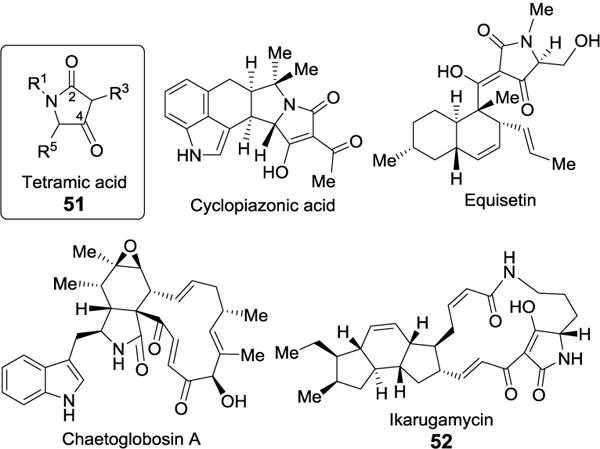
Tetramic acid-containing natural products.
The genes responsible for the biosynthesis of these tetramic acid-containing compounds have been identified. In all cases, a hybrid PKS-NRPS is believed to be responsible for the production of the tetramic acid moiety. Based on bioinformatic analysis, the C-terminal domain of this PKS-NRPS appears to encode a putative NAD(P)H-dependent thioester reduction (R) domain. The R domain has been proposed to catalyze the reductive hydrolysis of the thioester linkage joining the precursor to PKS-NRPS to yield an aldehyde intermediate, which then undergoes aldol-type cyclization to generate tetramic acid (Figure 14a, route A). However, studies the biosynthesis of equisetin,[43] cyclopiazonic acid[44] and HSAF[45] revealed that the suspected R domain actually catalyzes a NAD(P)H independent cyclization to form tetramic acid and its concomitant release from the PKS-NRPS. The fact that the reaction is NAD(P)H independent supports the turnover via a Dieckmann mechanism (Figure 14a, route B). Similar Dieckmann cyclases have later been discovered in other tetramate and pyridine-based natural products biosynthesis.[46] Likewise, the biosynthesis of tetronic acid moiety, an oxygen analog of tetramic acid, has been proposed to proceed via an analogous pathway as that of tetramic acid. However, Sun et al. has recently reconstituted the biosynthetic pathway of RK-682 in vitro and demonstrated that the formation of tetronate does not involve Dieckmann reaction (Figure 14b).[47]
Figure 14.
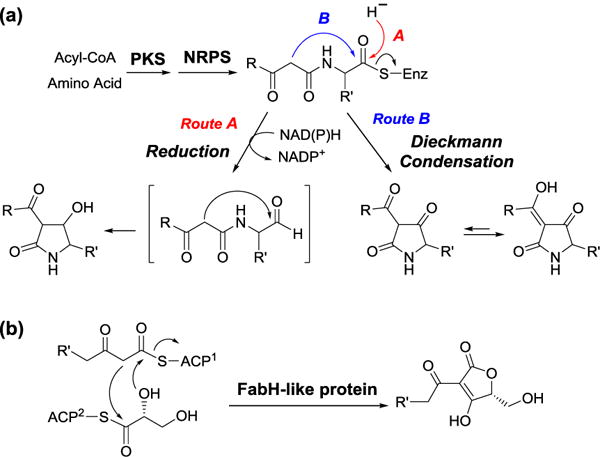
Proposed biosynthesis of (a) tetramic and (b) tetronic acid.
2.3.2. Reaction catalyzed by 1,4-dihydroxynaphthoyl-CoA synthase (MenB)
Vitamin K2 (menaquinone, 53) is a redox cofactor in the electron transport chain of most Gram-positive and a few Gram-negative bacteria (Figure 15).[48, 49] It is a polyisoprenylated naphthoquinone. Menaquinone is derived from chorismate in nine biosynthetic steps.[50–52] During the sixth step, the aromatic naphthoquinone ring in 1,4-dihydroxynaphthoyl-CoA (DHNA-CoA, 55) is formed from o-succinylbenzoyl-CoA (OSB-CoA, 54) via a Dieckmann cyclization catalyzed by DHNA-CoA synthase (MenB). A mechanism for the MenB reaction has been proposed based on the X-ray crystal structure of the E. coli enzyme in complex with the substrate analog, o-succinylbenzoylamino-CoA (Figure 15).[53]
Figure 15.
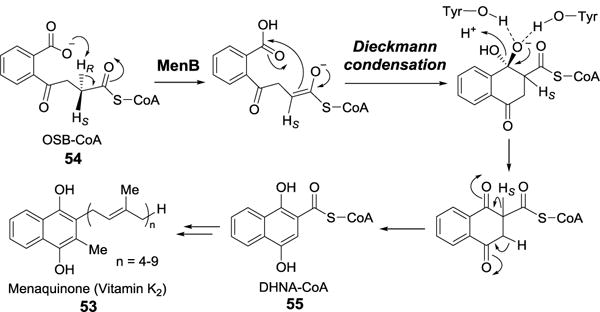
MenB-catalyzed Dieckmann condensation.
2.3.3. Reaction catalyzed by (S)-2-ketocyclohexanecarboxyl-CoA hydrolase (BadI)
Benzoate (56) is a common intermediate in the anaerobic breakdown of aromatic compounds including aromatic hydrocarbons, phenols, halogenated aromatics, and phenylpropanoids by bacteria.[54–56] Benzoate is further catabolized to pimelyl-CoA (59), which is ultimately degraded to acetyl-CoA (41, Figure 16).[57–59] The key step leading to the formation of pimelyl-CoA in the catabolic process is the ring opening of (1S)-2-ketocyclohexanecarboxyl-CoA ((S)-KC-CoA, 58) catalyzed by BadI. Despite good sequence homology to MenB, BadI actually catalyzes a reverse-Dieckmann reaction. A study using recombinant BadI from Rhodopseudomonas palustris demonstrated that BadI utilizes (S)-KC-CoA (58) as the substrate and incorporates a solvent-derived proton into the 2-pro-S position of pimelyl-CoA (59), consistent with the proposed mechanism (Figure 16).[60]
Figure 16.

BadI-catalyzed reverse Dieckmann condensation involved in the anaerobic degradation of aromatic compounds.
2.4. Pictet-Spengler Condensation
The Pictet-Spengler reaction was first discovered in 1911 by Amé Pictet and Theodore Spengler who observed formation of 1,2,3,4-tetrahydroisoquinoline (THIQ, 61) when heating β-phenylethylamine (60) and formaldehyde (24) together with hydrochloric acid (Figure 17a).[61] Seventeen years later, Tatsui reacted tryptamine (62) with acetaldehyde (63) in the presence of sulfuric acid and noted the production of 1-methyl-1,2,3,4-tetrahydro-β-carboline (THBC, 64, Figure 17b).[62] THIQ and THBC comprise the core structures of numerous naturally occurring isoquinoline and indole alkaloids. Many of them are medicinally valuable, such as morphine, the muscle relaxants papaverine and turbocurarine, the antimicrobials berberine and sanguinarine, the anti-cancer drugs vincristine and vinblastine, the antihypertensive drug ajmalicine, and the toxin strychnine.[63]
Figure 17.

Pictet-Spengler reactions for the synthesis of (a) THIQ and (b) THBC.
It is interesting that Pictet-Spengler reaction is in fact the method-of-choice for the biosynthesis of benzylisoquinoline and monoterpenoid indole alkaloids in nature. Representative enzymes capable of catalyzing Pictet-Spengler reactions include strictosidine synthase (STR) and norcoclaurine synthase (NCS), which have been biochemically characterized. The catalyses of these enzymes are discussed below. Pictet-Spenglerases have also been recognized in the biosynthesis of β-carboline (72) and saframycin A (81).
2.4.1. Strictosidine synthase
STR catalyzes the condensation between tryptamine (62) and the monoterpenoid aldehyde secologanin (65) to produce 3α-(S)-strictosidine (66). The STR activity was first detected over 35 years ago when radiolabeled precursors were added to the cell-free extracts of Catharanus roseus leading to radiolabel incorporation into the indole alkaloid, ajmalicine.[64] Soon thereafter, STR was partially purified from C. roseus,[65] and the Rauvolfia serpentina enzyme, designated STR1, was isolated and purified to homogeneity nine years later.[66] The gene encoding STR1 was identified allowing recombinant expression of the enzyme in E. coli[67] and in insect cells.[68] X-ray crystal structures containing bound tryptamine or secologanin,[69] and bound strictosidine[70] were also obtained. An active site glutamate residue is within H-bonding distance to the amine group of tryptamine.[69] These results implied that Glu309 may deprotonate the amine allowing the nucleophilic attack on the secologanin aldehyde carbonyl to form a Schiff base (67).
A schematic representation of the proposed mechanism for the STR1 reaction is depicted in Figure 18. After formation of the Schiff base, an electrophilic attack of C-2 of the tryptamine indole ring to the iminium carbon of the Schiff base may occur to yield the intermediate 69 (Figure 18a, route A). It is also plausible that the electrophilic attack takes place at C-3 position, resulting in a spiro-intermediate (68), which then undergoes a 1,2-alkyl shift to give 69 (Figure 18a, route B). However, ab-initio calculations suggest that the proposed 1,2-alkyl shift is energetically unfavorable, implying that 68 is unlikely a productive intermediate.[71] Further experiments revealed a kinetic isotope effect of ~2.7 when [2-2H]-tryptamine was used as the reactant in the STR1 reaction. The results indicate that re-aromatization, rather than ring closure, is the rate limiting step for both (although only the enzymatic reaction is stereospecific) and that all steps prior to re-aromatization are reversible. It was also proposed that Glu309 protonates the Schiff base to give the iminium intermediate and that, after cyclization, it deprotonates the resulting carbocation intermediate (69) resulting in re-aromatization.
Figure 18.
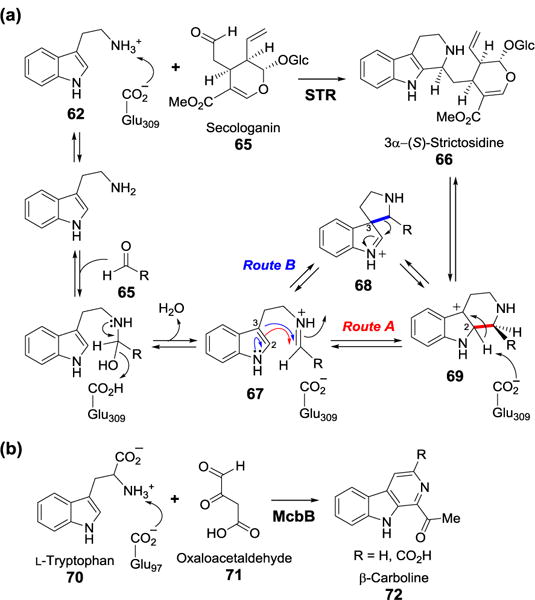
(a) Proposed mechanism of the STR-catalyzed reaction. (b) The microbial McbB-catalyzed Pictet-Spengler reaction.
In addition to STR1, a new class of Pictet-Spenglerase was recently found in the microbial biosynthesis of indole alkaloids (Figure 18b).[72] McbB from Marinactinospora thermotolerans catalyzes the condensation between L-tryptophan (70) and oxaloacetaldehyde (71) to produce β-carboline (72). Although McbB displays a different protein fold from that of strictosidine synthase, site-directed mutagenesis studies of McbB identified Glu97 as the catalytic residue.[73] This structural evidence supports that both McbB and strictosidine synthase likely catalyze the condensation reactions via a similar mechanism.
2.4.2. Norcoclaurine synthase
NCS represents another well-documented Pictet-Spenglerase catalyzing the stereospecific condensation of dopamine (73) and 4-hydroxyphenylacetaldehyde (4-HPAA, 74) to yield (S)-norcoclaurine (76). NCS was originally named as “norlaudanosoline synthase” based on the early studies in which the partially purified enzyme from various plant extracts was capable of converting dopamine (73) and 3,4-dihydroxyphenylacetaldehyde (75) to norlaudanosoline (77, Figure 19).[74] Further studies on the biosynthesis of reticuline, a precursor to a number of benzylisoquinoline alkaloids, demonstrated that norcoclaurine (76), rather than norlaudanosoline (77), is the central precursor for this class of compounds. The identification of the gene encoding NCS in Thalictrum flavum led to its recombinant expression in E. coli facilitating further structural and mechanistic studies.[75, 76] Interestingly, no significant amino acid sequence homology is detected between STR and NCS, indicating that the two enzymes are not evolutionarily related.
Figure 19.

Proposed mechanism of the NCS-catalyzed reaction.
The X-ray crystal structure of NCS containing bound dopamine and 4-hydroxybenzaldehyde, a non-reactive 4-HPAA homolog, was solved in 2009.[77] The crystal structure, along with prior mechanistic studies,[76] allowed a mechanism to be proposed (Figure 19). As with STR1, the NCS reaction could also proceed through a spirocyclic intermediate (78, Figure 19, route A) or not (route B). Lys122 is thought to stabilize the negative charge on the 4-hydroxyphenylacetaldehyde carbonyl oxygen during the formation of the Schiff base with dopamine. Mutation of Lys122 to Ala led to enzyme inactivation. Glu110 is proposed to abstract a proton after cyclization resulting in re-aromatization. Earlier mechanistic experiments with [2,5,6-2H]dopamine revealed an isotope effect of 1.7 ± 0.1 on kcat/KM, which implicated that re-aromatization is at least partially rate limiting and is the first irreversible step in the reaction.[76] This result is similar to that observed for the STR1 reaction. Both 4-deoxydopamine and 4-methoxydopamine serve as substrates for the NCS reaction but neither 3-deoxydopamine nor 3-methoxydopamine are accepted. These results strongly disfavor the spiro-species (78) as a productive intermediate.
2.4.3. Deacetylipecoside synthase
An enzyme from Alangium lamarckii that catalyzes a Pictet-Spengler condensation between dopamine (73) and secologanin (65) to give deacetylipecoside (79, Figure 20) has been purified and partially characterized.[78] The enzyme is designated deacetylipecoside synthase. Deacetylipecoside and its C1-epimer, deacetylisoipecoside (80), are the biosynthetic precursors for ipecac alkaloids. The gene encoding diacetylipecoside synthase has not been identified.
Figure 20.

Biosynthetic pathway of the alkaloids in Alangium lamarckii.
2.4.4. Saframycin biosynthesis: Pictet-Spenglerase activity in a non-ribosomal peptide synthase
Recently, an interesting example of a biological Pictet-Spengler condensation was uncovered in the biosynthesis of the non-ribosomal peptide antitumor antibiotic saframycin A (81, Figure 21).[79, 80] Saframycin isolated from Streptomyces lavendulae belongs to a group of THIQ antibiotics. The primary saframycin biosynthetic machinery consists of the NRPS modules: SfmA, B, and C (Figure 21). Notably, the R-domain of SfmC catalyzes the NADPH-dependent reductive release of NRPS-bound thioester intermediates to yield their corresponding aldehydes (e.g., 82 and 84). The C-domain of SfmC is responsible for two consecutive Pictet-Spengler condensations: the first between modified, NRPS-bound tyrosine (83) and a fatty-acyl-dipeptidyl aldehyde (82) produced by SfmA and B, and the second between the resulting bicyclic aldehyde (84) and a second modified, NRPS-bound tyrosine. The C13-fatty acyl chain is required for substrate recognition by the Pictet-Spenglerase and it is removed at a later stage of the pathway. The mechanism by which the C-domain catalyzes the Pictet-Spenglerase condensation reactions remains unclear so additional research is required for a complete understanding of this fascinating system.
Figure 21.

Proposed biosynthetic pathway of saframycin A.
2.5. Unnatural Condensation Reactions Catalyzed by Enzymes with Broad Substrate Specificity
2.5.1. Knoevenagel condensation
The Knoevenagel condensation is an aldol reaction involving the attack of a carbanion species to the carbonyl carbon of a ketone or an aldehyde followed by dehydration to give a conjugated enone product (Figure 22a). This reaction is named after Emil Knoevenagel who first reported this reaction in 1898.[81] Two enzymes, porcine pancreatic lipase[82] and Bacillus licheniformis alkaline protease,[83] have been shown to catalyze Knoevenagel condensation. The reaction catalyzed by porcine lipase was demonstrated with a series of benzaldehyde derivatives (85) and methylcyanoacetate (86) in ethanol (Figure 22b). During these reactions, the methyl ester was transesterified with solvent ethanol to yield ethyl esters (87). The alkaline protease-catalyzed Knoevenagel condensation was conducted with a series of salicylaldehyde (88) and β-keto-ethyl esters (89). The reactions were followed by transesterification to yield 2H-1-benzopyran-2-one derivatives (90), which are the core structures in a number of biologically active compounds with anticoagulant and antitumor activity (Figure 22b).
Figure 22.

(a) Knoevenagel reaction and (b) enzymatic Knoevenagel reactions.
2.5.2. Friedländer quinoline synthesis
The Friedländer quinoline synthesis involves condensation between 2-aminobenzaldehydes and the α-anion of a ketone under acid or Lewis acid catalyzed conditions to form a quinoline product (91, Figure 23). This reaction is named after Paul Friedländer who first reported this useful quinoline synthesis in 1880s,[84] when the scientific interests and commercial activities focused on the utilization of quinolines as dyes or medicines. Two mechanisms are conceivable. The distinction lies at whether the C-C bond (92) or the C-N bond (93) is formed first (route A versus route B). However, in both cases the key step is an aldol condensation reaction. Interestingly, lipase from porcine pancreatic was found to be able to catalyze the Friedländer quinoline synthesis when using 2-amino-3,5-dibromo-benzaldehyde and cyclopentanone as substrates.[85] This discovery may provide a useful tool to generate quinoline derivatives under mild conditions.
Figure 23.

Friedländer quinoline synthesis.
3. Nucleophilic Addition to C-C and C-Hetero Multiple Bonds
3.1. Michael Addition
The Michael addition which was originally reported by Arthur Michael in 1887 is the addition of an enolate of a ketone or aldehyde to an α,β-unsaturated carbonyl compound at the β carbon.[86] The definition of Michael has now been generalized to include addition of any nucleophile to a conjugated π-system. Thus, 1,4-addition of a heteroatom nucleophile to form a carbon-heteroatom bond is also considered to be a Michael addition reaction. Because numerous enzyme-catalyzed reactions in which Michael addition is a key step, it is considered to be one of the most prevalent C-C bond formation reactions, along with the aldol reaction, in biological system. For example, biosynthesis of lanthionine (Lan, 96) and methyllanthionine (MeLan, 97), which are the unifying structural motifs present in lantibiotics, includes a nucleophilic conjugate addition step (Figure 24a).[87] The thioether linkage in lantibiotics is generated by intramolecular Michael addition of a cysteine thiolate to a dehydroalanine (Dha, 94) or dehydrobutyrine (Dhb, 95) residue catalyzed by lantibiotic cyclase (LanC). The proposed mechanism illustrated in Figure 24a is based on the biochemical characterization and the X-ray crystal structure of NisC, the cyclase involved in nisin biosynthesis.[88] Although dehydro amino acids are less electrophilic than common Michael acceptors due to the build-in enamine substituent, nucleophilic addition to dehydro amino acids proceeds relatively fast. The cis-trans isomerization between maleylacetoacetate (98) and fumarylacetoacetate (99) catalyzed by maleylacetoacetate isomerase also involves a Michael addition of glutathione (GSH) to generate an enolate adduct (100), which after bond rotation and elimination of glutathione completes the isomerization (Figure 24b).[89] A few other examples where participation of coenzyme is needed are discussed below.
Figure 24.

Proposed mechanism for (a) LanC-catalyzed Michael addition and (b) the reaction catalyzed by maleylacetoacetate isomerase.
3.1.1. Tryptophan synthase and cystathionine γ-synthase
Both tryptophan synthase[90] and cystathionine γ-synthase[91] are PLP-dependent enzymes catalyzing the coupling of serine (22) and indole (101) to form tryptophan (70) and the replacement of the succinate group of O-succinyl-L-homoserine (104) with cysteine (105) to form cystathionine (106), respectively. Tryptophan synthase exists as an α2β2 tetramer. The β-subunits, where PLP resides, catalyze the conversion of serine (22) to an aminoacrylate-PLP intermediate (102) in an β-elimination reaction (Figure 25). Subsequently, indole (101), channeled from the α subunit, acts as nucleophile and attacks the highly conjugated intermediate (102) in a Michael addition manner to give 103, which after deprotonation and regeneration of the PLP coenzyme yields the final product tryptophan. In this case, a PLP-mediated 1,8-conjugate addition instead of classical 1,4-Michael addition is involved. The γ-replacement reaction catalyzed by cystathionine γ-synthase[91] follows a similar mechanism involving the formation of an extended conjugated intermediate (107), which serves as Michael acceptor for cysteine (105, Figure 26).
Figure 25.

Proposed mechanism for tryptophan synthase.
Figure 26.
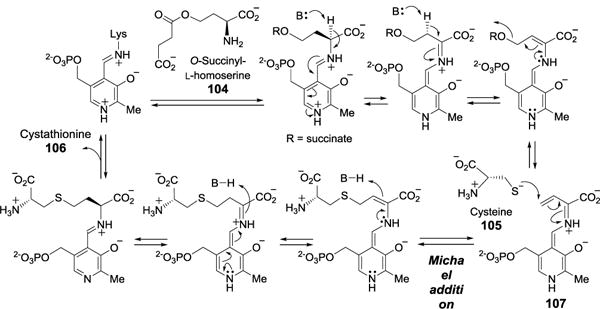
Proposed mechanism for cystathionine γ-synthase.
3.1.2. Nitroalkane oxidase and alkyldihydroxyacetone phosphate synthase
The nitroalkane oxidase is a flavoprotein catalyzing the oxidation of nitroalkanes (109) to the corresponding aldehydes or ketones with concomitant elimination of a nitrite anion and the consumption of O2 to give H2O2 (Figure 27).[92, 93] The reaction is initiated by deprotonation of the Cα-H of the nitroalkane substrate, and the resulting carbanion (110) adds to the N5 of FAD (108) in a Michael addition to form a covalent adduct (111). Subsequent elimination of the nitrite anion leads to an N5-iminium dihydroflavin intermediate (112), which yields the aldehyde product (114) after hydrolysis. Re-oxidation of the reduced coenzyme (113) by O2 produces H2O2 as a byproduct.
Figure 27.

Proposed mechanism for nitroalkane oxidase.
The replacement of a long chain fatty acid in diacylglycerol phospholipids (115) by a long chain fatty alcohol to generate the monoacyl, monoalkyl ether phosphate lipids (119) catalyzed by alkyldihydroxyacetone phosphate synthase (ADPS) also involves the intermediacy of an N5-substrate adduct with the bound flavin coenzyme (Figure 28).[94] The catalytic cycle is believed to proceed with C1 deprotonation of the substrate followed by addition of the resulting carbanion (116) to N5 of FAD to give 117. Cleavage of the ester linkage to expel the acyl product leads to a N5-flavin iminium intermediate (118), which is then reacted with the fatty alcohol substrate. Re-oxidation of the flavin coenzyme along with C–N bond cleavage affords the alkyldihydroxyacetone phosphate product (119). In the mechanisms for both nitroalkane oxidase and ADPS, formation of the N5-adduct (112 and 117) results from addition of a carbon nucleophile to the conjugated diimine moiety in the oxidized flavin cofactor, which serves as the Michael acceptor.
Figure 28.

Proposed mechanism for alkyldihydroxyacetone phosphate synthase.
3.1.3. Branching module in rhizoxin biosynthesis
Polyketide biosynthesis was briefly discussed in the previous section. While chain growth proceeds linearly, branched polyketides also exist and are usually resulted from the incorporation of α-branched building blocks during chain extension or post-chain-extension modification of the linear backbone at the α-position. Another branching mechanism involves a β-aldol reaction catalyzed by hydroxymethylglutaryl-CoA synthase (HCS) cassette.[95] Recently, a novel β-branching PKS module catalyzing Michael-addition was identified in the biosynthetic pathway of rhizoxin (120), a phytotoxin produced by Burkholderia rhizoxinica (Figure 29).[96, 97] Using synthetic thioester (121) as surrogate substrate, Bretschneider et al. demonstrated the activity of the β-branching module, which is consisted of a KS domain, an ACP domain and a branching (B) domain.[97] The chain branching mechanism was proposed to involve a Michael addition of the extender unit to the α,β-unsaturated thioester bound to the KS domain. The nucleophilic hydroxyl group can either attack the KS-bound or the ACP-bound thioester to form δ-lactone (Figure 29). In order to distinguish these two mechanisms, 13C-labeled malonyl-CoA was used as the extender unit to trace the lactonization reaction. Characterization of the ACP-bound product by nuclear magnetic resonance (NMR) experiments showed that the labeled atoms were incorporated in the polyketide chain (122) instead of the lactone ring (123) and therefore supported route A.
Figure 29.

Michael addition and lactonization catalyzed by a branching module.
3.2. Morita-Baylis-Hillman Reaction
The Morita–Baylis–Hillman (MBH) reaction is a synthetically useful transformation named after Ken-ichi Morita, Anthony Baylis and Melville Hillman. It is a carbon-carbon bond forming reaction between the α-position of an activated alkene and an aldehyde (or an imine).[98, 99] The reaction requires a nucleophilic catalyst, such as tertiary amine or phosphine, and produces a densely functionalized product (Figure 30a).
Figure 30.

(a) Morita-Baylis-Hillman reaction. (b) Rauhut-Currier reaction.
The transformation catalyzed by thymidylate synthase is the biological precedent of the MBH reaction. Thymidylate synthase facilitates the methylation of 2′-deoxyuridine 5′-monophosphate (dUMP, 124) to thymidine 5′-monophosphate (TMP, 128) using N5,N10-methylene-THF (28) as the methyl donor (Figure 31). The mechanism of thymidylate synthase-catalyzed reaction has been extensively studied.[100] This one-carbon transfer reaction is initiated by the addition of an active site cysteine to dUMP (124) to generate an enolate intermediate (125), which is used to attack N5,N10-methylene-THF (28) to form a new C-C bond (126). Subsequent C-5 deprotonation triggers β-elimination of THF to form an enone intermediate (127), which after reduction by THF (20) yields the TMP product (128). The activation of deoxyuridylate by cysteine as a nucleophilic catalyst, and the addition of the resulting enolate to the iminium group of N5,N10-methylene-THF are all characteristics for a MBH reaction.
Figure 31.

Proposed reaction mechanism for thymidylate synthase.
3.3. Rauhut-Currier Reaction
Rauhut-Currier reaction is a nucleophilic conjugated coupling of one active alkene/latent enolate to a second Michael acceptor, not an aldehyde or an imine, to form a new C–C bond between the α-position of the activated alkene and the β-position of the second alkene under the influence of a nucleophilic catalyst (Figure 30b). While it is also known as a vinylogous MBH reaction, the Rauhut-Currier reaction reported by Rauhut and Currier in 1963 is actually predated the MBH reaction.[101]
SpnL, in the biosynthetic pathway of spinosyns, has been proposed to catalyze a Rauhut-Currier-type C-C bond formation reaction. The spinosyns are a complex of polyketide insecticides produced by Saccharopolyspora spinosa. The aglycone core of spinosyns has a tetracyclic architecture in which a 12-membered lactone ring is fused with a perhydro-as-indacene moiety (129, Figure 32).[102] The biosynthetic route for the formation of the macrolactone can be largely inferred by analysis of genes encoding the polyketide biosynthetic modules.[103] All of the post-PKS tailoring steps including cyclization of the macrolactone to form the tetracyclic core have been elucidated (Figure 32).[104, 105] SpnF catalyzes a [4+2] cycloaddition reaction resulting in the formation of a cyclohexene ring (130 → 131). This may represent an example of a biological Diels-Alder reaction, a possibility that will be discussed in detail later. The SpnF reaction is followed by a second cross-bridging reaction catalyzed by SpnL (132 → 135), which has been proposed to proceed via a Rauhut-Currier mechanism. Accordingly, coupling between C-3 and C-14 likely results from the nucleophilic addition of an enolate to a Michael acceptor (133 → 134) facilitated by a nucleophilic residue in the active site (Figure 32). In the case of SpnL, the nucleophilic catalyst has yet to be identified.
Figure 32.

The biosynthetic pathway of spinosyn A.
Iridoid alkaloids are bicyclic monoterpenes found in plants[106] and insects[107] to defend themselves against predators. Rauhut-Currier reaction has been proposed for the cyclization of 8-oxogeranial (136) to iridodial (137), a key intermediate for the biosynthesis of iridoids, in Chrysomelina species (Figure 33a).[108] However, the biosynthesis of iridoids in beetles may be quite different from what happens in plants. In fact, mechanistic studies of plant iridoid synthase from Catharanthus roseus (CrISY) suggested that the cyclization is a Michael reaction and occurs after the reduction of 136 by NADPH (Figure 33b).[109, 110] Moreover, the hypothetic Rauhut-Currier mechanism is unlikely for CrISY due to the absence of suitably placed nucleophilic residues (cysteine or histidine) in the active site.[111]
Figure 33.

Proposed mechanisms of the conversion of 8-oxogeranial to iridodial. (a) Rauhut-Currier reaction covalently assisted by a nucleophilic residue occurs first followed by reduction. (b) Reduction occurs priot to Michael reaction.
3.4. Mannich Reaction
The Mannich reaction, named after Carl Mannich, is a three-component condensation between an amine and an aldehyde to give an electrophilic iminium ion, which then joins with the α-carbon of an enolate to give a β-amino-carbonyl compound (Figure 34).[112] Mannich reaction has been established to be an important transformation in the biosynthesis of plant alkaloids. This subject has been the focus of several reviews.[113–116] Here, tropane alkaloid is selected as an example to illustrate the involvement of Mannich reaction in alkaloid biosynthesis. Tropane alkaloids, such as hyoscyamine (142), scopolamine (143) and cocaine, have been popular targets for organic synthesis and used in the development of clinically useful pharmaceutics.[117] The synthesis of tropinone (140) from succindialdehyde, methylamine and acetonedicarboxylate was first reported by Robert Robinson in 1917.[118] Robinson later proposed that the biosynthesis of the bicyclic ring of tropane may follow a similar route involving an amino acid and an acetone equivalent.[119] This hypothesis is supported by many lines of evidence collected over 60 years of research. Our current understanding of the tropane alkaloids biosynthesis is summarized in Figure 34.[114, 117] The coupling between the enolate of an acetone equivalent and the pyrrolinium cation (139) proceeds through a Mannich-like mechanism. Formation of the tropane skeleton in the subsequent step is likely accomplished by an aldol reaction which may be considered as an intramolecular Mannich reaction (139 → 140).
Figure 34.

(a) Mannich reaction. (b) The proposed biosynthetic pathway of hyoscyamine and scopolamine.
3.5. Stetter Reaction and Benzoin Condensation
The Stetter reaction is a coupling reaction between an aldehyde and an α,β-unsaturated ketone to yield a 1,4-dicarbonyl product.[120, 121] The reaction is catalyzed by cyanide ion or a safer surrogate, the thiazolium salt, which is analogous to the biological cofactor thiamine diphosphate (TPP, 144). This reaction was first reported by Hermann Stetter in 1973.[120] Under the reaction conditions, the aldehyde undergoes polarity inversion (umpolung)[122] to become a nucleophile instead of an electrophile. The mechanism of Stetter reaction is related to that of benzoin condensation. However, the latter reaction occurs in 1,2-addition manner, whereas the Stetter reaction proceeds with 1,4-conjugate addition (Figure 35). The reactions catalyzed by MenD and PigD are the biological equivalents of Stetter reactions.
Figure 35.
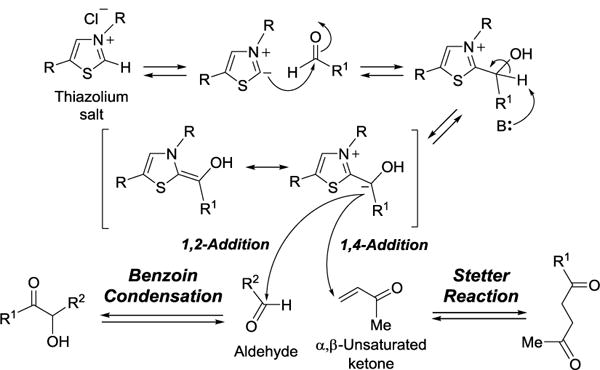
Stetter reaction and benzoin condensation.
MenD is a TPP-dependent enzyme responsible for the second step of menaquinone (53) biosynthesis. Its catalysis consists of two reactions, the decarboxylation of α-ketoglutarate (α-KG, 145) to generate a succinic semialdehyde-TPP anion (146), and the addition of this anion to isochorismate (147) to yield (1R,2S,5S,6S)-2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate (SEPHCHC, 148) as the product (Figure 36).[51] The TPP stabilized ketol fragment (146) is an α-imine anion equivalent and serves as the nucleophile to attack the β carbon of the conjugated olefin in 146 in a Michael addition to form a new C-C bond. In this reaction, the thiazolium cofactor has to be deprotonated first to be catalytic. The absence of an appropriate general acid/base residue in the active site led to the proposal that the cofactor N4′ may serve this role in the mechanism of MenD (Figure 36).[123]
Figure 36.

Proposed mechanism of MenD-catalyzed reaction.
PigD from Serratia marcescens, has been postulated to catalyze the decarboxylating condensation of (2E)-octenal (149) with pyruvate during the biosynthesis of the antibiotic prodigiosin. Its catalytic function is assigned based on gene knockout experiments and sequence annotation as a TPP-dependent protein (Figure 37).[124] However, the recombinant PigD enzyme was shown to catalyze the 1,2-addition of acetaldehyde (the decarboxylated product of pyruvate, 63) instead of the predicted 1,4-addition with 2-octenal (149). The 1,4-selectivity was only observed when the (2E)-octenoyl-N-acetyl cysteamine thioester ((2E)-octenoyl-SNAC, 150) was used as substrate.[125] These observations suggested that either an acyl thioester or CoA derivative is the substrate for PigD in prodigiosin biosynthesis.
Figure 37.

PigD-catalyzed Stetter reactions.
The reaction catalyzed by transketolase is another example in which coenzyme TPP (144) is required to stabilize an anion intermediate used in the aldol reaction.[126] The overall transformation is the reversible transfer of a two-carbon fragment from D-xylulose 5-phosphate (151) to D-ribose 5-phosphate (154) to give D-sedoheptulose 7-phosphate (155) and glyceraldehyde 3-phosphate (3) as products (Figure 38). This is an important reaction in the pentose phosphate pathway for glucose metabolism. Since the nucleophilic attack occurs in a 1,2-addition manner, this reaction is a biological example of benzoin condensation. Although benzoin condensation is not a so-called “name reaction”, it is perhaps the oldest reaction which has given a name based on the product from condensation of two molecules of benzaldehyde.[127]
Figure 38.

Reaction mechanism of transketolase.
3.6. Retro-Prins Reaction
In 1919, Hendrik Jacobus Prins reported the acid-catalyzed coupling of aldehydes to olefinic compounds to make 1,3-diols.[128] Depending on the reaction conditions, different products can also be produced (Figure 39). The reverse process called retro-Prins reaction is a thermal elimination reaction and has proved synthetically useful.[129] The cyclization and fragmentation of farnesyl diphosphate (FPP, 156) to geosmin catalyzed by geosmin synthase from Streptomyces coelicolor (ScGS) has been proposed to involve retro-Prins reaction. Geosmin is a volatile metabolite responsible for the characteristic smell of moist soil. Geosmin synthase has been shown to be a bifunctional enzyme.[130] The N-terminal domain catalyzes the conversion of FPP to germacradienol (156 → 157), while the C-terminal domain catalyzes the subsequent cyclization of 157 to form geosmin (158) and acetone via a retro-Prins reaction. The two domains function independently and there is no obvious channel between the two active sites.[131] It thus remains unclear that germacradienol intermediate (157) generated in the N-terminal domain is processed by the C-terminal domain of the same or a different protein molecule.
Figure 39.

(a) Prins reaction. (b) Proposed reaction mechanism catalyzed by geosmin synthase.
3.7. Unnatural Addition Reactions Catalyzed by Enzymes with Broad Substrate Specificity
It is well known that some enzymes can catalyze conversions of small molecules distinct from their natural substrates to give new products in stereospecific manner. Enzymes that accept alternative substrates are said to be “promiscuous” with regard to substrate specificity. The activity of an enzyme can also be altered by site-specific mutagenesis to catalyze unnatural reactions. There are too many examples of these types of applications to list them all here. This section will provide a sampling of non-substrate specific condensations with an emphasis on name reactions. Several reviews provide a more comprehensive overview of the subject.[132–134]
3.7.1. Henry reaction
The nitroaldol reaction, also known as Henry reaction, is a variation of the aldol reaction in which the carbonyl carbon of a ketone or an aldehyde substrate undergoes nucleophilic attack by a deprotonated nitroalkane to yield a β-nitro alcohol product (Figure 40). This reaction was first reported by M. Louis Henry in 1895 and has been used widely in organic synthesis.[135] Transglutaminase (TGase) from Streptoverticillum griseoverticillatum,[136] D-aminoacylase from Mucor javanicus,[137] D-glucoamylase from Aspergillus niger,[138] and bovine serum albumin,[139] have been shown to catalyze Henry reaction. These observations are intriguing since the physiological role of TGase is to catalyze the acyl transfer from glutamate to the ε-amino group of a lysine or another primary amine, and that of D-aminoacylase is to hydrolyze N-acyl-D-amino acids. Likewise, D-glucoamylase catalyzes the degradation of starch, and serum albumin is a protein that binds lipids circulating in blood and is not an enzyme. Since the observed activities on nitroaldol reaction of these enzymes are unrelated to their physiological functions, how they catalyze Henry reactions remains elusive. While none of the above mentioned Henry reactions are enantioselective, hydroxynitrile lyase (HNL) from Hevea brasiliensis (the rubber tree) can catalyze stereoselective nitroaldol reaction using aldehydes and nitroalkanes,[140] although the physiological role of HNL is to catalyze the degradation of cyanohydrins to the corresponding aldehyde with release of hydrogen cyanide (Figure 40).[141] The cyanide released acts as a toxin to deter predators and parasites.
Figure 40.

Henry nitroaldol reaction and hydroxylnitrile lyase-catalyzed reversible cyanohydrin formations.
3.7.2. Ugi reaction
The conventional Ugi reaction, named after Ivar Karl Ugi, is a multi-component condensation of a carboxylic acid, an isocyanide, an amine and a carbonyl compound.[142] The reaction mechanism is believed to proceed via a nitrilium ion intermediate (159), which is attacked by the nucleophilic carboxylic anion (Figure 41). The following Mumm rearrangement[143] generates the diamide compound and water as the sole by-product. Based on the mechanism, Pan and List have proposed a catalytic three-component Ugi reaction, in which the nitrilium intermediate (159) is reacted with a water molecule instead of carboxylate ion. This designed reaction was later shown to be as efficient as the classic Ugi reaction when using the acid as catalyst.[144] In addition to the chemical catalyst, Novozym 435 was found to be an effective lipase and is capable of catalyzing multi-component Ugi condensation of ethyl isocyanoacetate, isovaleric aldehyde and 2 equivalents of benzylamine (Figure 41).[145] Whether the addition of the second amine is due to the involvement of enzyme or the chemical nature of isocyanide remains unclear. However, this enzymatic Ugi reaction removes the requirement of heat in the acid-catalyzed Ugi reaction and may become useful in applied chemistry.
Figure 41.

Ugi reaction.
4. Oxidation and Reduction Reactions
4.1. Baeyer-Villiger Reaction
The Baeyer-Villiger (BV) reaction is an oxidative C-C bond cleavage reaction of an aldehyde or a ketone by insertion an oxygen atom into the C-C bond next to the carbonyl group to form the corresponding formate, lactone or ester upon treatment with suitable oxidants. The first example of BV reaction is the lactonization of menthone with potassium monopersulfate reported by Adolf von Baeyer and Victor Villiger in 1899.[146] The reaction proceeds by nucleophilic addition of peroxide on the carbonyl to form a tetrahedral intermediate (160), also known as Criegee intermediate,[147] followed by a concerted migration of one of the adjacent carbons to oxygen to afford the rearrangement product (Figure 42).[148]
Figure 42.

Baeyer-Villiger reaction and Criegee intermediate.
Enzymes catalyzing BV reaction (known as BV monooxygenases, BVMOs) are widely distributed in nature with at least 1000 putative homologs in the database.[5] The first biological example of BV reaction was discovered in the microbial degradation of steroids in 1948.[149] Later, BVMO enzymes were purified to homogeneity from Nocardia globerula CL1 and Acinetobacter sp. strain NCIB 9871 and shown to catalyze the lactonization of cyclohexanone.[150] Most BVMOs characterized thus far are flavin-dependent enzymes. They generally consume one equivalent of NAD(P)H and oxygen for each turnover and most of them can be classified into three different subsets. For type 1 BVMOs, each is a single polypeptide with FAD and NADPH bound to separate domains. Type 2 BVMOs utilize FMN and either NADPH or NADH, and are composed of two distinct polypeptides: one binds FMN and the second binds NAD(P)H.[151] Type “O” (means odd) BVMOs are evolutionarily distinct from types 1 and 2. This group of enzymes consist of a single polypeptide utilizing a surface bound FAD and either NADH or NADPH as the reducing agent.[152] BVMOs that are not flavin-dependent have also been identified. Toxoflavin lyase and AflM catalyze BV oxidation by utilizing manganese and heme-iron as cofactors, respectively.
The identification of novel BVMOs is of increasing importance, as the reactions they catalyze are generally regio-, chemo-, and enantiospecific. By using BVMOs in organic synthesis, one obviates the use of potentially dangerous oxidants and the disposal or recycling of the corresponding carboxylic acid from the peroxyacid reagent, because the only byproduct in BVMO-catalyzed reaction is water. A few selected BVMOs and the proposed catalytic mechanisms are described below. For an overview of the current state of BVMOs in organic synthesis please see the review by Leisch et al.[5]
4.1.1. The prototype of flavin-dependent BVMOs: cyclohexanone monooxygenase
BVMOs play important roles in the microbiological catabolic pathways of steroids, terpenoids, and aliphatic ketones. As the prototypical example, cyclohexanone monooxygenase (CHMO) from Acinetobacter NCIB 9871 has been extensively studied. CHMO is a type I BVMO and is the first BVMO of which the encoding gene was cloned.[153, 154] Under physiological conditions, CHMO catalyzes an oxygen insertion reaction on cyclohexanone (161) to form a seven-membered cyclic lactone (162), a key step in the degradation pathway of cyclohexanol (Figure 43).[150] Oxygen insertion catalyzed by CHMO has been proposed to proceeds through a Criegee intermediate (165) in the same manner as the non-enzymatic BV reactions.[147] Early experiments using [2,2,6,6-2H4]cyclohexanone as the substrate demonstrated that no solvent proton was incorporated into the product. Therefore, the reaction unlikely proceeds through an enolate intermediate.[155] Further spectroscopic studies on the protonation state of the oxygenated flavin intermediate revealed that, although both flavin C4a-OO– (163) and flavin C4a-OOH (164) co-exist in equilibrium (Figure 43), it is the deprotonated species (163) that reacts with the substrate.[156]
Figure 43.

Proposed mechanism of cyclohexanone monooxygenase (CHMO).
4.1.2. BVMOs in natural products biosynthesis
In addition to the catabolic BVMOs found in the degradation pathways, BVMOs have also been identified in the biosynthetic pathways of polyketides, aminoglycosides, cofactors and many other secondary metabolites. Knockout experiments in Streptomyces sp. LZ35 indicated that gene hgc3 encodes a flavin-dependent enzyme that catalyzes a lactonization step during the biosynthesis of hygrocin antibiotics (Figure 44a).[157] Although the catalytic activity of the purified recombinant Hgc3 has not yet been demonstrated in vitro, the Hgc3 protein is dissimilar from the categorized BVMOs known so far based on sequence alignment.
Figure 44.
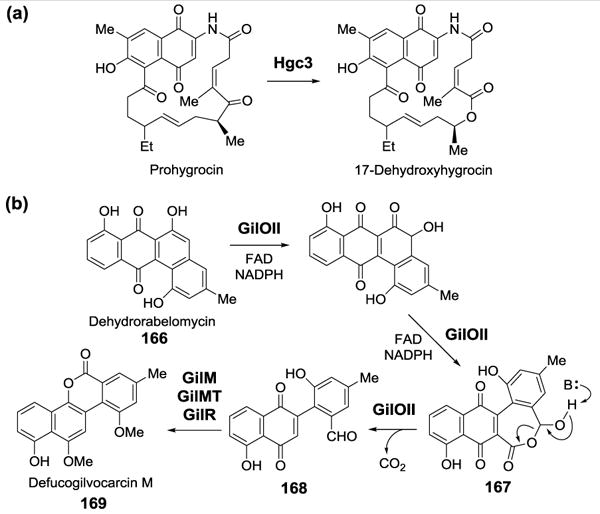
(a) Proposed lactonization catalyzed by Hgc3. (b) The GilOII-catalyzed C-C bond cleavage reaction.
A BVMO involved in the biosynthesis of the gilvocarcins produced by various strains of Streptomyces was identified and its activity was confirmed by in vitro assays using purified, recombinant protein.[158] This enzyme, GilOII catalyzes the hydroxylation and the lactonization of dehydrorabelomycin (166), an intermediate in the biosynthesis of gilvocarcins (Figure 44b). The reaction only occurred in the presence of FAD, which was reduced in situ with NADPH and E. coli flavodoxin reductase. Based on sequence analysis, it is not obvious that GilOII is a flavoprotein. Rather, it displays sequence homology to the cofactor-free anthrone oxygenases, TcmH[159] and AknX,[160] which are involved in the biosynthesis of tetracinomycin C in Streptomyces glaucescens and aklavinone in Streptomyces galilaeus, respectively. The lactone product (167) is further hydrolyzed, decarboxylated, and converted to defucogilvocarcin M (169) through the actions of the downstream biosynthetic enzymes GilMT, GilM, and GilR.
The dimethylbenzimidazole (DMB, 171) is a key structural component of coenzyme B12 (170), where a nitrogen atom of DMB serves as an axial ligand to the cobalt atom. Flavin mononucleotide (FMN, 172) has been shown to be the precursor for DMB and the responsible enzyme, BluB, has recently been identified in Sinorhizobium meliloti.[161, 162] BluB is a flavin protein catalyzing the contraction of the tricyclic isoalloxazine to form DMB (Figure 45). A flavin-dependent reductase is needed to generate the reduced flavin (FMNH2, 173) in the active site of BluB before each catalytic event. The mechanism of BluB likely involves a FMN-4a-OOH intermediate (174), which undergoes an intramolecular Baeyer–Villiger reaction to yield the 7-membered lactone (175).[163] Subsequent hydrolyses occurring at both the expanded pyrimidine and the central pyrazine ring lead to the release of oxalic acid, urea and CO2. The final transformations to DMB include oxidation, retro-aldol cleavage of the ribityl side chain to liberate erythrose 4′-phosphate, cyclization and aromatization. The reaction catalyzed by BluB is unusual since the BV reaction takes place on the flavin coenzyme itself.
Figure 45.

The proposed mechanism of BluB-catalyzed reaction.
Recently, a BVMO was shown to catalyze the formation of carbonate group in the biosynthesis of cytochalasins, which are fungal natural products isolated from Aspergillus clavatus.[164] In the presence of FAD, NADPH and a flavin reductase, the recombinant CcsB converts ketocytochalasin (176) to a lactone iso-precytochalasin (178) and a carbonate-containing compound cytochalasin Z16 (180, Figure 46).[165] Iso-precytochalasin (178) is likely a side-product resulting from the rapid isomerization of the nascent product (177) from the first oxidation, which is a Baeyer-Villiger reaction. The second oxidation converting an ester to a carbonate moiety through a pseudo-BV mechanism is an intrinsically challenging transformation because the increased electron density on the ester carbonyl renders it less susceptible toward nucleophilic attack. Since the sequence analysis of CcsB reveals only moderate homology with most canonical type I BVMOs, its capability to catalyze carbonate formation has been speculated to rely on substrate activation inflicted by the functional groups adjacent to the ester carbonyl group: the vinylogous 1,5-diketo moiety, the α,β-unsaturated ester unit and the acidic proton at the γ-position (Figure 46). Such a chemical environment may allow the flavin C4a-OO– to insert through either a direct attack at the carbonyl group (Figure 46, route A) or a Michael addition to the β-carbon (Figure 46, route B). Instead of the classic BV migratory mechanism, the second oxidation reaction is proposed to form an epoxide intermediate (179), which rearranges to the carbonate product via the aid of the distal vinylogous ketone.
Figure 46.
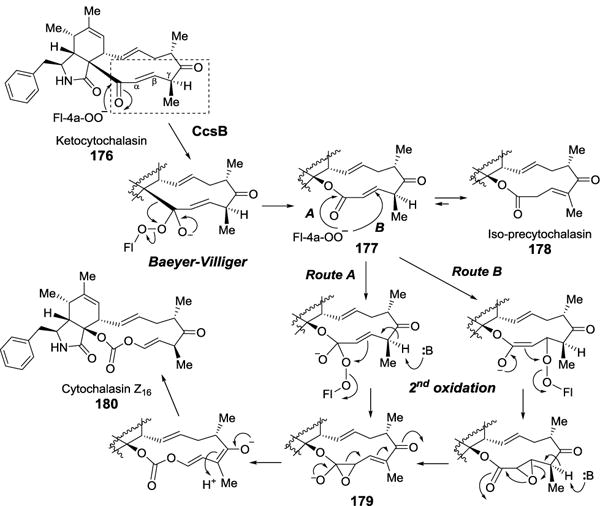
The proposed mechanism of CcsB-catalyzed carbonate formation.
BVMO also plays a crucial role in carbamate group formation in the biosynthesis of bacterial pyrrolizidine alkaloids (PAs). It has been shown that legonmycins (181, Figure 47) biosynthetic gene cluster from Streptomyces sp.MA37 contains three NRPS genes (lgnABD) and one putative BVMO gene (lgnC).[166] The purified flavoprotein LgnC was shown to catalyze an oxygen insertion reaction of legonindolizidins (182) to form a carbamate intermediate (183), which undergoes hydrolysis followed by decarboxylative recyclization to generate 185 (Figure 47). LgnC is also expected to catalyze the second hydroxylation reaction leading to the production of legonmycins (181, Figure 47). Similar biosynthetic route has also been proposed for the formation of pyrrolizixenamides in Xenorhabdus stockiae.[167]
Figure 47.

Proposed biosynthetic pathway for legonmycins.
4.1.3. Cytochrome P450 BVMO
Although the BVMOs described above are all flavin-dependent, several cytochrome P450 enzymes catalyzing Baeyer-Villiger reactions have also been identified. For example, there exist two Bayer-Villiger lactonization reactions in the biosynthesis of aflatoxins (186), a class of environmental carcinogens produced by the fungi of the genus Aspergillus (Figure 48). Early studies of aflatoxin biosynthesis in Aspergillus parasiticus suggested that the conversion of hydroxyversicolorone (187) to versiconal acetate (188) is catalyzed by a BVMO (Figure 48).[168] Subsequent gene deletion experiments led to the identification of moxY, which encodes a cytochrome P450 monooxygenase, as the gene responsible for this reaction.[169] In separate experiments, the genes aflN and aflM, which encode a cytochrome P450 monooxygenase and an NADPH-dependent reductase, respectively, were shown to be associated with the subsequent conversion of versicolorin A (189) to demethylsterigmatocystin (191) in aflatoxin biosynthesis in Aspergillus nidulans. The gene aflN from A. nidulans is also known as stcS.[170, 171] Using chemically prepared intermediates, AflM has been verified to catalyze the reduction of the dienone intermediate (190) generated from 189 by AflN. AflN also catalyzes the following Baeyer-Villiger monooxygenation reaction leading to ring opening, re-cyclization, and decarboxylation to give 191 (Figure 48).[172, 173]
Figure 48.

The proposed biosynthetic pathway of aflatoxin B1 and mechanisms of key enzymes involved.
Brassinosteroids are a class of polyhydroxylsteroids hormones important for cell expansion, cell elongation, vascular differentiation, and many other growth processes in plants.[174] Many brassinosteroids, such as brassinolide (193), contain a seven-membered lactone ring which is formed via a Baeyer-Villiger oxidation reaction from the corresponding 5-keto precursor (192, Figure 49). This reaction is catalyzed by brassinolide synthase which is a cytochrome P450 BVMO. The brassinolide synthases have been isolated from tomato and also from Arabidopsis thaliana.[175, 176]
Figure 49.

The lactonization catalyzed by brassinolide synthase.
4.1.4. Metal ion-dependent BVMO: toxoflavin lyase
Toxoflavin (194), is a plant toxin produced by several bacterial species that causes food poisoning.[177] The gene tflA, encoding an enzyme that catabolizes toxoflavin, was identified in a toxoflavin resistant strain of Paenibacillus polymyxa.[178] The activity of purified TflA was only observed in the presence of dithiothreitol (DTT) and Mn2+.[179] An X-ray crystal structure of TflA containing bound toxoflavin revealed that Mn2+ is coordinated to two glutamate residues and one histidine residue in the enzyme active site.[179] The products resulting from the degradation of toxoflavin by TflA were identified by a combination of mass spectrometry and NMR spectroscopy (Figure 50).[180] Based on the identity of these products, a Bayer-Villiger monooxygenation mechanism was proposed, in which O2 is activated for electrophilic attack on reduced toxoflavin (195) by coordination with Mn2+. In this scheme, DTT is required for the reduction of toxoflavin. The proposed mechanism has no precedent in biology.
Figure 50.

Mechanistic proposal for the degradation of toxoflavin.
4.2. Prilezhaev Reaction
The Prilezhaev reaction is a convenient method to prepare oxiranes from alkenes using peracid (Figure 51a). The reaction is named after Nikolaus Prilezhaev who first reported this reaction in 1909.[181] Such epoxidation of olefins is a common transformation in living systems and can be catalyzed by a variety of monooxygenases including flavoproteins and P450 enzymes. While the reactive oxygen species in these enzymes, the flavin-4a-hydroperoxo (164) and ferric-peroxo (compound 0, 196), respectively, are not peracids per se, they are clearly peroxide-containing reagents (Figure 51b). Hence, their catalyzed epoxidation may be classified as pseudo-Prilezhaev reaction. Most epoxidation of double bonds in the biosynthesis of secondary metabolites are carried out by cytochrome P450 enzymes, such as PimD in the pimaricin pathway[182] and EpoK in the epothilone A pathway[183] (Figure 51c).
Figure 51.

(a) Prilezhaev reaction. (b) Biological equivalents of peracids. (c) Proposed mechanism and examples of P450 enzyme-catalyzed epoxidation.
A well-known flavoprotein capable of catalyzing such epoxidation is squalene epoxidase which epoxidizes the terminal double bond of squalene (198) to yield squalene-2,3-oxide (199), the common precursor of many sterols (Figure 52a).[184] A more recent example is the FAD-dependent fungal indole epoxidase Af1206095, which converts fumiquinazoline F (FQF, 200) to an unstable epoxyindole intermediate (201) that is further processed by an alanine-activating NRPS module Af12050 to yield fumiquinazoline A (FQA, 202) (Figure 52b).[185, 186]
Figure 52.

Epoxidation reactions catalyzed by flavoproteins, (a) squalene 2,3-epoxidase (b) Af1206095.
4.3. Nef Reaction
The Nef reaction is the conversion of a primary or secondary nitroalkane (203) to the corresponding aldehyde or ketone (204) (Figure 53a).[187] This reaction was first reported in 1894 by John U. Nef and the original experiment was the acid hydrolysis of nitroethane to yield acetaldehyde and nitrous oxide. The Nef reaction can also occur under reductive condition using catalysts such as titanium trichloride, and hydrolysis of the oxime intermediate (205) leads to a carbonyl product (Figure 53b). [188] A number of enzymes related to “old yellow enzyme” (OYE) had been demonstrated to be involved in the metabolism of electrophilic, and potentially toxic, molecules such as α,β-unsaturated carbonyl compounds and aliphatic and aromatic nitro esters.[189] These enzymes utilize FMN as coenzyme with NAD(P)H as the likely physiological reductant. The following OYE-type enzymes had been shown to have the capability to catalyze the conversion of (E/Z)-1-nitro-2-phenyl-1-propene (206) to the corresponding oxime and aldehyde (Figure 53c): xenobiotic reductases A and B from Pseudomonas putida, 12-oxophytodienoic acid reductase (OPR3) from Lycopersicon esculentum, pentaerythritol teranitrate reductase (PETN-R) from Enterobacter cloacae, glycerol teranitrate reductase (NerR) from Agrobacterium radiobacter, and morphinone reductase (MorR) as well as quinone reductase (YhdA) from unnamed sources.[190] These enzyme-catalyzed degradations of nitroalkanes have been proposed to proceed via the mechanism of Nef reaction, involving the formation of an oxime intermediate.
Figure 53.

(a) Nef reaction. (b) Nef reaction via reductive methods. (c) Nef reaction catalyzed by old yellow enzymes.
4.4. Cannizzaro Reaction
The Cannizzaro reaction is a base-induced disproportionation between two aldehydes to produce an alcohol and a carboxylic acid (Figure 54a).[191] If the starting material is an α-keto aldehyde, an intramolecular Cannizzaro disproportionation can proceed as well.[192] The aldehyde used should not have an acidic α-hydrogen. This reaction was discovered by Stanislao Cannizzaro in 1853. The transformation of methylglyoxal (207), which is a toxic side product derived from the non-enzymatic breakdown of intermediates formed in several primary metabolic pathways, to lactate (210) catalyzed by the sequential actions of glyoxalases I and II was considered to be a biological equivalent of intramolecular Cannizzaro reaction (Figure 54b).[193] Glyoxalase I catalyzes the conversion of methylglyoxal to the thioester of lactate (208), utilizing glutathione (GSH) as a co-substrate. Glyoxalase II hydrolyzes this thioester intermediate to lactate (210). Unlike most other biological redox reactions, the reaction catalyzed by glyoxalase I is coenzyme-independent. When the enzymatic reaction was carried out in 3H2O, the lactate product contained less than 4% of the expected tritium. This observation supported the hypothesis that glyoxalase I operates via a Cannizzaro mechanism, which is involves a 1,2-hydride transfer step (Figure 54b, route A).[194] However, subsequent experiments using [1-2H]-fluoromethylglyoxal as the substrate led to the observation of defluorination event, which strongly supported an alternative mechanism that proceeds via an enediol intermediate (209). The fact that incorporation of solvent hydrogen was also noted in the 3-fluorolactate product lent further credence to the enediol mechanism (Figure 54b, route B).[195, 196] Thus, the reaction catalyzed by glyoxalase I may not be a Cannizzaro reaction. It is mentioned here to highlight the ambiguity surrounding enzyme mechanisms. Several of the biological name reactions described here are a matter of ongoing dispute and some have yet to be confirmed.
Figure 54.

(a) Cannizzaro reaction. (b) The conversion of methylglyoxal to lactate catalyzed by glyoxalase I and II.
4.5. Birch Reduction
4.5.1. Birch reduction in the anaerobic catabolism of aromatic compounds
Microbes growing under anaerobic conditions utilize unique reductive pathways for the breakdown of aromatic compounds. This is in contrast to many oxidative metabolic pathways where oxygen serves as a co-substrate to facilitate the cleavage of the aromatic ring.[197] While some bacteria can degrade hydroxoquinone[198] with NADPH, and resorcinol with ferredoxin,[199] others use benzoyl-CoA (57) as a central intermediate for the reductive degradation of aromatic compounds, such as phenols, toluenes or benzoates.[200] A good example is phenol metabolism in the denitrifying bacterium Thauera aromatica, which is initiated by phosphorylation of phenol (211) to give phenylphosphate (212, Figure 55).[201] Subsequent carboxylation of 212 to generate 4-hydroxybenzoate (4-HBA, 213) is catalyzed by phenylphosphate carboxylase, which represents a biological example of Kolbe-Schmitt reaction.[202] 4-Hydroxybenzoate is then ligated with coenzyme A, followed by dehydroxylation by hydroxybenzoyl-CoA reductase (HBCR) to give benzoyl-CoA (57).[203, 204] Direct carboxylation of benzene to give benzoic acid has also been inferred in select microbial systems.[205, 206] Break down of benzoyl-CoA via the addition of two hydride equivalents by the action of benzoyl-CoA reductase (BCR) yields cyclohexa-1,5-diene-1-carboxyl-CoA (dienoyl-CoA, 215), which can then be catabolized to primary metabolites (Figure 55).[197]
Figure 55.
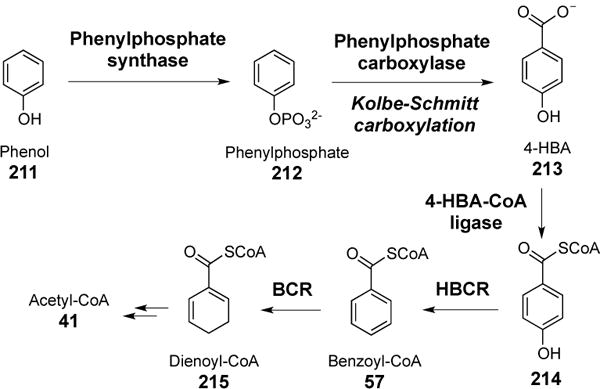
Bacterial anabolism of phenols.
Both HBCR and BCR operate via “Birch-like” reduction mechanisms. The Birch reaction, which was first reported by Arthur Birch in 1944, is a 1,4-reduction of aromatic compounds to their 1,4-hexadienes by alkali metals in ammonia in the presence of alcohol (Figure 56).[207] The alkali metal serves as a source of electrons and the alcohol as a source of protons. Two hydride equivalents are transferred to the reactant and the reaction proceeds via a radical anion intermediate (216). Given the harsh conditions required for the chemical Birch reaction, it is remarkable that such reduction reactions actually occur in biological systems. Although both HBCR and BCR operate via a similar mechanism, the subunit architecture and cofactor composition of these two enzymes are dissimilar. The most characterized examples of these enzymes are from the facultative anaerobe T. aromatica. The T. aromatica BCR will be described first because its mechanism necessitates prior dehydroxylation by HBCR, which will be discussed later.
Figure 56.

Birch reduction.
Benzoyl-CoA reductase (BCR) from T. aromatica
T. aromatica BCR is composed of four subunits: A-D. Subunits C and D, each harboring a single [4Fe-4S] cluster, are the sites where substrate reduction likely takes place.[208–210] In contrast, subunits A and D bind a single [4Fe-4S] cluster at the subunit interface with each subunit contributes two cysteine residues to coordinate the metal cluster. Subunits A and D also contain one ATP-binding site each. The two-electron reduction of benzoyl-CoA is coupled to the hydrolysis of ATP, which transfers its γ-phosphate to a lysine or histidine residue on subunit C.[211] Hydrolysis of ATP can occur in the absence of substrate but the rate is slower. Ferredoxin serves as the ultimate electron source for the BCR reaction but the pathway that the electrons transfer from ferredoxin, through the iron-sulfur clusters, to the substrate remains obscure. The exergonic hydrolysis of ATP plays a central role in driving the endergonic reduction of benzoyl-CoA. EPR spectra recorded 30 sec after the mixing of BCR and ATP revealed the conversion of a low-spin S = 1/2 [4Fe-4S] species to a high-spin S = 7/2 [4Fe-4S] species. It is conceivable that this high-spin species, which has a calculated redox potential ~ 400–500 mV lower than that of the low spin [4Fe-4S], donates electrons to the substrate thus playing the role of metallic sodium in the non-biological Birch reaction.
During the Birch reaction, the first electron transfer to form a radical anion intermediate (e.g., 216) is rate-limiting. To study the mechanism of BCR, a comparison of kcat and Km for various ortho-substituted substrates showed that both kcat and Km increased with the escalation of the electron-acceptor strength of the substituents (F > Cl = H > OH > NH2).[212] A Hammett plot of kcat/Km of the substituted substrate analogs versus kcat of unsubstituted benzoyl-CoA as a function of the pKa of the corresponding benzoic acids was constructed. The slope of the correlation between these two parameters is several orders of magnitude too small for the reaction to proceed via a radical anion mechanism. Thus, in contrast to the chemical Birch reaction, a radical anion (217) is unlikely an intermediate formed during the BCR reaction because proton transfer to the substrate appears to occur concurrently with electron transfer (proton coupled electron transfer, PCET) (57 → 218), drastically lowering the energy barrier for the reaction (Figure 57a). For this reason, the BCR reaction mechanism has been referred to as “Birch-like”. In addition, the p-fluoro substituted substrate analog was not utilized by BCR, suggesting that the para position is the site of the initial proton transfer. This result explains why it is necessary for HBCR to dehydroxylate 4-HBA-CoA (214) to benzoyl-CoA (57) prior to dearomatization by BCR.
Figure 57.

Benzoyl-CoA reductases. (a) Anabolic metabolism of phenols in T. aromatica. (b) BamBC-catalyzed reduction of benzoyl-CoA in G. metallireducens.
Benzoyl-CoA reductase (BCR) from Geobacter metallireducens
A second class of BCR enzymes was recently identified.[213] The genome of the obligate anaerobe Geobacter metallireducens, which can grow on benzoic acid, includes an operon encoding a set of proteins, BamBCDEFGHI (named after benzoic acid metabolism).[214] The gene cluster is conserved in several other members of the genus Geobacter. Initial studies showed that the active enzyme is an α2β2 complex composed of BamB and BamC. The in vitro activity of BamBC was first demonstrated by following the reverse reaction, the oxidation of dienoyl-CoA (215) to benzoyl-CoA (57) in the presence of the electron acceptors. The activity in the forward direction was not successfully reconstituted until Kung et al. utilized a bridged 2,2′-bipyridyl dye as the electron donor instead of sodium dithionite or titanium(III) citrate.[215] Sequence alignment indicated that BamB contains a tungstopterin and a [4Fe-4S] cluster, while subunit C holds two [4Fe-4S] and one [3Fe-4S] clusters. It was also proposed that BamD-I form a protein complex shuttling electrons to the active site of BamBC in vivo.[216] Unlike BCR from T. aromatica, the activity of BCR from G. metallireducens does not involve the hydrolysis of ATP.
Further studies of this system demonstrated the reversible reduction of benzoyl-CoA (57) to dienoyl-CoA (215) when the G. metallireducens BCR was reduced with dienoyl-CoA in the absence of additional electron donors or acceptors (Figure 57b).[215] The midpoint potential for the reversible reduction of benzoyl-CoA was calculated to be ~ 622 mV. This value is far lower than those of other redox couples in biological systems and contrasts with the chemical Birch reaction, which is essentially irreversible. Recently, the crystal structures of BamBC complex in Zn2+-bound inactive state and CoA ester-bound active state were characterized.[217] The structural analyses revealed that the C4 of the substrate aligns with the electron-rich tungsten ligand over a short distance in a solvent-shielded environment. This finding led to the Birch-reduction like mechanism involving protein-assisted electron transfer from metal to the aromatic ring.
Hydroxybenzoyl-CoA reductase (HBCR) from T. aromatica
The dehydroxylating enzyme HBCR is a molybdopterin-mononucleotide-dependent enzyme composed of three subunits with an overall (αβγ)2 architecture.[214, 218, 219]- Based on its similarity to the molybdoproteins with known structures,[220] the α-subunits of T. aromatica HBCR likely bind the molybdopterin cofactor, where catalysis takes place. The β-subunits should bind FAD and the two γ-subunits should each bind two [2Fe-2S] clusters that mediate electron transfer from FAD to the molybdenum cofactor. Unlike similar enzymes, each of the β-subunits of HBCR also includes an additional loop of 40 amino acids long that binds a [4Fe-4S] cluster. As with T. aromatica BRC, the source of electrons for HBCR is from ferredoxin.
Another distinction between HBCR and related enzymes is that HBCR catalyzes the reduction of its substrate while the others catalyze oxidation reactions using oxygen or NAD+ as the ultimate electron acceptor.[220] Moreover, HBCR reaction probably proceeds via a radical mechanism while other related enzymes reduce their substrates two electrons at a time. Based on the redox potentials measured experimentally for the electron mediators in HBCR, electrons are thought to flow sequentially from ferredoxin to the [4Fe-4S] clusters in the γ-subunit, to FAD and to each of the two [2Fe-2S] clusters in the β-subunit, to molybdenum(V), and then to the substrate in the α-subunit. This scheme is the reverse of the electron transfer pathway in related enzymes.[220] The proposed catalytic cycle for the reduction of 4-HBA-CoA (214) beginning with molybdenum(IV) is depicted in Figure 58. It is possible that a molybdenum-coordinated sulfhydryl group serves as the proton donor to protonate the resulting unstable carbanion radical intermediate since the corresponding deprotonated sulfur atom has been shown to function as a proton acceptor in similar enzymes.[220]
Figure 58.

Proposed catalytic cycle of hydroxybenzoyl-CoA reductase.
4.5.2. Birch reduction in isoprene biosynthesis
Terpenes are derived from the sequential coupling of isopentylpyrophosphate (IPP, 220) and its configurational isomer dimethylallylpyrophosphate (DMAPP, 221). Over 55,000 terpenes have been identified and they are ubiquitous in nature.[221] For decades, the mevalonate (MVA) pathway was thought to be the only pathway for the biosynthesis of IPP and DMAPP. But inconsistencies in labeling experiments aimed at elucidating the biosynthesis of terpenes in select organisms eventually led to the discovery of a second pathway for the biosynthesis of IPP and DMAPP in most bacteria, green algae and the plastids of higher plants: the MEP (36) pathway.[222] The ultimate step in the MEP pathway is the conversion of 4-hydroxy-3-methyl-butenyl-1-pyrophosphate (HMBPP, 219) to IPP and DMAPP catalyzed by the enzyme IspH, which contains a [4Fe-4S] cluster that serves as an electron conduit for the reductive dehydration of the substrate (Figure 59a).
Figure 59.

(a) Reactions catalyzed by IspH. (b) Proposed Birch-like reduction mechanism. (c) Proposed organometallic mechanism.
A Birch-like mechanism is one of several that have been proposed for the IspH reaction (Figure 59b).[223] Another similarly viable mechanism under consideration is the organometallic mechanism (Figure 59c).[224] The proposed Birch-like reduction entails two, sequential single electron transfers to the substrate. The first would generate a radical anion (222) and result in the elimination of water to yield an allylic radical, which would then be reduced either directly or with an electron from the [4Fe-4S] cluster, followed by protonation to give IPP and DMAPP.[223] The Birch-like reduction has been challenged because the predicted radical anion intermediate has never been directly observed. On the other hand, a paramagnetic intermediate trapped in an inactive mutant of IspH has been detected and interpreted as a metallocyclopropane species.[224] Moreover, feeding experiments using a series of deuterated isotopologs of 1-deoxy-D-xylulose, the biosynthetic precursor of HMBPP (219), have traced the stereochemical course of the IspH-catalyzed transformation by the deuterium labeling pattern of downstream pentalenene product. The results also favors the organometallic mechanism (Figure 59c).[225]
5. Rearrangement
5.1. Amadori Rearrangement
The series of tautomerizations that result in the conversion of an aldosylamine (223) to a ketosylamine (224) is known as an Amadori rearrangement (Figure 60). The rearrangement is part of the Maillard reaction, which occurs between reducing carbohydrates and amino compounds and is responsible for the aroma, taste, and appearance of processed food.[226] This rearrangement was first reported by Mario Amadori in 1912,[227] and has since been identified as part of a number of enzymatic reaction mechanisms. A few examples of enzyme-catalyzed Amadori rearrangements are described below.
Figure 60.
Amadori rearrangement.
5.1.1. GTP cyclohydrolase in folic acid biosynthesis
The conversion of GTP (13) to 7,8-dihydroneopterin triphosphate (14) catalyzed by GTP cyclohydrolase I (GCHI) is the first step in the biosynthesis of tetrahydrofolate (20, Figure 4).[14] The GCHI reaction mechanism begins with nucleophilic attack by water on guanine C-8 facilitated by Zn2+ leading to loss of C-8 as formate to yield an aminopyrimidine intermediate (225, Figure 61a). Subsequent opening of the ribose is followed by a series of tautomerizations to give the corresponding 2-keto sugar (226). This part of the GCHI mechanism is analogous to Amadori rearrangement.[228, 229] Final ring closure to yield the pterin ring system is achieved by nucleophilic attack of the pyridine amino group upon the C2´ carbonyl followed by dehydration. The same reaction is also involved in the biosynthesis of tetrahydrobiopterin (227), a cofactor required for phenylalanine hydroxylase and nitric oxide synthase, and 7-deazapurine (228), which is a key component of hypermodified tRNA nucleosides (such as queuosine, 229) and nucleoside antibiotics (such as toyocamycin, 230, Figure 61b).[230]
Figure 61.

(a) The Amadori rearrangement in the proposed catalytic mechanism of GCHI. (b) The natural products which share GCHI-catalyzed reaction in their biosynthesis.
5.1.2. ThiG in prokaryotic thiamine biosynthesis
The biosynthesis of the thiamine thiazole ring-containing precursor (231) in Bacillus subtilis requires five enzymes: glycine oxidase (ThiO), which oxidizes glycine (27) to the corresponding imine (232); ThiF, which adenylates the carboxy terminus of the sulfur-carrier protein ThiS to prime it for accepting the sulfur atom from cysteine mediated by a cysteine desulfurase; and thiazole synthase (ThiG), which catalyzes the actual C-S bond formation reaction with the DXP (29) substrate (Figure 62).[231] The reaction is initiated by imine formation between a lysine residue in the active site of ThiG with DXP. The adduct then undergoes tautomerization via Amadori rearrangement to give a 3-keto intermediate (233), which is then subject to nucleophilic attack by the thiocarboxyl-sulfur from ThiS to achieve sulfur incorporation (Figure 62). The final steps include acyl migration, elimination of ThiS, and coupling with 232 to yield thiazole pyrophosphate (231).
Figure 62.

Biosynthesis of thiazole phosphate in Bacillus subtilis.
5.1.3. BexX in the biosynthesis of antibiotic BE-7585A
The angucycline, polyketide antibiotic BE-7585A (234) produced by Amycolatopsis orientalis subsp. vinearia BA-07585 is decorated with three sugars including a disaccharide appendage, 2-thiotrehalose (235, Figure 63). The BE-7585A biosynthetic gene cluster was found to encode an enzyme, designated BexX, which bearing sequence similarity to thiamin synthase (ThiG).[232] Further experiments showed that the mechanisms of these two enzymes are similar as well (Figure 63). The reaction of BexX, which has been named as 2-thioglucose synthase, is initiated by the formation of an imine bond of Lys110 with glucose-6-phosphate (236). This is followed by an Amadori rearrangement to yield a 2-keto sugar intermediate (237), which serves as an acceptor for a nucleophilic sulfur group donated by a sulfur-carrier protein.[233] Interestingly, it was found that the operational sulfur transfer machinery to deliver a sulfur atom to produce the 2-thiosugar 6-phosphate (238) in this case is assembled from components of the sulfur-carrier systems of primary metabolism.[234] The co-opting the sulfur-delivery machinery of primary metabolism for the biosynthesis of sulfur-containing natural products may be a general strategy employed by nature.
Figure 63.

Proposed mechanism for 2-thiosugar formation in BE-7585A biosynthesis.
5.2. Bamberger-like Rearrangement during the Catabolism of Nitrobenzene
The Bamberger rearrangement, named after Eugen Bamberger for his discovery of this reaction in 1890s, is an acid-mediated conversion of N-phenylhydroxylamines (239) to 4-aminophenols (240, Figure 64a).[235] This reaction has been proposed to be a key step in the catabolism of nitrobenzene in a species of Pseudomonas pseudocaligenes which can utilize nitrobenzene as a sole carbon, nitrogen, and energy source (Figure 64b).[236] This is based on the observation that conversion of hydroxylaminobenzene to 2-aminophenol could be achieved using cell-free lysate of P. pseudocaligenes, suggesting a “Bamberger-like” rearrangement. Similar Bamberger-like rearrangements are thought to occur in the catabolism of chloronitrobenzene in bacterial strain LW1 of the family Comamonadaceae,[237] 4-nitrotoluene in Mycobacterium sp. strain HL-4-NT-1,[238] 2,4,6-trinitrotoluene in Clostridium acetobutylicum,[239] and 2-nitrobenzoate in Pseudomonas fluorescens strain KU-7.[240] Two genes encoding enzymes that catalyze the conversion of hydroxylaminobenzene (239) to 2-aminophenol (241), hydroxylaminobenzene (HAB) mutases HabA and HabB, had been identified in a genomic library of P. pseudocaligenes JS45.[241] The recombinantly expressed HAB mutases show no cofactor requirement and are active under anaerobic conditions. When the mutase reaction is carried out in buffer prepared with 18O-labeled water, the resulting product, 2-aminophenol (241), is unlabeled. This result suggests that the hydroxyl group in the product is not derived from solvent but rather from the intramolecular transfer of the hydroxyl group on the hydroxylamino moiety of the substrate (Figure 64b).[242] In contrast to the Bamberger-like reactions described above, in which the ortho position of the hydroxylamino substrate is hydroxylated, 3-hydroxylaminophenol mutase from Ralstonia eutropha JMP134 catalyzes a bona-fide Bamberger rearrangement.[243] The reaction catalyzed by the purified enzyme produced both the ortho (Bamberger-like) and para (Bamberger) hydroxylaminobenzene.
Figure 64.

(a) Bamberger rearrangement. (b) Proposed pathway for the enzyme-catalyzed biodegradation of nitrobenzene.
5.3. Bergman and Myers-Saito Cyclization of Enediyne Antibiotics
Enediyne antibiotics including calicheamicins (242) and neocarzinostatin (243) represent a highly cytotoxic class of polyketides that are characteristic for the presence of two acetylenic groups conjugated to a C-C double bond in their macrocyclic scaffolds (Figure 65).[244] The macrocyclic enediyne core is composed of either nine carbons, as is the case for the neocarzinostatin, or ten carbons in the case of the calicheamicins. The C-C triple bonds in enediyne antibiotics are key to their cytotoxicity. Enediynes typically undergo Bergman cyclization to yield a 1,4-benzene diradical species (244, Figure 65). This is a cycloaromatization reaction named after Robert Bergman who reported the intermediacy of the diradical species in thermal isomerization of enediynes in 1972.[245] However, neocarzinostatin (243) undergoes a different type of rearrangement which yields a 1,4-toluene biradical species (246) via a highly reactive cumulene intermediate (245, Figure 65). Two research groups led by Andrew Myers and Isao Saito discovered this new cyclization independently during their studies of the DNA-cleavage process of neocarzinostatin and the reaction was later named as Myers-Saito cyclization.[246, 247] The substrate for Myers-Saito cyclization is an allenyl enyne and the reaction occurs at a much lower temperature compared to Bergman cyclization. The formation of biradical can be initiated by nucleophilic attack of a thiol nucleophile or photoactivation.[248–251] Once formed, the biradical can abstract hydrogen from the ribose moieties of DNA leading to oxidative single and double strand cleavage.[4]
Figure 65.

Nucleophilic activation of enediynes via Bergman or Myers-Saito cyclizations.
5.4. Fries Rearrangement
The Lewis-acid catalyzed rearrangement of phenol esters and lactams to 2- or 4-ketophenols is known as the Fries rearrangement which was discovered by Karl T. Fries in early 1900s (Figure 66a, route A).[252] In addition to the carbocation-mediated Fries rearrangement, the photo-Fries rearrangement involving radical intermediates was later observed in 1960 (Figure 66a, route B).[253] Although both reactions do not occur in biological systems, the enzyme soybean peroxidase (SBP) was reported to affect a radical-mediated Fries rearrangement of hydroquinone that was linked to a solid-phase support resulting in a 2-ketophenol radical (249, Figure 66b).[254] This radical could be quenched by the addition of apocynin radical (248) to form an ortho-methoxyphenol dimer (250).
Figure 66.
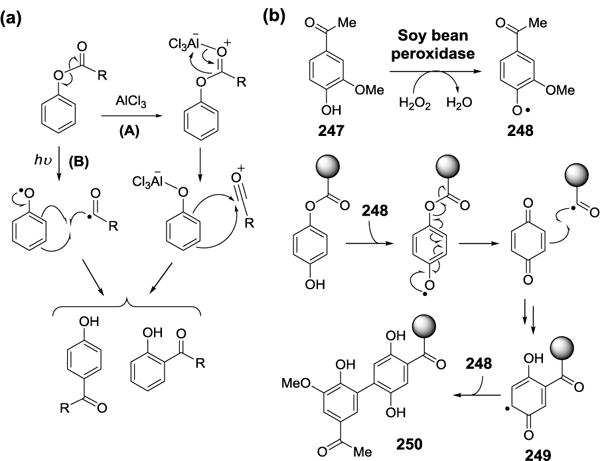
(a) Fries rearrangement and photo-Fries rearrangement. (b) Proposed Fries rearrangement catalyzed by soy bean peroxidise.
5.5. Lossen Rearrangement in Glucosinolate Degradation
Glucosinolates (253) are a class of thiosugar containing plant metabolites produced almost exclusively by the order Capparales, which includes the family Brassicaceae. Broccoli, cabbage, cauliflower, mustard, horseradish, and wasabi are all belong to this family.[255] Upon leaf damage by predators, abrasion, or freeze-thawing, the enzyme myrosinase is released from the vacuole, where it is normally sequestered, and hydrolyzes the C-S glycosidic bond in glucosinolates resulting in the production of variously substituted thiohydroximate-O-sulfates (254, Figure 67b). These compounds then undergo spontaneous desulfonation leading to the production of isothiocyanates (255), thiocyanates, or nitriles depending on the substituents.[256] The release of thiohydroximate-O-sulfate breakdown products is thought to be toxic to plant predators. Thiohydroximate-O-sulfates (254) are most commonly degraded to isothiocyanates (255) via a non-enzymatic Lossen rearrangement (Figure 67b).[257] The Lossen rearrangement was first identified as the base-catalyzed conversion of O-acylated hydroxamic acids (251) to isocyanates (252) by Wilhelm C. Lossen in 1872 (Figure 67a).[258]
Figure 67.

(a) Lossen rearrangement. (b) Non-enzymatic Lossen rearrangement in glucosinolate degradation.
5.6. Criegee Rearrangement
The Criegee rearrangement is a 1,2-rearrangement of an alkyl hydroperoxide (256) involving ionic migration of a carbon substituent onto the neighboring electron-deficient oxygen with loss of the second oxygen atom via heterolytic O–O cleavage (Figure 68).[147, 259] The reaction is named after Rudolf Criegee who first reported this reaction in 1940s. This type of 1,2-shift is similar to the one observed in Baeyer-Villiger oxidation of ketones as described above (Figure 42). In organic chemistry, the alkyl hydroperoxide is formed through the reaction of a tertiary alcohol and a peracid. Similar alky hydroperoxide intermediates are also proposed as part of the catalytic mechanism for several non-heme iron-dependent dioxygenases, such as catechol intradiol and extradiol dioxygenases.[260, 261] Although the ways of alkyl hydroperoxides formation in organic and enzymatic reactions are different, the downstream migration of a C–C bond onto an electron deficient oxygen upon acid or enzyme catalysis clearly represent the key feature of Criegee rearrangement.
Figure 68.
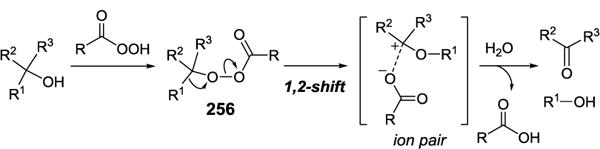
Criegee rearrangement.
The intradiol dioxygenases, represented by catechol 1,2-dioxygenase, require Fe(III) as a cofactor and catalyze the cleavage of the C–C bond between the phenolic hydroxyl groups of catechol (257) to yield muconic acid (261) as product (Figure 69a).[260, 261] The extradiol dioxygenases, exemplified by catechol 2,3-dioxygenase, are Fe(II)-dependent and cleave the C–C bond adjacent to the phenolic hydroxyl groups of catechol to yield 2-hydroxymuconaldehyde (267) as product (Figure 69b). For intradiol cleavage, formation of the Fe(III)-catecholate binary complex (258) introduces semiquinonate radical character to the bound catechol rendering it susceptible to O2 attack to generate an alkylperoxoiron(III) intermediate (259). The alkylperoxo intermediate (259) then undergoes a Criegee-type migration of the adjacent acyl group to yield muconic anhydride (260). The Fe(III)-OH species thus formed acts as the nucleophile to convert 260 to the ring-opened product (261).[260]
Figure 69.

(a) Catechol intradiol dioxygenase. (b) Catechol extradiol dioxygenase.
An analogous alkylperoxo intermediate (265) is also proposed for enzymes catalyzing extradiol cleavage (Figure 69b). In this case, O2 binding to the bound Fe(II)-catechol (262) results in a transient Fe(III)-superoxide complex (263), which is transformed to the proximal alkylperoxo intermediate (265) via a semiquinonato-iron(II)-superoxide species (264). The next step is the migration of the adjacent alkene to give an α-ketolactone intermediate (266), which is hydrolyzed by Fe(II)-hydroxide to give 2-hydroxymuconate semi-aldehyde product (267).[260] The fate of the alkylperoxo intermediate to undergo acyl or alkenyl migration, which ultimately determines the reaction products, is likely governed by stereoelectronic factors, notably the positioning of the alkylperoxo O–O bond.
A Criegee rearrangement may also be a key step for the oxidative cleavage reaction catalyzed by carotenoid dioxygenase. This protein is a non-heme iron-dependent enzyme which generates the substrate radical (269) as a result of direct addition of the Fe(III)-superoxo species (268) to the carotenoid substrate (Figure 70). It is possible that single electron transfer to Fe(III) would yield an allylic carbocation intermediate (270). Subsequent H2O attack followed by a Criegee rearrangement leads to another carbocation intermediate (271 → 272), which is quenched by Fe(II)-OH and decomposed to two aldehyde products.[262]
Figure 70.

Possible mechanism for the cleavage reaction catalyzed by carotenoid cleavage dioxygenase.
5.7. Favorskii Rearrangement
The transformation of enolizable α-haloketones to esters, carboxylic acids or amides via alkoxide-, hydroxide-, or amine-catalyzed rearrangements, respectively, are known as Favorskii rearrangements (Figure 71a). This reaction was first reported in 1894 by Alexei Favorskii.[263] Early labeling experiments have led to the proposal of Favorskii-like rearrangements during the biosynthesis of a handful of polyketide antibiotics including aspyrone from Aspergillus melleus,[264] okadaic acid from Prorocentrum lima,[265] lankacidins from Streptomyces rochei,[266] and enterocin (273) produced by several species of Streptomyces bacteria[267] (Figure 71b). Recent feeding experiments with S. rochei suggested that the proposed Favorskii-type cyclization in lankacidin biosynthesis may be unlikely. In addition, a linear compound was isolated from an lkcE knock-out strain. These results indicated that LkcE, a flavin-dependent amine oxidase, may be responsible for the formation of the 17-membered macrocyclic moiety in lankacidin.[268]
Figure 71.

(a) Favorskii rearrangement. (b) Biosynthetic pathway of enterocin and proposed mechanism for EncM.
The gene cluster responsible for the biosynthesis of enterocin in Streptomyces maritimus had been identified and the genes were designated encA-N.[269] Gene knockout experiments in S. maritimus[270] and heterologous expression of enterocin biosynthetic genes in Streptomyces lividans[271] showed that EncM is the enzyme responsible for an oxidative Favorskii rearrangement (274→275→276) which is the key step in the pathway (Figure 71b). In addition, the EncM also catalyzes two aldol reactions and the formation of two heterocycles (276→277). The purified EncM is a FAD-dependent enzyme. X-ray crystal structures of EncM revealed that flavin is covalently bound to His78 in EncM’s active site. However, study of its in vitro activity was hampered due to the instability of its ACP-bound substrate (274). In a recent report, EncM activity was demonstrated in vivo using a C7,O4-dihydrotetraketide analog as substrate (278).[272] Experiments with 18O2 established that the oxygen at C-4 in the product is derived from molecular oxygen. Interestingly, spectroscopic data suggests that flavin-N5-oxide, rather than flavin C4a-peroxide, is the oxygen donor adding oxygen to C-4 of the substrate.[273]
5.8. Pinacol and Semipinacol Rearrangement
In organic chemistry, pinacol rearrangement is an acid-catalyzed dehydration of a vicinal diol, such as pinacol (279), to generate a carbonyl product, such as pinacolone (280, Figure 72a). This reaction was first described by Wilhelm Rudolph Fittig in 1860s[274] and the rearrangement mechanism has been shown involving formation of a carbocation intermediate followed by a 1,2-migration of the adjacent β–alkyl group. Several variations of pinacol rearrangement have been developed over years, and the term “semipinacol rearrangement” coined by Marc Tiffeneau has been widely used to describe reactions that are related to or similar as pinacol rearrangement (Figure 72b).[275] Compared to the original acid-catalyzed pinacol rearrangement, the reactants of semipinacol rearrangement do not limit to 1,2-diols and the reactions can proceed under acidic, basic or neutral conditions. Two recent examples of pinacol and semipinacol rearrangements found in the biosynthesis of tropolone and aurachin, respectively, are discussed below.
Figure 72.

(a) Pinacol rearrangement. (b) General semipinacol rearrangement.
5.8.1. Pinacol rearrangement in tropolone biosynthesis
Tropolones are seven-membered aromatic ring-containing compounds found in many natural products from bacteria, fungi and plants.[276] Early feeding experiments performed in different organisms revealed that the biosynthetic precursors of tropolones could derive from polyketide, terpene, alkaloid or shikimate intermediates and the ring expansion of an alkylated six-membered ring to tropolone, is the common step in all proposed biosynthetic pathways (Figure 73). One representative example of tropolones, stipitatic acid (282), is a fungal metabolite isolated from Talaromyces stipitatus.[277, 278] An aromatic polyketide, 3-methylorcinaldehyde (281), has been identified as the precursor based on radiotracing study.[279] Two oxidative rearrangement mechanisms for the tropolone ring formation have been proposed. The first one proceeds with a monooxygenase-mediated pinacol rearrangement (Figure 73, route A) and the second mechanism involves dioxygenase-mediated cleavage of the benzene ring, followed by ring-closure via Claisen condensation (Figure 73, route B). To investigate the ring expansion mechanism, T. stipitatus was grown under 18O2 and the isolated stipitatic acid (281) showed the incorporation of a single oxygen atom at the C-1 position. This labeling pattern supports that the ring formation involves a pinacol rearrangement.[280]
Figure 73.

Proposed isotopic labeling patterns in stipitatic acid biosynthesis.
Recent genome sequencing of T. stipitatus led to the discovery of the stipitatic acid biosynthetic gene cluster.[281] Subsequent gene deletion experiments and biochemical characterization showed that three enzymes are required for the tropolone core biosynthesis. TropA, a non-reducing polyketide synthase, is responsible for the construction of the precursor 3-methylorcinaldehyde (281), which is hydroxylated by a FAD-dependent monooxygenase, TropB. The ring expansion of 283 to form the tropolone core is catalyzed by a non-heme iron(II) dioxygenase, TropC. As shown in Figure 74, the mechanism of TropC is likely initiated by the hydroxylation of 283 via an iron(IV) oxo species and the resulting enzyme-bound intermediate 284 then undergoes a pinacol rearrangement to yield the ring expansion product (285).
Figure 74.

Proposed mechanism of tropolone formation catalyzed by TropC.
5.8.2. Semipinacol rearrangement in aurachin biosynthesis
Aurachins are quinoline alkaloids isolated from myxobacterium Stigmatella aurantiaca Sg a15.[282] The discovery of the biosynthetic gene cluster of aurachin derivatives and the following gene-deletion experiments have established that AuaA-E are responsible for the assembly of aurachin D (288, Figure 75),[283] which is the biosynthetic precursor for other aurachin derivatives based on early feeding studies.[284] The N-hydroxylation of aurachin D (288) to aurachin C (289) may be mediated by AuaF (a Rieske [2Fe-2S] protein).[285] Two enzymes, AuaG (a flavin–dependent monooxygenase) and AuaH (a NADH-dependent reductase), are speculated to catalyze the oxidative migration of the prenyl group from C3 to C4 (289→290). It was later demonstrated that the recombinant AuaG can convert aurachin C (289) to an intermediate 292 (Figure 76). This intermediate is then reduced and dehydrated by AuaH to aurachin B (290) in the presence of NADH.[286] A type IV semipinacol rearrangement was proposed to account for AuaG-catalyzed migration of the farnesyl substituent (Figure 76, route A).
Figure 75.

Proposed biosynthetic pathway of aurachin A-D.
Figure 76.

Proposed reaction mechanisms of AuaG/AuaH-catalyzed prenyl group migration.
However, an unexpected ether product (298) was identified when a short-chained analog of aurachin C (297) was used as substrate for AuaG.[287] This ether product (298) is likely resulted from a retro-[2,3]-Wittig rearrangement of 297 (Figure 76). The anionic sigmatropic [2,3]-Wittig rearrangement was observed during the mechanistic studies of the classic [1,2]-Wittig rearrangement.[288] Similar to Claisen or Cope rearrangements, [2,3]-Wittig rearrangement is a concerted, pericyclic reaction proceeding via a five-membered transition state (Figure 76). The finding of the ether product (298 is equivalent to 295 with R=Me) led to an alternative mechanism for the conversion of aurachin C to aurachin B (Figure 76, route B). In this case, the migration of farnesyl group may proceed via a retro-[2,3]-Wittig rearrangement to yield the ether intermediate (293→295). An additional Claisen rearrangement follows to form 294, which is then reduced by AuaH to produce aurachin B (290).
5.9. Wagner-Meerwein Rearrangement in Terpene Biosynthesis
The assembly of terpenes is catalyzed by terpene synthases that are well established to operate via carbocation intermediates. The key features of the catalysis of terpene synthases are the 1,2-rearrangement of carbocation intermediates from which the migratory group, such as alkyl, aryl, or vinyl group, rearranges from a neighboring carbon to the carbocation center to form a more stable carbocation or to alleviate the ring strain. Such 1,2-migraton reactions are also known as Wagner-Meerwein rearrangements. This reaction was first reported by Wagner in 1899,[289] and was subsequently extended to include photo illumination induced carbocation migration reported by Meerwein[290] to become the Wagner-Meerwein rearrangement. A terpene synthase is sometimes capable of generating multiple products from a single substrate resulting in the tremendous structural diversity observed in nature for this class of compounds, which includes over 55,000 members.[221] The number of examples is far too vast to enumerate here. Two pathways shown in Figure 77 illustrate the Wagner-Meerwein rearrangements (299→300 and 302→303) involved in the biosynthesis of the monoterpene camphene (301),[291] and the sesquiterpene vetispiradiene (304).[292]
Figure 77.

Proposed mechanisms for (a) monoterpene and (b) sesquiterpene synthases.
5.10. Pericyclic Reactions: Claisen Rearrangement
The great majority of enzymatic reaction mechanisms are ionic/polar, however, radical mechanisms, including several of the name reactions mentioned above, are also observed to a lesser extent. Pericyclic reactions are another type of organic reactions that are concerted, proceeding via a single transition state with cyclic geometry, and are often rearrangement reactions. Although there exist a few biological pericyclic hydrogen sigmatropic rearrangement reactions, enzyme-catalyzed name reactions of the pericyclic type are exceedingly rare. Thus far, only three are found: the Claisen and Cope rearrangements, and the Diels-Alder cycloaddition.
The Claisen rearrangement is the [3,3]-sigmatropic rearrangement of an allyl vinyl ether to yield a γ,δ-unsaturated carbonyl product. This reaction was discovered by Rainer Ludwig Claisen in 1912[293] and is the first example of a [3,3]-sigmatropic rearrangement. The Cope rearrangement, which was developed by Arthur C. Cope in 1940s, is a [3,3]-sigmatropic rearrangement of 1,5-dienes.[294] The Diels-Alder reaction is a [4+2] cycloaddition between a conjugated diene and an electrophilic alkene (the dienophile) to form a cyclohexene system. (Figure 78). It was named after Otto Paul Hermann Diels and Kurt Alder who first described this reaction in 1928.[295] Except for a few examples, many enzymes that are hypothesized to catalyze pericyclic reactions are only partially or not at all characterized. Hence, their designated functions and the proposed mechanisms remain to be verified.
Figure 78.

Pericyclic name reactions catalyzed by enzymes.
5.10.1. Chorismate mutase
Chorismate mutase catalyzes the conversion of chorismate (305) to prephenate (307) in bacteria, fungi, and plants during the biosynthesis of aromatic amino acids (Figure 79). The overall transformation is a Claisen rearrangement, and the enzyme accelerates the conversion of the chorismate to prephenate by more than 2–3×106-fold than the spontaneous reaction.[296] Early mechanistic studies showed that the cyclization proceeds with the substrate adapts a chair conformation in the transition state for both enzyme-catalyzed and the spontaneous reaction.[297] Although the mechanism is expected to be concerted, kinetic isotope experiments indicated that C5-O bond cleavage occurs to a greater extent than C1-C9 bond formation during the transition state (306).[298] So the transition state has a dipole moment and the reaction is referred to as a “concerted-asynchronous” transformation.
Figure 79.

Transition state of the reaction catalyzed by chorismate mutase.
5.10.2. A Claisen rearrangement during the biosynthesis of the non-ribosomal peptide antibiotic
Cyanobactins (such as 308) are a class of ribosomally encoded peptides produced by ~30% of cyanobacteria.[299, 300] Their structures can be post-translationally modified by cyclization to give oxazolines and thiazolines, and by C- and O-prenylation (Figure 80a). A comparison of known cyanobactin biosynthetic gene clusters had led to the assignment of TruF family of proteins as putative prenyltransferases despite the absence of the conserved prenyltransferase motif.[299] The enzyme LynF from Lyngbya aestuarii is a TruF homolog found in the biosynthetic pathway of aestuaramides. It was exogenously expressed in E. coli,[301] and then incubated with a substrate analog (309) to test its proposed prenyltransferase activity. The results demonstrated that LynF indeed catalyzes prenylation of the tyrosyl residue. Interestingly, the reaction occurs first at the hydroxyl group of tyrosine followed by a non-enzymatic Claisen rearrangement resulting in the observed prenylation at the meta-tyrosyl carbon in the product (310→311, Figure 80b).
Figure 80.

(a) Proposed lyn biosynthetic pathway. (b) LynF-catalyzed O-prenylation and the spontaneous Claisen rearrangement of the analog of aestuaramide B.
5.11. Pericyclic Reactions: Cope Rearrangement
Prenylated indole alkaloids comprise a large class of natural products derived, in part, from tryptophan (70) and DMAPP (221).[302]. A key step in the biosynthesis of the prenylated indole alkaloids is the prenylation of tryptophan at C4 position by dimethylallyltryptophan synthase (DMATS). The X-ray crystal structure of the enzyme from Aspergillus fumigatus has been elucidated and a catalytic mechanism involving initial C-4 prenylation followed by C-4 deprotonation of 312 by Lys174 leading to rearomatization of the indole ring to give dimethylallyltryptophan (DMAT, 313) has been proposed (Figure 81, route A).[303] However, to further investigate the roles of active-site residues in DMATS, it was found that the K174A mutant produced a tricyclic compound (315) prenylated at C-3 as the major product (9-fold excess over 313).[304] This result suggested that the wild-type enzyme may initially prenylate tryptophan at C-3 and the resulting C-3 prenyl group is transferred to C-4 via a reversible Cope rearrangement (Figure 81 route B). Cyclization of 314 to give the tricyclic compound 315 was hypothesized to take place outside of the enzyme active site after abortive attempts to deprotonate the product at C-4.
Figure 81.
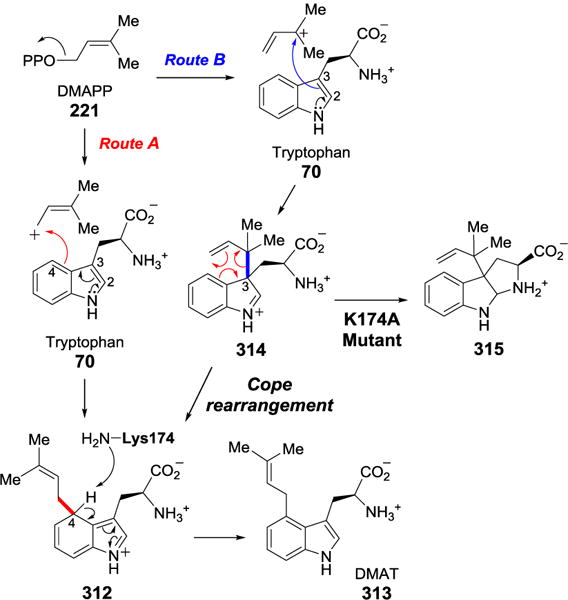
Proposed mechanisms for the reaction catalyzed by DMAT.
5.12. Pericyclic Reactions: Diels-Alder Cycloaddition
The Diels-Alder reaction is a specific type of [4+2] cycloaddition reaction and is an important synthetic strategy for the construction of cyclohexene ring.[305] This ring formation process has been proposed as the key step in the biosynthesis of numerous natural products, including polyketides, isoprenoids and alkaloids.[306] Although many enzyme candidates have been speculated to catalyze [4+2] cycloaddition reactions, only a few of them have been purified and biochemically characterized to catalyze the proposed [4+2]-cycloaddition reactions. However, whether they are indeed naturally evolved Diels-Alderase depends on whether the cyclizations being catalyzed are concerted. Investigation of the catalytic properties of these carbocyclases is a challenging task because most substrates are structurally complex and are difficult to synthesize. Moreover, many of these enzymes, such as macrophomate synthase,[307, 308] riboflavin synthase,[309] solanapyrone synthase,[310] and lovastatin nonaketide synthase,[311, 312] have dual functions rendering validation of the concertness of the cycloaddition reaction more intricate. Because the pursuit to clinch the existence of naturally selected Diels-Alderases has been summarized in several reviews,[313–315] this section will focus on the recent progress in biochemical characterization and mechanistic investigation of several new examples of biosynthetic Diels-Alder reactions.
5.12.1. The aza-Diels-Alder cycloaddition during thiopeptides biosynthesis
Similar to above-mentioned cyanobactins, thiopeptides (such thiocillin, 316) are ribosomally synthesized cyclic oligopeptides with extensive post-translational modifications. One unique component shared among all thiopeptide antibiotics is a pyridine ring (317) formed by a cyclization reaction between two regions of the peptide chain, which can be present in various states of oxidation (Figure 82). An early proposal for the origin of the pyridine ring is a cycloaddition between a diene, consisting of a dehydroalanine residue and an adjacent enol tautomer of the amide backbone, and a dienophilic dehydroalanine residue (318, Figure 82).[316] Identification of a number of thiopeptide biosynthetic gene clusters along with comparative genomic analysis have led to the hypothesis that the proteins encoded by tclM and its homologs including nosO, tsrE, sioL, tpdD, tpdD, and ctlG, could be responsible for the assembly of the pyridine ring despite the fact that these gene products show little homology to any known enzyme families.[317] As predicted, the knock-out strain, Bacillus cereus ATCC 14579 ΔtclM, was unable to achieve pyridine ring cyclization in thiocillin.[318] The purified TclM was later demonstrated to catalyze the intramolecular formation of the pyridine ring (320) when incubated with a substrate analog (319) prepared by chemoenzymatic synthesis. Although TclM and it homologous proteins clearly represent a new class of enzyme catalyzing aza-[4+2] cycloadditions, it remains to be determined whether their catalytic mechanisms are stepwise or concerted.[319]
Figure 82.

Proposed mechanism for TclM-catalyzed aza-[4+2] cycloaddition.
5.12.2. The Diels-Alder cycloaddition during spirotetronates, spirotetramates and PTMs biosynthesis
The spirotetronate polyketide antibiotics contain a cyclohexene ring that is spiro-fused to tetronic acid (322, Figure 83a).[320] Some of the spirotetronates identified also contain a trans-decalin ring system (323). Both the cyclohexene and trans- decalin rings have been proposed to be assembled via Diels-Alder cycloaddition reactions.[321] Although the gene clusters responsible for the biosynthesis of chlorothricin,[322] kijanimicin,[323] tetrocarcin A,[324] abyssomicin C,[325] lobophorin,[326], quartromicin,[327] and other spirotetronates had been identified, no gene could be assigned to encode a putative “Diels-Alderase” in each cluster. A breakthrough was made when Hashimoto et al. discovered a small protein, VstJ, which catalyzes the stereoselective [4+2] cycloaddition to form the spirotetronate structure in versipelostatin (321).[328] When the gene vstJ was inactivated, the resulting transformant accumulated an unknown metabolite which was later identified as the acyclic intermediate (324). This intermediate, upon incubation with VstJ, underwent [4+2] cycloaddition to yield the spirotetronate moiety (325). Genes encoding VstJ homologs, ChlL, TcaU4, LobU2, QmnH, Kijcyc and AbyU are also found in the biosynthetic gene clusters of other spirotetronate-containing natural products. Further biochemical characterizations and structural studies of AbyU by Byrne et al. revealed that this type of Diels-Alderase is cofactor-independent with a β-barrel scaffold.[329] The linear substrate chains are likely arranged in the active site to expedite a transannular pericyclic reaction.
Figure 83.

(a) Versipelostatin and the VstJ-catalyzed reaction. (b) Pyrroindomycin and PryE3-catalyzed decalin formation.
Pyrroindomycin (326) belongs to the family of spirotetramate-containing natural products (Figure 83b). Based on the apparent structural similarity to spirotetronate polyketides, the spirotetramate (327) and decalin (328) moieties of pyrroindomycin were also proposed to be generated via Diels-Alder cycloaddition.[330] Right after the discovery of VstJ as the cyclase for the formation of spiro-conjugate in versipelostatin biosynthesis, Tian et al. reported PyrI4 as the VstJ equivalent catalyzing the formation of the spirotetramate moiety.[328] The X-ray structure of PyrI4 was recently resolved and showed that the N-terminal 22 amino acids of PyrI4 form an α-helix lid which seals the active site in the binary complex with its product.[331] Tien et al also identified PyrE3 as the cyclase catalyzing the cyclization of decalin in pyrroindomycin biosynthesis.[332] Accumulation of intermediate 329 was observed in the ΔpyrE3 mutant (Figure 83b). Results from biochemical experiments using the linear acyclic intermediate 329 as substrate clearly showed that PyrE3 can catalyze the formation of decalin moiety. The counterparts of PyrE3, ChlE3, TcaE1, LbP3 and KijA, can also be found in the biosynthetic pathways of spirotetronates. Although PyrE3 was annotated as a flavoenzyme, the NAD(P)-independence of the cyclization reaction indicated that flavin cofactor does not function as a redox mediator, but plays a structural role instead adjusting the enzyme conformation for [4+2] cycloaddition.
Polycyclic tetramate natural products, such as ikarugamycin (58) and HSAF, have been briefly mentioned in the Dieckmann condensation section. Recent biochemical characterizations of enzymes encoded in the corresponding gene clusters revealed that the tricyclic skeleton is formed in a series of tandam reactions catalyzed by two reductases, IkaB and IkaC, with the cyclohexene core assembled through either a Michael addition or a Diels-Alder reaction (Figure 84).[333, 334]
Figure 84.

Proposed biosynthetic pathway of ikarugamycin.
5.12.3. The Diels-Alder cycloaddition catalyzed by SpnF during spinosyn biosynthesis
The best characterized, naturally occurring [4+2]-cyclase is SpnF, which affects an intramolecular [4+2]-cycloaddition reaction (130→131) during the biosynthesis of spinosyn A, a phytotoxin produced by Saccharopolyspora spinosa (Figure 32).[105] The biosynthetic pathway of spinosyn has been briefly discussed in the Rauhut-Currier section. Although this cyclization can occur non-enzymatically, catalysis by SpnF increases the reaction rate by 500-fold. The rate enhancement may result from the binding and re-orientation of the polyketide macrocycle mediated by SpnF to reach optimal geometry for the cycloaddition reaction to occur in its active site. SpnF structure with the consensus docking of its substrate and product revealed an active cavity forming favorable interactions between SpnF and the macrolactone substrate.[335] However, as all aforementioned [4+2]-cyclases, it remains unclear whether the SpnF reaction proceeds via a pericyclic transition state. Although a concerted, albeit asynchronous, Diels-Alder mechanism for SpnF-catalyzed cyclization is supported by the theoretical study by Hess and Smentek,[336] recent quantum mechanical computations and dynamic simulations show that this cycloaddition is not well described as either a concerted or stepwise process.[283] The transition state for the reaction is ambimodal and leads to both the observed Diels-Alder and an unobserved [6+4] cycloadduct (Figure 85).[337] A rapid Cope rearrangement converts the [6+4] adduct into the observed [4+2] adduct. Thus, the cycloaddition appears to occur by dynamically stepwise modes featuring an “entropic intermediate”. A recently developed chemoenzymatic synthesis of spinosyn[334] is useful for preparing labeled substrates required for the study of the kinetic isotope effects of the cyclization reaction to address the key mechanistic question of SpnF.
Figure 85.

The [4+2] cycloaddition reaction catalyzed by SpnF may be a step process involving [6+4] cycloaddition reaction followed by Cope rearrangement.
6. Miscellaneous
6.1. Friedel-Crafts Alkylation
The Friedel-Crafts reactions are aromatic electrophilic substitution reactions which were first developed by Charles Friedel and James Crafts in late 1870s.[338] In a typical Friedel-Crafts alkylation reaction, the electrophile is often an acyl halide activated by a strong Lewis acid catalyst such as aluminum trichloride.
In the reactions of aromatic amino acid prenyltransferases
The reactions catalyzed by prenyltransferases, which mediated the coupling of prenyl moieties with different chain lengths from different prenyl donors to various acceptors, have been demonstrated to be biological Friedel-Drafts reactions.[302, 339] The best-characterized prenyltransferases are from the DMATS superfamily, such as FgaPT2, 5-DMATS, 6-DMATSSv and 7-DMATS. This enzyme superfamily is also mentioned in the Cope rearrangement section and known to catalyze regiospecific C-prenylation of tryptophan (70) in the presence of DMAPP (221, Figure 86a).[340] Studies of several DMATS have shown that the prenyl transfer is initiated by the formation of a dimethylallyl carbocation,[303, 304] followed by nucleophilic attack by the electron-rich indole ring to form a non-aromatic intermediate, which is re-aromatized by elimination of a proton to generate the prenylated product (Figure 86b). Since the appended prenyl chain can be further modified by cyclization, oxidation, and rearrangement, prenyltransferases play an important role in generating structural diversity of secondary metabolites.[341]
Figure 86.

(a) Selected reactions catalyzed by dimethylallyltryptophan synthases. (b) Proposed mechanisms for the reaction catalyzed by dimethylallyltryptophan synthase.
In the reactions of aromatic amino acid lyases and mutases
Histidine, phenylalanine, and tyrosine ammonia lyases (HAL, PAL, and TAL) are a class of enzyme that catalyze the elimination of the amine group from their respective amino acid substrates (Figure 87a).[342–348] On the other hand, phenylalanine and tyrosine aminomutase (PAM and TAM) catalyze the conversion of phenylalanine and tyrosine to their corresponding β-amino acids (Figure 87a).[349–352] Each of the enzymes described above contains 5-methylene-3,5-dihydroimidazole-4-one (MIO, 338) as a cofactor, which is formed in a spontaneous post-translational modification process involving cyclization of the backbone tripeptide Ala-Ser-Gly (337)(Figure 87b).[353, 354]
Figure 87.

(a) The MIO-based amino acids lyases and mutases. (b) Formation of MIO by post-translational modification.
Two mechanisms have been proposed for the role of MIO during catalysis. The first mechanism is an E1cB (elimination unimolecular conjugate base) reaction in which MIO forms a covalent adduct with the α-amino group of the substrate (Figure 88, route A). This mechanism was supported by early studies on HAL, in which conversion of urocanate (332) to histidine (331) proceeds without incorporation of exogenously added 15NH3. This result suggested the intermediacy of a covalently bound amine, which undergoes internal return in the reverse reaction.[355] The problem with the E1cB mechanism for HAL is that it requires the removal of a non-acidic β-proton (pKa > 40) in the subsequent step (339→332). However, this is not an issue for PAL/PAM or TAL/PAM, since the proton being abstracted is a benzylic proton in these cases. An alternative mechanism was also proposed in which covalent bond formation occurs between the imidazole (in HAL case) and the benzene ring (in the case of PAL/PAM and TAL/TAM) of the substrate and MIO (Figure 88, route B).[356] Considering that the imidazole moiety is also an aromatic system, if this mechanism is operative, it would be an example of a biological Friedel-Crafts alkylation reaction.
Figure 88.

Proposed mechanisms for HAL-catalyzed reaction.
Several lines of evidence support the Friedel-Crafts mechanism for this class of enzymes. For example, 5-nitrohistidine can still be turned over by the dehydroalanine-less HAL mutants.[357] These results indicated that the primary role of MIO is to acidify the β-proton since, in the absence of MIO, a nitro substituent on the imidazole ring can assume the same role to activate the β-proton. Likewise, m-tyrosine (3-hydroxyphenylalanine) was shown to be a slightly better substrate for PAL than phenylalanine.[358] This observation also supported the Friedel-Crafts mechanism because the m-hydroxyl group would increase the nucleophilicity at the ortho position of the phenyl ring thereby enhancing covalent bond formation with MIO.
However, the Friedel-Crafts mechanism has been called into question when the structure of Streptomyces globisporus TAM co-crystallized with the product analog α,α-difluoro-β-tyrosine became available.[359] The crystal structure clearly shows that the amino group is covalently bound to the methylene carbon in MIO as predicted by the E1cB mechanism. Interestingly, Bornscheuer and Bartch recognized that a conserved asparagine residue in TAL and TAM is substituted by glutamate residue in PAL and PAM.[360] Using molecular dynamics simulations, they predicted a strong interaction between MIO and the carboxylate group of glutamate residue in Petroselinum crispum PAL. Such interaction renders the amino group distant from MIO, making the E1cB mechanism unlikely in this case.[360] Yet, the recently reported crystal structure of PAL from Pantoea agglomerans revealed the presence of a N-linked covalent adduct with MIO in the active site, thus sustaining the E1cB mechanism.[361] While more recent computational studies also favor the formation of a covalent MIO-amine adduct during the HAL,[362] TAL,[363] and PAM reactions,[364] it remains feasible that these MIO-dependent aromatic amino acid ammonia lyases and mutases are capable of utilizing both mechanisms.
6.2. Kolbe-Schmitt Carboxylation
The carboxylation of sodium phenolate with carbon dioxide to yield salicylic acid is known as the Kolbe-Schmitt reaction which is named after Hermann Kolbe and Rudolf Schmitt for their discovery of this reaction in the late 1800s (Figure 89a).[365, 366] The carboxylation of phenylphosphate (212) to yield 4-HBA (213) catalyzed by phenylphosphate carboxylase is an example of biological Kolbe-Schmitt reaction (Figure 89b).[201, 202] As mentioned in the section of Birch reduction, this carboxylation reaction is the second step of the anaerobic degradation of phenol (211) in facultative anaerobic bacteria (Figure 55).[201, 202] Phenylphosphate carboxylase from T. aromatica has been characterized to be composed of four subunits:[367] a heterononameric “core” (αβγ)3 complex and a homotrimeric phosphorylase (δ)3 complex. The α and β subunits display 50% sequence similarity to E. coli 3-octaprenyl-4-hydroxybenzoate carboxy-lyase (UbiD), which is involved in ubiquinone biosynthesis.[368] Interestingly, the (αβγ)3 complex can catalyze the reversible decarboxylation of 4-HBA (213) to phenol (211), but the (δ)3 complex is required for the carboxylation of phenyl-phosphate (Figure 89b). In this system, dephosphorylation by the (δ)3 complex is coupled to the carboxylation of the substrate, thus conserving ATP consumed during the first step of the pathway.
Figure 89.

(a) Kolbe-Schmitt carboxylation reaction. (b) Possible roles of αβγ and δ subunits in the phenylphosphate carboxylation reaction.
The bacterial UbiD and UbiX, and its fungal equivalents Fdc1 and Pad1, catalyze the non-oxidative reversible decarboxylation of aromatic compounds, such as cinnamic acid (334, Figure 90a).[200, 369] Although both enzymes show distant homology to flavoproteins and have been annotated as decarboxylases, their proposed activities have not been reconstituted successfully in vitro.[370, 371] Recently, an in-depth structural study combined with biochemical characterization revealed that the real function of UbiX/Pad1 is actually an unusual flavin prenyltransferase, catalyzing the condensation of DMAPP (221) and FMNH2 (173) to yield a prenylated FMN (prFMN, 341, Figure 90b).[372] A neutral iminium form of this prenylated flavin (prFMNiminium, 342), an oxidized product of prFMN (341) generated in the Fdc1-catalyzed maturation, was shown to be catalytically relevant for the reversible decarboxylation activity of UbiD/Fdc1.[373] Since the substrate of UbiD/Fdc1 is an α,β-unsaturated acid, the reaction mechanism of UbiD/Fdc1 unlikely follows the Kolbe-Schmitt reaction. It is thus hypothesized that the substrate (334) and the modified flavin cofactor (342) may form a pyrrolidine adduct (343) via 1,3-dipolar cycloaddition, which then undergoes Grob-type fragmentation to release the carbon dioxide (Figure 90c).[373] While the mechanistic details of UbiD/Fdc1-mediated reaction require further investigation, the proposed mechanism suggests the likely involvement of a brand new type of flavin-derived cofactor in the reversible decarboxylation.
Figure 90.
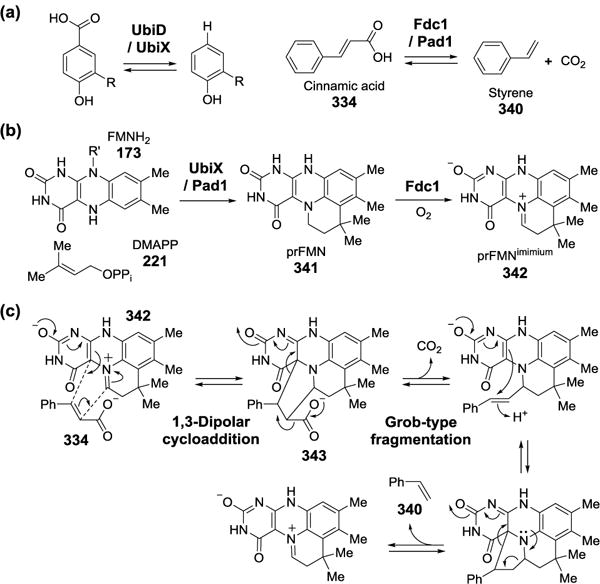
(a) The reactions catalyzed by bacterial UbiD/UbiX and fungal Fdc1/Pad1. (b) FMNH2 prenylation catalyzed by UbiX/Pad1. (c) Proposed mechanism for UbiD/Fdc1-catalyzed reversible decarboxylation.
6.3. Grob Fragmentation in Triterpene Biosynthesis
Over 100 triterpene skeletons leading to compounds including steroids and many triterpene natural products are derived from a common precursor, oxidosqualene (199).[374–376] An array of terpene synthases called oxidosqualene cyclases are responsible for the structural diversity of the products derived from this single molecule through catalytic variability in cation-π annulation, 1,2-methyl and hydride shifts, ring expansion, and deprotonation. Recently, the gene At5g42600 in Arabidopsis Thaliana was determined to encode an oxidosqualene cyclase that catalyzes a Grob fragmentation to yield marneral (344) and the enzyme was designated marneral synthase (Figure 91b).[377] Prior to its identification, marneral had been proposed as a central precursor for the biosynthesis of iridals (345), a class of triterpenoids from the plant genus Iridaceae[378] that display diverse biological activities.[379–381] The Grob fragmentation named after Cyril A. Grob is a concerted C-C bond cleavage involving a five-atom system (Figure 91a).[382]
Figure 91.
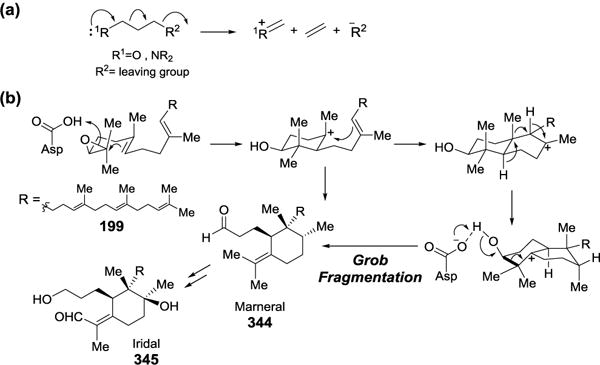
(a) Grob fragmentation. (b) Proposed mechanism for marneral synthase.
6.4. Hunsdiecker-type Halogenation in Polybrominated Aromatic Compounds Biosynthesis
The Hunsdiecker reaction, also known as Hunsdiecker-Borodin reaction, is a reaction between metal carboxylates and halogens to produce organic halides. It was first reported by Aleksandr P. Borodin in a synthesis of methyl bromide from silver acetate and bromine in 1861.[383] It was further demonstrated by Cläre and Heinz Hunsdiecker to be a general and useful halogenation method (Figure 92a).[384, 385] The mechanism of the reaction involves homolytic cleavage of the O−X bond of the acyl hypohalite intermediate (346) followed by decarboxylation and radical recombination to give the alkyl halide product.[386] Many variations of Hunsdiecker reaction had been developed over the years. A metal-free version starting from α,β-unsaturated carboxylic acids (347) is also known and the reaction is believed to proceed with an ionic mechanism (Figure 92b).[387] A flavoenzyme Bmp5 functioning as a phenol brominase in the biosynthesis of marine polybrominated natural products is speculated as a biological precedent of the Hunsdiecker-type halogenation.[388] Biochemical experiments showed that the conversion of 4-HBA (213) to 2,4-dibormophenol (348) catalyzed by Bmp5 likely involves two sequential electrophilic brominium additions followed by decarboxylation. Further studies are needed to verify the mechanism of this biological halogenation reaction.
Figure 92.
(a) The Hunsdiecker halogenation. (b) A metal-free version of Hunsdiecker reaction. (c) Proposed mechanism for Bmp5-catalyzed decarboxylative bromination.
7. Conclusion
Nature has been a rich source for new chemical reactions because enzymatic transformations are continuously evolved via the random process of genetic mutation and tested through natural selection. Hence, we identified dozens of name reactions that occur in biological systems. Many of these reactions have physiological and medicinal importance. The decarboxylating Claisen condensation, for example, is the reaction used for C-C bond formation in fatty acid biosynthesis as well as polyketides biosynthesis in many organisms. One of the steps in the biosynthesis of menaquinone (vitamin K) is a Dieckmann condensation. The Dieckmann condensation is also responsible for the release of tetramic acid-containing PK-NRPs from their corresponding PK-NRP synthases. Likewise, the Mannich reaction and Pictet-Spengler condensation are highly important in the biosynthesis of plant alkaloids. The Bergman and Meyers-Seito cyclization are required for the activation of enediyne antibiotics. More recently, enzymatic Diels-Alder reactions have been in vitro demonstrated to occur in the biosynthesis of a number of bioactive compounds including spirotetronate and spirotetramate antibiotics, thiopeptides and spinosyn.
Additional enzymes that catalyze name reactions have been increasingly employed during organic synthesis owing to the regio- and enantiospecificity of the reactions catalyzed by the responsible enzymes. These include the Bayer-Villiger monooxygenases and TPP-dependent enzymes capable of catalyzing stereospecific oxidation and Stetter reactions, respectively. A number of ‘promiscuous’ enzymes that can accept a variety of substrates have also been used to catalyze organic reactions. For example, hydroxynitrile lyase can effect Henry (nitroaldol) reactions, lipases can catalyze Knoevenagel condensations, and soybean peroxidase reportedly can catalyze Fries rearrangement. Still many other enzymes catalyzing name reactions can be utilized for the bioremediation of polluted soil and water. These include members of the OYE family of enzymes, which catalyze the denitration of aromatic compounds via the Nef reaction. A number of enzymatically catalyzed name reactions are specifically involved in the anaerobic degradation of aromatic pollutants. These include the Kolbe-Schmitt reaction catalyzed by phenylphosphate carboxylase and 4-hydroxybenzoic acid decarboxylase, and the Birch reaction catalyzed by hydroxybenzoyl-CoA reductase and the benzoyl-CoA reductases.
Our understanding of the biosynthesis of complex natural products, including antibiotics, and the degradation of xenobiotic compounds is expanding at a rapid pace. Although nature’s repertoire of chemical reaction types is less than that achieved through synthetic chemistry efforts, ongoing efforts will certainly discover new enzymatic reactions that have been explored in organic chemistry, or develop biomimetic synthetic methods inspired by natural catalysts. Interestingly, some atypical biological reactions have been described as “example[s] of life imitating the art of synthetic chemistry”;[389] however, one could just as easily argue the converse of this statement. While such existential debates are probably best reserved for the pub and over beers, it is nevertheless instructive to explore the parallels between the synthetic paradigms that have been developed in the laboratory and those which have arisen naturally in living organisms. Lessons learned from such analyses can help one to understand the chemistry underlying enzyme catalysis while at the same time providing inspiration for the discovery of new synthetic methods. The present review is thus intended as a study in this very spirit with an emphasis on named synthetic reactions that have established biological counterparts. It is hoped that the synthetic organic chemist and biochemist alike will both come away with a deeper appreciation of just how much their fields in fact overlap and the insights each has to offer the other.
Acknowledgments
We thank Mark Ruszczycky and Jon Gengler for their comments on this paper. This work was supported in part by grants from the National Institutes of Health (GM035906 and GM040541) and the Welch Foundation (F-1511). R.M.M was a recipient of the Ruth L. Kirschstein National Research Service Award (GM103181).
Biographies

Hung-wen (Ben) Liu was born in Taipei (Taiwan). He received a PhD under Professor Koji Nakanishi from Columbia University and did his postdoctoral training under Professor Chris Walsh at MIT. He joined the faculty of Chemistry at the University of Minnesota in 1984. He was promoted through the ranks becoming the Distinguished McKnight University Professor in 1999. In 2000, he moved to the University of Texas at Austin, where he now holds the George H. Hitchings Regents Chair in Drug Design and is Professor in the Division of Chemical Biology and Medicinal Chemistry, College of Pharmacy, and the Department of Chemistry. His research lies at the crossroads of organic and biological chemistry, with particular emphasis on enzymatic mechanisms, natural-product biosynthesis, carbohydrate chemistry, protein function regulation, inhibitor design and synthesis, and metabolic pathway engineering.

Chia-I Lin grew up in Taitung (Taiwan) and earned both Bachelors and Master degrees in Chemistry from the National Taiwan University. She performed doctoral research in Professor Hung-wen Liu’s group at the University of Texas, Austin, where she focused on elucidation of the biosynthetic pathways of heteroatom-containing unusual sugars. Chia-I is currently a postdoctoral scientist in the laboratory of Professor Michelle Chang at the University of California, Berkeley.

Reid M. McCarty received his bachelor’s degree in chemistry from the University of Arizona, where he also received his Ph.D. in biochemistry under the guidance of Prof. Vahe Bandarian. He was a recipient of the Ruth L. Kirschstein National Research Service Award and worked as a postdoctoral fellow in the laboratory of Prof. Hung-wen Liu at the University of Texas, Austin. His research was focused on the mechanistic study of radical SAM enzymes. He passed away in June of 2014.
Footnotes
C.-I L. and R. M. M. contributed equally to this work
References
- 1.Pelouze J. Ann Pharm. 1834;10:249. [Google Scholar]
- 2.Tu Y, Wang ZX, Shi Y. J Am Chem Soc. 1996;118:9806. [Google Scholar]
- 3.Kallmeyer J, Pockalny R, Adhikari RR, Smith DC, D’Hondt S. Proc Natl Acad Sci U S A. 2012;109:16213. doi: 10.1073/pnas.1203849109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith AL, Nicolaou KC. J Med Chem. 1996;39:2103. doi: 10.1021/jm9600398. [DOI] [PubMed] [Google Scholar]
- 5.Leisch H, Morley K, Lau PCK. Chem Rev. 2011;111:4165. doi: 10.1021/cr1003437. [DOI] [PubMed] [Google Scholar]
- 6.Wurtz CA. Bull Soc Chim Fr. 1872;17:436. [Google Scholar]
- 7.Claisen RL. Ber Dtsch Chem Ges. 1887;20:655. [Google Scholar]
- 8.Dieckmann W. Ber Dtsch Chem Ges. 1894;27:102. [Google Scholar]
- 9.Machajewski TD, Wong CH. Angew Chem Int Ed. 2000;39:1352. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 10.Brovetto M, Gamenara D, Mendez P Saenz, Seoane GA. Chem Rev. 2011;111:4346. doi: 10.1021/cr100299p. [DOI] [PubMed] [Google Scholar]
- 11.Jaffe E. J Bioenerg Biomembr. 1995;27:169. doi: 10.1007/BF02110032. [DOI] [PubMed] [Google Scholar]
- 12.Jaffe EK, Hanes D. J Biol Chem. 1986;261:9348. [PubMed] [Google Scholar]
- 13.Stork G, Dowd SR. J Am Chem Soc. 1963;85:2178. [Google Scholar]
- 14.Wang Y, Li Y, Yan H. Biochemistry. 2006;45:15232. doi: 10.1021/bi060949j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews RG, Drummond JT. Chem Rev. 1990;90:1275. [Google Scholar]
- 16.Takahashi S, Kuzuyama T, Watanabe H, Seto H. Proc Natl Acad Sci U S A. 1998;95:9879. doi: 10.1073/pnas.95.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munos JW, Pu X, Mansoorabadi SO, Kim HJ, Liu H-w. J Am Chem Soc. 2009;131:2048. doi: 10.1021/ja807987h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manning KA, Sathyamoorthy B, Eletsky A, Szyperski T, Murkin AS. J Am Chem Soc. 2012;134:20589. doi: 10.1021/ja310353c. [DOI] [PubMed] [Google Scholar]
- 19.Al-Mestarihi A, Romo A, Liu H-w, Bachmann BO. J Am Chem Soc. 2013;135:11457. doi: 10.1021/ja404987r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plowman KM, Cleland WW. J Biol Chem. 1967;242:4239. [PubMed] [Google Scholar]
- 21.Salam WH, Bloxham DP. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1986;873:321. doi: 10.1016/0167-4838(86)90079-8. [DOI] [PubMed] [Google Scholar]
- 22.Phillips RS, Dua RK. J Am Chem Soc. 1991;113:7385. [Google Scholar]
- 23.Rock CO, Cronan JE. Biochim Biophys Acta. 1996;1302:1. doi: 10.1016/0005-2760(96)00056-2. [DOI] [PubMed] [Google Scholar]
- 24.Smith S. FASEB J. 1994;8:1248. [PubMed] [Google Scholar]
- 25.White SW, Zheng J, Zhang YM, Rock CO. Annu Rev Biochem. 2005;74:791. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 26.Cantu DC, Chen Y, Reilly PJ. Protein Sci. 2010;19:1281. doi: 10.1002/pro.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modis Y, Wierenga RK. J Mol Biol. 2000;297:1171. doi: 10.1006/jmbi.2000.3638. [DOI] [PubMed] [Google Scholar]
- 28.Mathieu M, Modis Y, Zeelen JP, Engel CK, Abagyan RA, Ahlberg A, Rasmussen B, Lamzin VS, Kunau WH, Wierenga RK. J Mol Biol. 1997;273:714. doi: 10.1006/jmbi.1997.1331. [DOI] [PubMed] [Google Scholar]
- 29.Hopwood DA, Sherman DH. Annu Rev Genet. 1990;24:37. doi: 10.1146/annurev.ge.24.120190.000345. [DOI] [PubMed] [Google Scholar]
- 30.Yu D, Xu F, Zeng J, Zhan J. IUBMB Life. 2012;64:285. doi: 10.1002/iub.1005. [DOI] [PubMed] [Google Scholar]
- 31.Austin MB, Noel JP. Nat Prod Rep. 2003;20:79. doi: 10.1039/b100917f. [DOI] [PubMed] [Google Scholar]
- 32.Austin MB, Bowman ME, Ferrer JL, Schroder J, Noel JP. Chem Biol. 2004;11:1179. doi: 10.1016/j.chembiol.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Moore BS, Hertweck C. Nat Prod Rep. 2002;19:70. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- 34.Chan YA, Podevels AM, Kevany BM, Thomas MG. Nat Prod Rep. 2009;26:90. doi: 10.1039/b801658p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khosla C. J Org Chem. 2009;74:6416. doi: 10.1021/jo9012089. [DOI] [PubMed] [Google Scholar]
- 36.Hertweck C. Angew Chem Int Ed. 2009;48:4688. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 37.Schobert R, Schlenk A. Bioorg Med Chem. 2008;16:4203. doi: 10.1016/j.bmc.2008.02.069. [DOI] [PubMed] [Google Scholar]
- 38.Sims JW, Fillmore JP, Warner DD, Schmidt EW. Chem Commun. 2005;186 doi: 10.1039/b413523g. [DOI] [PubMed] [Google Scholar]
- 39.Tokuoka M, Seshime Y, Fujii I, Kitamoto K, Takahashi T, Koyama Y. Fungal Genet Biol. 2008;45:1608. doi: 10.1016/j.fgb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Schumann J, Hertweck C. J Am Chem Soc. 2007;129:9564. doi: 10.1021/ja072884t. [DOI] [PubMed] [Google Scholar]
- 41.Yu F, Zaleta-Rivera K, Zhu X, Huffman J, Millet JC, Harris SD, Yuen G, Li XC, Du L. Antimicrob Agents Chemother. 2007;51:64. doi: 10.1128/AAC.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jomon K, Kuroda Y, Ajisaka M, Sakai H. J Antibiot. 1972;25:271. doi: 10.7164/antibiotics.25.271. [DOI] [PubMed] [Google Scholar]
- 43.Sims JW, Schmidt EW. J Am Chem Soc. 2008;130:11149. doi: 10.1021/ja803078z. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Walsh CT. Biochemistry. 2009;48:8746. doi: 10.1021/bi901123r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lou L, Chen H, Cerny RL, Li Y, Shen Y, Du L. Biochemistry. 2012;51:4. doi: 10.1021/bi2015025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gui C, Li Q, Mo X, Qin X, Ma J, Ju J. Org Lett. 2015;17:628. doi: 10.1021/ol5036497. [DOI] [PubMed] [Google Scholar]
- 47.Sun Y, Hahn F, Demydchuk Y, Chettle J, Tosin M, Osada H, Leadlay PF. Nat Chem Biol. 2010;6:99. doi: 10.1038/nchembio.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bishop DHL, Pandya KP, King HK. Biochem J. 1962;83:606. doi: 10.1042/bj0830606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collins MD, Goodfellow M, Minnikin DE, Alderson G. J Appl Bacteriol. 1985;77:86. doi: 10.1111/j.1365-2672.1985.tb01431.x. [DOI] [PubMed] [Google Scholar]
- 50.Meganathan R. In: Cofactor Biosynthesis. Litwack TB Gerald., editor. Vol. 61. Academic Press; 2001. p. 173. [Google Scholar]
- 51.Jiang M, Cao Y, Guo ZF, Chen M, Chen X, Guo Z. Biochemistry. 2007;46:10979. doi: 10.1021/bi700810x. [DOI] [PubMed] [Google Scholar]
- 52.Jiang M, Chen X, Guo ZF, Cao Y, Chen M, Guo Z. Biochemistry. 2008;47:3426. doi: 10.1021/bi7023755. [DOI] [PubMed] [Google Scholar]
- 53.Li HJ, Li X, Liu N, Zhang H, Truglio JJ, Mishra S, Kisker C, Garcia-Diaz M, Tonge PJ. Biochemistry. 2011;50:9532. doi: 10.1021/bi200877x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fuchs G, Mohamed MES, Altenschmidt U, Koch J, Lack A, Brackmann R, Lochmeyer C, Oswald B. In: Kluwer Academic Publishers. Ratledge C, editor. 1994. p. 513. [Google Scholar]
- 55.Colberg PJS, Young LY. In: Wiley-Liss. Young LY, Cerniglia CE, editors. 1995. p. 307. [Google Scholar]
- 56.Gibson J, Harwood CS. In: Kluwer Academic Publishers. Blankenship RE, Madigan MT, Bauer CE, editors. 1995. p. 991. [Google Scholar]
- 57.Harwood CS, Gibson J. J Bacteriol. 1997;179:301. doi: 10.1128/jb.179.2.301-309.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pelletier DA, Harwood CS. J Bacteriol. 1998;180:2330. doi: 10.1128/jb.180.9.2330-2336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pelletier DA, Harwood CS. J Bacteriol. 2000;182:2753. doi: 10.1128/jb.182.10.2753-2760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eberhard ED, Gerlt JA. J Am Chem Soc. 2004;126:7188. doi: 10.1021/ja0482381. [DOI] [PubMed] [Google Scholar]
- 61.Pictet Ae, Spengler T. Ber Dtsch Chem Ges. 1911;44:2030. [Google Scholar]
- 62.Tatsui G. J Pharm Soc Jpn. 1928;48:453. [Google Scholar]
- 63.Stöckigt J, Antonchick AP, Wu F, Waldmann H. Angew Chem Int Ed. 2011;50:8538. doi: 10.1002/anie.201008071. [DOI] [PubMed] [Google Scholar]
- 64.Scott AI, Lee SL. J Am Chem Soc. 1975;97:6906. doi: 10.1021/ja00856a073. [DOI] [PubMed] [Google Scholar]
- 65.Treimer JF, Zenk MH. Eur J Biochem. 1979;101:225. doi: 10.1111/j.1432-1033.1979.tb04235.x. [DOI] [PubMed] [Google Scholar]
- 66.Hampp N, Zenk MH. Phytochemistry. 1988;27:3811. [Google Scholar]
- 67.Kutchan TM, Hampp N, Lottspeich F, Beyreuther K, Zenk MH. FEBS Lett. 1988;237:40. doi: 10.1016/0014-5793(88)80167-4. [DOI] [PubMed] [Google Scholar]
- 68.Kutchan TM, Dittrich H, Bracher D, Zenk MH. Tetrahedron. 1991;47:5945. [Google Scholar]
- 69.Ma X, Panjikar S, Koepke J, Loris E, Stöckigt J. The Plant Cell. 2006;18:907. doi: 10.1105/tpc.105.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loris EA, Panjikar S, Ruppert M, Barleben L, Unger M, Helmut S, Joachim S. Chem Biol. 2007;14:979. doi: 10.1016/j.chembiol.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 71.Maresh JJ, Giddings L-A, Friedrich A, Loris EA, Panjikar S, Trout BL, Stöckigt J, Peters B, O’Connor SE. J Am Chem Soc. 2008;130:710. doi: 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen Q, Ji C, Song Y, Huang H, Ma J, Tian X, Ju J. Angew Chem Int Ed. 2013;52:9980. doi: 10.1002/anie.201303449. [DOI] [PubMed] [Google Scholar]
- 73.Mori T, Hoshino S, Sahashi S, Wakimoto T, Matsui T, Morita H, Abe I. Chem Biol. 2015;22:898. doi: 10.1016/j.chembiol.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 74.Rueffer M, El-Shagi H, Nagakura N, Zenk MH. FEBS Lett. 1981;129:5. [Google Scholar]
- 75.Samanani N, Liscombe DK, Facchini PJ. The Plant Journal. 2004;40:302. doi: 10.1111/j.1365-313X.2004.02210.x. [DOI] [PubMed] [Google Scholar]
- 76.Luk LYP, Bunn S, Liscombe DK, Facchini PJ, Tanner ME. Biochemistry. 2007;46:10153. doi: 10.1021/bi700752n. [DOI] [PubMed] [Google Scholar]
- 77.Ilari A, Franceschini S, Bonamore A, Arenghi F, Botta B, Macone A, Pasquo A, Bellucci L, Boffi A. J Biol Chem. 2009;284:897. doi: 10.1074/jbc.M803738200. [DOI] [PubMed] [Google Scholar]
- 78.De-Eknamkul W, Suttipanta N, Kutchan TM. Phytochemistry. 2000;55:177. doi: 10.1016/s0031-9422(00)00260-0. [DOI] [PubMed] [Google Scholar]
- 79.Koketsu K, Watanabe K, Suda H, Oguri H, Oikawa H. Nat Chem BIol. 2010;6:408. doi: 10.1038/nchembio.365. [DOI] [PubMed] [Google Scholar]
- 80.Koketsu K, Minami A, Watanabe K, Oguri H, Oikawa H. Curr Opin Chem Biol. 2012;16:142. doi: 10.1016/j.cbpa.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 81.Knoevenagel E. Ber Dtsch Chem Ges. 1898;31:2596. [Google Scholar]
- 82.Lai YF, Zheng H, Chai SJ, Zhang PF, Chen XZ. Green Chem. 2010;12:1917. [Google Scholar]
- 83.Wang CH, Guan Z, He YH. Green Chem. 2011;13:2048. [Google Scholar]
- 84.Friedlaender P. Ber Dtsch Chem Ges. 1882;15:2572. [Google Scholar]
- 85.Zheng H, Liu J, Mei YJ, Shi QY, Zhang PF. Catal Lett. 2012;142:573. [Google Scholar]
- 86.Michael A. J Prakt Chem. 1887;35:349. [Google Scholar]
- 87.Chatterjee C, Paul M, Xie L, van der Donk WA. Chem Rev. 2005;105:633. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 88.Li B, Yu JPJ, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Science. 2006;311:1464. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 89.Seltzer S, Lin M. J Am Chem Soc. 1979;101:3091. [Google Scholar]
- 90.Yanofsky C, Crawford IP, Paul DB. The Enzymes. Vol. 7. Academic Press; 1972. p. 1. [Google Scholar]
- 91.Banerjee R, Evande R, Kabil M, Ojha S, Taoka S. Biochim Biophys Acta, Proteins Proteomics. 2003;1647:30. doi: 10.1016/s1570-9639(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 92.Fitzpatrick PF, Orville AM, Nagpal A, Valley MP. Arch Biochem Biophys. 2005;433:157. doi: 10.1016/j.abb.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 93.Heroux A, Bozinovski DM, Valley MP, Fitzpatrick PF, Orville AM. Biochemistry. 2009;48:3407. doi: 10.1021/bi8023042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Razeto A, Mattiroli F, Carpanelli E, Aliverti A, Pandini V, Coda A, Mattevi A. Structure. 2007;15:683. doi: 10.1016/j.str.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 95.Hertweck C. Trends Biochem Sci. 2015;40:189. doi: 10.1016/j.tibs.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 96.Kusebauch B, Busch B, Scherlach K, Roth M, Hertweck C. Angew Chem Int Ed. 2009;48:5001. doi: 10.1002/anie.200900277. [DOI] [PubMed] [Google Scholar]
- 97.Bretschneider T, Heim JB, Heine D, Winkler R, Busch B, Kusebauch B, Stehle T, Zocher G, Hertweck C. Nature. 2013;502:124. doi: 10.1038/nature12588. [DOI] [PubMed] [Google Scholar]
- 98.Morita K-i, Suzuki Z, Hirose H. Bull Chem Soc Jpn. 1968;41:2815. [Google Scholar]
- 99.Baylis AB, Hillman MED. Chem Abstr. 1972;77:34174q. [Google Scholar]
- 100.Carreras CW, Santi DV. Annu Rev Biochem. 1995;64:721. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 101.Currier H, Rauhut MM. Vol US3074999. American Cyanamid Co.; United States: 1963. [Google Scholar]
- 102.Kirst HA, Michel KH, Martin JW, Creemer LC, Chio EH, Yao RC, Nakatsukasa WM, Boeck LD, Occolowitz JL, Paschal JW, Deeter JB, Jones ND, Thompson GD. Tetrahedron Lett. 1991;32:4839. [Google Scholar]
- 103.Waldron C, Matsushima P, Broughton PRRMC, Jr, Turner J, Madduri K, Crawford KP, Merlo DJ, Baltz RH. Chem Biol. 2001;8:487. doi: 10.1016/s1074-5521(01)00029-1. [DOI] [PubMed] [Google Scholar]
- 104.Kim HJ, Pongdee R, Wu Q, Hong L, Liu H-w. J Am Chem Soc. 2007;129:14582. doi: 10.1021/ja076580i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim HJ, Ruszczycky MW, Choi S-h, Liu Y-n, Liu H-w. Nature. 2011;473:109. doi: 10.1038/nature09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dobler S, Petschenka G, Pankoke H. Phytochemistry. 2011;72:1593. doi: 10.1016/j.phytochem.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 107.Birkett MA, Pickett JA. Phytochemistry. 2003;62:651. doi: 10.1016/s0031-9422(02)00568-x. [DOI] [PubMed] [Google Scholar]
- 108.Kunert M, Rahfeld P, Shaker KH, Schneider B, David A, Dettner K, Pasteels JM, Boland W. Chem Bio Chem. 2013;14:353. doi: 10.1002/cbic.201200689. [DOI] [PubMed] [Google Scholar]
- 109.Geu-Flores F, Sherden NH, Courdavault V, Burlat V, Glenn WS, Wu C, Nims E, Cui Y, O’Connor SE. Nature. 2012;492:138. doi: 10.1038/nature11692. [DOI] [PubMed] [Google Scholar]
- 110.Lindner S, Geu-Flores F, Brase S, Sherden Nathaniel H, O’Connor SE. Chem Biol. 2014;21:1452. doi: 10.1016/j.chembiol.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kries H, Caputi L, Stevenson CEM, Kamileen MO, Sherden NH, Geu-Flores F, Lawson DM, O’Connor SE. Nat Chem Biol. 2016;12:6. doi: 10.1038/nchembio.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mannich C, Krosche W. Arch Pharm. 1912;250:647. [Google Scholar]
- 113.Stöckigt J, Panjikar S. Nat Prod Rep. 2007;24:1382. doi: 10.1039/b711935f. [DOI] [PubMed] [Google Scholar]
- 114.Ziegler J, Facchini PJ. Annu Rev Plant Biol. 2008;59:735. doi: 10.1146/annurev.arplant.59.032607.092730. [DOI] [PubMed] [Google Scholar]
- 115.Dewick PM. Medicinal natural products: a biosynthetic approach. John Wiley & Sons; 2002. [Google Scholar]
- 116.Croteau R, Davis EM, Hartmann T, Hemscheidt T, Sanz-Cervera JF, Shen B, Stocking EM, Williams RM, Leeper FJ, Vederas JC. Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids. Vol. 209. Springer; 2003. [Google Scholar]
- 117.Humphrey AJ, O’Hagan D. Nat Prod Rep. 2001;18:494. doi: 10.1039/b001713m. [DOI] [PubMed] [Google Scholar]
- 118.Robinson R. J Chem Soc Trans. 1917;111:762. [Google Scholar]
- 119.Robinson R. The Structural Relations of Natural Products. Clarendon Press; 1955. [Google Scholar]
- 120.Stetter H, Schreckenberg M. Angew Chem. 1973;85:89. [Google Scholar]
- 121.Stetter H. Angew Chem Int Ed. 1976;15:639. [Google Scholar]
- 122.Seebach D. Angew Chem Int Ed. 1979;18:239. [Google Scholar]
- 123.Dawson A, Fyfe PK, Hunter WN. J Mol Biol. 2008;384:1353. doi: 10.1016/j.jmb.2008.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Williamson NR, Simonsen HT, Ahmed RAA, Goldet G, Slater H, Woodley L, Leeper FJ, Salmond GPC. Mol Microbiol. 2005;56:971. doi: 10.1111/j.1365-2958.2005.04602.x. [DOI] [PubMed] [Google Scholar]
- 125.Dresen C, Richter M, Pohl M, Lüdeke S, Müller M. Angew Chem Int Ed. 2010;49:6600. doi: 10.1002/anie.201000632. [DOI] [PubMed] [Google Scholar]
- 126.Heinrich PC, Steffen H, Janser P, Wiss O. Eur J Biochem. 1972;30:533. doi: 10.1111/j.1432-1033.1972.tb02124.x. [DOI] [PubMed] [Google Scholar]
- 127.Wöhler, Liebig Ann Pharm. 1832;3:249. [Google Scholar]
- 128.Prins HJ. Chem Weekbl. 1919;16:1072. [Google Scholar]
- 129.Schulte‐Elte KH, Pamingle H. Helv Chim Acta. 1989;72:1158. [Google Scholar]
- 130.Jiang J, He X, Cane DE. Nat Chem Biol. 2007;3:711. doi: 10.1038/nchembio.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Harris GG, Lombardi PM, Pemberton TA, Matsui T, Weiss TM, Cole KE, Koksal M, Murphy FV, Vedula LS, Chou WKW, Cane DE, Christianson DW. Biochemistry. 2015;54:7142. doi: 10.1021/acs.biochem.5b01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Humble MS, Berglund P. Eur J Org Chem. 2011;2011:3391. [Google Scholar]
- 133.Wu Q, Liu BK, Lin XF. Curr Org Chem. 2010;14:1966. [Google Scholar]
- 134.Kapoor M, Gupta MN. Process Biochem. 2012;47:555. [Google Scholar]
- 135.Henry L. CR Hebd Acad Sci. 1895;120:1265. [Google Scholar]
- 136.Tang RC, Guan Z, He YH, Zhu W. J Mol Catal B: Enzym. 2010;63:62. [Google Scholar]
- 137.Wang JL, Li X, Xie HY, Liu BK, Lin XF. J Biotechnol. 2010;145:240. doi: 10.1016/j.jbiotec.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 138.Gao N, Chen YL, He YH, Guan Z. RSC Adv. 2013;3:16850. [Google Scholar]
- 139.Busto E, Gotor-Fernández V, Gotor V. Org Process Res Dev. 2010;15:236. [Google Scholar]
- 140.Purkarthofer T, Gruber K, Gruber-Khadjawi M, Waich K, Skranc W, Mink D, Griengl H. Angew Chem Int Ed. 2006;45:3454. doi: 10.1002/anie.200504230. [DOI] [PubMed] [Google Scholar]
- 141.Wajant H, Effenberger F. Biol Chem. 1996;377:611. [PubMed] [Google Scholar]
- 142.Ugi I. Angew Chem Int Ed. 1962;1:8. [Google Scholar]
- 143.Mumm O. Ber Dtsch Chem Ges. 1910;43:886. [Google Scholar]
- 144.Pan SC, List B. Angew Chem Int Ed. 2008;47:3622. doi: 10.1002/anie.200800494. [DOI] [PubMed] [Google Scholar]
- 145.Klossowski S, Wiraszka B, Berlozecki S, Ostaszewski R. Org Lett. 2013;15:566. doi: 10.1021/ol3033829. [DOI] [PubMed] [Google Scholar]
- 146.Baeyer A, Villiger V. Ber Dtsch Chem Ges. 1899;32:3625. [Google Scholar]
- 147.Criegee R. Justus Liebigs Ann Chem. 1948;560:127. [Google Scholar]
- 148.Li JJ, Corey EJ. John Wiley & Sons. 2007;1:160. [Google Scholar]
- 149.Turfitt GE. Biochem J. 1948;42:376. [PubMed] [Google Scholar]
- 150.Donoghue NA, Norris DB, Trudgill PW. Eur J Biochem. 1976;63:175. doi: 10.1111/j.1432-1033.1976.tb10220.x. [DOI] [PubMed] [Google Scholar]
- 151.Willetts A. Trends Biotechnol. 1997;15:55. doi: 10.1016/S0167-7799(97)84204-7. [DOI] [PubMed] [Google Scholar]
- 152.Gibson M, Nur-e-alam M, Lipata F, Oliveira MA, Rohr J. J Am Chem Soc. 2005;127:17594. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- 153.Chen YC, Peoples OP, Walsh CT. J Bacteriol. 1988;170:781. doi: 10.1128/jb.170.2.781-789.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Iwaki H, Hasegawa Y, Teraoka M, Tokuyama T, Bergeron H, Lau PCK. Appl Environ Microbiol. 1999;65:5158. doi: 10.1128/aem.65.11.5158-5162.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ryerson CC, Ballou DP, Walsh C. Biochemistry. 1982;21:2644. doi: 10.1021/bi00540a011. [DOI] [PubMed] [Google Scholar]
- 156.Sheng D, Ballou DP, Massey V. Biochemistry. 2001;40:11156. doi: 10.1021/bi011153h. [DOI] [PubMed] [Google Scholar]
- 157.Li S, Wang H, Li Y, Deng J, Lu C, Shen Y, Shen Y. Chem Bio Chem. 2014;15:94. doi: 10.1002/cbic.201300599. [DOI] [PubMed] [Google Scholar]
- 158.Tibrewal N, Pahari P, Wang G, Kharel MK, Morris C, Downey T, Hou Y, Bugni TS, Rohr J. J Am Chem Soc. 2012;134:18181. doi: 10.1021/ja3081154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shen B, Hutchinson CR. Biochemistry. 1993;32:6656. doi: 10.1021/bi00077a019. [DOI] [PubMed] [Google Scholar]
- 160.Chung J-y, Fujii I, Harada S, Sankawa U, Ebizuka Y. J Bacteriol. 2002;184:6115. doi: 10.1128/JB.184.22.6115-6122.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Maggio-Hall LA, Dorrestein PC, Escalante-Semerena JC, Begley TP. Org Lett. 2003;5:2211. doi: 10.1021/ol034530m. [DOI] [PubMed] [Google Scholar]
- 162.Taga ME, Larsen NA, Howard-Jones AR, Walsh CT, Walker GC. Nature. 2007;446:449. doi: 10.1038/nature05611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Walsh CT, Wencewicz TA. Nat Prod Rep. 2013;30:175. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Steyn PS, van Heerden FR, Rabie CJ. J Chem Soc, Perkin Trans 1. 1982:541. [Google Scholar]
- 165.Hu Y, Dietrich D, Xu W, Patel A, Thuss JAJ, Wang J, Yin WB, Qiao K, Houk KN, Vederas JC, Tang Y. Nat Chem Biol. 2014;10:552. doi: 10.1038/nchembio.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang S, Tabudravu J, Elsayed SS, Travert J, Peace D, Tong MH, Kyeremeh K, Kelly SM, Trembleau L, Ebel R, Jaspars M, Yu Y, Deng H. Angew Chem Int Ed. 2015;54:12697. doi: 10.1002/anie.201502902. [DOI] [PubMed] [Google Scholar]
- 167.Schimming O, Challinor VL, Tobias NJ, Adihou H, Grün P, Pöschel L, Richter C, Schwalbe H, Bode HB. Angew Chem Int Ed. 2015;54:12702. doi: 10.1002/anie.201504877. [DOI] [PubMed] [Google Scholar]
- 168.Townsend CA, Christensen SB, Davis SG. J Am Chem Soc. 1982;104:6154. [Google Scholar]
- 169.Wen Y, Hatabayashi H, Arai H, Kitamoto HK, Yabe K. Appl Environ Microbiol. 2005;71:3192. doi: 10.1128/AEM.71.6.3192-3198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Keller NP, Kantz NJ, Adams TH. Appl Environ Microbiol. 1994;60:1444. doi: 10.1128/aem.60.5.1444-1450.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Keller NP, Segner S, Bhatnagar D, Adams TH. Appl Environ Microbiol. 1995;61:3628. doi: 10.1128/aem.61.10.3628-3632.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Henry KM, Townsend CA. J Am Chem Soc. 2005;127:3724. doi: 10.1021/ja0455188. [DOI] [PubMed] [Google Scholar]
- 173.Henry KM, Townsend CA. J Am Chem Soc. 2005;127:3300. doi: 10.1021/ja045520z. [DOI] [PubMed] [Google Scholar]
- 174.Bajguz A, Tretyn A. Phytochemistry. 2003;62:1027. doi: 10.1016/s0031-9422(02)00656-8. [DOI] [PubMed] [Google Scholar]
- 175.Nomura T, Kushiro T, Yokota T, Kamiya Y, Bishop GJ, Yamaguchi S. J Biol Chem. 2005;280:17873. doi: 10.1074/jbc.M414592200. [DOI] [PubMed] [Google Scholar]
- 176.Kim TW, Hwanga JY, Kim YS, Joo SH, Chang SC, Lee JS, Takatsuto S, Kim SK. The Plant Cell. 2005;17:2397. doi: 10.1105/tpc.105.033738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.van Damme PA, Johannes AG, Cox HC, Berends W. Recl Trav Chim Pays-Bas. 1960;79:255. [Google Scholar]
- 178.Hwang IG, Moon JS, Jwa NS. Vol US20100269215. Snu R&Db Foundation; 2007. [Google Scholar]
- 179.Fenwick MK, Philmus B, Begley TP, Ealick SE. Biochemistry. 2011;50:1091. doi: 10.1021/bi101741v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Philmus B, Abdelwahed S, Williams HJ, Fenwick MK, Ealick SE, Begley TP. J Am Chem Soc. 2012;134:5326. doi: 10.1021/ja211759n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Prileschajew N. Ber Dtsch Chem Ges. 1909;42:4811. [Google Scholar]
- 182.Kells PM, Ouellet H, Santos-Aberturas J, Aparicio JF, Podust LM. Chem Biol. 2010;17:841. doi: 10.1016/j.chembiol.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nagano S, Li H, Shimizu H, Nishida C, Ogura H, de Montellano PRO, Poulos TL. J Biol Chem. 2003;278:44886. doi: 10.1074/jbc.M308115200. [DOI] [PubMed] [Google Scholar]
- 184.Nagumo A, Kamei T, Sakakibara J, Ono T. J Lipid Res. 1995;36:1489. [PubMed] [Google Scholar]
- 185.Ames BD, Liu X, Walsh CT. Biochemistry. 2010;49:8564. doi: 10.1021/bi1012029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gao X, Chooi YH, Ames BD, Wang P, Walsh CT, Tang Y. J Am Chem Soc. 2011;133:2729. doi: 10.1021/ja1101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nef JU. Justus Liebigs Ann Chem. 1894;280:263. [Google Scholar]
- 188.McMurry JE, Melton J. J Org Chem. 1973;38:4367. [Google Scholar]
- 189.Williams RE, Bruce NC. Microbiology. 2002;148:1607. doi: 10.1099/00221287-148-6-1607. [DOI] [PubMed] [Google Scholar]
- 190.Durchschein K, Fabian WMF, Macheroux P, Zangger K, Trimmel G, Faber K. Org Biomol Chem. 2011;9:3364. doi: 10.1039/c0ob01216e. [DOI] [PubMed] [Google Scholar]
- 191.Cannizzaro S. Justus Liebigs Ann Chem. 1853;88:129. [Google Scholar]
- 192.Russell AE, Miller SP, Morken JP. J Org Chem. 2000;65:8381. doi: 10.1021/jo0010734. [DOI] [PubMed] [Google Scholar]
- 193.Inoue Y, Kimura A. Adv Microb Physiol. 1995;37:177. doi: 10.1016/s0065-2911(08)60146-0. [DOI] [PubMed] [Google Scholar]
- 194.Silverman RB. The organic chemistry of enzyme-catalyzed reactions. Academic Press; 2002. [Google Scholar]
- 195.Chari RV, Kozarich JW. J Biol Chem. 1981;256:9785. [PubMed] [Google Scholar]
- 196.Kozarich JW, Chari RVJ, Wu JC, Lawrence TL. J Am Chem Soc. 1981;103:4593. [Google Scholar]
- 197.Fuchs G. Annals of the New York Academy of Sciences. 2008;1125:82. doi: 10.1196/annals.1419.010. [DOI] [PubMed] [Google Scholar]
- 198.Reichenbecher W, Philipp B, Suter MJ, Schink B. Arch Microbiol. 2000;173:206. doi: 10.1007/s002039900130. [DOI] [PubMed] [Google Scholar]
- 199.Kluge C, Tschech A, Fuchs G. Arch Microbiol. 1990;155:68. [Google Scholar]
- 200.Boll M. Biochim Biophys Acta, Bioenerg. 2005;1707:34. doi: 10.1016/j.bbabio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 201.Boll M, Fuchs G. Biol Chem. 2005;386:989. doi: 10.1515/BC.2005.115. [DOI] [PubMed] [Google Scholar]
- 202.Lack A, Fuchs G. J Bacteriol. 1992;174:3629. doi: 10.1128/jb.174.11.3629-3636.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Heider J, Fuchs G. Eur J Biochem. 1997;243:577. doi: 10.1111/j.1432-1033.1997.00577.x. [DOI] [PubMed] [Google Scholar]
- 204.Breese K, Fuchs G. Eur J Biochem. 1998;251:916. doi: 10.1046/j.1432-1327.1998.2510916.x. [DOI] [PubMed] [Google Scholar]
- 205.Laban N Abu, Selesi D, Rattei T, Tischler P, Meckenstock RU. Environ Microbiol. 2010;12:2783. doi: 10.1111/j.1462-2920.2010.02248.x. [DOI] [PubMed] [Google Scholar]
- 206.Holmes DE, Risso C, Smith JA, Lovley DR. Appl Environ Microbiol. 2011;77:5926. doi: 10.1128/AEM.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Birch AJ. J Chem Soc. 1944;430 [Google Scholar]
- 208.Boll M, Fuchs G. Eur J Biochem. 1995;234:921. doi: 10.1111/j.1432-1033.1995.921_a.x. [DOI] [PubMed] [Google Scholar]
- 209.Boll M, Albracht SSP, Fuchs G. Eur J Biochem. 1997;244:840. doi: 10.1111/j.1432-1033.1997.00840.x. [DOI] [PubMed] [Google Scholar]
- 210.Boll M, Fuchs G, Meier C, Trautwein A, Lowe DJ. J Biol Chem. 2000;275:31857. doi: 10.1074/jbc.M001508200. [DOI] [PubMed] [Google Scholar]
- 211.Unciuleac M, Boll M. Proc Natl Acad Sci U S A. 2001;98:13619. doi: 10.1073/pnas.241375598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mobitz H, Boll M. Biochemistry. 2002;41:1752. doi: 10.1021/bi0113770. [DOI] [PubMed] [Google Scholar]
- 213.Kung JW, Löffler C, Dörner K, Heintz D, Gallien S, Van Dorsselaer A, Friedrich T, Boll M. Proc Natl Acad Sci U S A. 2009;106:17687. doi: 10.1073/pnas.0905073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wischgoll S, Heintz D, Peters F, Erxleben A, Sarnighausen E, Reski R, Van Dorsselaer A, Boll M. Mol Microbiol. 2005;58:1238. doi: 10.1111/j.1365-2958.2005.04909.x. [DOI] [PubMed] [Google Scholar]
- 215.Kung JW, Baumann S, von Bergen M, Müller M, Hagedoorn P-L, Hagen WR, Boll M. J Am Chem Soc. 2010;132:9850. doi: 10.1021/ja103448u. [DOI] [PubMed] [Google Scholar]
- 216.Löffler C, Kuntze K, Vazquez JR, Rugor A, Kung JW, Böttcher A, Boll M. Environ Microbiol. 2011;13:696. doi: 10.1111/j.1462-2920.2010.02374.x. [DOI] [PubMed] [Google Scholar]
- 217.Weinert T, Huwiler SG, Kung JW, Weidenweber S, Hellwig P, Stark HJ, Biskup T, Weber S, Cotelesage JJH, George GN, Ermler U, Boll M. Nat Chem Biol. 2015;11:586. doi: 10.1038/nchembio.1849. [DOI] [PubMed] [Google Scholar]
- 218.Brackmann R, Fuchs G. Eur J Biochem. 1993;213:563. doi: 10.1111/j.1432-1033.1993.tb17795.x. [DOI] [PubMed] [Google Scholar]
- 219.Boll M, Fuchs G, Meier C, Trautwein A, Kasmi A El, Ragsdale SW, Buchanan G, Lowe DJ. J Biol Chem. 2001;276:47853. doi: 10.1074/jbc.M106766200. [DOI] [PubMed] [Google Scholar]
- 220.Kisker C, Schindelin AH, Rees DC. Annu Rev Biochem. 1997;66:233. doi: 10.1146/annurev.biochem.66.1.233. [DOI] [PubMed] [Google Scholar]
- 221.Ajikumar PK, Tyo K, Carlsen S, Mucha O, Phon TH, Stephanopoulos G. Mol Pharmaceutics. 2008;5:167. doi: 10.1021/mp700151b. [DOI] [PubMed] [Google Scholar]
- 222.Rohmer M. Pure Appl Chem. 2007;79:739. [Google Scholar]
- 223.Zhao L, Chang W-c, Xiao Y, Liu H-w, Liu P. Annu Rev Biochem. 2013;82:497. doi: 10.1146/annurev-biochem-052010-100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wang W, Wang K, Span I, Jauch J, Bacher A, Groll M, Oldfield E. J Am Chem Soc. 2012;134:11225. doi: 10.1021/ja303445z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Citron CA, Brock NL, Rabe P, Dickschat JS. Angew Chem Int Ed. 2012;51:4053. doi: 10.1002/anie.201201110. [DOI] [PubMed] [Google Scholar]
- 226.Hellwig M, Henle T. Angew Chem Int Ed. 2014;53:10316. doi: 10.1002/anie.201308808. [DOI] [PubMed] [Google Scholar]
- 227.Amadori M. Atti Accad Lincei. 1925;2:337. [Google Scholar]
- 228.Wolf WA, Brown GM. Biochim Biophys Acta, Gen Subj. 1969;192:468. doi: 10.1016/0304-4165(69)90396-1. [DOI] [PubMed] [Google Scholar]
- 229.Bracher A, Eisenreich W, Schramek N, Ritz H, Götze E, Herrmann A, Gütlich M, Bacher A. J Biol Chem. 1998;273:28132. doi: 10.1074/jbc.273.43.28132. [DOI] [PubMed] [Google Scholar]
- 230.Tobias G, Fischer M, Bacher A. IUBMB Life. 2013;65:310. doi: 10.1002/iub.1153. [DOI] [PubMed] [Google Scholar]
- 231.Dorrestein PC, Huili, Taylor SV, McLafferty FW, Begley TP. J Am Chem Soc. 2004;126:3091. doi: 10.1021/ja039616p. [DOI] [PubMed] [Google Scholar]
- 232.Sasaki E, Ogasawara Y, Liu H-W. J Am Chem Soc. 2010;132:7405. doi: 10.1021/ja1014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sasaki E, Liu H-W. J Am Chem Soc. 2010;132:15544. doi: 10.1021/ja108061c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sasaki E, Zhang X, Sun HG, Lu M-YJ, Liu T-L, Ou A, Li J-Y, Chen Y-H, Ealick SE, Liu H-W. Nature. 2014;510:427. doi: 10.1038/nature13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bamberger E. Ber Dtsch Chem Ges. 1894;27:1548. [Google Scholar]
- 236.Nishino SF, Spain JC. Appl Environ Microbiol. 1993;59:2520. doi: 10.1128/aem.59.8.2520-2525.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Katsivela E, Wray V, Pieper DH, Wittich RM. Appl Environ Microbiol. 1999;65:1405. doi: 10.1128/aem.65.4.1405-1412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Spiess T, Desiere F, Fischer P, Spain JC, Knackmuss HJ, Lenke H. Appl Environ Microbiol. 1998;64:446. doi: 10.1128/aem.64.2.446-452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hughes JB, Wang C, Yesland K, Richardson A, Bhadra R, Bennett G, Rudolph F. Environ Sci Technol. 1998;32:494. [Google Scholar]
- 240.Hasegawa Y, Muraki T, Tokuyama T, Iwaki H, Tatsuno M, Lau PCK. A novel degradative pathway of 2-nitrobenzoate via 3-hydroxyanthranilate in Pseudomonas fluorescens strain KU-7. 190:2000. doi: 10.1111/j.1574-6968.2000.tb09284.x. [DOI] [PubMed] [Google Scholar]
- 241.Davis JK, Paoli GC, He Z, Nadeau LJ, Somerville CC, Spain JC. Appl Environ Microbiol. 2000;66:2965. doi: 10.1128/aem.66.7.2965-2971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.He Z, Nadeau LJ, Spain JC. Eur J Biochem. 2000;267:1110. doi: 10.1046/j.1432-1327.2000.01107.x. [DOI] [PubMed] [Google Scholar]
- 243.Schenzle A, Lenke H, Spain JC, Knackmuss HJ. J Bacteriol. 1999;181:1444. doi: 10.1128/jb.181.5.1444-1450.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Van Lanen SG, Shen B. Curr Top Med Chem. 2008;8:448. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Jones RR, Bergman RG. J Am Chem Soc. 1972;94:660. [Google Scholar]
- 246.Myers AG, Proteau PJ, Handel TM. J Am Chem Soc. 1988;110:7212. [Google Scholar]
- 247.Nagata R, Yamanaka H, Murahashi E, Saito I. Tetrahedron Lett. 1990;31:2907. [Google Scholar]
- 248.Myers AG. Tetrahedron Lett. 1987;28:4493. [Google Scholar]
- 249.De Voss JJ, Hangeland JJ, Townsend CA. J Am Chem Soc. 1990;112:4554. [Google Scholar]
- 250.Uesawa Y, Kuwahara J, Sugiura Y. Biochem Biophys Res Commun. 1989;164:903. doi: 10.1016/0006-291x(89)91544-1. [DOI] [PubMed] [Google Scholar]
- 251.Shiraki T, Sugiura Y. Biochemistry. 1990;29:9795. doi: 10.1021/bi00494a006. [DOI] [PubMed] [Google Scholar]
- 252.Fries K, Finck G. Ber Dtsch Chem Ges. 1908;41:4284. [Google Scholar]
- 253.Bellus D. Adv Photochem. 1971;8:109. [Google Scholar]
- 254.Antoniotti S, Dordick JS. Adv Synth Catal. 2005;347:1119. [Google Scholar]
- 255.Fahey JW, Zalcmann AT, Talalay P. Phytochemistry. 2001;56:5. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 256.Cole RA. Phytochemistry. 1976;15:759. [Google Scholar]
- 257.Ettlinger MG, Lundeen AJ. J Am Chem Soc. 1957;79:1764. [Google Scholar]
- 258.Lossen W. Justus Liebigs Ann Chem. 1872;161:347. [Google Scholar]
- 259.Criegee R. Ber Dtsch Chem Ges. 1945;77:722. [Google Scholar]
- 260.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 261.Bugg TDH, Lin G. Chem Commun. 2001;941 [Google Scholar]
- 262.Harrison PJ, Bugg TDH. Arch Biochem Biophys. 2014;544:105. doi: 10.1016/j.abb.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 263.Faworsky A. J Prakt Chem. 1895;51:533. [Google Scholar]
- 264.Simpson TJ, Holker JSE. Tetrahedron Lett. 1975;16:4693. [Google Scholar]
- 265.Wright JLC, Hu T, McLachlan JL, Needham J, Walter JA. J Am Chem Soc. 1996;118:8757. [Google Scholar]
- 266.Kakinuma K, Uzawa J, Uramoto M. Tetrahedron Lett. 1982;23:5303. [Google Scholar]
- 267.Seto H, Sato T, Urano S, Uzawa J, Yonehara H. Tetrahedron Lett. 1976;17:4367. [Google Scholar]
- 268.Arakawa K, Sugino F, Kodama K, Ishii T, Kinashi H. Chem Biol. 2005;12:249. doi: 10.1016/j.chembiol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 269.Piel JR, Hertweck C, Shipley PR, Hunt DM, Newman MS, Moore BS. Chem Biol. 2000;7:943. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 270.Xiang L, Kalaitzis JA, Nilsen G, Chen L, Moore BS. Org Lett. 2002;4:957. doi: 10.1021/ol0255155. [DOI] [PubMed] [Google Scholar]
- 271.Xiang L, Kalaitzis JA, Moore BS. Proc Natl Acad Sci U S A. 2004;101:15609. doi: 10.1073/pnas.0405508101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Teufel R, Miyanaga A, Michaudel Q, Stull F, Louie G, Noel JP, Baran PS, Palfey B, Moore BS. Nature. 2013;503:552. doi: 10.1038/nature12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Teufel R, Stull F, Meehan MJ, Michaudel Q, Dorrestein PC, Palfey B, Moore BS. J Am Chem Soc. 2015;137:8078. doi: 10.1021/jacs.5b03983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Fittig R. Justus Liebigs Ann Chem. 1860;114:54. [Google Scholar]
- 275.Song ZL, Fan CA, Tu YQ. Chem Rev. 2011;111:7523. doi: 10.1021/cr200055g. [DOI] [PubMed] [Google Scholar]
- 276.Cox RJ, Al-Fahad A. Curr Opin Chem Biol. 2013;17:532. doi: 10.1016/j.cbpa.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 277.Birkinshaw JH, Chambers AR, Raistrick H. Biochem J. 1942;36:242. doi: 10.1042/bj0360242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Dewar MJS. Nature. 1945;155:50. [Google Scholar]
- 279.Bentley R. J Biol Chem. 1963;238:1895. [PubMed] [Google Scholar]
- 280.Osullivan MC, Schwab JM. Bioorg Chem. 1995;23:131. [Google Scholar]
- 281.Davison J, Fahad AAL, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Proc Natl Acad Sci U S A. 2012;109:7642. doi: 10.1073/pnas.1201469109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kunze B, Höfle G, Reichenbach H. J Antibiot. 1987;40:258. doi: 10.7164/antibiotics.40.258. [DOI] [PubMed] [Google Scholar]
- 283.Sandmann A, Dickschat J, Jenke-Kodama H, Kunze B, Dittmann E, Müller R. Angew Chem Int Ed. 2007;46:2712. doi: 10.1002/anie.200603513. [DOI] [PubMed] [Google Scholar]
- 284.Hofle G, Kunze B. J Nat Prod. 2008;71:1843. doi: 10.1021/np8003084. [DOI] [PubMed] [Google Scholar]
- 285.Pistorius D, Li Y, Sandmann A, Muller R. Mol BioSyst. 2011;7:3308. doi: 10.1039/c1mb05328k. [DOI] [PubMed] [Google Scholar]
- 286.Katsuyama Y, Harmrolfs K, Pistorius D, Li Y, Müller R. Angew Chem Int Ed. 2012;51:9437. doi: 10.1002/anie.201204138. [DOI] [PubMed] [Google Scholar]
- 287.Katsuyama Y, Li X-W, Müller R, Nay B. ChemBioChem. 2014;15:2349. doi: 10.1002/cbic.201402373. [DOI] [PubMed] [Google Scholar]
- 288.Cast J, Stevens TS, Holmes J. J Chem Soc. 1960;3521 [Google Scholar]
- 289.Wagner G, Brickner W. Ber Dtsch Chem Ges. 1899;32:2302. [Google Scholar]
- 290.Meerwein H, van Emster K. Ber Dtsch Chem Ges. 1922;55:2500. [Google Scholar]
- 291.Croteau R, Gershenzon J, Wheeler CJ, Satterwhite DM. Arch Biochem Biophys. 1990;277:374. doi: 10.1016/0003-9861(90)90593-n. [DOI] [PubMed] [Google Scholar]
- 292.Back K, Chappell J. Proc Natl Acad Sci U S A. 1996;93:6841. doi: 10.1073/pnas.93.13.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Claisen L. Ber Dtsch Chem Ges. 1912;45:3157. [Google Scholar]
- 294.Cope AC, Hardy EM. J Am Chem Soc. 1940;62:441. [Google Scholar]
- 295.Diels O, Alder K. Justus Liebigs Ann Chem. 1928;460:98. [Google Scholar]
- 296.Gray JV, Eren D, Knowles JR. Biochemistry. 1990;29:8872. doi: 10.1021/bi00489a051. [DOI] [PubMed] [Google Scholar]
- 297.Sogo SG, Widlanski TS, Hoare JH, Grimshaw CE, Berchtold GA, Knowles JR. J Am Chem Soc. 1984;106:2701. [Google Scholar]
- 298.Gustin DJ, Mattei P, Kast P, Wiest O, Lee L, Cleland WW, Hilvert D. J Am Chem Soc. 1999;121:1756. [Google Scholar]
- 299.Donia MS, Ravel J, Schmidt EW. Nat Chem Biol. 2008;4:341. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Mander L, Liu HW. Comprehensive Natural Products II: Chemistry and Biology: 10 Volume Set. Vol. 1. Newnes; 2010. [Google Scholar]
- 301.McIntosh JA, Donia MS, Nair SK, Schmidt EW. J Am Chem Soc. 2011;133:13698. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Li SM. Nat Prod Rep. 2010;27:57. doi: 10.1039/b909987p. [DOI] [PubMed] [Google Scholar]
- 303.Metzger U, Schall C, Zocher G, Unsold I, Stec E, Li SM, Heide L, Stehle T. Proc Natl Acad Sci U S A. 2009;106:14309. doi: 10.1073/pnas.0904897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Luk LYP, Qian Q, Tanner ME. J Am Chem Soc. 2011;133:12342. doi: 10.1021/ja2034969. [DOI] [PubMed] [Google Scholar]
- 305.Huisgen R. Angew Chem Int Ed. 1968;7:321. [Google Scholar]
- 306.Stocking EM, Williams RM. Angew Chem Int Ed. 2003;42:3078. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]
- 307.Ose T, Watanabe K, Mie T, Honma M, Watanabe H, Yao M, Oikawa H, Tanaka I. Nature. 2003;422:185. doi: 10.1038/nature01454. [DOI] [PubMed] [Google Scholar]
- 308.Serafimov JM, Gillingham D, Kuster S, Hilvert D. J Am Chem Soc. 2008;130:7798. doi: 10.1021/ja8017994. [DOI] [PubMed] [Google Scholar]
- 309.Kim RR, Illarionov B, Joshi M, Cushman M, Lee CY, Eisenreich W, Fischer M, Bacher A. J Am Chem Soc. 2010;132:2983. doi: 10.1021/ja908395r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, Ebizuka Y, Fujii I. Chem Bio Chem. 2010;11:1245. doi: 10.1002/cbic.201000173. [DOI] [PubMed] [Google Scholar]
- 311.Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson C Richard. Science. 1999;284:1368. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- 312.Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Oikawa H, Tokiwano T. Nat Prod Rep. 2004;21:321. doi: 10.1039/b305068h. [DOI] [PubMed] [Google Scholar]
- 314.Kelly WL. Org Biomol Chem. 2008;6:4483. doi: 10.1039/b814552k. [DOI] [PubMed] [Google Scholar]
- 315.Kim HJ, Ruszczycky MW, Liu H-W. Curr Opin Chem Biol. 2012;16:124. doi: 10.1016/j.cbpa.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Mocek U, Knaggs AR, Tsuchiya R, Nguyen T, Beale JM, Floss HG. J Am Chem Soc. 1993;115:7557. [Google Scholar]
- 317.Li C, Kelly WL. Nat Prod Rep. 2010;27:153. doi: 10.1039/b922434c. [DOI] [PubMed] [Google Scholar]
- 318.Bowers AA, Walsh CT, Acker MG. J Am Chem Soc. 2010;132:12182. doi: 10.1021/ja104524q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC, Bowers AA. J Am Chem Soc. 2015;137:3494. doi: 10.1021/jacs.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Lacoske MH, Theodorakis EA. J Nat Prod. 2015;78:562. doi: 10.1021/np500757w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Vieweg L, Reichau S, Schobert R, Leadlay PF, Sussmuth RD. Nat Prod Rep. 2014;31:1554. doi: 10.1039/c4np00015c. [DOI] [PubMed] [Google Scholar]
- 322.Jia XY, Tian ZH, Shao L, Qu XD, Zhao QF, Tang J, Tang GL, Liu W. Chem Biol. 2006;13:575. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 323.Zhang H, White-Phillip JA, Melançon CE, Kwon H-J, Yu W-L, Liu H-W. J Am Chem Soc. 2007;129:14670. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Fang J, Zhang Y, Huang L, Jia X, Zhang Q, Zhang X, Tang G, Liu W. J Bacteriol. 2008;190:6014. doi: 10.1128/JB.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Gottardi EM, Krawczyk JM, von Suchodoletz H, Schadt S, Mühlenweg A, Uguru GC, Pelzer S, Fiedler H-P, Bibb MJ, Stach JEM, Süssmuth RD. Chem Bio Chem. 2011;12:1401. doi: 10.1002/cbic.201100172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Li S, Xiao J, Zhu Y, Zhang G, Yang C, Zhang H, Ma L, Zhang C. Org Lett. 2013;15:1374. doi: 10.1021/ol400342e. [DOI] [PubMed] [Google Scholar]
- 327.He HY, Pan HX, Wu LF, Zhang BB, Chai HB, Liu W, Tang GL. Chem Biol. 2012;19:1313. doi: 10.1016/j.chembiol.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 328.Hashimoto T, Hashimoto J, Teruya K, Hirano T, Shin-ya K, Ikeda H, Liu H-W, Nishiyama M, Kuzuyama T. J Am Chem Soc. 2015;137:572. doi: 10.1021/ja510711x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Byrne MJ, Lees NR, Han LC, van der Kamp MW, Mulholland AJ, Stach JEM, Willis CL, Race PR. J Am Chem Soc. 2016;138:6095. doi: 10.1021/jacs.6b00232. [DOI] [PubMed] [Google Scholar]
- 330.Wu Q, Wu Z, Qu X, Liu W. J Am Chem Soc. 2012;134:17342. doi: 10.1021/ja304829g. [DOI] [PubMed] [Google Scholar]
- 331.Zheng Q, Guo Y, Yang L, Zhao Z, Wu Z, Zhang H, Liu J, Cheng X, Wu J, Yang H, Jiang H, Pan L, Liu W. Cell Chem Biol. 2016;23:352. doi: 10.1016/j.chembiol.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 332.Tian Z, Sun P, Yan Y, Wu Z, Zheng Q, Zhou S, Zhang H, Yu F, Jia X, Chen D, Mandi A, Kurtan T, Liu W. Nat Chem Biol. 2015;11:259. doi: 10.1038/nchembio.1769. [DOI] [PubMed] [Google Scholar]
- 333.Zhang G, Zhang W, Zhang Q, Shi T, Ma L, Zhu Y, Li S, Zhang H, Zhao YL, Shi R, Zhang C. Angew Chem Int Ed. 2014;53:4840. doi: 10.1002/anie.201402078. [DOI] [PubMed] [Google Scholar]
- 334.Antosch J, Schaefers F, Gulder TAM. Angew Chem Int Ed. 2014;53:3011. doi: 10.1002/anie.201310641. [DOI] [PubMed] [Google Scholar]
- 335.Fage CD, Isiorho EA, Liu Y, Wagner DT, Liu H-W, Keatinge-Clay AT. Nat Chem Biol. 2015;11:256. doi: 10.1038/nchembio.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Hess BA, Smentek L. Org Biomol Chem. 2012;10:7503. doi: 10.1039/c2ob25827g. [DOI] [PubMed] [Google Scholar]
- 337.Patel A, Chen Z, Yang Z, Gutiérrez O, Liu H-W, Houk KN, Singleton DA. J Am Chem Soc. 2016;138:3631. doi: 10.1021/jacs.6b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Friedel C, Crafts JM. Compt rend. 1877;84:1392. [Google Scholar]
- 339.Winkelblech J, Fan A, Li SM. Appl Microbiol Biotechnol. 2015;99:7379. doi: 10.1007/s00253-015-6811-y. [DOI] [PubMed] [Google Scholar]
- 340.Fan A, Xie X, Li SM. Org Biomol Chem. 2015;13:7551. doi: 10.1039/c5ob01040c. [DOI] [PubMed] [Google Scholar]
- 341.Yu X, Li SM. Methods Enzymol. 2012;516:259. doi: 10.1016/B978-0-12-394291-3.00005-8. [DOI] [PubMed] [Google Scholar]
- 342.Mehler AH, Tabor H. J Biol Chem. 1953;201:775. [PubMed] [Google Scholar]
- 343.Hahlbrock K, Scheel D. Annu Rev Plant Biol. 1989;40:347. [Google Scholar]
- 344.Appert C, Logemann E, Hahlbrock K, Schmid J, Amrhein N. Eur J Biochem. 1994;225:491. doi: 10.1111/j.1432-1033.1994.00491.x. [DOI] [PubMed] [Google Scholar]
- 345.Anson JG, Gilbert HJ, Oram JD, Minton NP. Gene. 1987;58:189. doi: 10.1016/0378-1119(87)90375-1. [DOI] [PubMed] [Google Scholar]
- 346.Kyndt JA, Meyer TE, Cusanovich MA, Van Beeumen JJ. FEBS Lett. 2002;512:240. doi: 10.1016/s0014-5793(02)02272-x. [DOI] [PubMed] [Google Scholar]
- 347.Zhu Y, Liao S, Ye J, Zhang H. Biotechnol Lett. 2012;34:269. doi: 10.1007/s10529-011-0755-9. [DOI] [PubMed] [Google Scholar]
- 348.Schöner TA, Fuchs SW, Schönau C, Bode HB. Microb Biotechnol. 2014;7:232. doi: 10.1111/1751-7915.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Walker KD, Klettke K, Akiyama T, Croteau R. J Biol Chem. 2004;279:53947. doi: 10.1074/jbc.M411215200. [DOI] [PubMed] [Google Scholar]
- 350.Magarvey NA, Fortin PD, Thomas PM, Kelleher NL, Walsh CT. ACS Chem Biol. 2008;3:542. doi: 10.1021/cb800085g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Christenson SD, Liu W, Toney MD, Shen B. J Am Chem Soc. 2003;125:6062. doi: 10.1021/ja034609m. [DOI] [PubMed] [Google Scholar]
- 352.Nicolaou KC, Smith AL, Yue EW. Proc Natl Acad Sci U S A. 1993;90:5881. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Poppe L. Curr Opin Chem Biol. 2001;5:512. doi: 10.1016/s1367-5931(00)00253-2. [DOI] [PubMed] [Google Scholar]
- 354.Retéy J. Biochim Biophys Acta, Proteins Proteomics. 2003;1647:179. doi: 10.1016/s1570-9639(03)00091-8. [DOI] [PubMed] [Google Scholar]
- 355.Peterkofsky A. J Biol Chem. 1962;237:787. [PubMed] [Google Scholar]
- 356.Poppe L, Rétey J. Angew Chem Int Ed. 2005;44:3668. doi: 10.1002/anie.200461377. [DOI] [PubMed] [Google Scholar]
- 357.Langer M, Pauling A, Rétey J. Angew Chem Int Ed. 1995;34:1464. [Google Scholar]
- 358.Schuster B, Rétey J. Proc Natl Acad Sci U S A. 1995;92:8433. doi: 10.1073/pnas.92.18.8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Christianson CV, Montavon TJ, Festin GM, Cooke HA, Shen B, Bruner SD. J Am Chem Soc. 2007;129:15744. doi: 10.1021/ja0762689. [DOI] [PubMed] [Google Scholar]
- 360.Bartsch S, Bornscheuer UT. Angew Chem Int Ed. 2009;48:3362. doi: 10.1002/anie.200900337. [DOI] [PubMed] [Google Scholar]
- 361.Strom S, Wanninayake U, Ratnayake ND, Walker KD, Geiger JH. Angew Chem Int Ed. 2012;124:2952. doi: 10.1002/anie.201108525. [DOI] [PubMed] [Google Scholar]
- 362.Seff AL, Pilbak S, Silaghi-Dumitrescu I, Poppe L. J Mol Model. 2011;17:1551. doi: 10.1007/s00894-010-0849-7. [DOI] [PubMed] [Google Scholar]
- 363.Pilbák S, Farkas Ö, Poppe L. Chem-Eur J. 2012;18:7793. doi: 10.1002/chem.201103662. [DOI] [PubMed] [Google Scholar]
- 364.Wang K, Hou Q, Liu Y. J Mol Graphics Modell. 2013;46:65. doi: 10.1016/j.jmgm.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 365.Kolbe H, Kolbe H, Schmitt R, Lindsey AS, Jeskey H, Markovic Z, Engelbrecht JP, Markovic S. Liebigs Ann. 1860;113:125. [Google Scholar]
- 366.Schmitt R. J Prakt Chem. 1885;31:397. [Google Scholar]
- 367.Schuhle K, Fuchs G. J Bacteriol. 2004;186:4556. doi: 10.1128/JB.186.14.4556-4567.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Meganathan R. Ubiquinone biosynthesis in microorganisms. 203:2001. doi: 10.1111/j.1574-6968.2001.tb10831.x. [DOI] [PubMed] [Google Scholar]
- 369.Gulmezian M, Hyman KR, Marbois BN, Clarke CF, Javor GT. Arch Biochem Biophys. 2007;467:144. doi: 10.1016/j.abb.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Rangarajan ES, Li Y, Iannuzzi P, Tocilj A, Hung LW, Matte A, Cygler M. Protein Sci. 2004;13:3006. doi: 10.1110/ps.04953004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Jacewicz A, Izumi A, Brunner K, Schnell R, Schneider G. PLoS One. 2013;8:1. doi: 10.1371/journal.pone.0063161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.White MD, Payne KAP, Fisher K, Marshall SA, Parker D, Rattray NJW, Trivedi DK, Goodacre R, Rigby SEJ, Scrutton NS, Hay S, Leys D. Nature. 2015;522:502. doi: 10.1038/nature14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Payne KAP, White MD, Fisher K, Khara B, Bailey SS, Parker D, Rattray NJW, Trivedi DK, Goodacre R, Beveridge R, Barran P, Rigby SEJ, Scrutton NS, Hay S, Leys D. Nature. 2015;522:497. doi: 10.1038/nature14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Wendt KU, Schulz GE, Corey EJ, Liu DR. Angew Chem Int Ed. 2000;39:2812. [PubMed] [Google Scholar]
- 375.Xu R, Fazio GC, Matsuda SPT. Phytochemistry. 2004;65:261. doi: 10.1016/j.phytochem.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 376.Wendt KU. Angew Chem Int Ed. 2005;44:3966. doi: 10.1002/anie.200500804. [DOI] [PubMed] [Google Scholar]
- 377.Xiong Q, Wilson WK, Matsuda SPT. Angew Chem Int Ed. 2006;45:1285. doi: 10.1002/anie.200503420. [DOI] [PubMed] [Google Scholar]
- 378.Marner FJ. Curr Org Chem. 1997;1:153. [Google Scholar]
- 379.Yoshida T. Curr Top Phytochem. 2000;4:135. [Google Scholar]
- 380.Bonfils JP, Pinguet F, Culine S, Sauvaire Y. Planta Med. 2001;67:79. doi: 10.1055/s-2001-10625. [DOI] [PubMed] [Google Scholar]
- 381.Shao L, Lewin NE, Lorenzo PS, Hu Z, Enyedy IJ, Garfield SH, Stone JC, Marner FJ, Blumberg PM, Wang S. J Med Chem. 2001;44:3872. doi: 10.1021/jm010258f. [DOI] [PubMed] [Google Scholar]
- 382.Grob CA, Baumann W. Helv Chim Acta. 1955;38:594. [Google Scholar]
- 383.Borodine A. Justus Liebigs Ann Chem. 1861;119:121. [Google Scholar]
- 384.Hunsdiecker H, Hunsdiecker C. Ber Dtsch Chem Ges. 1942;75:291. [Google Scholar]
- 385.Olson JA, Shea KM. Acc Chem Res. 2011;44:311. doi: 10.1021/ar100114m. [DOI] [PubMed] [Google Scholar]
- 386.Stein RA, Kayama M. J Org Chem. 1961;26:5231. [Google Scholar]
- 387.Das JP, Roy S. J Org Chem. 2002;67:7861. doi: 10.1021/jo025868h. [DOI] [PubMed] [Google Scholar]
- 388.Agarwal V, Gamal AA El, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, Moore BS. Nat Chem Biol. 2014;10:640. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Kelly WL. Nature. 2011;473:35. doi: 10.1038/473035a. [DOI] [PubMed] [Google Scholar]