

SUMMARY
Neutrophils are the first line of defense against bacterial infections, and the generation of reactive oxygen species is a key part of their arsenal. Pathogens use detoxification systems to avoid the bactericidal effects of reactive oxygen species. Here we demonstrate that the Gram-negative pathogen Pseudomonas aeruginosa is susceptible to reactive oxygen species but actively blocks the reactive oxygen species burst using two type III secreted effector proteins, ExoS and ExoT. ExoS ADP-ribosylates Ras and prevents it from interacting with and activating phosphoinositol-3-kinase (PI3K), which is required to stimulate the phagocytic NADPH-oxidase that generates reactive oxygen species. ExoT also affects PI3K signaling via its ADP-ribosyltransferase activity but does not act directly on Ras. A non-ribosylatable version of Ras restores reactive oxygen species production and results in increased bacterial killing. These findings demonstrate that subversion of the host innate immune response requires ExoS-mediated ADP-ribosylation of Ras in neutrophils.
Keywords: type III secretion, T3SS, effector function, keratitis
eTOC Blurb
Reactive oxygen species (ROS) production by neutrophils is a key antimicrobial defense. Vareechon et al. show that two type III secreted effectors of Pseudomonas aeruginosa, ExoS and ExoT, independently block ROS production by neutrophils. ExoS ADP-ribosylates Ras, which prevents binding to PI3K, thereby blocking NADPH oxidase activation and ROS production.
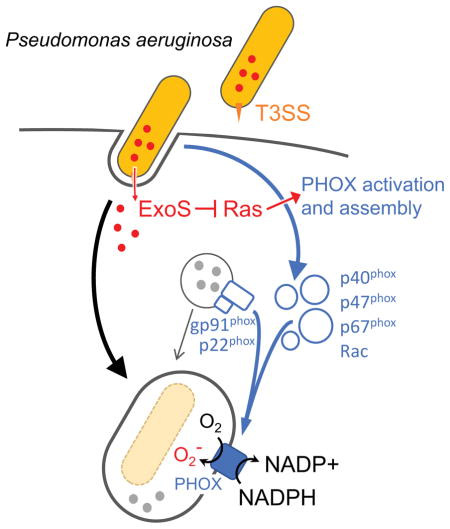
INTRODUCTION
Neutrophils are essential immune cells that are rapidly recruited to sites of bacterial infection and are critical for host defense (Dohrmann et al., 2016). Bacteria avoid killing by neutrophils, by inhibiting phagocytosis (Andersson et al., 1996; Rooijakkers et al., 2005), escaping the phagosome, detoxifying reactive oxygen species (Aussel et al., 2011), resisting antimicrobial peptides (Kraus and Peschel, 2006), degrading NETs (Buchanan et al., 2006), or killing neutrophils recruited to the site of infection (Sun et al., 2012).
Pseudomonas aeruginosa is a major cause of acute, hospital-acquired infections and microbial keratitis, as well as chronic lung infections in cystic fibrosis patients (Lyczak et al., 2002; Roy-Burman et al., 2001; Stapleton and Carnt, 2012). P. aeruginosa has a type III secretion system (T3SS), which is a molecular syringe that allows the bacterium to directly inject effector proteins into the cytoplasm of host cells. Type III secretion is linked to increased patient morbidity and mortality in ventilator associated pneumonia and blood stream infections (El-Solh et al., 2012; Hauser et al., 2002). The T3SS is likewise a crucial virulence factor in animal models of pulmonary and corneal infections, and primarily targets neutrophils (Diaz and Hauser, 2010; Finck-Barbancon et al., 1997; Lee et al., 2005; Sun et al., 2012).
P. aeruginosa has four effector proteins at its disposal. Almost all P. aeruginosa strains produce ExoT, whereas ExoS and ExoU are for the most part distributed in a mutually exclusive manner, with the majority of strains producing ExoS (Feltman et al., 2001; Toska et al., 2014). A fourth effector, ExoY, appears to play only a minimal role in infection (Lee et al., 2005; Sun et al., 2012). ExoS and ExoT are closely related (76% amino acid identity), hetero-bifunctional enzymes, with amino-terminal Rho-GAP and C-terminal ADP-ribosyltransferase activities (Barbieri and Sun, 2004). In animal models of infection, the survival benefit of having a type III secretion system can be attributed almost entirely to the ADP-ribosyltransferase activities of these two effector proteins (Shaver and Hauser, 2004; Sun et al., 2012). However, the molecular mechanism by which ExoS and ExoT prevent clearance by neutrophils remains an open question.
Here we demonstrate that ExoS and ExoT disrupt the signaling pathway responsible for activation and assembly of the phagocytic NADPH oxidase (PHOX). Blocking ROS production is linked to survival in neutrophils in vitro, and in a mouse model of corneal infection. Moreover, we present evidence that ExoS interferes with ROS production by ADP-ribosylating Ras. This modification prevents binding of Ras to, and activation of phosphoinositol-3-kinase (PI3K), which is required ROS production.
RESULTS
NADPH oxidase is required for P. aeruginosa clearance
Neutrophils are the predominant immune cells in P. aeruginosa infections (Diaz et al., 2008). A key feature of their antimicrobial arsenal is the generation of reactive oxygen species (ROS). We assessed the role of ROS in clearing P. aeruginosa infections using mice that are unable to generate ROS due to a mutation in the NADPH oxidase gp91phox subunit, a mouse model of chronic granulomatous disease (Pollock et al., 1995). Corneas of C57BL/6 and gp91phox−/− mice were infected with wild-type P. aeruginosa (PAO1) that produce ExoS, ExoT, and ExoY, or with a mutant strain that lacks the essential T3SS inner membrane component PscD (ΔpscD), and therefore cannot assemble a functional T3SS. Corneal opacification resulting from infiltration of neutrophils into the cornea (Sun et al., 2012), and bacterial load (colony forming units, CFU) were quantified after 24h.
Figure 1A shows pronounced corneal opacification in representative C57BL/6 mice infected with the parental P. aeruginosa strain PAO1, but not in mice infected with the ΔpscD T3SS null mutant bacteria. In contrast, corneas of gp91phox−/− mice infected with the ΔpscD mutant strain exhibited severe corneal opacification (Figure 1A and B). Consistent with the corneal opacification data, we recovered significantly more PAO1 than the ΔpscD mutant bacteria from infected C57BL/6 mouse corneas (Figure 1C), indicating that the ΔpscD mutant bacteria were being cleared. However, in infected gp91phox−/− corneas, CFU of both the PAO1 and ΔpscD strains were equivalent (no statistical difference). Similar results were obtained with an exoST(A-) strain in which the ADPRT activities of both ExoS and ExoT are inactivated (Figure S1). Figures 1D and E show the presence of neutrophils in the corneal stroma of infected gp91phox−/− mice, indicating that there is no defect in neutrophil recruitment in these mice. ROS production is therefore required for bacterial clearance, and the data suggest that the T3SS promotes bacterial survival by inhibiting ROS production by neutrophils.
Figure 1. NADPH oxidase mediates ROS production by neutrophils and facilitates clearance of P. aeruginosa during bacterial keratitis.

(A) Representative images of corneal opacification 24 h post-infection of C57BL/6 and gp91phox −/− (CGD) mice infected with 1x105 CFU PAO1 (WT) or with the ΔpscD (T3SS null) mutant strain. (B) Quantification of corneal opacity by determining average pixel intensity of corneas described previously (Sun et al., 2012)(n = 5 mice). (C) Colony forming units (CFU) recovered from infected corneas 24 h post-infection (n = 9 mice). (D) Corneal sections were stained with hematoxylin and eosin, or (E) 4’,6-Diamidino-2-Phenylindole dye (DAPI, Blue) and an antibody to Ly6G (NIMP-R14, FITC, green) (Epi: epithelium, Str: stroma, End: corneal endothelium, AC: anterior chamber). B, C: Data points represent individual corneas. Median and interquartile range are indicated. Significance was calculated using the Kruskal-Wallis test, with Dunn’s multiple comparison correction. * p<0.05, **p<0.01, ***p<0.001, or n.s., not significant. See also Figure S1.
Neutrophil ROS inhibition is mediated by the ADPRT activities of ExoS and ExoT
To assess if the P. aeruginosa T3SS inhibits neutrophil ROS production, peripheral blood neutrophils from healthy volunteers were infected with PAO1 or with the T3SS null mutant strain (ΔpscD), and ROS production was measured. Neutrophils infected with the ΔpscD mutant strain exhibited robust production of reactive oxygen species compared with wild type PAO1 (Figure 2A), indicating that P. aeruginosa is able to directly inhibit ROS production in neutrophils in a T3SS-dependent manner. The decrease in ROS production was not due to a defect in phagocytic uptake of PAO1, since uptake of wild-type and T3SS-null mutant P. aeruginosa was equivalent under the conditions of this assay (Figure S2A). In addition, the T3SS did not induce neutrophil lysis over the course of the experiment (Figure S2B).
Figure 2. ExoS and ExoT ADPRT activities inhibit ROS production in human neutrophils.

ROS production was measured using a chemiluminescent substrate (relative light units, RLU). Neutrophils were infected with: wild type (PAO1), a T3SS null mutant (ΔpscD), as well as: (A) a strain lacking the translocation apparatus (ΔpopBD), a strain lacking all 3 effectors (Δ3TOX), and a strain lacking exoS and exoT effector genes (ΔexoST). (B) a strain lacking exoS (ΔexoS), a strain lacking exoT(ΔexoT), or (C) strains with chromosomal point mutations inactivating the Rho-GAP (G-) or ADP-ribosyltransferase activities (A-) of ExoS and/or ExoT. A time course representative of at least three independent experiments is shown. See also Figure S2.
We also examined neutrophils infected with a strain that has the intact needle structure, but lacks the effector molecules ExoS, ExoT, and ExoY (Δ3TOX). While initial ROS production by neutrophils infected with the Δ3TOX or ΔexoST strain was similar to that seen in neutrophils infected by the ΔpscD mutant, ROS production decreased more rapidly (Figure 2A), suggesting that formation of pores in the phagosome membrane by the translocation apparatus interferes with ROS production. Similarly, the pore-forming toxin streptolysin O was shown to blunt ROS production in Streptococcus pyogenes infected neutrophils (Uchiyama et al., 2015). In support of this, a strain lacking the pore-forming translocator proteins PopB and PopD (ΔpopBD) replicated the ΔpscD mutant phenotype. We had previously observed that ExoS and ExoT, but not ExoY, are required for PAO1 virulence in a murine model of P. aeruginosa keratitis, and for survival of P. aeruginosa in neutrophils in vitro (Sun et al., 2012). Consistent with those data, a strain lacking only ExoS and ExoT induced ROS production to similar levels as the Δ3TOX mutant strain (Figure 2A). Deletion of exoS or exoT individually did not prevent the block in ROS production (Figure 2B). Taken together, these data indicate that ExoS and ExoT independently block ROS production by infected neutrophils, and that there is no role for ExoY.
ExoS and ExoT are highly homologous effectors with a similar domain structure. They each have an N-terminal rho-GAP and C-terminal ADPRT domain. The Rho GAP domains of these two proteins target Rho, Rac, and CDC42, while the ADPRT domains of ExoS and ExoT have different target specificities. ExoS ADP-ribosylates multiple proteins, including low molecular weight GTPases, ERM (Ezrin-Radixin-Moesin) proteins, and vimentin, whereas the only known targets of ExoT are CrkI, CrkII, and phosphoglycerate kinase (Barbieri and Sun, 2004). To assess the individual contributions of these activities to blocking ROS production, we infected neutrophils with P. aeruginosa strains in which either the rho-GAP activity (G-) or the ADPRT activity (A-) were inactivated by point mutations. Whereas Rho-GAP mutations had no effect on the ability of P. aeruginosa to block ROS production by human neutrophils, inactivating the ADPRT activities of both ExoS and ExoT [exoST(A-)] resulted in elevated ROS production by infected neutrophils (Figure 2C). As with the whole-gene deletions, inactivating the ADPRT-activities of ExoS or ExoT individually had no effect. ExoS and ExoT therefore act independently to block ROS production in an ADPRT-dependent fashion.
ExoS and ExoT block NADPH Oxidase activity by inhibiting PI3K signaling
Activation and assembly of NADPH oxidase in neutrophils involves the phosphoinositide 3-kinase (PI3K) signaling pathway (Hawkins et al., 2010). Activation of PI3Kγ leads to phosphorylation and activation of Akt and protein kinase C (PKC), which are needed to phosphorylate the p47phox and p40phox cytosolic components of NADPH oxidase. Once activated, p-p47phox, p-p40phox, and p67phox translocate to the membrane and, in conjunction with activated Rac, interact with p22phox/gp91phox to form the active NADPH oxidase complex (Groemping and Rittinger, 2005). Activation of NADPH oxidase in P. aeruginosa infected neutrophils similarly depends on the PI3K signaling pathway, as ROS production was blocked in the presence of PI3K inhibitors (Figure S3).
To determine whether P. aeruginosa T3SS-effectors interfere with the PI3K signaling pathway, human neutrophils were infected with PAO1 or ΔpscD mutant bacteria, and phosphorylation of Akt and p40phox was examined. Infection of neutrophils with the ΔpscD mutant strain induced Akt and p40phox phosphorylation within 15 minutes, which was sustained over 60 minutes (Figure 3A). In marked contrast, PAO1 only induced phosphorylation of Akt and p40phox at the 15-minute time point, and to a lesser extent than the ΔpscD mutant bacteria. Together, these findings demonstrate that P. aeruginosa-induced ROS production by human neutrophils requires PI3K, and that the T3SS inhibits phosphorylation of Akt and the p40phox subunit of NADPH oxidase, both of which are signaling events downstream of PI3K.
Figure 3. ExoS and ExoT ADP-ribosyltransferase activities interfere with PI3K signaling in neutrophils.

(A) Cell lysates from uninfected human neutrophils, or from neutrophils infected with wild type PAO1, or a ΔpscD mutant strain were probed by Western blot for Akt, P-Akt (Thr308), p40phox, P-p40phox (Thr154), and Ras. The experiments were repeated 3 times with similar results. (B) Cell lysates from uninfected human neutrophils, or neutrophils infected for 30 minutes with PAO1, ΔpscD, or with strains in which the ADP-ribosyltransferase activity was inactivated in ExoS only (exoS(A-)), ExoT only (exoT(A-)), or both (exoST(A-)). P-Akt (Thr308), Akt, P-p40phox (Thr154), p40phox, total Ras, GTP-bound Ras, and Grb2 (loading control) were detected by western blot. The experiments were repeated 5 times with similar results. (C) Model of Ras (gray) bound to the Ras-binding domain (light blue) of PI3K (dark blue) based on the structure PDB:1he8 (Pacold et al., 2000). Residue Arg 41 of Ras is highlighted red. (D) Purified ExoS was used to ADP-ribosylate HA-tagged versions of Ras, or Ras(R41K), in vitro and subsequently mixed with purified PI3Kγ. The interaction between Ras and PI3Kγ was probed by immunoprecipitating Ras using an anti-HA-tag antibody. PI3Kγ, as well as unmodified and ADP-ribosylated Ras were detected by western blot. The experiments were repeated 3 times with similar results. Input and output levels of PI3Kγ were determined by densitometry. The input/output ratio for the untreated control sample was set to 100% and compared to the corresponding ExoS-treatment condition (mean and standard deviation of three independent replicates are noted below each lane). Results were compared by 1-way ANOVA with Bonferroni correction (**** p<0.0001). See also Figures S3.
The ADPRT activities of ExoS and ExoT disrupt PI3K signaling and NADPH Oxidase activation
To examine the role of ExoS and ExoT ADPRT activities on PI3K signaling, human neutrophils were infected with the exoT(A-), exoS(A-) or exoST(A-) mutants and after 30 min, cell lysates were assayed for phosphorylation of Akt and p40phox. Infection of neutrophils with the exoST(A-) double mutant strain resulted in phosphorylation of Akt and p40phox, akin to the T3SS-null mutant strain. Consistent with the redundant role of ExoS and ExoT in blocking ROS production, strains in which the ADPRT activity of only one of these two effectors had been inactivated, exoS(A-) or exoT(A-), blocked phosphorylation of Akt and p40phox (Figure 3B). As with ROS production, the GAP activities of ExoS and ExoT do not affect the PI3K pathway or the NADPH oxidase complex.
Ras is critical for activation of PI3Kγ mediated ROS production in neutrophils (Pacold et al., 2000; Suire et al., 2006), and is a known target of ExoS in epithelial cells. The addition of ADP-ribose to Ras by ExoS, both in vitro and in vivo, results in a gel mobility shift (Coburn and Gill, 1991; Ganesan et al., 1999). We found that Ras in lysates from neutrophils infected with wild-type P. aeruginosa exhibited a shift in its mobility, indicating that Ras is ADP-ribosylated (Figure 3A, B). This mobility shift was uniquely dependent on the ADPRT activity of ExoS, since ADP-ribosylation of Ras was evident in neutrophils infected with the exoT(A-) mutant strain, but not neutrophils infected with the exoS(A-) strain (Figure 3B). Using purified proteins, we confirmed that the mobility shift depends both on the presence of ExoS, and residue Arg41 in Ras (Figure 3D). These data are also in agreement with previous studies in epithelial cells, which indicated that Ras is not a target of ExoT (Sun and Barbieri, 2003).
ADP-ribosylation of Ras at Arg41, in vitro, results in a 3-fold slower rate of GDP/GTP exchange compared to unmodified Ras, which led to the proposal that ExoS interferes with Ras signaling by reducing guanine nucleotide exchange (Ganesan et al., 1999). To determine if ExoS reduces the amount of active Ras in infected human neutrophils, the same lysates probed in Figure 3B were also used to purify GTP-bound Ras using the immobilized Ras-binding domain of Raf1. ADP-ribosylation of Ras does not interfere with binding to Raf1 (Ganesan et al., 1999). Total- and GTP-bound Ras were detected by western blot. ADP-ribosylation of Ras did not significantly affect the relative amount of GTP-bound Ras in infected neutrophils (averages of five independent experiments are reported in Figure S3). Our finding that all GTP-bound Ras in neutrophils infected with ExoS+ bacteria is ADP-ribosylated was highly reproducible in five experiments. We speculate that this reflects the activation state of Ras near the site of bacterial phagocytosis (and injection of ExoS).
In summary, injected ExoS and ExoT, through their ADPRT-activities, disrupt the PI3K signaling pathway in human neutrophils. ADP-ribosylation of Ras is a likely candidate for the block elicited by ExoS, but not by affecting the level of GTP-bound Ras in infected neutrophils.
ADP-ribosylation of Ras interferes with binding to PI3Kγ
We hypothesized that instead of interfering with GDP/GTP exchange, ADP-ribosylation could be blocking the Ras-PI3K interaction that is required for activation of PI3K. In fact, arginine 41 of Ras is close to the Ras-binding domain of PI3K, and is oriented towards PI3K in the published crystal structure of the Ras-PI3K complex (Figure 3C)(Pacold et al., 2000).
To test this hypothesis, we used recombinant, HA-tagged versions of human, full-length Ras, or Ras(R41K), where Arg41 is replaced by lysine, to monitor binding of PI3K by affinity chromatography using purified proteins. PI3Kγ co-purified with Ras (Figure 3D, lane 2). ADP-ribosylation of Ras by ExoS significantly reduced binding of wild type Ras to PI3Kγ (Figure 3D, lane 3). The R41K mutation did not interfere with Ras binding to PI3Kγ. However, in the presence of ExoS, there was no ADP-ribosylation of Ras, and PI3Kγ binding was significantly restored (Figure 3D, lane 5). These results provide evidence that ExoS mediated ADP-ribosylation of Ras at arginine 41 impedes Ras binding to PI3Kγ.
Intracellular delivery of R41K Ras protein into neutrophils rescues ROS production in the presence of ExoS
If ADP-ribosylation of Ras by ExoS interferes with activation of the PI3K signaling pathway, we should be able to reverse the ExoS-dependent block in ROS production by introducing Ras(R41K) into neutrophils. We used Tat peptide-mediated cellular delivery to introduce Ras(R41K) into primary human neutrophils. The HIV Tat-derived peptide is a cell penetrating peptide (CPP) that can deliver proteins into cells (Zhao and Weissleder, 2004).
We purified the recombinant Tat-Ras(R41K) fusion protein containing an HA tag from E. coli, and introduced it into human neutrophils. To examine if Tat-Ras(R41K) was delivered into the cells, we used a proteinase K protection assay, which degrades extracellular, but not intracellular proteins unless the cells are permeabilized. The Tat-Ras(R41K) fusion protein was taken up in a dose-dependent manner, and Ras was not detected in Triton-X100 treated neutrophils (Figure 4A), indicating that the Tat-Ras(R41K) fusion protein was protected from proteolysis, and therefore intracellular.
Figure 4. Tat-Ras(R41K) rescues ROS production in human neutrophils, resulting in increased killing of P. aeruginosa.

(A) Human neutrophils were treated with increasing concentrations of Tat-Ras(R41K) for 30 minutes, and extracellular protein was degraded by proteinase K. Western blots of cell lysates were probed with antibodies to Ras (which detect endogenous and Tat-Ras(R41K)) or the HA tag (which detects only Tat-Ras(R41K)). (B) ROS production by human neutrophils infected with PAO1, exoT(A-), or exoST(A-) P. aeruginosa was measured by chemiluminescence. Neutrophils were incubated with increasing amounts of Tat-Ras (R41K) thirty minutes prior to infection with an exoT(A-) strain. The experiment was repeated 4 times with similar results. (C) Human neutrophils were incubated with Tat-Ras(R41K) (red) or Tat-Ras (blue) (3 μM final concentration) for 30 minutes prior to infection with PAO1, ΔpscD, exoS(A-), or exoST(A) 15 min (MOI30). Extracellular bacteria were killed with gentamicin for 30 minutes. Each point represents an individual human donor (n = 4 donors). Statistical significance was measured by 1-way ANOVA with Bonferroni correction. **p<0.01, ***p<0.001, n.s. not significant. See also Figure S4.
Delivery of Tat-Ras(R41K) into human neutrophils prior to infection with the exoT(A-) strain resulted in a dose-dependent increase in ROS production, indicating that Tat-Ras(R41K) was able to reverse the ExoS-dependent block in ROS production (Figure 4B, S4A). ROS production did not reach the level induced by the exoST(A-) double mutant. This could be due to the presence of endogenous wild-type Ras, or indicate that a second target of ExoS also contributes to the block in ROS production. Delivery of either Tat-Ras(R41K) on its own, or the unrelated Tat-fusion protein, Tat-GFP, did not induce ROS production (Figure S4B). Tat-GFP, unlike Tat-Ras(R41K), did not reverse the ExoS-dependent block in ROS production (Figure S4C). While delivery of wild-type Tat-Ras also resulted in increased ROS production (Figure S4D), this was only significant at the highest concentration tested, arguing that preventing ADP-ribosylation of Ras on Arg41 specifically interfered with the ExoS-dependent block in ROS production.
To determine if the increased ROS production by Tat-Ras(R41K)-treated neutrophils results in increased bacterial killing, we assessed survival using a gentamicin protection assay. As shown in Figure 4C, we recovered about a log fewer CFU from neutrophils infected with the ΔpscD or exoST(A-) mutant strains, compared to the PAO1 or exoT(A-) strains. However, CFU recovered from TAT-Ras(R41K)-, but not Tat-Ras treated neutrophils infected with the exoT(A-) strain were significantly reduced compared to untreated neutrophils, which is consistent with increased ROS production in the presence of Ras(R41K). Recovered CFU for the exoST(A-) strain was not affected by Tat-Ras or Tat-Ras(R41K), indicating that the effect of the Tat-Ras(R41K) fusion protein on survival of phagocytosed P. aeruginosa is specific to the ADPRT activity of ExoS. Taken together our data demonstrate that ADP-ribosylation of Ras at Arg41 blocks ROS production and allows P. aeruginosa to survive intracellularly in human neutrophils.
DISCUSSION
ROS production by neutrophils plays a major role in pathogen clearance, which is highlighted by chronic granulomatous disease patients (CGD), who have genetic defects that inactivate the phagocytic NADPH oxidase (Heyworth et al., 2003). While P. aeruginosa is not among the most common pathogens afflicting CGD patients, epidemiological data suggests that this patient group is also more susceptible to P. aeruginosa infections (Liese et al., 2000; Soler-Palacin et al., 2007; Winkelstein et al., 2000). Using CGD mice that do not express gp91, we found that ROS production is needed to control P. aeruginosa replication in the cornea. Moreover, we demonstrate here that the T3SS is not required for survival in the cornea in CGD mice, despite massive influx of neutrophils to the site of infection. This finding indicates that an important function of the T3SS is to prevent ROS production by infiltrating neutrophils. Indeed, we found that P. aeruginosa ExoS and ExoT can each, independently block ROS production in neutrophils. Inhibition or ROS production depends on the ADP-ribosyltransferase activities of ExoS and ExoT, which block the NADPH oxidase signaling cascade.
This efficient inhibition of neutrophil ROS production is unusual. Most pathogens survive the effect of ROS by producing superoxide dismutases, catalases, and peroxidases to detoxify ROS (Andisi et al., 2012; Aussel et al., 2011; Karavolos et al., 2003). Inhibition or reduction of ROS production have been described (McCaffrey et al., 2010; Smirnov et al., 2014), but the molecular mechanism is unknown. Salmonella enterica sv. Typhimurium uses the SPI2 secretion system to not actually block ROS production, but instead prevent localization of ROS production to the phagosome (van der Heijden et al., 2015; Vazquez-Torres et al., 2000), thereby reducing the exposure of intracellular bacteria to ROS. This reduction in exposure to ROS may work hand-in-hand with periplasmic superoxide dismutase (De Groote et al., 1997), and a set of cytoplasmic catalases, peroxidases, and thiol-reducing systems that protect Salmonella from ROS-mediated oxidative damage and are needed for survival in animal models of infection (Aussel et al., 2011; Bjur et al., 2006). S. pyogenes streptolysin O inhibits ROS production (Uchiyama et al., 2015), but the reduction is comparatively minor. It is possible that pore-formation in the phagosome membrane is sufficient to partially interfere with ROS production, which could also explain the translocation-pore dependent reduction in ROS production that we observed. However, P. aeruginosa strains lacking all effectors are as defective in mouse models of infection as strains lacking the translocon or the T3SS entirely (Lee et al., 2005; Shaver and Hauser, 2004; Sun et al., 2012), arguing that the effector-mediated block in ROS production is the key factor that leads to virulence.
In the current study, we examined the molecular mechanism by which P. aeruginosa type III secreted effectors inhibit ROS production. Specifically, we demonstrate that ExoS interferes with the signaling cascade that mediates NADPH oxidase assembly by ADP-ribosylating Ras on arginine 41. Delivery of the ribosylation resistant Ras(R41K) into primary human neutrophils restored ROS production and resulted in increased killing of P. aeruginosa. This result highlights the intimate relationship between the effector-mediated block in ROS production and the ability of P. aeruginosa to survive in neutrophils. We also demonstrate that ADP-ribosylation of Ras interferes with its ability to bind to PI3K, a step that is critical for activation of PI3K.
Ras(R41K) reversed the ExoS-dependent block in ROS production incompletely. This may be a consequence of endogenous Ras, which is still susceptible to ADP-ribosylation by ExoS, or the presence of a second target of ExoS. Possible candidates are Ezrin-Radixin-Moesin (ERM) proteins as they are high affinity targets for ExoS and regulate phagosome maturation in both macrophages and dendritic cells (Erwig et al., 2006; Maresso et al., 2007). Rab5, another known target of ExoS, directs the intracellular fusion of granules with pathogen-containing phagosomes in neutrophils (Perskvist et al., 2002). ExoS could also target an as yet uncharacterized, neutrophil-specific protein. The target of ADP-ribosylation by ExoT in neutrophils is similarly unclear. While Crk proteins are targets of ExoT, and activate Rac via the Elmo1-/DOCK180 complex, this would not explain the defect in PI3K signaling we observed, suggesting that more targets of ExoT-ADP-ribosylation remain to be discovered.
The block in ROS production is clearly important for mediating the survival of P. aeruginosa in neutrophils. However, ExoS and ExoT likely contribute to P. aeruginosa pathogenesis in addition to blocking ROS production. For example, the ADP-ribosyltransferase activities of ExoS and ExoT induce apoptosis in infiltrating neutrophils (Sun et al., 2012). Also, both effectors are anti-phagocytic (Frithz-Lindsten et al., 1997), which was recently observed in vivo, in a mouse model of lung infection (Rangel et al., 2014). While we saw no effect on uptake in our in vitro experiments, our infection period was very short. A plausible explanation is that the first bacteria that encounter the infiltrating neutrophils are in fact phagocytosed. T3SS-mediated injection of ExoS and ExoT might then block phagocytosis of subsequently attaching bacteria. The combined activities of inhibiting phagocytosis and inducing apoptosis could contribute to the overall survival of the infecting population of bacteria. Clearly however, neither mechanism contributes much to the ability of P. aeruginosa to survive if the host is incapable of mounting an effective reactive oxygen species burst. Our data therefore argue that blocking ROS production is a critical function of ExoS and ExoT in subverting the anti-microbial activity of neutrophils.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and reagent requests may be directed to the lead contact, Arne Rietsch, arne.rietsch@case.edu
Bacterial strains and Culture Conditions
All strains and plasmids used in this study have been described previously and are listed in the Key Resources Table.P. aeruginosa was cultured in high salt LB to mid-log (10 g of tryptone, 5 g of yeast extract, and 11.7 g of NaCl per L, supplemented with 10 mM MgCl2 and 0.5 mM CaCl2) with 5mM EGTA to induce production of the T3SS (Rietsch and Mekalanos, 2006).
In vivo model of corneal infection
Five week old female C57BL/6 and Cybbtm1Din (gp91−/−) mice were purchased from Jackson Laboratory. Mice were anesthetized by i.p. injection of 0.4 ml 2,2,2-tribromoethanol (1.2%). Three parallel 1-mm–long abrasions in the central cornea were applied using a 26-gauge needle, and a 2.5-μl aliquot containing 105 bacteria was placed on the corneal surface as described (Sun et al., 2010). Images of corneal opacity were taken at 24hr post infection. At 24hrs post infection whole eyes were homogenized using a Mixer Mill MM300 (Retsch) at 33 Hz for 4 min. Serial log dilutions were plated onto brain heart infusion agar plates (BD Biosciences), and CFU were determined after overnight incubation at 37°C. Eyes from control mice were homogenized at 2 hours post infection to determine the starting inoculum. Quantification of corneal opacity, histology- and immunohistochemistry methods are outlined below.
Corneal opacity quantification
Corneal opacity was quantified as previously described (Leal et al., 2010; Sun et al., 2012). Mouse corneas were illuminated using a gooseneck fiber optic light source and constant light levels were maintained during image acquisition. Twenty-four-bit color images were captured with a SPOT RTKE camera (Diagnostic Instruments) connected to a Leica MZF III stereo microscope. Image analysis was performed using Metamorph Imaging software (Molecular Devices). All images were captured using the same exposure time. Naïve mice were used to acquire images of the iris and these images were used to generate a color threshold for the iris. This iris color threshold was then applied to the experimental images and set to an intensity of zero thus effectively eliminating the iris from the subsequent analysis process. To eliminate areas of reflective glare on experimental images saturated pixels were identified and then set to zero thus eliminating these pixels from the subsequent analysis. A circular region of constant area was applied to the modified experimental image and centered on the cornea. Integrated intensity values were then obtained from within the circular region and recorded as the opacity value for each mouse cornea. More opaque corneas display a greater value of integrated intensity when compared to less opaque corneas.
Histology and immunohistochemistry
Whole eyes were fixed in 10% phosphate buffered formalin, paraffin embedded, and sectioned. For immunohistochemistry, sections were treated with proteinase K (DakoCytomation) and blocked in 1.5% serum. Corneal sections were stained with anti-mouse neutrophil antibody NIMP-R14 (20 μg/ml), followed by staining with Alexa Fluor 488 goat anti-rat IgG (1:1000, Life Technologies), and DAPI (Life Technologies). Hematoxylin and eosin (H&E) staining was performed by the Case Western Reserve University Visual Science Research Center histology core.
ROS measurement
Human neutrophils were incubated with 500μM luminol (Sigma), 50U of superoxide dismutase (SOD) (Sigma), and 2,000U catalase (Milipore) for 15 minutes. Cell were then dispensed into black-wall 96 well plates with an optically clear bottom (CoStar 3720) and infected with Pseudomonas aeruginosa at MOI 30. Chemiluminescence was measured every 2 minutes for 90 minutes (Synergy HT; Biotek).
Isolation of peripheral blood neutrophils
Human neutrophils were isolated from normal, healthy donors by Ficoll-Paque Plus (GE Healthcare) density centrifugation. Peripheral blood (20 ml) was obtained and layered onto 3% dextran in PBS (Sigma-Aldrich). Red blood cells (RBCs) were separated from whole blood via incubation at 1xg for 20 minutes. The top clear layer containing leukocytes was overlaid onto 10ml of Ficoll-Paque Plus in a fresh 50-ml conical tube. The cell suspension was centrifuged at 500xg for 20 minutes at 4°C to separate mononuclear cell s from neutrophils and the remaining RBCs. The overlying plasma and monocyte layers were aspirated, and the neutrophil/RBC pellet was re-suspended in RBC Lysis Buffer (eBioscience) (8.3 g NH4Cl, 1 g KHCO3, 0.09 g EDTA/1 l ddH2O), incubated at 37°C for 10 minutes to lyse remain ing RBCs, and spun at 300xg for 5 minutes at 4°C. The lysis procedure was repeated as needed to obtain sufficient rbc lysis in cell preparations. Subsequently, cells were washed twice in PBS and re-suspended in RPMI1640 plus l-glutamine without phenol red (Hyclone). The neutrophil cell suspension was counted using a hemocytometer, and samples were collected by Cytospin and stained by Wright-Giemsa (Fisher). Using this approach, neutrophils were routinely found to be greater than 97% of the final cell preparation. Donor population is composed of 60% female and 40% male with ages ranging from 22–60 years old.
Western Blot analysis
Cells were lysed in ice-cold lysis buffer (Cell Signaling Technology), 1mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail (Cell Signaling Technology). Cell lysate were cleared by rapid centrifugation for 5 min at 4° C. Equal amounts of proteins were separated by SDS-PAGE on a 10% or 12% polyacrylamide gel (Bio-Rad), transferred to polyvinylidene difluoride (PVDF) membrane, blocked with 5% BSA (Fisher, Pittsburgh PA) in TBS-T (25 mM Tris, 0.15M NaCl, 0.05% Tween-20, pH 7.5). Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies and a chemiluminescent detection reagent (Western Bright Quantum [Advansta]). Antibodies to pAkt, Akt, p-p40phox, Ras and GRB2 were purchased from Cell Signaling Technology and p40phox from Santa Cruz Biotechnology. Blots were imaged using a GE ImageQuant LAS 4000 digital imaging system. Scanned images were processed for brightness and contrast using only the levels function of Adobe Photoshop applied to the entire image before cropping.
Ras activity assay of neutrophils
GTP-loaded Ras was quantified by GST-Raf-RBD pull-down assay (Cell Signaling Technology). Human neutrophils (1x107 cells) were incubated with P. aeruginosa (MOI 30) for 25 min in serum-free RPMI 1640 at 37 ºC. Cells were then centrifuged and lysed in ice-cold lysis buffer (Cell Signaling Technology), 1mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail (Cell Signaling Technology). The cell lysate was obtained following rapid centrifugation for 5 min at 4° C. Equal amounts of cell lysate were incubated with GST-Raf-RBD and glutathione agarose beads for 1.5 h at 4ºC with light shaking. Bound proteins were washed 3x with ice-cold lysis buffer, eluted with SDS sample buffer, boiled at 95 C for 5 min, and detected by Western blot.
Tat fusion protein production
BL21 pET28b-Tat-Ras (strain RE9356) and BL21 pET28b-Tat-Ras (R41K) (strain RE9357) were grown overnight in 5ml LB (10 g of tryptone, 5 g of yeast extract, 10g NaCl per liter) with kanamycin (50μg/ml) and chloramphenicol (30μg/ml). The overnight cultures were diluted 1:250 into 1L of 2xYT (16g tryptone, 10g yeast extract, 5g NaCl, and 1ml 1M NaOH per liter) and grown at 37°C with shaking to mid-logarithmic phase . Expression was then induced with 100μM IPTG and cultures were incubated overnight at room temperature with shaking. Bacteria were pelleted (10 minutes at 7000 RPM) and re-suspended in 100ml sonication buffer (20mM Tris pH 7.4, 5mM MgCl2, 200mM NaCl, 0.5mM DTT, and 1mM PMSF). 50ml aliquots were centrifuged again, the supernatant discarded, and each cell pellet was re-suspended in 20ml chilled sonication buffer. An aliquot was divided into four 15ml conical tubes and sonicated on ice for 8 minutes (30 sec on, 30sec off).
Pooled lysates were centrifuged for 10 minutes at 7000RPM and supernatants were moved to 20ml BioRad Econo-Pac chromatography column (BioRad). After washing 3x with sonication buffer, 500μl of Ni-NTA beads (Qiagen) were added to the supernatant, and place on a rocker at 4°C for 2hrs. The flow through fraction was collected and beads were washed 3x with wash buffer (20mM Tris pH 7.4, 5mM MgCl2, 200mM NaCl, 0.5mM DTT, and 10% glycerol). Bound protein was eluted with 3x1ml elution buffer (wash buffer supplemented with 200mM Imidazole + 1mM GDP), and then 1ml elution buffer containing 500mM Imidazole.
Purified Tat-fusion proteins were dialyzed in G-Bioscience Tube-O-Lyzers (Medi, 8K MWCO) against RPMI1640 plus L-glutamine without phenol red (Hyclone) at 4°C with stirring. For each 4ml of eluted protein, the first dialysis step was against 500mL RPMI1640 for one hour after which the tubes were moved to a second, overnight dialysis step against fresh 500ml RPMI1640. The protein solution was removed from the Tube-O-Lyzer, syringe filtered, and 1mM GDP was added. The protein sample was then moved to an Amicon Ultra-4 Centrifugal Filter unit (Regenerated cellulose, 3,000 NMWL, Milipore) and centrifuged at 4500RPM at 4C until the sample was concentrated to 200μl. The protein concentration was determined by Bradford protein assay (BioRad) and samples stored at 4°C. All Tat-fusion proteins contain a human influenza hemagglutinin (HA) tag.
GTP-Loading of Ras
Purified HA-Tat-Ras and HA-Tat-R41K Ras were diluted in 100ul GTP buffer (10mM Tris pH 7.5, 20mM NaCl) to a final concentration of 10μM. 2mM of non-hydrolysable GTP was added and incubated for 45 minutes at room temperature. GTP loaded Ras was stored at 4°C.
In vitro ADP-ribosylation of Ras
HA-agarose beads (Sigma Aldrich) were blocked in 5% BSA Kinase buffer (50mM HEPES pH 7.4, 150mM NaCl, 5mM EDTA, 5mM dithiothreitol, 10mM MgCl2, 0.01% Triton X-100) for one hour at 4°C. 1.6μM purified GTP-HA-Tat -Ras or GTP-HA-Tat-R41K Ras were added and incubated for two hours at 4°C with rocking. HA-agarose beads were centrifuged at 1000 RPM for one minute, washed with 1ml kinase buffer, centrifuged again, and the supernatant was removed using a 30-gauge needle. Beads were then re-suspended in 80μl 5% BSA kinase buffer and divided into two separate tubes. 0.8μM purified ExoS, 200μM NAD (Sigma-Aldrich), and 3.2μM human recombinant 14-3-3 zeta (Sigma-Aldrich) were added to ADP-ribosylate Ras where indicated. Samples of un-modified Ras and ADP-ribosylated Ras were brought up to a final volume of 100ul with kinase buffer. The tubes were then placed in the dark with end over end mixing and incubated with for 6.5 hours at room temperature. After incubation, tubes were stored in 4°C.
Cell-free PI3Kγ affinity assay
Unmodified and ADP-ribosylated Ras and R41K Ras were mixed with 0.08μM purified PI3Kγ (SignalChem) in a total volume of 500μl. Tubes were then placed on a rocker to incubate for 30 minutes at room temperature, then at in 4°C for an additional 30 minutes. Mixtures were washed 5x at 1000RPM for one minute with 1ml ice cold kinase buffer. After washing, residual kinase buffer was removed with a 30-gauge needle, beads were re-suspended in 1x sample buffer, and boiled at 95°C for five minutes.
Proteinase K protection assay
Human neutrophils (1x106) were incubated in 1ml serum-free RPMI 1640 with 0.01, 0.03, 0.1, 0.3, 1, or 3μM Tat-Ras(R41K) for 30 min. Cells were then pelleted at 300g for five minutes and supernatants were aspirated. Samples were re-suspended in 100ul of 1xPBS and incubated for 15 minutes at room temperature in the presence of 250μg/ml proteinase K and, where indicated, 0.1% triton X-100 (final). Proteinase K was inactivated through the addition of phenylmethane sulfonyl fluoride (PMSF, 1mM for 5 min at room temperature), samples were mixed with SDS sample buffer and boiled.
In vitro neutrophil survival assay
Human neutrophils (1x107) were incubated in 2ml serum-free RPMI 1640 at 37ºC with 3x108 bacteria for 15 min, media was replaced with RPMI1640 containing 400μg/ml gentamicin and incubated for an additional 30 minutes to kill extracellular bacteria. Cells were subsequently washed twice with 1xPBS and immediately lysed using 0.1% Triton X-100 (Sigma-Aldrich). Surviving CFU were quantified by serial log dilutions, plated on LB plates.
Bacterial Uptake by Human Neutrophils
GFP+ P. aeruginosa were cultured in high salt LB (10 g of tryptone, 5 g of yeast extract, and 11.7 g of NaCl per L, supplemented with 10 mM MgCl2 and 0.5 mM CaCl2) with 5mM EGTA to induce production of the T3SS. Bacteria were then pelleted and re-suspended in 1ml PBS containing 0.5mg/ml sulfosuccinimidyl-6-(biotinamido) hexanoate (Sulfo-NHS-LC-Biotin, Fisher) to a final concentration of 2x109 cells/ml. P aeruginosa were incubated at room temperature with end over end mixing for one hour, after which, the bacteria were pelleted at 14,000 RPM for 5 minutes, washed twice with 1xPBS, and re-suspended in 1xPBS. Human neutrophils (1x106) were incubated with 3x107 bacteria for 15 min in 2ml serum-free RPMI at 37ºC and then washed 3x with PBS. Neutrophils were fixed with 4% paraformaldehyde, stained with a 1:20 dilution of streptavidin-APC (eBioscience) for one hour, washed 3x with PBS, and re-suspended in incubation buffer (0.5% BSA in 1xPBS). Cells were analyzed on an Accuri C6 flow cytometer (BD Bioscience). Gating was based upon uninfected human neutrophils subjected to the same staining protocol.
Quantification and Statistical Analysis
Experimental data analyzed for significance were from at least three independent experiments using GraphPad Prism. The statistical test used is indicated in the figure legends. Statistical significance was defined as p<0.05. N represents animals, human donor, or experimental replicates, and is specified in the figure legends. We assumed a Gaussian distribution for our data. Accordingly, statistical significance was determined by using 1-way ANOVA with Bonferroni correction for multiple comparisons, and data are reported as mean with standard deviation. The one exception to this are the animal experiments, which can include outliers and therefore not follow a Gaussian distribution. These experiments were analyzed using a non-parametric, Kruskal-Wallis test, with Dunn’s multiple comparison correction. In these experiments, we reported median and interquartile range, since these are less vulnerable to skewing by outliers.
Experimental Model and Subject Details
Mice were housed and maintained according to institutional guidelines and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. The corneal infection protocol was approved by the CWRU Institutional Animal Care and Use Committee [protocols #2012-0105 (E.P.) and #2013-0055 (A.R.)], as well as at the University of California, Irvine, [protocol #2016-3200-0 (E.P.)]. The protocol for the use of human peripheral blood from normal healthy volunteers was approved by the Institutional Review Board of University Hospitals of Cleveland (protocol 01-15-43). Informed consent was obtained from each volunteer.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Akt rabbit polyclonal | Cell Signaling Technology | 9272 |
| Phospho-Akt rabbit polyclonal | Cell Signaling Technology | 9275 |
| p40phox | Santa Cruz Biotechnology | Sc-48388 |
| Phospho-p40phox rabbit polyclonal | Cell Signaling Technology | 4311 |
| Ras rabbit polyclonal | Cell Signaling Technology | 3965 |
| GRB2 rabbit polyclonal | Cell Signaling Technology | 3972 |
| Streptavidin APC | eBioscience | 17-4317-82 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor® 488 | Life Technologies | A-11006 |
| Bacterial Strains | ||
| PAO1F (wild-type PAO1) | (Bleves et al., 2005) | RP1831 |
| PAO1F ΔpscD | (Sun et al., 2012) | RP1903 |
| PAO1F ΔexoS | (Sun et al., 2012) | RP1883 |
| PAO1F ΔexoT | (Sun et al., 2012) | RP1945 |
| PAO1F ΔexoST | (Sun et al., 2012) | RP1947 |
| PAO1F ΔexoS ΔexoT ΔexoY (Δ3TOX) | (Cisz et al., 2008) | RP1949 |
| PAO1F ΔpopBD | (Sun et al., 2012) | RP2750 |
| PAO1F exoS(ADPR-) | (Sun et al., 2012) | RP5481 |
| PAO1F exoT(ADPR-) | (Sun et al., 2012) | RP6202 |
| PAO1F exoS(GAP-) exoT(GAP-) | (Sun et al., 2012) | RP6203 |
| PAO1F exoS(ADPR-) exoT(ADPR-) | (Sun et al., 2012) | RP6205 |
| pP25-GFPo constitutive GFP producing plasmid, CarbR | (Goodman et al., 2004) | N/A |
| BL21/pET28b-TAT-Ras | This study | RE9356 |
| BL21/ pET28b-TAT-Ras(R41K) | This study | RE9357 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Luminol | Sigma-Aldrich | 123072 |
| Superoxide dismutase from human erythrocytes | Sigma-Aldrich | S9636 |
| Catalase from human erythrocytes | Millipore | 219008 |
| Ficoll-Paque Plus (density 1.077 g/ml) | GE Healthcare | 17-1440-03 |
| Dextran from Leuconostoc spp. (MR 450,00-650,000) | Sigma-Aldrich | 31392 |
| 1x RBC Lysis Buffer | eBioscience | 00-4333-57 |
| 100x Protease Inhibitor Cocktail | Cell Signaling Technology | 5871 |
| 10x Cell Lysis Buffer | Cell Signaling Technology | 9803 |
| DAPI Solution 1mg/mL | Pierce | 62248 |
| Human PI3K (p120γ), Active full length recombinant protein expressed in Sf9 cells | SignalChem | P29-10H |
| NAD | Sigma-Aldrich | N1636 |
| Human 14-3-3 Zeta, histidine tagged | Sigma-Aldrich | Z3402 |
| Sulfo-NHS-LC-Biotin | ThermoFisher | 21327 |
| PI3K inihibitor (AS-605240) | Selleckchem | S1410 |
| PI3K inhibitor (GDC-0941) | Selleckchem | S1065 |
| Critical Commercial Assays | ||
| Active Ras Detection Kit | Cell Signaling Technology | 8821 |
| Experimental Models: Cell Lines | ||
| Human Peripheral Blood Neutrophils | Case Western Reserve University: Hematopoietic Biorepository Core | |
| C57BL/6J mice | Jackson Laboratory | 000664 |
| B6.129S-Cybbtm1Din/J (gp91phox−/−) mice | Jackson Laboratory | 002365 |
| Software and Algorithms | ||
| Metamorph Microscopy Automation and Image Analysis Software | Molecular Devices | |
| Prizm | GraphPad Software | |
Supplementary Material
HIGHLIGHTS.
P. aeruginosa inhibits ROS production by neutrophils
Inhibition depends on the ADP-ribosyltransferase activities of ExoS and ExoT
ExoS blocks ROS production by ADP-ribosylating Ras on Arg41
ADP-ribosylation of Ras blocks interaction with PI3K
Acknowledgments
The work was supported by grants for the National Institutes of Health, R01 EY022052 (to A.R.), R01 EY14362 (to E.P.). Chairut Vareechon was supported by the Visual Sciences Training Program (T32 EY007157). Image analysis was performed in the visual sciences imaging core supported by the Center Core Grant for Vision Research (P30 EY011373).
Footnotes
AUTHOR CONTRIBUTIONS
C.C.V., S.Z., and M.K., performed all experiments. C.C.V., E.P., and A.R., designed the experiments, analyzed, and interpreted the data, and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson K, Carballeira N, Magnusson KE, Persson C, Stendahl O, Wolf-Watz H, Fallman M. YopH of Yersinia pseudotuberculosis interrupts early phosphotyrosine signalling associated with phagocytosis. Molecular microbiology. 1996;20:1057–1069. doi: 10.1111/j.1365-2958.1996.tb02546.x. [DOI] [PubMed] [Google Scholar]
- Andisi VF, Hinojosa CA, de Jong A, Kuipers OP, Orihuela CJ, Bijlsma JJ. Pneumococcal gene complex involved in resistance to extracellular oxidative stress. Infection and immunity. 2012;80:1037–1049. doi: 10.1128/IAI.05563-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aussel L, Zhao W, Hebrard M, Guilhon AA, Viala JP, Henri S, Chasson L, Gorvel JP, Barras F, Meresse S. Salmonella detoxifying enzymes are sufficient to cope with the host oxidative burst. Molecular microbiology. 2011;80:628–640. doi: 10.1111/j.1365-2958.2011.07611.x. [DOI] [PubMed] [Google Scholar]
- Barbieri JT, Sun J. Pseudomonas aeruginosa ExoS and ExoT. Reviews of physiology, biochemistry and pharmacology. 2004;152:79–92. doi: 10.1007/s10254-004-0031-7. [DOI] [PubMed] [Google Scholar]
- Bjur E, Eriksson-Ygberg S, Aslund F, Rhen M. Thioredoxin 1 promotes intracellular replication and virulence of Salmonella enterica serovar Typhimurium. Infect Immun. 2006;74:5140–5151. doi: 10.1128/IAI.00449-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleves S, Soscia C, Nogueira-Orlandi P, Lazdunski A, Filloux A. Quorum sensing negatively controls type III secretion regulon expression in Pseudomonas aeruginosa PAO1. Journal of bacteriology. 2005;187:3898–3902. doi: 10.1128/JB.187.11.3898-3902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Current biology : CB. 2006;16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- Cisz M, Lee PC, Rietsch A. ExoS controls the cell contact-mediated switch to effector secretion in Pseudomonas aeruginosa. Journal of bacteriology. 2008;190:2726–2738. doi: 10.1128/JB.01553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn J, Gill DM. ADP-ribosylation of p21ras and related proteins by Pseudomonas aeruginosa exoenzyme S. Infection and immunity. 1991;59:4259–4262. doi: 10.1128/iai.59.11.4259-4262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groote MA, Ochsner UA, Shiloh MU, Nathan C, McCord JM, Dinauer MC, Libby SJ, Vazquez-Torres A, Xu Y, Fang FC. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13997–14001. doi: 10.1073/pnas.94.25.13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MH, Hauser AR. Pseudomonas aeruginosa cytotoxin ExoU is injected into phagocytic cells during acute pneumonia. Infection and immunity. 2010;78:1447–1456. doi: 10.1128/IAI.01134-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MH, Shaver CM, King JD, Musunuri S, Kazzaz JA, Hauser AR. Pseudomonas aeruginosa induces localized immunosuppression during pneumonia. Infection and immunity. 2008;76:4414–4421. doi: 10.1128/IAI.00012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrmann S, Cole JN, Nizet V. Conquering Neutrophils. PLoS pathogens. 2016;12:e1005682. doi: 10.1371/journal.ppat.1005682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Solh AA, Hattemer A, Hauser AR, Alhajhusain A, Vora H. Clinical outcomes of type III Pseudomonas aeruginosa bacteremia. Critical care medicine. 2012;40:1157–1163. doi: 10.1097/CCM.0b013e3182377906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwig LP, McPhilips KA, Wynes MW, Ivetic A, Ridley AJ, Henson PM. Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin-radixin-moesin (ERM) proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12825–12830. doi: 10.1073/pnas.0605331103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltman H, Schulert G, Khan S, Jain M, Peterson L, Hauser AR. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology. 2001;147:2659–2669. doi: 10.1099/00221287-147-10-2659. [DOI] [PubMed] [Google Scholar]
- Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, Wu C, Mende-Mueller L, Frank DW. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Molecular microbiology. 1997;25:547–557. doi: 10.1046/j.1365-2958.1997.4891851.x. [DOI] [PubMed] [Google Scholar]
- Frithz-Lindsten E, Du Y, Rosqvist R, Forsberg A. Intracellular targeting of exoenzyme S of Pseudomonas aeruginosa via type III-dependent translocation induces phagocytosis resistance, cytotoxicity and disruption of actin microfilaments. Molecular microbiology. 1997;25:1125–1139. doi: 10.1046/j.1365-2958.1997.5411905.x. [DOI] [PubMed] [Google Scholar]
- Ganesan AK, Vincent TS, Olson JC, Barbieri JT. Pseudomonas aeruginosa exoenzyme S disrupts Ras-mediated signal transduction by inhibiting guanine nucleotide exchange factor-catalyzed nucleotide exchange. The Journal of biological chemistry. 1999;274:21823–21829. doi: 10.1074/jbc.274.31.21823. [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. The Biochemical journal. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AR, Cobb E, Bodi M, Mariscal D, Valles J, Engel JN, Rello J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Critical care medicine. 2002;30:521–528. doi: 10.1097/00003246-200203000-00005. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Current topics in microbiology and immunology. 2010;346:183–202. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Current opinion in immunology. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology. 2003;149:2749–2758. doi: 10.1099/mic.0.26353-0. [DOI] [PubMed] [Google Scholar]
- Kraus D, Peschel A. Molecular mechanisms of bacterial resistance to antimicrobial peptides. Current topics in microbiology and immunology. 2006;306:231–250. doi: 10.1007/3-540-29916-5_9. [DOI] [PubMed] [Google Scholar]
- Leal SM, Jr, Cowden S, Hsia YC, Ghannoum MA, Momany M, Pearlman E. Distinct roles for Dectin-1 and TLR4 in the pathogenesis of Aspergillus fumigatus keratitis. PLoS pathogens. 2010;6:e1000976. doi: 10.1371/journal.ppat.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VT, Smith RS, Tummler B, Lory S. Activities of Pseudomonas aeruginosa effectors secreted by the Type III secretion system in vitro and during infection. Infection and immunity. 2005;73:1695–1705. doi: 10.1128/IAI.73.3.1695-1705.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, Gahr M, Belohradsky BH. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. The Journal of pediatrics. 2000;137:687–693. doi: 10.1067/mpd.2000.109112. [DOI] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresso AW, Deng Q, Pereckas MS, Wakim BT, Barbieri JT. Pseudomonas aeruginosa ExoS ADP-ribosyltransferase inhibits ERM phosphorylation. Cellular microbiology. 2007;9:97–105. doi: 10.1111/j.1462-5822.2006.00770.x. [DOI] [PubMed] [Google Scholar]
- McCaffrey RL, Schwartz JT, Lindemann SR, Moreland JG, Buchan BW, Jones BD, Allen LA. Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. Journal of leukocyte biology. 2010;88:791–805. doi: 10.1189/jlb.1209811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- Perskvist N, Roberg K, Kulyte A, Stendahl O. Rab5a GTPase regulates fusion between pathogen-containing phagosomes and cytoplasmic organelles in human neutrophils. Journal of cell science. 2002;115:1321–1330. doi: 10.1242/jcs.115.6.1321. [DOI] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- Rangel SM, Logan LK, Hauser AR. The ADP-ribosyltransferase domain of the effector protein ExoS inhibits phagocytosis of Pseudomonas aeruginosa during pneumonia. mBio. 2014;5:e01080–01014. doi: 10.1128/mBio.01080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietsch A, Mekalanos JJ. Metabolic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Molecular microbiology. 2006;59:807–820. doi: 10.1111/j.1365-2958.2005.04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nature immunology. 2005;6:920–927. doi: 10.1038/ni1235. [DOI] [PubMed] [Google Scholar]
- Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. The Journal of infectious diseases. 2001;183:1767–1774. doi: 10.1086/320737. [DOI] [PubMed] [Google Scholar]
- Shaver CM, Hauser AR. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infection and immunity. 2004;72:6969–6977. doi: 10.1128/IAI.72.12.6969-6977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A, Daily KP, Criss AK. Assembly of NADPH oxidase in human neutrophils is modulated by the opacity-associated protein expression state of Neisseria gonorrhoeae. Infection and immunity. 2014;82:1036–1044. doi: 10.1128/IAI.00881-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Palacin P, Margareto C, Llobet P, Asensio O, Hernandez M, Caragol I, Espanol T. Chronic granulomatous disease in pediatric patients: 25 years of experience. Allergologia et immunopathologia. 2007;35:83–89. doi: 10.1157/13106774. [DOI] [PubMed] [Google Scholar]
- Stapleton F, Carnt N. Contact lens-related microbial keratitis: how have epidemiology and genetics helped us with pathogenesis and prophylaxis. Eye (Lond) 2012;26:185–193. doi: 10.1038/eye.2011.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303–1309. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- Sun J, Barbieri JT. Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. The Journal of biological chemistry. 2003;278:32794–32800. doi: 10.1074/jbc.M304290200. [DOI] [PubMed] [Google Scholar]
- Sun Y, Karmakar M, Roy S, Ramadan RT, Williams SR, Howell S, Shive CL, Han Y, Stopford CM, Rietsch A, et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J Immunol. 2010;185:4272–4283. doi: 10.4049/jimmunol.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Karmakar M, Taylor PR, Rietsch A, Pearlman E. ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J Immunol. 2012;188:1884–1895. doi: 10.4049/jimmunol.1102148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toska J, Sun Y, Carbonell DA, Foster AN, Jacobs MR, Pearlman E, Rietsch A. Diversity of virulence phenotypes among type III secretion negative Pseudomonas aeruginosa clinical isolates. PloS one. 2014;9:e86829. doi: 10.1371/journal.pone.0086829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama S, Dohrmann S, Timmer AM, Dixit N, Ghochani M, Bhandari T, Timmer JC, Sprague K, Bubeck-Wardenburg J, Simon SI, et al. Streptolysin O Rapidly Impairs Neutrophil Oxidative Burst and Antibacterial Responses to Group A Streptococcus. Frontiers in immunology. 2015;6:581. doi: 10.3389/fimmu.2015.00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden J, Bosman ES, Reynolds LA, Finlay BB. Direct measurement of oxidative and nitrosative stress dynamics in Salmonella inside macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:560–565. doi: 10.1073/pnas.1414569112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, Fang FC. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 2000;287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Zhao M, Weissleder R. Intracellular cargo delivery using tat peptide and derivatives. Medicinal research reviews. 2004;24:1–12. doi: 10.1002/med.10056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.