

ABSTRACT
Methicillin-resistant Staphylococcus aureus (MRSA) is responsible for large numbers of postsurgical nosocomial infections across the United States and worldwide. Propofol anesthesia is widely used in surgery and in intensive care units, and recent evidence indicates that even brief exposure to propofol can substantially increase host susceptibility to microbial infection. Here, we delineate the impact of propofol sedation on MRSA bloodstream infections in mice in the presence and absence of prophylactic antibiotic treatment. Consistent with previous reports, brief periods of anesthesia with propofol were sufficient to significantly increase bacterial burdens and kidney pathology in mice infected with MRSA. Propofol exposure increased neutrophilic infiltrates into the kidney and enhanced bacterial dissemination throughout kidney tissue. Propofol sedation reduced populations of effector phagocytes and mature dendritic cells within the kidney and led to the apparent expansion of myeloid-derived suppressor cell-like populations. When propofol was coadministered with vancomycin prophylaxis, it dramatically increased kidney abscess formation and bacterial dissemination throughout kidney tissue at early times post-S. aureus infection compared to antibiotic-treated but nonsedated animals. Taken together, our data indicate that short-term sedation with propofol significantly increases the severity of bloodstream MRSA infection, even when administered in conjunction with vancomycin prophylaxis.
KEYWORDS: MRSA, anesthesia, bacterial pathogenesis, immune suppression, kidney abscess, macrophages, monocytes, vancomycin
INTRODUCTION
Nosocomial infections are common complications that follow surgery or prolonged stays in intensive care units (ICU) of hospitals (1–5) and which result in large increases in patient morbidity and mortality in addition to elevated health care costs (6). The causative agents of nosocomial infection are often bacteria (4, 7), and Staphylococcus aureus is one of the most frequently isolated pathogens in this regard (8). Antibiotic treatment of S. aureus infections can be complicated by the prevalence of methicillin-resistant S. aureus (MRSA) strains in hospitals and increasingly in community settings (9, 10). Patients often require multiple antibiotics or use of broad-spectrum antibiotics to eradicate infection (11). Despite numerous measures taken to ensure sterility and limit spread of MRSA from health care workers to patients in hospitals (12, 13), the rate of infection remains high (14, 15). While significant effort is generally focused on enforcing aseptic technique as a method to prevent infection, there may be additional factors that influence the frequency and/or severity of nosocomial infections (16).
Propofol is one of the most common anesthetic agents used in the induction and maintenance of anesthesia prior to surgery, both in the intensive care unit (ICU) and in routine outpatient procedures (17–20). This is in part due to its low incidence of anesthesia-related side effects and the short half-life of the drug (21–23), but some studies have suggested that exposure to propofol can also negatively impact host immune defenses (23–28). Studies in cell culture have shown that propofol exposure is associated with decreased secretion of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), decreased expression of inducible nitric oxide synthase (iNOS) gene expression (a potent mediator of antimicrobial defenses), and decreased macrophage phagocytosis (29, 30). While a number of in vitro reports have demonstrated that propofol can alter immune cell effector function, less information is available regarding the possible effects of propofol on host immunity using in vivo models of microbial infection. Recent studies from our laboratory and others have shown that exposure to propofol contributes to increased mortality in a rat model of sepsis (31) as well as increased mortality and pathology following systemic bacterial infections (28). Given the extensive use of propofol in hospital settings and the prevalence of MRSA as a causative agent of nosocomial infections, we sought to determine the effects of propofol anesthesia on host susceptibility to bloodstream MRSA infections in the presence and absence of antibiotic vancomycin prophylaxis.
RESULTS
Propofol exposure inhibits the clearance of MRSA from infected mice and increases tissue pathology.
In vitro cell culture-based studies have indicated that exposure to propofol can inhibit macrophage phagocytosis as well as the induction of antimicrobial mediators such as iNOS (29, 30, 32, 33). We therefore examined propofol anesthesia and host susceptibility to microbial infection in vivo using a well-established murine model of MRSA bloodstream infection. Adolescent female Swiss Webster outbred mice were infected with 3 × 106 CFU of MRSA strain USA300 LAC via tail vein injection in the presence or absence of intravenous (i.v.) propofol administration. The dose of propofol used was comparable to anesthetic induction doses used in humans based on body weight and metabolic rate, as determined by the FDA, and resulted in approximately 5 min of sedation (34). Control animals were given an Intralipid vehicle solution, the solvent for commercially available propofol, in place of the anesthetic. Animals were sacrificed at 7, 10, 14, or 32 days postinfection, and livers, spleens, and both kidneys were isolated to determine viable CFU. While propofol exposure did not alter detectable bacterial burdens within the livers or spleens (see Fig. S1 in the supplemental material), differences in burdens were detected at specific time points postinfection for the kidneys. At 7 days postinfection, anesthetized animals exhibited bacterial burdens in the kidneys that were significantly higher than those of infected controls (Fig. 1A). At 10 days postinfection (dpi), propofol-treated animals and infected controls had similar bacterial burdens in the kidney; however, as the infection progressed from 14 to 32 days, control animals exhibited some evidence of bacterial clearance from infected kidneys, whereas anesthetized animals maintained bacterial burdens from 10 dpi to 32 dpi (Fig. 1A).
FIG 1.
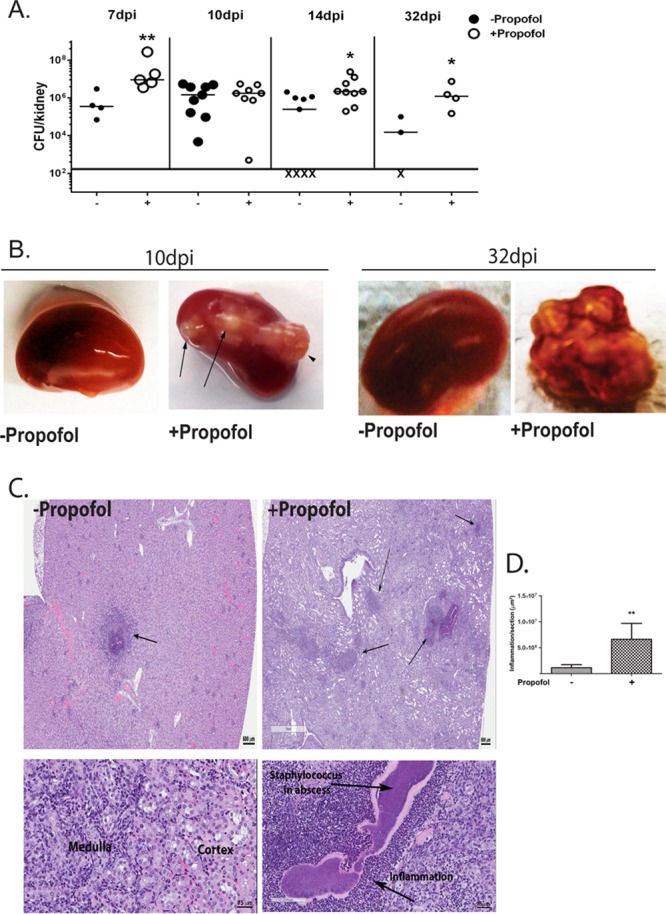
Propofol sedation results in increased bacterial burdens and kidney pathology during MRSA bloodstream infection. (A) Animals were infected with 3 × 106 CFU of the USA300 strain of methicillin-resistant S. aureus via tail vein injection. Animals were sacrificed at the indicated time points, and both kidneys were harvested, homogenized together, and plated to determine viable CFU. Propofol treatment significantly increased bacterial burdens relative to controls at 7, 14, and 32 days postinfection. Data are compiled from 2 independent experiments; X represents undetectable bacterial burdens. (B) Mice were intravenously infected with 3 × 106 CFU of S. aureus in the presence and absence of propofol sedation. Kidneys were isolated from infected animals at the indicated times, and gross kidney pathology is shown. Propofol treatment induced enlarged abscesses during MRSA infection up to 32 days postinfection. Data are representative of 2 independent experiments with n = 4 to 5 mice/treatment group. (C and D) Animals were infected as described for panel A in the presence or absence of propofol, and kidneys were harvested at 7 days postinfection. Kidneys were fixed, sectioned, and processed for hematoxylin and eosin (H&E) staining. Anesthetized animals displayed larger areas of inflammation and necrosis than control animals. Images were taken at 1× (top) and 10× (bottom) magnification; scale bar, 500 μm (top) and 75 μm (bottom). *, P < 0.05; **, P < 0.01.
Examination of the kidneys of infected animals indicated that propofol treatment significantly increased abscess formation during infection. At 10 dpi, while infected controls displayed very little, if any, pathology (Fig. 1B, left), animals exposed to propofol presented with numerous abscesses extending through the kidney capsule (Fig. 1B, left middle). The overall extent of abscess formation was even more striking at 32 dpi, when anesthetized animals developed several large abscesses such that healthy kidney tissue was not readily visible (Fig. 1B, right). Histological examination of infected kidneys at earlier time points postinfection (7 dpi) revealed extensive tissue damage and pathology in propofol-treated animals (Fig. 1C and D). Animals exposed to propofol exhibited a significantly increased proportion of kidney tissue associated with inflammation and abscess formation in comparison to non-drug-treated animals (Fig. 1C and D), suggesting that even brief exposure to propofol induced significant kidney damage during MRSA bloodstream infection, even under conditions where the overall bacterial burdens in the kidney were similar between control and anesthetized mice (Fig. 1A).
Propofol increases both neutrophil infiltration and MRSA dissemination throughout the kidney.
Kidney pathology during sepsis is generally associated with immune-mediated pathology (35). Therefore, we decided to investigate whether propofol treatment affected neutrophil recruitment to sites of infection within the kidney. Immunofluorescence-based microscopy was used to examine the distribution of Ly-6G+ neutrophils and S. aureus within kidney sections in the presence and absence of propofol sedation. At early times postinfection (7 days postinfection), areas of kidney sections containing both S. aureus and neutrophil infiltrate appeared similar with respect to the presence and distribution of neutrophils as well as infiltrating bacteria (Fig. 2A). Total neutrophil percentages in the kidney were also not significantly different between treatment groups at 7 dpi (Fig. S3b). However, by 14 days postinfection, kidneys from animals treated with propofol showed a markedly increased presence of both MRSA dissemination and inflammatory neutrophils (Fig. 2B and 3B; see also Fig. S3b). Interestingly, inflammation in kidneys isolated from control animals was limited to the kidney cortex at 14 days postinfection (Fig. 3A, left), with bacteria efficiently walled off within eosinophilic pseudocapsules (36) (Fig. 2B, second row). In contrast, kidneys from propofol-treated animals contained large amounts of bacteria in the renal pelvis and medulla, as well as in multiple abscesses in the kidney cortex (Fig. 2B, fourth row, and 3A, right). Bacteria were found disseminated throughout kidney tissue in anesthetized mice (Fig. S2), suggesting that propofol exposure inhibits the ability of animals to contain MRSA in the kidney in discrete and limited foci of infection.
FIG 2.
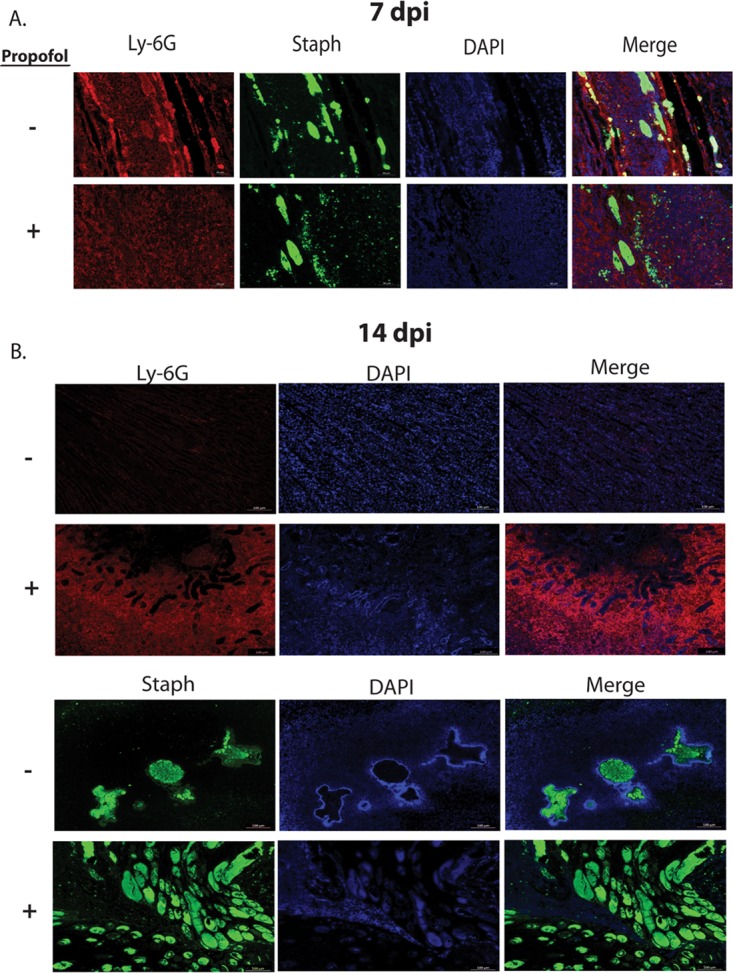
Propofol increases neutrophilic infiltrate and S. aureus dissemination into the kidney during acute infection. (A) Animals were infected i.v. with 3 × 106 CFU of S. aureus in the presence or absence of propofol. Animals were sacrificed at 7 days postinfection, and kidneys were harvested, fixed, sectioned, and stained with fluorescent antibodies against neutrophils (anti-Ly-6G; red), S. aureus (anti-S. aureus; green), or a nuclear dye (DAPI; blue). There were no apparent differences in the distribution of neutrophils or presence of S. aureus between control animals (top) and propofol-treated animals (bottom) at this time point. Images were taken at ×10 magnification; scale bar, 100 μm. (B) Animals were infected as described for panel A and sacrificed at 14 days postinfection. Kidneys were harvested and stained with fluorescent antibodies against neutrophils and stained with DAPI as described above. Mice treated with propofol (bottom) exhibited significantly enlarged neutrophilic abscesses compared with infected controls (top). Images were taken at ×10 magnification; scale bar, 100 μm. (C) Animals infected as described for panel A were sacrificed 14 days postinfection, and kidney sections were stained for the presence of S. aureus. Kidney sections from anesthetized mice contained significantly more bacteria (bottom) than control animals (top), and infected controls were able to contain infection more efficiently than propofol-treated mice. Images were taken at ×10 magnification; scale bar, 100 μm. Images are representative from 2 independent experiments with 5 animals per treatment group per time point.
FIG 3.

Neutrophilic abscesses are not confined to the kidney cortex in propofol-treated mice. (A) Kidney sections from animals were obtained at 14 days postinfection, as described in the legend to Fig. 2. Sections were stained with antibodies against neutrophils (anti-Ly-6G; red) and with DAPI (blue). Propofol treatment induces formation of multiple abscesses in the kidney cortex as well as the central medullary region. Arrows indicate neutrophilic abscesses. Images were taken at 0.6× magnification; scale bar, 500 μm. Images are representative from 2 independent experiments with 5 animals per treatment group. (B) Quantitation of area of kidney section staining positive for presence of S. aureus or neutrophils at 7 days postinfection (top) and 14 days postinfection (bottom). *, P < 0.05.
Propofol exposure leads to reduced numbers of mature phagocytes within the kidney during MRSA infection.
Abscess formation can occur in response to invading pathogens and may serve to contain microorganisms in discrete compartments separate from healthy tissue (37, 38). In the kidney, macrophages have been shown to be recruited in large numbers after neutrophils have established a swarm around sites of injury or infection, acting as a cap to limit the size of abscess formation in tissues (39). We have previously reported that exposure to propofol appears to reduce the numbers of mature phagocytes present within the spleen in response to L. monocytogenes infection (28). We therefore used flow cytometry to determine whether propofol affects monocyte and macrophage populations within the kidney during MRSA bloodstream infections.
At 10 days postinfection, F4/80+ CD11bint monocyte populations were present in similar numbers in infected controls and propofol-treated animals, with high expression of the β2 integrin CD11b, indicative of immature monocytes (40–42) (Fig. 4A). However, by 14 days postinfection, control animals showed a marked decrease in this cell population, while there was essentially no change in propofol-treated animals (Fig. 4B). This difference persisted up to 32 days postinfection, such that in control animals, the F4/80+ CD11bint population was nonexistent in the kidney, whereas animals exposed to propofol continued to have large numbers of these cells (Fig. 4B). Interestingly, despite the low numbers of F4/80+ CD11bint monocytes in unanesthetized mice at 14 days postinfection, these mice had significant numbers of F4/80+ CD11b− cells, indicative of mature macrophages within the kidney (Fig. 4C, top left). In contrast, almost all cells that were positive for F4/80 in anesthetized mice simultaneously expressed CD11b on the cell surface, suggesting that these cells were monocytes (Fig. 4C, top right, and D, top). At 32 days postinfection, a time point at which resolution of acute S. aureus infection would result in few immune cells present in the kidney (43), propofol-treated animals displayed large amounts of immature monocytes (Fig. 4D).
FIG 4.

Propofol treatment inhibits differentiation of monocytes into macrophages. (A) Animals were infected with 3 × 106 CFU of S. aureus via tail vein in the presence or absence of propofol. Mice were sacrificed at 10 days postinfection, and kidneys were isolated and processed for FACS analysis. While kidneys from naive mice contained negligible numbers of immune cells expressing the integrin CD11b or the macrophage marker F4/80 (left two images), kidneys from both propofol-treated mice (right two images) and infected controls (middle two images) contained similarly large numbers of CD11bint F4/80+ cells (green cell populations). (B) Animals were infected as described for panel A and sacrificed at 14 days postinfection. Kidneys were isolated and processed for FACS analysis. While kidneys from infected animals not given propofol showed a marked decrease in the CD11bint F4/80+ cell population (in green; top and middle left), kidneys from propofol-treated mice showed no significant change in this cell population from 10 days postinfection (top and middle right). This phenotype persisted up to 32 days postinfection, when kidneys from infected controls contained few CD11bint F4/80+ cells (bottom left), while those from anesthetized mice contained significant numbers (bottom right). (C) Control animals displayed significant numbers of F4/80+ cells that have lost CD11b cell surface expression at 14 days postinfection (top left). In contrast, kidneys from propofol-treated mice contained mostly CD11bint F4/80+ monocyte populations (top right). At 32 days postinfection, control animals exhibited far fewer F4/80+ cells in the kidney, whereas CD11bint F4/80+ cells persisted in significant numbers in kidneys from anesthetized mice. (D) Kidneys from propofol-treated animals contained significantly larger numbers of CD11bint F4/80+ monocytes than infected controls at 14 days postinfection. Conversely, control animals displayed considerably more F4/80+ macrophages than anesthetized mice at this same time point (top). At 32 days postinfection, propofol-treated mice contained substantial numbers of immature CD11bint F4/80+ MHC-II− monocytes in the kidney, whereas control animals had negligible numbers of this cell type. Data are representative of 2 independent experiments with 4 to 5 animals per treatment group per time point. *, P < 0.05; ****, P < 0.0001.
Propofol treatment expands myeloid-derived suppressor cell-like populations in MRSA-infected kidneys.
Neutrophils play a crucial role in the clearance of S. aureus from infected tissue. In addition to neutrophil phagocytosis of S. aureus, release of antimicrobial proteins (44), and oxidative killing of bacteria (45), neutrophil recruitment to sites of infection serves to wall off bacteria from healthy tissue, resulting in the formation of abscesses (46). Based on the importance of neutrophils to clearance of S. aureus infection and the immunofluorescence data suggesting that exposure to propofol dramatically increased neutrophil recruitment to the kidney during MRSA infection (Fig. 3A), we used flow cytometry to characterize neutrophil populations in the kidney during the course of infection. At 14 days postinfection, kidneys from control mice contained significantly fewer cells expressing the Ly-6G+ neutrophil cell surface marker (47) in the kidney than animals treated with propofol (Fig. S2). Further characterization of Ly-6G+ cell populations in animals exposed to propofol indicated that the cells were simultaneously CD11bhi NOS2+ (Fig. 5A), consistent with the extracellular and intracellular phenotype associated with myeloid-derived suppressor cells (MDSCs) (48). At 32 days postinfection, propofol-treated animals displayed 100-fold more MDSC-like cells in the kidney than control animals (Fig. 5A and B). MDSCs are a heterogeneous cell population, and studies have shown that they are potent suppressors of T cell activation (49); thus, their potential expansion during infection could represent one mechanism by which propofol contributes to bacterial persistence within the kidney.
FIG 5.
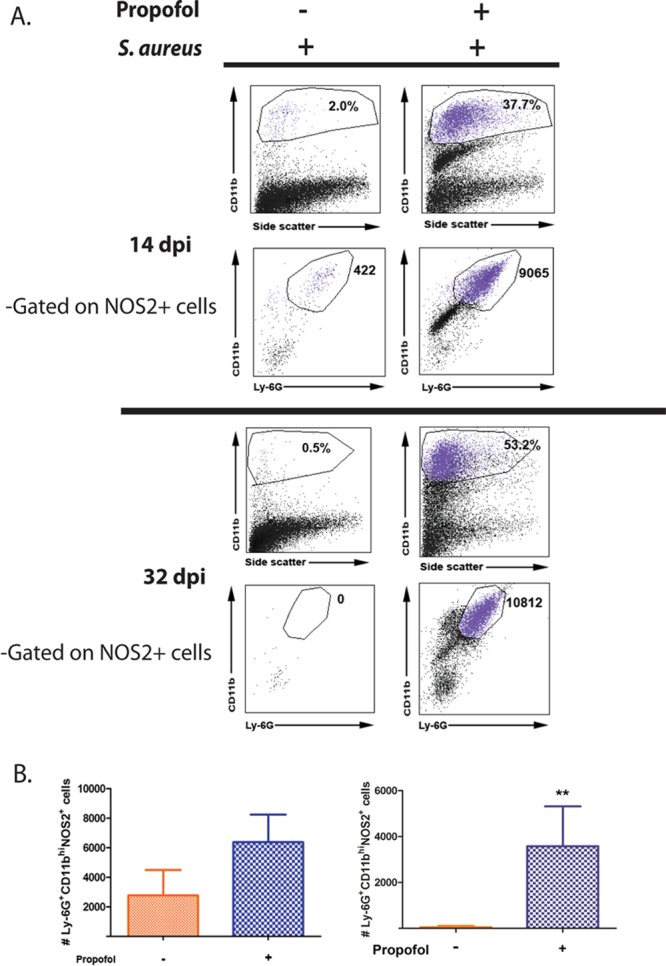
Anesthetized animals display significantly more CD11bhi Ly-6G+ NOS2+ cells in the kidney during MRSA bloodstream infection. (A) Animals were infected with 3 × 106 CFU of S. aureus via tail vein. Animals were sacrificed at 14 days (top) or 32 days (bottom) postinfection. Kidneys were isolated and processed for FACS analysis. (Right) At both 14 and 32 days postinfection, propofol-treated animals contained notably large numbers of CD11bhi Ly-6G+ NOS2+ cells that resembled myeloid-derived suppressor cells (MDSCs) (cell populations in purple). (Left) This cell population was largely absent from the kidneys of control mice. (B) Propofol-treated mice contained more MDSC-like cells at 14 days (in purple; left) and 32 days (right) postinfection. Data are representative of 2 independent experiments with 5 animals/treatment group/time point. **, P < 0.01.
Prophylaxis with vancomycin prior to infection and propofol anesthesia exacerbates MRSA-induced kidney pathology.
Because the risk of hospital-acquired infection increases as a result of surgery or prolonged stay (such as in intensive care units), patients generally receive prophylactic doses of antibiotics prior to undergoing any procedure (50–53). Vancomycin is a broad-spectrum antibiotic that is sometimes given as prophylaxis before surgery (54, 55). To assess whether the administration of vancomycin could effectively reduce the pathologies observed in animals exposed to propofol and infected with MRSA, mice were treated with vehicle alone, propofol alone, or vehicle or propofol plus a prophylactic single intravenous dose of vancomycin 24 h prior to tail vein infection with MRSA. Interestingly, during the course of these experiments it became necessary to euthanize infected animals treated with vancomycin and propofol by 5 days postinfection as the animals became moribund (data not shown). We observed that mice that were given vancomycin prior to infection in the presence or absence of propofol sedation generally exhibited more variability in bacterial burdens recovered, in that some animals exhibited bacterial burdens equivalent to animals not treated with vancomycin, whereas other animals had minimal burdens (Fig. 6A). This variability in bacterial burdens recovered from the kidneys was somewhat less apparent in animals given vancomycin followed by propofol sedation and infection, but the overall differences in bacterial burdens between the groups given vancomycin and those that were not were not statistically significant. However, despite the comparable numbers of bacteria recovered from the kidneys of mice, especially those given vancomycin followed by propofol sedation and infection with those not receiving vancomycin at all, the kidneys from vancomycin plus propofol-treated mice displayed significantly more inflammation, abscess formation, and necrosis at 4 days postinfection than any other treatment group (Fig. 6B and C). While animals sedated with propofol prior to infection exhibited increased kidney pathology by 7 days postinfection, these differences were not apparent at 4 days postinfection, except in sedated animals given vancomycin (Fig. 6B and C). Animals given vancomycin prior to propofol sedation and infection exhibited extensive areas of necrosis and abscess formation in comparison to the other treatment groups (Fig. 6B and C). In fact, infected animals given vehicle solution alone or propofol alone displayed no abnormalities in kidney structure or inflammation at 4 days postinfection (Fig. 6B, bottom). Uninfected animals given either propofol or vancomycin alone also did not have abnormal kidney architecture or pathology (Fig. S3). When kidney sections were analyzed for the presence of Ly-6G+ neutrophils and S. aureus, it was found that vancomycin treatment combined with propofol resulted in extensive areas of the kidney infiltrated by bacteria (Fig. 6D and E). These data strongly suggest that the prophylactic administration of vancomycin prior to bacterial infection and propofol sedation lead to an increase in MRSA dissemination and increased severity of associated kidney pathology.
FIG 6.
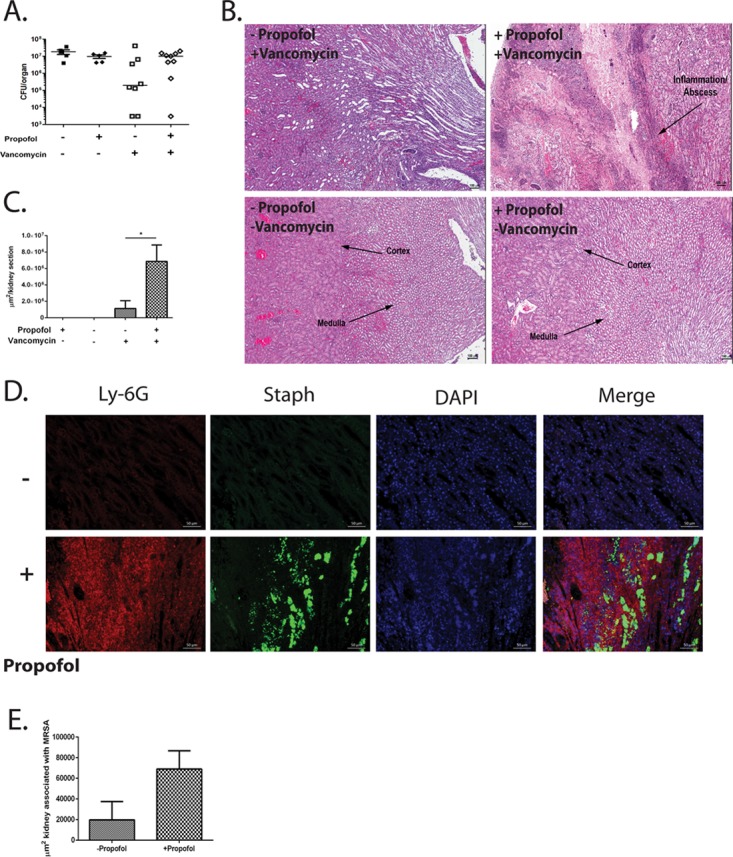
Prophylaxis with vancomycin concurrent with propofol anesthesia increases kidney pathology associated with MRSA infection. (A) Mice were intravenously injected with vehicle alone, propofol alone, vehicle plus vancomycin, or propofol plus vancomycin 24 h prior to i.v. infection with 3 × 106 CFU of S. aureus. Mice were sacrificed at 4 days postinfection, and kidneys were harvested, homogenized, and plated to determine viable CFU. Vancomycin treatment prior to infection decreased bacterial burdens at 4 days postinfection in control mice compared with anesthetized mice. Data are representative of 2 independent experiments. (B) Animals were infected as described for panel A and sacrificed at 4 days postinfection. Kidneys were harvested, fixed, and processed for H&E staining. Propofol treatment resulted in significantly larger areas of kidney sections affected by inflammation and necrosis (arrow, top right). Infected animals given vehicle solution and vancomycin (top left), vehicle alone (bottom left), or propofol alone (bottom right) displayed no signs of inflammation. Images were taken at ×4 magnification; scale bar, 100 μm. Data are representative of 5 animals per treatment group. (C) Area per kidney section associated with inflammation and/or necrosis. (D) Animals were infected as described for panel A, and kidneys were processed for immunofluorescent staining. Kidneys from propofol-treated animals (bottom) contained significantly greater amounts of Ly-6G+ inflammatory neutrophilic infiltrate (red) and S. aureus (green). Images were taken at ×20 magnification; scale bar, 50 μm. Data are representative of 2 independent experiments with 3 to 4 animals per treatment group. (E) Kidneys from propofol-treated animals contained larger areas infected with S. aureus than did control animals. *, P < 0.05.
DISCUSSION
The data presented in this study demonstrate that even brief periods of propofol sedation can significantly increase the severity of infections associated with S. aureus, even in the presence of limited prophylactic antibiotic treatment. Mice sedated for approximately 5 min with propofol exhibited dramatic increases in kidney pathology and inflammation following infection with MRSA, and these differences were evident up to 32 days postinfection despite the fact that the sedative effects of propofol are short-acting. Propofol sedation resulted in fewer mature immune effector cells and more extensive bacterial dissemination throughout the kidneys. Prophylactic treatment with vancomycin 1 day prior to sedation and infection did not reduce the severity of MRSA infection, but instead it appeared to increase kidney pathology and mortality. These data suggest that during an active infection, propofol sedation seriously impairs the ability of the host to mount an effective immune response that limits pathogen dissemination while minimizing tissue damage.
Pharmacokinetic studies on propofol have shown that while its sedative effects are relatively short acting, metabolites of propofol are detectable within the urine for several days after the initial administration (56). Propofol is lipophilic and is rapidly distributed into tissues and metabolized following the cessation of intravenous administration, a fact that explains why sedation is short-lived (22, 57, 58). It is possible that metabolites of propofol that persist within tissues for several days lead to the suppression of host immunity against microbial infection. We have previously reported that mice that have been briefly anesthetized with propofol and allowed to recover for 4 days prior to infection with the bacterial pathogen Listeria monocytogenes exhibit 100- to 1,000-fold increases in bacterial burdens in target organs in comparison to controls (28). Thus, propofol's ability to decrease host resistance to infection is not limited to a single pathogen but can be generalized to include pathogens that inhabit very different host niches.
Using mouse models of L. monocytogenes infection, we have previously reported that propofol exposure reduces the numbers of mature immune effector cells at sites of bacterial infection (28). Monocytes and macrophages are important for the efficient formation of abscesses in response to tissue injury or infection, and studies have shown that monocytes direct interstitial migration of neutrophils through tissues to effectively form abscesses (59). Additional studies have suggested that macrophage recruitment occurs after inflammatory neutrophilic infiltrate has formed focal abscesses surrounding sites of infection, and these macrophages serve to limit the size of the abscess (39) and the associated tissue damage through phagocytosis of necrotic neutrophil cell debris (36). At early time points following acute MRSA infection in the presence of propofol, monocyte/macrophage populations did not appear significantly different in absolute numbers between drug-treated and non-drug-treated control animals; however, by 14 days postinfection anesthetized animals exhibited distinct phenotypic differences in immune effector cell populations (Fig. 4A and B) as well as increased abscess size and formation. These differences in cell populations could reflect a propofol-associated impairment of monocyte-to-macrophage maturation. Monocytes recruited to tissues can be identified by cell surface expression of CD11b (60). Studies have shown that antibody blockade of CD11b results in uncontrolled bacterial growth when initiated at early times postinfection but not after infection has been established (61). This suggests that CD11b is far more important in monocyte recruitment to infected tissues than it is to controlling infection within tissues, when cells have differentiated into functional myeloid-derived effector cells. As monocytes are not as efficient in performing the effector functions of macrophages, including phagocytosis of dead cells (62, 63), it is possible that propofol exposure resulted in the uncontrolled expansion of neutrophilic abscesses as a result of reduced macrophage populations and the associated reduction in the phagocytosis of neutrophil corpses. In addition to the drug's potential effects on monocyte differentiation, in vitro studies have shown that exposure to propofol decreases phagocytosis of latex beads in human microglial cell culture (64) and in human THP-1 macrophage cell lines (26). We speculate that the combined effects of propofol on inhibition of monocyte differentiation and impaired phagocytosis contribute to the diffuse and extensive abscess formation observed in the kidneys of drug-treated animals.
Host immunity to both local and systemic MRSA infections depends on neutrophil chemotaxis to sites of infection (45, 65); in the kidney, abscess formation results in the “walling off” of discrete foci of pathogenic infiltrate by initially surrounding bacteria with an eosinophilic pseudocapsule (66) with subsequent neutrophil recruitment to surround the pseudocapsule (36). The overall size of the abscess can be limited by fibrin deposits that serve to partition areas of inflammation from healthy tissue (67). This process serves to constrain and localize inflammatory damage while preventing unrestricted enlargement of the inflammatory abscess. Our data indicate that propofol administration resulted in a much greater proportion of kidney tissue affected by inflammatory abscesses (Fig. 1D, 2B, and 3A), suggesting that exposure to propofol results in unrestricted inflammation during MRSA kidney infection. Overall, these findings suggest propofol exposure increases the severity and frequency of hospital-acquired infections through suppression of host defense mechanisms that limit inflammatory damage.
Significantly increased numbers of Ly-6G+ neutrophils were detected at sites of MRSA infection in drug-treated animals (Fig. 2 and 3). Based on cell surface expression of CD11b and Ly-6G, as well as intracellular expression of NOS2 (48), a significant fraction of the cells present within the abscesses appeared to be myeloid-derived suppressor cell (MDSC)-like cells (Fig. 5). MDSCs are a heterogeneous cell population and can be comprised of granulocyte-like or monocyte-like cells (68). Granulocytic MDSCs are known to expand in the context of infection in response to increased gamma interferon (IFN-γ) production (69), and they act as suppressors of antigen-specific T cell responses (70). MDSCs thus represent immature myeloid cells that have failed to differentiate into mature granulocytes. It has been shown in healthy individuals that immature myeloid cells generated in the bone marrow quickly differentiate in circulation to DCs, macrophages, and granulocytes (48). However, under pathological conditions, including sepsis, differentiation of these immature myeloid cells into mature effector cells is partially blocked (71). Numerous recent studies have shown that MDSCs can play a pathological role during S. aureus infection. One study used splenocytes from S. aureus-infected mice cocultured in vitro with OT-II-specific T cells to show that S. aureus infection induced MDSCs that blocked T cell activation and proliferation in an antigen-specific, contact-dependent manner (72). Another study using an S. aureus skin infection model showed that cutaneous sensing of S. aureus through TLR2-6 pathways induced MDSC expansion, which negatively regulated T cell responses in the skin (73). It is possible that propofol administration in conjunction with MRSA infection represents such a pathological condition wherein differentiation of immature myeloid cells is partially blocked, resulting in MDSC expansion and a potential decrease in T cell activation.
Importantly, though limited, our experimental data strongly suggest that prophylactic antibiotic treatment with vancomycin prior to anesthesia and infection does not alleviate and may in fact enhance the severe kidney pathology that results from propofol exposure. Single-dose antibiotic prophylaxis is part of routine patient care prior to surgery or admission into the ICU (55, 74–77). Studies have shown that administering antibiotic prophylaxis reduces postsurgical infections by up to 50% (78) and is similarly effective in reducing ICU-acquired infections (79). However, in the context of the systemic model of MRSA infection used in these studies, vancomycin prophylaxis failed to consistently reduce bacterial burdens in infected animals and instead resulted in increased kidney pathology compared to propofol-treated animals not treated with vancomycin or animals given vancomycin alone (Fig. 6). Vancomycin is a broad-spectrum antibiotic known to be effective against the USA300 strain of MRSA (80), the same strain used in our studies. It is used selectively prior to surgery (54), as it has been suggested to induce renal toxicity; however, this is not known to happen after single-dose administration (81). Vancomycin has also been shown to increase mast cell degranulation (82), which is associated with delayed-type hypersensitivity and renal toxicity (83). However, it is unlikely that this contributes to the increased inflammatory damage we observe in the presence of propofol, because studies have also shown that intravenous general anesthetics, including propofol, inhibit mast cell degranulation (84). It is possible that the combined administration of propofol and vancomycin increases the risk of kidney pathology following vancomycin exposure in infected animals. The mechanistic basis of the observed increase in pathology is not known but warrants further investigation to determine its possible relevance to patient care.
Our work has shown that brief periods of propofol anesthesia significantly increase susceptibility to bloodstream MRSA infections, with particularly striking effects on kidney pathology. Based on our working model speculating that the monocyte-to-macrophage transition (Fig. 4), as well as neutrophil maturation (Fig. 5), is disrupted in the presence of propofol, it is possible that propofol or its metabolites act as signaling molecules that disrupt differentiation programs of myeloid progenitor cells (Fig. 7). It is also possible that propofol does not so much orchestrate the alterations in immune cell signaling and differentiation as it takes advantage of redundant pathways. As there are master regulators of immune cell transcription (i.e., NF-κB and AP-1), propofol may somehow target these factors to induce profound changes in host responses to infection. After affecting differentiation of one cell type, for example, all subsequent immune signaling that depends on that cell type may be altered to the detriment of the host. Future studies could address whether propofol enacts large-scale transcriptional changes in innate immune effector cells during infection as a means of accounting for its significant impact on host susceptibility to microbial infection.
FIG 7.

Potential effects of propofol on host immunity against S. aureus infection. Upon infection with S. aureus, propofol treatment increases bacterial replication and abscess formation in the kidney. In the case of bloodstream S. aureus infection, while propofol does not affect the recruitment of blood monocytes into the kidney, it may prevent differentiation of monocytes to macrophages, allowing for greatly expanded inflammatory abscesses in the kidney. Propofol may also allow for the expansion of MDSCs, which can lead to reduced T cell activation.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
Intravenous S. aureus infections were performed using the methicillin-resistant USA300 LAC strain (courtesy of Victor Torres and Francis Alonzo). USA300 was grown with shaking overnight in tryptic soy agar (TSA) broth (MP Biomedicals, Solon, OH) at 37°C, 220 rpm.
Intravenous infections of mice.
Animal procedures were IACUC approved by the UIC Animal Care Committee and performed in the Biological Resources Laboratory at the University of Illinois at Chicago. S. aureus overnight cultures were diluted 1:100 and subsequently grown to mid-log phase (∼3 h) with shaking at 37°C, 220 rpm. Immediately prior to infection, 100 μl of bacterial suspension was mixed with 100 μl of vehicle solution (Intralipid [Sigma-Aldrich, St. Louis, MO] plus 5% dextrose or propofol [Abbot Laboratories, North Chicago, IL]), used at a concentration of 18.75 mg/kg of body weight. This dose is comparable with that used to induce anesthesia in humans based on FDA guidelines accounting for differences in metabolism (34), and it resulted in approximately 3 to 5 min of mouse sedation. No differences were observed with respect to bacterial growth or viability following suspension in either propofol or vehicle solution for at least 2 h at room temperature (data not shown). Adolescent female Swiss Webster mice (Charles River Laboratories, Chicago, IL) were infected with 3 × 106 CFU via tail vein injection. For experiments using vancomycin prophylaxis, mice were given 15 mg/kg vancomycin i.v. This was done 24 h prior to intravenous infection via the tail vein with S. aureus in the presence or absence of propofol. At the indicated times, organs were harvested and homogenized. Homogenates were plated on TSA plates to determine viable CFU.
Histological examination of infected tissues.
Mice infected with S. aureus with or without propofol were euthanized at the indicated times, and livers, spleens, and kidneys were isolated. Organs were suspended in 10% phosphate-buffered saline (PBS)-buffered formalin (Sigma-Aldrich, St. Louis, MO) overnight, prepared for histological analysis by the UIC Research Resource Center Histology Core, and stained with hematoxylin and eosin (H&E). Images were obtained using a Zeiss Axio Imager manual upright research microscope or an Aperio Technologies Scanscope. Quantification was performed using ImageScope software from Aperio Technologies (Vista, CA).
Immunofluorescent staining.
Paraffin-embedded organs were sectioned into 5-μm sections and mounted onto glass slides by the UIC Research Resource Center Histology Core. Samples were then deparaffinized with xylene and rehydrated with graded ethanol washes. Antigen retrieval was performed using 10 mM sodium citrate buffer in a pressure cooker at high pressure for 15 min. Slides were cooled and then incubated in a cold solution of 10% H2O2 to quench endogenous peroxidases and limit nonspecific red blood cell staining. Slides were then washed in Tris-buffered saline (TBS), and samples were marked with PAP pen circles. Background Buster blocking solution (Innovex Biosciences, Richmond, CA) was applied to each sample for 30 min prior to incubation in primary antibody. Samples were washed in TBS and then incubated with anti-human IgG to block nonspecific binding of anti-S. aureus antibody for 1 h at room temperature. Slides were then incubated sequentially with anti-S. aureus-fluorescein isothiocyanate and anti-Ly-6G for 1 h at room temperature or overnight at 4°C. Slides were washed in TBS and then incubated sequentially with goat anti-rat AF-594 (Abcam, Cambridge, MA) for 1 h at room temperature. Slides were once again washed in TBS and coverslips were mounted using ProLong gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Grand Island, NY). Slides were imaged at 25°C, with ×10 or ×20 magnification, using a Zeiss Axio Imager A2 manual upright research microscope and analyzed by Zen 2012 imaging software.
Flow cytometry.
Mice were infected i.v. via tail vein injection with S. aureus and euthanized at the indicated time points. Kidneys were isolated and processed for flow cytometry. Single-cell suspensions were obtained by first mechanically disrupting kidneys using a Stomacher 80 Biomaster (Seward International, West Sussex, United Kingdom) and then passing the homogenate through a 70-μm cell strainer. The following fluorescent and nonfluorescent primary antibodies were used: anti-Mac-3 (BD Pharmingen, San Diego, CA); anti-Ly-6G, CD-11b, NOS2, CD3 (EBioscience, San Diego, CA); anti-TNF-α (clone MP6-XT22; BioLegend, San Diego, CA); and anti-IFN-γ, IL-4, CD4, F4/80, CD11c, I-A/I-E (mouse major histocompatibility complex class II [MHC-II]) (Tonbo Biosciences, San Diego, CA). Fluorescence-activated cell sorting (FACS) was performed with a Cyan ADP flow cytometer (Beckman Coulter, Inc., Brea, CA), and data were analyzed with Summit software.
Statistical analysis.
All statistical analyses for in vivo infection assays were performed using Student's t test when data followed a normal distribution or a Wilcoxon or Mann-Whitney nonparametric test when data fell outside Gaussian distributions. X symbols denoting bacterial burdens below the limit of detection (i.e., 100 CFU) were input with a numerical value of 99 for the basis of all statistical analyses. All analyses for immunofluorescence quantitation were carried out using Student's t test. All statistical analyses for flow cytometry were performed using two-way analysis of variance with a Bonferroni posttest.
Supplementary Material
ACKNOWLEDGMENTS
We thank Francis Alonzo for helpful advice and members of the Freitag laboratory and the UIC Positive Thinking Group for helpful discussions.
This work was supported by NIH grant R03 AI097160 to N.E.F. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding source. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00097-17.
REFERENCES
- 1.Aaronson DS, Wu AK, Blaschko SD, McAninch JW, Garcia M. 2011. National incidence and impact of noninfectious urethral catheter related complications on the Surgical Care Improvement Project. J Urol 185:1756–1760. doi: 10.1016/j.juro.2010.12.041. [DOI] [PubMed] [Google Scholar]
- 2.Dudeck MA, Horan TC, Peterson KD, Allen-Bridson K, Morrell GC, Pollock DA, Edwards JR. 2011. National Healthcare Safety Network (NHSN) report, data summary for 2009, device-associated module. Am J Infect Control 39:349–367. doi: 10.1016/j.ajic.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Engels EA, Ellis CA, Supran SE, Schmid CH, Barza M, Schenkein DP, Koc Y, Miller KB, Wong JB. 1999. Early infection in bone marrow transplantation: quantitative study of clinical factors that affect risk. Clin Infect Dis 28:256–266. doi: 10.1086/515103. [DOI] [PubMed] [Google Scholar]
- 4.Giard M, Lepape A, Allaouchiche B, Guerin C, Lehot JJ, Robert MO, Fournier G, Jacques D, Chassard D, Gueugniaud PY, Artru F, Petit P, Robert D, Mohammedi I, Girard R, Cetre JC, Nicolle MC, Grando J, Fabry J, Vanhems P. 2008. Early- and late-onset ventilator-associated pneumonia acquired in the intensive care unit: comparison of risk factors. J Crit Care 23:27–33. doi: 10.1016/j.jcrc.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Holtz TH, Wenzel RP. 1992. Postdischarge surveillance for nosocomial wound infection: a brief review and commentary. Am J Infect Control 20:206–213. doi: 10.1016/S0196-6553(05)80148-8. [DOI] [PubMed] [Google Scholar]
- 6.Perencevich EN, Sands KE, Cosgrove SE, Guadagnoli E, Meara E, Platt R. 2003. Health and economic impact of surgical site infections diagnosed after hospital discharge. Emerg Infect Dis 9:196–203. doi: 10.3201/eid0902.020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pofahl WE, Ramsey KM, Nobles DL, Cochran MK, Goettler C. 2011. Importance of methicillin-resistant Staphylococcus aureus eradication in carriers to prevent postoperative methicillin-resistant Staphylococcus aureus surgical site infection. Am Surg 77:27–31. [PubMed] [Google Scholar]
- 8.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 9.Casey JA, Cosgrove SE, Stewart WF, Pollak J, Schwartz BS. 2012. A population-based study of the epidemiology and clinical features of methicillin-resistant Staphylococcus aureus infection in Pennsylvania, 2001-2010. Epidemiol Infect 141:1166–1179. doi: 10.1017/S0950268812001872:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matouskova I, Janout V. 2008. Current knowledge of methicillin-resistant Staphylococcus aureus and community-associated methicillin-resistant Staphylococcus aureus. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 152:191–202. doi: 10.5507/bp.2008.030. [DOI] [PubMed] [Google Scholar]
- 11.Lee WS, Chen YC, Chen HP, Chen TH, Cheng CY. 2013. Vertebral osteomyelitis caused by vancomycin-tolerant methicillin-resistant Staphylococcus aureus bacteremia: experience with teicoplanin plus fosfomycin combination therapy. J Microbiol Immunol Infect 49:600–603. doi: 10.1016/j.jmii.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Iacobelli S, Colomb B, Bonsante F, Astruc K, Ferdynus C, Bouthet MF, Neuwirth C, Aho Glele LS, Chavanet P, Gouyon JB. 2013. Successful control of a methicillin-resistant Staphylococcus aureus outbreak in a neonatal intensive care unit: a retrospective, before-after study. BMC Infect Dis 13:440. doi: 10.1186/1471-2334-13-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Tawfiq JA, Abed MS, Al-Yami N, Birrer RB. 2013. Promoting and sustaining a hospital-wide, multifaceted hand hygiene program resulted in significant reduction in health care-associated infections. Am J Infect Control 41:482–486. doi: 10.1016/j.ajic.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Harris AD, Pineles L, Belton B, Johnson JK, Shardell M, Loeb M, Newhouse R, Dembry L, Braun B, Perencevich EN, Hall KK, Morgan DJ, Shahryar SK, Price CS, Gadbaw JJ, Drees M, Kett DH, Munoz-Price LS, Jacob JT, Herwaldt LA, Sulis CA, Yokoe DS, Maragakis L, Lissauer ME, Zervos MJ, Warren DK, Carver RL, Anderson DJ, Calfee DP, Bowling JE, Safdar N. 2013. Universal glove and gown use and acquisition of antibiotic-resistant bacteria in the ICU: a randomized trial. JAMA 310:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, Lessa FC, Lynfield R, Nadle J, Petit S, Ray SM, Schaffner W, Townes J, Fridkin S. 2013. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med 173:1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlapfer M, Piegeler T, Dull RO, Schwartz DE, Mao M, Bonini MG, Z'Graggen BR, Beck-Schimmer B, Minshall RD. 2015. Propofol increases morbidity and mortality in a rat model of sepsis. Crit Care 19:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryson HM, Fulton BR, Faulds D. 1995. Propofol. An update of its use in anaesthesia and conscious sedation. Drugs 50:513–559. [DOI] [PubMed] [Google Scholar]
- 18.Heuss LT, Inauen W. 2004. The dawning of a new sedative: propofol in gastrointestinal endoscopy. Digestion 69:20–26. doi: 10.1159/000076543. [DOI] [PubMed] [Google Scholar]
- 19.Miner JR, Burton JH. 2007. Clinical practice advisory: emergency department procedural sedation with propofol. Ann Emerg Med 50:182–187. doi: 10.1016/j.annemergmed.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 20.Ostermann ME, Keenan SP, Seiferling RA, Sibbald WJ. 2000. Sedation in the intensive care unit: a systematic review. JAMA 283:1451–1459. doi: 10.1001/jama.283.11.1451. [DOI] [PubMed] [Google Scholar]
- 21.Lucendo AJ, Olveira A, Friginal-Ruiz AB, Guagnozzi D, Angueira T, Fernandez-Fuente M, Cruz-Campos M, Serrano-Valverde M, Sanchez-Cazalilla M, Tenias JM, Gonzalez-Castillo S. 2012. Nonanesthesiologist-administered propofol sedation for colonoscopy is safe and effective: a prospective Spanish study over 1000 consecutive exams. Eur J Gastroenterol Hepatol 24:787–792. doi: 10.1097/MEG.0b013e328353fcbc. [DOI] [PubMed] [Google Scholar]
- 22.Favetta P, Degoute CS, Perdrix JP, Dufresne C, Boulieu R, Guitton J. 2002. Propofol metabolites in man following propofol induction and maintenance. Br J Anaesth 88:653–658. doi: 10.1093/bja/88.5.653. [DOI] [PubMed] [Google Scholar]
- 23.Marik PE. 2005. Propofol: an immunomodulating agent. Pharmacotherapy 25:28S–33S. doi: 10.1592/phco.2005.25.5_Part_2.28S. [DOI] [PubMed] [Google Scholar]
- 24.Inada T, Kubo K, Ueshima H, Shingu K. 2011. Intravenous anesthetic propofol suppresses prostaglandin E2 production in murine dendritic cells. J Immunotoxicol 8:359–366. doi: 10.3109/1547691X.2011.620036. [DOI] [PubMed] [Google Scholar]
- 25.Lin MC, Chen CL, Yang TT, Choi PC, Hsing CH, Lin CF. 2012. Anesthetic propofol overdose causes endothelial cytotoxicity in vitro and endothelial barrier dysfunction in vivo. Toxicol Appl Pharmacol 265:253–262. doi: 10.1016/j.taap.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Shiratsuchi H, Kouatli Y, Yu GX, Marsh HM, Basson MD. 2009. Propofol inhibits pressure-stimulated macrophage phagocytosis via the GABAA receptor and dysregulation of p130cas phosphorylation. Am J Physiol Cell Physiol 296:C1400–C1410. doi: 10.1152/ajpcell.00345.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsing CH, Lin MC, Choi PC, Huang WC, Kai JI, Tsai CC, Cheng YL, Hsieh CY, Wang CY, Chang YP, Chen YH, Chen CL, Lin CF. 2011. Anesthetic propofol reduces endotoxic inflammation by inhibiting reactive oxygen species-regulated Akt/IKKbeta/NF-kappaB signaling. PLoS One 6:e17598. doi: 10.1371/journal.pone.0017598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Visvabharathy L, Xayarath B, Weinberg G, Shilling RA, Freitag NE. 2015. Propofol increases host susceptibility to microbial infection by reducing subpopulations of mature immune effector cells at sites of infection. PLoS One 10:e0138043. doi: 10.1371/journal.pone.0138043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen RM, Wu CH, Chang HC, Wu GJ, Lin YL, Sheu JR, Chen TL. 2003. Propofol suppresses macrophage functions and modulates mitochondrial membrane potential and cellular adenosine triphosphate synthesis. Anesthesiology 98:1178–1185. doi: 10.1097/00000542-200305000-00021. [DOI] [PubMed] [Google Scholar]
- 30.Chiu WT, Lin YL, Chou CW, Chen RM. 2009. Propofol inhibits lipoteichoic acid-induced iNOS gene expression in macrophages possibly through downregulation of toll-like receptor 2-mediated activation of Raf-MEK1/2-ERK1/2-IKK-NFkappaB. Chem Biol Interact 181:430–439. doi: 10.1016/j.cbi.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Schlapfer M, Piegeler T, Dull RO, Schwartz DE, Mao M, Bonini MG, Z'Graggen BR, Beck-Schimmer B, Minshall RD. 2015. Propofol increases morbidity and mortality in a rat model of sepsis. Crit Care 19:45. doi: 10.1186/s13054-015-0751-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen RM, Wu GJ, Tai YT, Sun WZ, Lin YL, Jean WC, Chen TL. 2003. Propofol reduces nitric oxide biosynthesis in lipopolysaccharide-activated macrophages by downregulating the expression of inducible nitric oxide synthase. Arch Toxicol 77:418–423. doi: 10.1007/s00204-003-0453-z. [DOI] [PubMed] [Google Scholar]
- 33.Heller A, Heller S, Blecken S, Urbaschek R, Koch T. 1998. Effects of intravenous anesthetics on bacterial elimination in human blood in vitro. Acta Anaesthesiol Scand 42:518–526. doi: 10.1111/j.1399-6576.1998.tb05160.x. [DOI] [PubMed] [Google Scholar]
- 34.FDA. 2005. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Office of Training and Communications, Division of Drug Information, HFD-240, Center for Drug Evaluation and Research Food and Drug Administration, Rockville, MD. [Google Scholar]
- 35.Zarjou A, Agarwal A. 2011. Sepsis and acute kidney injury. J Am Soc Nephrol 22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 36.Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 23:3393–3404. doi: 10.1096/fj.09-135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tzianabos AO, Kasper DL, Cisneros RL, Smith RS, Onderdonk AB. 1995. Polysaccharide-mediated protection against abscess formation in experimental intra-abdominal sepsis. J Clin Investig 96:2727–2731. doi: 10.1172/JCI118340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorbach SL. 1995. Good and laudable pus. J Clin Investig 96:2545. doi: 10.1172/JCI118316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, Germain RN. 2013. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498:371–375. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorczyca W, Sun ZY, Cronin W, Li X, Mau S, Tugulea S. 2011. Immunophenotypic pattern of myeloid populations by flow cytometry analysis. Methods Cell Biol 103:221–266. doi: 10.1016/B978-0-12-385493-3.00010-3. [DOI] [PubMed] [Google Scholar]
- 41.Patarroyo M. 1994. Adhesion molecules mediating recruitment of monocytes to inflamed tissue. Immunobiology 191:474–477. doi: 10.1016/S0171-2985(11)80453-5. [DOI] [PubMed] [Google Scholar]
- 42.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. 2004. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol 172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 43.Ziegler C, Goldmann O, Hobeika E, Geffers R, Peters G, Medina E. 2011. The dynamics of T cells during persistent Staphylococcus aureus infection: from antigen-reactivity to in vivo anergy. EMBO Mol Med 3:652–666. doi: 10.1002/emmm.201100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hurst JK. 2012. What really happens in the neutrophil phagosome? Free Radic Biol Med 53:508–520. doi: 10.1016/j.freeradbiomed.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rigby KM, DeLeo FR. 2012. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol 34:237–259. doi: 10.1007/s00281-011-0295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng AG, DeDent AC, Schneewind O, Missiakas D. 2011. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol 19:225–232. doi: 10.1016/j.tim.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ribes S, Regen T, Meister T, Tauber SC, Schutze S, Mildner A, Mack M, Hanisch UK, Nau R. 2013. Resistance of the brain to Escherichia coli K1 infection depends on MyD88 signaling and the contribution of neutrophils and monocytes. Infect Immun 81:1810–1819. doi: 10.1128/IAI.01349-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabrilovich DI, Nagaraj S. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. 2008. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 50.Heller A, McIff TE, Lai SM, Burton DC. 2013. Intrawound vancomycin powder decreases staphylococcal surgical site infections following posterior instrumented spinal arthrodesis. J Spinal Disord Tech 28:E584–E589. doi: 10.1097/BSD.0000000000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yavuz SS, Tarcin O, Ada S, Dincer F, Toraman S, Birbudak S, Eren E, Yekeler I. 2013. Incidence, aetiology, and control of sternal surgical site infections. J Hosp Infect 85:206–212. doi: 10.1016/j.jhin.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 52.Lusardi G, Lipp A, Shaw C. 2013. Antibiotic prophylaxis for short-term catheter bladder drainage in adults. Cochrane Database Syst Rev 7:CD005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oostdijk EA, Smits L, de Smet AM, Leverstein-van Hall MA, Kesecioglu J, Bonten MJ. 2013. Colistin resistance in gram-negative bacteria during prophylactic topical colistin use in intensive care units. Intensive Care Med 39:653–660. doi: 10.1007/s00134-012-2761-3. [DOI] [PubMed] [Google Scholar]
- 54.Crawford T, Rodvold KA, Solomkin JS. 2012. Vancomycin for surgical prophylaxis? Clin Infect Dis 54:1474–1479. doi: 10.1093/cid/cis027. [DOI] [PubMed] [Google Scholar]
- 55.Bratzler DW, Dellinger EP, Olsen KM, Perl TM, Auwaerter PG, Bolon MK, Fish DN, Napolitano LM, Sawyer RG, Slain D, Steinberg JP, Weinstein RA. 2013. Clinical practice guidelines for antimicrobial prophylaxis in surgery. Surg Infect (Larchmt) 14:73–156. doi: 10.1089/sur.2013.9999. [DOI] [PubMed] [Google Scholar]
- 56.Bleeker C, Vree T, Lagerwerf A, Willems-van Bree E. 2008. Recovery and long-term renal excretion of propofol, its glucuronide, and two di-isopropylquinol glucuronides after propofol infusion during surgery. Br J Anaesth 101:207–212. doi: 10.1093/bja/aen134. [DOI] [PubMed] [Google Scholar]
- 57.Kanto J, Gepts E. 1989. Pharmacokinetic implications for the clinical use of propofol. Clin Pharmacokinet 17:308–326. doi: 10.2165/00003088-198917050-00002. [DOI] [PubMed] [Google Scholar]
- 58.Al-Jahdari WS, Yamamoto K, Hiraoka H, Nakamura K, Goto F, Horiuchi R. 2006. Prediction of total propofol clearance based on enzyme activities in microsomes from human kidney and liver. Eur J Clin Pharmacol 62:527–533. doi: 10.1007/s00228-006-0130-2. [DOI] [PubMed] [Google Scholar]
- 59.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, Pless R, Gelman AE, Krupnick AS, Miller MJ. 2010. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A 107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Serbina NV, Shi C, Pamer EG. 2012. Monocyte-mediated immune defense against murine Listeria monocytogenes infection. Adv Immunol 113:119–134. doi: 10.1016/B978-0-12-394590-7.00003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosen H, Gordon S, North RJ. 1989. Exacerbation of murine listeriosis by a monoclonal antibody specific for the type 3 complement receptor of myelomonocytic cells. Absence of monocytes at infective foci allows Listeria to multiply in nonphagocytic cells. J Exp Med 170:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steigbigel RT, Lambert LH Jr, Remington JS. 1974. Phagocytic and bacterial properties of normal human monocytes. J Clin Investig 53:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gordon S. 2007. The macrophage: past, present and future. Eur J Immunol 37(Suppl 1):S9–S17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- 64.Yu G, Dymond M, Yuan L, Chaturvedi LS, Shiratsuchi H, Durairaj S, Marsh HM, Basson MD. 2011. Propofol's effects on phagocytosis, proliferation, nitrate production, and cytokine secretion in pressure-stimulated microglial cells. Surgery 150:887–896. doi: 10.1016/j.surg.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. 2010. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog 6:e1001036. doi: 10.1371/journal.ppat.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wallenfang T, Bohl J, Kretzschmar K. 1980. Evolution of brain abscess in cats formation of capsule and resolution of brain edema. Neurosurg Rev 3:101–111. [DOI] [PubMed] [Google Scholar]
- 68.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. 2008. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goni O, Alcaide P, Fresno M. 2002. Immunosuppression during acute Trypanosoma cruzi infection: involvement of Ly6G (Gr1+)CD11b+ immature myeloid suppressor cells. Int Immunol 14:1125–1134. doi: 10.1093/intimm/dxf076. [DOI] [PubMed] [Google Scholar]
- 70.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. 2001. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol 166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 71.Nagaraj S, Youn JI, Gabrilovich DI. 2013. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol 191:17–23. doi: 10.4049/jimmunol.1300654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tebartz C, Horst SA, Sparwasser T, Huehn J, Beineke A, Peters G, Medina E. 2015. A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J Immunol 194:1100–1111. doi: 10.4049/jimmunol.1400196. [DOI] [PubMed] [Google Scholar]
- 73.Skabytska Y, Wolbing F, Gunther C, Koberle M, Kaesler S, Chen KM, Guenova E, Demircioglu D, Kempf WE, Volz T, Rammensee HG, Schaller M, Rocken M, Gotz F, Biedermann T. 2014. Cutaneous innate immune sensing of Toll-like receptor 2-6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity 41:762–775. doi: 10.1016/j.immuni.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 74.Bonten MJ. 2011. Healthcare epidemiology: ventilator-associated pneumonia: preventing the inevitable. Clin Infect Dis 52:115–121. doi: 10.1093/cid/ciq075. [DOI] [PubMed] [Google Scholar]
- 75.Lipp A, Lusardi G. 2013. Systemic antimicrobial prophylaxis for percutaneous endoscopic gastrostomy. Cochrane Database Syst Rev 11:CD005571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwulst SJ, Mazuski JE. 2012. Surgical prophylaxis and other complication avoidance care bundles. Surg Clin North Am 92:285–305. doi: 10.1016/j.suc.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 77.Suzuki M, Inage E, Minowa K, Saito N, Naritaka N, Tsubahara M, Ohtsuka Y, Tokita A, Shimizu T. 2012. Prophylaxis for ribavirin-related anemia using eicosapentaenoic acid in chronic hepatitis C patients. Pediatr Int 54:528–531. doi: 10.1111/j.1442-200X.2012.03603.x. [DOI] [PubMed] [Google Scholar]
- 78.Platt R. 1998. Antibiotic prophylaxis in clean surgery: does it work? Should it be used if it does? New Horiz 6:S53–S57. [PubMed] [Google Scholar]
- 79.Liberati A, D'Amico R, Pifferi S, Telaro E. 2000. Antibiotic prophylaxis in intensive care units: meta-analyses versus clinical practice. Intensive Care Med 26(Suppl 1):S38–S44. [DOI] [PubMed] [Google Scholar]
- 80.Gardete S, Kim C, Hartmann BM, Mwangi M, Roux CM, Dunman PM, Chambers HF, Tomasz A. 2012. Genetic pathway in acquisition and loss of vancomycin resistance in a methicillin resistant Staphylococcus aureus (MRSA) strain of clonal type USA300. PLoS Pathog 8:e1002505. doi: 10.1371/journal.ppat.1002505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elyasi S, Khalili H, Dashti-Khavidaki S, Mohammadpour A. 2012. Vancomycin-induced nephrotoxicity: mechanism, incidence, risk factors and special populations. A literature review. Eur J Clin Pharmacol 68:1243–1255. doi: 10.1007/s00228-012-1259-9. [DOI] [PubMed] [Google Scholar]
- 82.Horinouchi Y, Abe K, Kubo K, Oka M. 1993. Mechanisms of vancomycin-induced histamine release from rat peritoneal mast cells. Agents Actions 40:28–36. [DOI] [PubMed] [Google Scholar]
- 83.Marik PE, Ferris N. 1997. Delayed hypersensitivity reaction to vancomycin. Pharmacotherapy 17:1341–1344. [PubMed] [Google Scholar]
- 84.Fujimoto T, Nishiyama T, Hanaoka K. 2005. Inhibitory effects of intravenous anesthetics on mast cell function. Anesth Analg 101:1054–1059. doi: 10.1213/01.ane.0000166955.97368.80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.