

Summary
The intricate connection between the circadian clock and metabolism remains poorly understood. We used high temporal resolution metabolite profiling to explore clock regulation of mouse liver and cell autonomous metabolism. In liver, ~50% of metabolites were circadian, with enrichment of nucleotide, amino acid, and methylation pathways. In U2 OS cells, 28% were circadian, including amino acids and NAD biosynthesis metabolites. Eighteen metabolites oscillated in both systems and a subset of these in primary hepatocytes. These 18 metabolites were enriched in methylation and amino acid pathways. To assess clock-dependence of these rhythms, we used genetic perturbation. BMAL1 knockdown diminished metabolite rhythms, while CRY1 or CRY2 perturbation generally shortened/lengthened rhythms, respectively. Surprisingly, CRY1 knockdown induced 8 h rhythms in amino acid, methylation, and vitamin metabolites, decoupling metabolite from transcriptional rhythms, with potential impact on nutrient sensing in vivo. These results provide the first comprehensive views of circadian liver and cell autonomous metabolism.
Blurb
Using high temporal resolution metabolite profiling, XXX et al show that over 50% of liver metabolites are circadian, with a significant overlap of cycling metabolites between mouse and human liver, especially those involved in epigenetic regulation. Coupling of metabolite with transcriptional rhythms is regulated by core clock genes.
Introduction
Circadian rhythms in physiology and behavior are critical to most organisms. In mammals, disruption of circadian rhythms leads to many pathophysiological conditions such as cognitive dysfunction, psychiatric disorders, cancer, obesity, insulin resistance, metabolic syndrome, and inflammation (Albrecht, 2013; Altman et al., 2015; Antunes et al., 2010; Bechtold et al., 2010; Kawachi et al., 1995; Parkes, 2002; Reppert and Weaver, 2002; Sharifian A, 2005). Recent work shows the circadian clock is intimately connected to metabolism and sheds light on metabolic pathways that are potentially under circadian control (Asher and Schibler, 2011; Bass, 2012; Eckel-Mahan and Sassone-Corsi, 2013; Green et al., 2008; Papagiannakopoulos, 2016; Zhang et al., 2014; Zwighaft et al., 2015)
Cell autonomous circadian rhythms are generated through a transcriptional regulatory network of clock genes. In the core feedback loop, the transcriptional activators BMAL1 and CLOCK regulate the Cryptochrome (Cry1 and Cry2) and Period (Per1 and Per2) transcriptional repressors. After translation and nuclear localization, the PER and CRY proteins inhibit BMAL1/CLOCK function, leading to rhythmic genome-wide gene expression (including their own). In many physiological pathways, rate-limiting steps are under circadian clock control. Genetic mutation or environmental perturbation (e.g. shift work or jet lag) disrupt clock function and can cause sleep disorders, cancer, cardiovascular, and metabolic diseases (Asher and Schibler, 2011; Bass and Takahashi, 2010; Green et al., 2008).
Over the last several years, a number of studies investigated oscillatory metabolism using a range of experimental designs. These varied from sampling every 4 or 6 hours for 24 h under light / dark (LD) conditions (most common) to every 2 h for 24 h in LD to every 4 h for 40 h in DD (See Table S1). While important for a basic understanding of oscillatory metabolism, these studies weren’t adequately designed to detect circadian oscillations for several reasons. First, the vast majority were done under driven (LD) conditions, not constant (LL or DD), conditions. Second, most of these studies were under-powered to comprehensively detect rhythms. In particular, sampling every 4 or 6 h for a 24 h day (even with replicates) limits the ability to detect truly rhythmic metabolites. These designs are also subject to false positive errors, as any monotonic noise can be interpreted as rhythmic. Further, in reporting these rhythms, most studies failed to account for multiple testing (false discoveries). While these studies provide valuable first insights, they were not designed to comprehensively detect circadian rhythms in metabolism.
We sought to characterize circadian regulation of metabolism to follow up our high temporal resolution transcriptomics studies (Zhang et al., 2014). Because we did not know the technical limitations imposed by the metabolomics platform and their impact on detecting rhythmicity (Hughes, 2007), we sampled mouse liver, in replicate, every 1 h for 48 h in DD. Further, as metabolite oscillations in a cell autonomous model have not been previously investigated, we sampled U2 OS cells every 2 h for 48 h under control conditions, and also after knockdown of the core clock genes BMAL1, CRY1, and CRY2. Surprisingly, in mouse liver, we found a higher percentage (>50%) of detected metabolites are clock-regulated than detected transcripts (~37%). This observation is much more striking in the U2 OS cell autonomous clock function, with 28% of metabolites detected as cycling compared with just 1.5% of transcripts using the same mathematical approaches. This suggests an amplification of the circadian drive between transcription, translation, and function. As predicted, genetic perturbations showed the vast majority of this cycling depends on an intact clock. Strikingly, knockdown of CRY1, short period in cells and mice, resulted in the appearance of both 8 h and 32 h rhythmic metabolites, decoupling the transcriptional and metabolic oscillations. Collectively, these results have important implications for experimental design as well as circadian regulation of metabolism in vitro and in vivo.
Results and Discussion
Clock regulation of liver metabolites
Early studies established the strong link between the circadian oscillator and metabolism (Panda et al., 2002; Storch et al., 2002; Ueda et al., 2002). These global analyses showed that many or even most liver metabolic pathways harbored clock-controlled steps, often key and rate limiting. Later, investigation of core clock genes confirmed this linkage, as mutation of Clock or Bmal1 led to profound metabolic abnormalities in mice (Rudic et al., 2004; Turek et al., 2005). More recently, several papers have elucidated mechanistic links between clock function and specific clock-regulated metabolism (Asher and Schibler, 2011; Bass, 2012; Gerhart-Hines and Lazar, 2015; Peek et al., 2013; Zwighaft et al., 2015) . These papers and the advent of high-throughput metabolite profiling platforms prompted a number of studies whose broad aim was to describe rhythmic metabolism (Table S1).
However, these experimental designs were not ideal to comprehensively investigate metabolite circadian rhythms. To address these limitations, we sought to understand the impact of time-sampling intervals, duration, and replicates on detection of circadian metabolites. We analyzed liver samples from mice maintained under constant darkness with 1 h time sampling over 48 hours in duplicate (Figure S1A). Extensive metabolite rhythms were observed, with ~50% of measured metabolites (90 of 189) deemed rhythmic by JTK_Cycle algorithm analysis (FDR controlled q-value < 0.05) (Figure 1A, S1C). This rhythmicity spanned many areas of metabolism, most notably nucleotide, energy, oxidative, and carbohydrate metabolism (Figure 1B). Interestingly, this number is similar to the percentage of rhythmic genes in transcription using a similar experimental design. However, the amplitudes of metabolite rhythms were lower than those found in transcription. Interestingly, like transcription, cycling metabolite peak phases were multi-modal and associated with subjective dawn. Unlike transcription, the high-resolution analyses here reveals that metabolite cycling has multiple additional daytime peaks near CT 3 and CT 7, and a nighttime peak at CT 17 (Figure 1C).
Figure 1. Mouse liver circadian metabolites uncovered using high temporal resolution profiling.

(A) Heat map demonstrating robust circadian metabolite cycling for 90/179 (~50%) metabolites measured in two replicates collected every hour for 48 hours with a JTK BH Q < 0.05. Compounds with low confidence identification are indicated by a numeric m/z transition. (B) Metabolite enrichment analysis of oscillating metabolites indicates largest pathway impact on arginine and proline metabolism; cysteine and methionine metabolism; glycine, serine and threonine metabolism and pyrimidine metabolism. Y-axis depicts significance of the over-representation compared to a random reference of known metabolites. (C) Distribution of circadian phases from metabolite cycling indicates discrete peaks anticipating dawn and through the circadian day. (D) Distribution of periodicity of cycling metabolites, with the majority of metabolites having circadian periods, a significant set with ~12 h periods and 3 metabolites with ~8 h periods. (E) “Leave some out” analysis of resolution required to adequately sample circadian metabolite data. Lines are labeled with time between samples (temporal resolution), and the most common reported experimental designs in literature noted with a red box. See also Figure S1, Table S1 and Dataset S1.
We further sought to determine the optimal experimental design for metabolite studies using “leave some out” analyses. In our transcriptional analyses, the selected design was every 2 h for 2 days, which captured 70% of the true rhythmic transcripts with a low false discovery rate at half the experimental burden. In contrast, and likely a reflection of the signal/noise of metabolite profiling, higher temporal resolution was even more important. The most common designs for these studies, every 4 or 6 h over 1 day, had high false negative rates (56/90, ~62% with 4 replicates), particularly when controlling for multiple testing (Figure 1E). As with the transcript data, single replicate 2-hour sampling over 2 days maximized true positive detection (49/90, ~54%) at the cost of a few false positives (4/90, ~4%) with a minimum number of samples required (24). As such we suggest that 1 h resolution is required when attempting to provide a ‘gold standard’ to detect cycling, but can be balanced with lower resolution analysis to overcome the experimental burden implied by such a design, so long as the error rates are acceptable for the question at hand.
Interpretation of acrophase modulation of metabolite rhythms gives rise to a number of biologically relevant ‘metabolic waves’ occurring through the circadian day. For example, diurnal glycogen content is known to peak between CT 0-2 in mice (Doi R, 2010; Ishikawa K, 1980; Roesler W.J, 1985). Our data suggests a wave by which conversion of glycogen to glucose and subsequent entry into the TCA cycle follows rapidly with glucose and citrate having peak phases of CT 2.5 h and CT 1 h respectively (Figure S1D). Subsequently, the other TCA cycle intermediate showing significant cycling, malate, has an acrophase at CT 5.5 h. Similarly, the metabolite cycling is reflective of changes in the oxidative status of the cell with shifts in oxidative metabolite peaks. For example, Figure S1E demonstrates opposing acrophases in reduced glutathione and ascorbate (CT 23.5 and CT 1.5) compared with FAD (CT 11.5). FAD is notable as one of only two metabolites with acrophase near dusk, the other being phenyllactate. UDP-glucose and the oxidized counterpart UDP-glucuronate also demonstrate opposing acrophases of CT 6 and CT 18.5 hours respectively. While some of these compounds have been noted to have circadian oscillations in liver, this is the first comprehensive atlas of temporal metabolic changes in liver tissue.
The 1 h resolution time sampling of liver metabolites allowed us to interrogate these data for other periodicities. Using JTK, we searched for metabolites cycling in a window between 4 and 40 h. We found ~15 metabolites cycling at the 12 h frequency, a 2nd harmonic of the circadian frequency. These metabolites were enriched for members of aromatic amino acid and nucleotide metabolism. Intriguingly, we also found 3 metabolites cycling at the 8 h frequency, a 3rd harmonic of circadian periodicity, 2-hydroxyglutarate, aconitate, and NADH, all involved in energy metabolism. These results are strikingly similar to earlier findings of transcription and protein regulation(Ramsey, 2009). Earlier, we described hundreds of transcripts expressed at the 12 h periodicity and dozens at the 8hr in liver (Hughes et al., 2009) . These harmonics depended on an intact clock (Hughes et al., 2012), as the 12 h rhythmic transcripts became 24 h when clock function was selectively restored to the brain. Gaschon and colleagues described 12 h rhythms in proteins levels for components of the unfolded protein response (Ern1/Ire1) and the endoplasmic reticulum, with similar dependence on an intact clock. Therefore, like transcription and translation, harmonics of the circadian frequency appear a hallmark of metabolite cycling (Cretenet et al., 2010).
Pathway analyses of these data shows an interconnected network consisting of methylation, one-carbon, creatine, and nitrogen metabolism (Figure 2). The majority of these metabolites cycle with acrophase near CT 18 (yellow color in figure) with the notable exceptions of 5’-methylthioadenosine (MTA), S-adenosylmethionine (SAM), and creatine that have acrophases near CT 4 (blue color). SAM and MTA are directly connected via 5-adenosylmethionamine (not measured in this study) and are precursors in methylation to S-adenosylhomocysteine (SAH). In a previous report, this pathway oscillates after high-fat diet challenge (Eckel-Mahan et al., 2013), which could be an amplification of the endogenous rhythm detected in our study. Methylation has been shown to be a key circadian regulatory process through chromatin transitions, for example trimethylation of Lys4 in histone H3 (Ripperger and Schibler, 2006).
Figure 2.

Network of pathways consisting of methylation, nitrogen and creatine metabolism. Metabolites with significant circadian oscillations are colored as a gradient with blue indicating peaking near CT 6 and yellow near CT 18. Outside panels show individual normalized mass spectrometry measurements at each time point (mean ± SEM), with a loess fit demonstrating the oscillatory metabolites. Data for all time-series plots was derived from two biological replicates and two analytical replicates. Error bars represent standard error of the mean.
Cell autonomous metabolite cycling
A distinguishing feature of circadian clocks is their ability to cycle independently of external cues (zeitgeibers) such as light, nutrients, hormones, or activity. The U2 OS osteosarcoma cell line has a robust circadian clock and has been used extensively in clock research (Baggs et al., 2009; Brown et al., 2008; Zhang et al., 2009) However, despite robust rhythms in luminescence assays (reflecting clock function), only several hundred genes cycle at the mRNA level (1.5% of measured genes, JTK q-value < 0.2, Figure 1A)(Hughes et al., 2009) compared to 500 to several thousands in almost every tissue in vivo (Zhang et al., 2014). Using 2-hour sampling over 48 hours (Figure S1B), we found 28% of measurable metabolites (39/137, Figure 3A; Figure S1C) demonstrate circadian rhythmicity (JTK q-value < 0.2). This observation was robust to the q-value used. In general, amplitudes were lower for cell autonomous rhythms, however the phases were surprisingly coherent with peaks near CT 6 and 10 (Figure 3B). These observations are consistent with the fact that cell autonomous rhythms, where cells are separated from rhythmic temperature, hormones, diet, and activity, are weaker than rhythms in vivo where these signals are integrated and coherent. The remarkable discrepancy between the percentage of transcripts and metabolites cycling suggests the powerful effect that a limited number of genes has on overall cellular metabolic output, suggesting an amplification of clock-control between transcription, translation, and function. While deeper metabolite coverage would be helpful to understand these relationships, it is clear that irrespective of the depth of coverage, there are a limited number of transcripts which give rise to significant metabolic rhythms. We note that metabolite levels are a function of multiple inputs via metabolite flux through a number of enzymatic pathways, both catabolic and anabolic. As such the relative contribution of phases in enzyme and metabolite levels as well as post-translational modifications remains to be determined.
Figure 3. Cell autonomous U2OS cells undergo large-scale rhythms in metabolism.

(A) 27% of measured metabolites (left panel) and 1.5% of measured genes (right panel, from Hughes et al, 2009) show circadian oscillations in the U2 OS cell system (assessed as periods between 20-28 hours, JTK BH.Q<0.2). Cells were sampled every 2 hours over two days starting 48 hours post-synchronization with dexamethasone. (B) Distribution of circadian phases from metabolite cycling indicates discrete peaks at CT 6 and CT 10. (C) Heat map showing wide range of cycling metabolites. Metabolites were ordered according to periodicity. (D, E) Cycling metabolites over the range of observed periods with a peak just less than 24 h, with a minor peak at 12 and 30 h. The lowest BH.Q values from JTK analysis corresponded to 22 and 24 hours. (F) Pathway enrichment analysis using MSEA highlights predominately one carbon, branched chain, glycerophospholipid and nicotinamide metabolism. See also Figure S1, Table S1 and Dataset S1.
Analysis of the metabolites themselves demonstrates pan-metabolome cycling (Figures 3C, F), with amino acids, energy and lipid metabolism, and enzyme cofactors being highly represented. Analysis of the range of periods demonstrates that the metabolites are cycling with a major peak slightly less than 24 hours (Figure 3D), with minor peaks at 12 and 30 hours. The amplitude of the metabolites at the extreme periods (i.e. 12 and 30 hours) was notably smaller than those with periods in the range of 20–26 hours (Figure 3E). The metabolites cycling with the lowest JTK q-values were also found in the 20–26 hour range. Overall, this suggests that in the presence of an intact transcriptional circadian clock, metabolism also cycles with a period near 24 hours in the absence of any external zeitgeibers with remarkable coherence in phase.
Cell autonomous metabolite rhythms depend on the circadian clock but can be frequency decoupled
The genetic tractability of the U2 OS system enabled us to investigate the relationship between the core clock and circadian metabolism. To see if metabolite rhythms required an intact oscillator and transcription, we knocked down BMAL1 and performed metabolite profiling every 2 h for 2 days (Figures S2A). Like behavioral rhythms in mice, knockdown of BMAL1 leads to progressive loss of amplitude in reporter gene activity in U2 OS cells (Baggs et al., 2009). Similar to these observations, ~70% knockdown of BMAL1 resulted in loss of rhythmicity in 36/39 metabolites, all but ATP, ADP, and nicotinamide ribotide (Figure 4A, Figure S2B). Interestingly, the glycolytic metabolites lactate and bisphosphoglycerate metabolites cycled in response to BMAL1 knockdown despite not cycling under control conditions. Together, these results suggest that cell metabolite rhythms largely depend on an intact transcriptional oscillator, but some independent metabolic oscillators may also exist.
Figure 4. Perturbation to core clock components BMAL1, CRY1 and CRY2 in U2OS cells alters metabolic cycling.

(A) BMAL1 knock down ablates cycling of most metabolites, with only 8/137 metabolites oscillating (JTK BH.Q < 0.2). Three metabolites (ATP, ADP, Nicotinamide ribotide) also oscillate with addition of the scrambled siRNA control, and the remaining 5 oscillate de novo. (B) Knockdown of CRY1 and CRY2 induce asymmetric shifts on the overall period of cycling metabolites. CRY1 knockdown creates a bimodal distribution of periods centered at ~8 and ~30 hours. CRY2 knockdown induces a shift in periods over a range of 20-40 hours with a peak near 34 hours. (C) Metabolites with well-defined 8 h oscillations in CRY1 knockdown cells (JTK BH.Q < 0.01), are primarily amino acids, including methylated species. The y-axis is the intensity following run-order correction as described in the methods. Data for time-series plots was derived from two analytical replicates, with error bars representing standard error of the mean. (D) Metabolite pathway mapping and trajectory created using Impala represents the impact knockdown of CRY1 and CRY2 have on oxidative stress and energy related pathways. Depletion of either CRY species increases the number of metabolites perturbed and extent of pathway coverage. (E) Effect of CRY1 and CRY2 knockdown on the 18 clock controlled metabolites identified as overlapping between U2 OS metabolism and liver metabolism. Top row of numbers in gray boxes are the observed period lengths with lowest BH.Q values from JTK_CYCLE analysis and the intensity is proportional to the amplitude (AMP). These metabolites follow overall trends in period shifting observed across all metabolites, however rhythmicity in methylation metabolites is lost following CRY1 knockdown (SAH) and in both CRY1 or CRY2 KD (5-methylthioadenosine). See also Figure S3.
In contrast to BMAL1, which is required for rhythms, CRY1 and CRY2 regulate the periodicity, with Cry1 knockdown inducing a short period (~22.5 h) and Cry2 a longer period (~24.6 h) in behavioral rhythms in mice (Okamura et al., 1999) and in transcriptional rhythms in U2 OS cells (Baggs et al., 2009) We hypothesized that the period of metabolite rhythms would decrease for CRY1 KD and increase for CRY2 KD. As expected, knockdown of CRY2 resulted in long period rhythms in metabolites The frequency of these oscillations were dispersed with a mean period of ~30 h and also increased the number of observed cycling metabolites compared to control (64/137, ~47%, Figures 3B, S2A, S2B and Dataset S1). This longer ~30 h period was in fact similar to that observed in transcriptional rhythms in U2 OS cells, but unique from behavioral rhythms in mice.
Unexpectedly, knockdown of CRY1 resulted in an increase in the number of cycling metabolites (69/137, ~50%; Figure S3 and Dataset S1) with a bi-modal distribution in periodicity. Two peaks were observed centered at approximately 8 and 32 hours. These period shifts are far greater than that observed in the transcriptional clock or behavioral rhythms and frequency decouple transcription from metabolite cycling following CRY1 perturbation. Interestingly, while there were a few 8 h harmonics under control conditions and no 12 h harmonics, CRY1 perturbation led to an increase in metabolites with an 8 h period (JTK q-value < 0.01 and Period ≤8 h, Figure 4C). These ultradian cycling metabolites included an abundance of amino acids (arginine, methionine, tyrosine, valine, tryptophan, leucine, and isoleucine), methylation products (dimethylarginine, dimethylglycine), and vitamin B1 (thiamine). Cycling in oxidative status of the cells was also more pronounced in the CRY1 and CRY2 knockdown cells compared to BMAL1 knockdown, as measured by the ion count ratio of NAD+/NADH (Figure S2C).
Pathway analysis of the control and CRY1/CRY2 KD cycling metabolites (Figure 4D) demonstrates that overall both Cry perturbations impact similar metabolic pathways compared to control, but exacerbate the cycling effect notably in pathways such as central carbon metabolism, glucose homeostasis, and glutathione metabolism. This result highlights the connection between the clock and energy and oxidative stress metabolism.
Metabolic consequences of Cry1 loss implicate effects on nutrient sensing
In order to further probe the decoupling of metabolite and transcript rhythms, we examined the U2 OS Cry1 KD metabolite data by first comparing the Cry1 KD with scrambled control at all timepoints. 55 compounds demonstrated significantly different concentrations, with eleven metabolites reduced under Cry1 KD and the remaining elevated (Figures 5A, S4A). We subsequently cross-referenced these differentially regulated compounds with those found to be significant cyclers (Figure 5B). Amongst this group, a number of amino acids were noted as elevated, including Arg, Ser, Asp, Val, Leu/Ile, Trp, Tyr, His, Thr, as well as amino acid derivatives such as N6-aceytllysine and dimethylarginine. This pattern matches an overrepresentation of amino acids amongst 8 h cyclers under Cry1 KD. There are 34 cycling metabolites with q< 0.05, of which 12 are amino acids, all of which have 8 h period.
Figure 5. Metabolic impact of CRY1 loss.

A) Global metabolic changes in U2 OS cells under CRY1 siRNA KD conditions. Samples from all timepoints were compared with control cells scrambled control siRNA to establish global metabolic defects. B) Volcano plot of differential changes in U2 OS cells under CRY1 KD. The annotated compounds correspond to those with significant cycling. C) Targeted analysis of amino acids and UMP involved in nutrient sensing via mToR signaling from male Cry1−/− animals and background controls (N=5–6 / group). Error bars represent standard error of the mean. See also Figure S4.
The over-representation of amino acid metabolism in both the cycling and differential concentration analysis led us to reason that Cry1 perturbation and the observed related transcriptional / metabolic uncoupling would have an impact in vivo on nutrient sensing. As the two most significant hits from JTK in the U2 OS experiment were dimethylarginine and Arginine, we performed a targeted analysis of livers from male Cry1−/− animals focusing on the relationship between Cry1 and specific metabolites known to be involved in this pathway (Figure 5C). Samples were collected at ZT10 and ZT22 corresponding to Cry1 acrophase and trough, respectively (Figure S4B). This metabolite analysis revealed that Arg was elevated in the Cry1−/− genotype (FDR<0.1), and that this elevation was robust to time. Similarly, Gln was noted to be reduced irrespective of time. Dimethylarginine demonstrated a time-dependent change, as did UMP, an end product of de novo nucleotide synthesis shown to be regulated by mTOR (Ben-Sahra, 2013; Robitaille, 2013). Alterations in Arg levels are known to impact mTOR signaling via CASTOR1 (Chantranupong, 2016), and thus we examined downstream mTOR targets (Figure S4B), and noted a consistent upregulation of mTOR pathway activation in CRY1−/− mouse livers compared to control mice (which were more heterogenenous overall), particularly at ZT10. Taken as a whole, we suggest that the Cry1-dependent coupling observed in U2OS cells has physiological consequences, particularly in time-dependent response to amino acids.
A set of ‘core clock metabolites’ links cell autonomous and in vivo metabolism
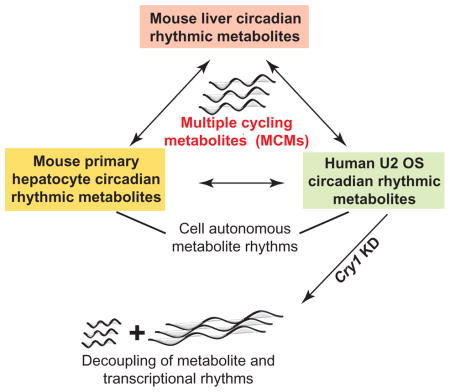
To further probe the interaction between cellular metabolism and the molecular clock, we examined the overlap between the cycling metabolites in human U2 OS cells and mouse liver (Figure 6A). We found a set of 18 cycling metabolites that oscillate between tissues/cell types and across humans and mice. We call these ‘multi-cycling metabolites’ (MCMs) and suggest that they are regulated by primary outputs of the clock. This set consists of amino acids (L-arginine, L-histidine, L-proline, L-methionine, L-glutamate, L-glycine) vitamins and co-factors (1-methylnicotinamide, nicotinamide), lipid related compounds (phosphocholine, glycerophosphocholine, butyrlcarnitine, L-malic acid), methylation compounds (SAH, MTA), oxidized glutathione, Uridine disphosphate glucose (UDP-glucose), taurine, and cytidine monophosphate (CMP). These MCMs were over-represented for components of cysteine and methionine metabolism (Figure 6B) in addition to glycerophospholipid, histidine, and nicotinate and nicotinamide metabolism. Four of the metabolites (L-arginine, MTA, SAH and L-methionine) were contained in the interconnected pathway represented in Figure 1F, with three of them closely related in the critical methylation step (Figure 6C). Some of these MCMs may have clock functions through actions on RNA methylation or chromatin, for example, UDP-glucose derived glycosylation or methylation at histone tails (Eckel-Mahan and Sassone-Corsi, 2013). This leads to the possibility that these metabolites could play a role in epigenetic regulation.
Figure 6. Rhythmic metabolites are under common clock control from U2 OS cells and mouse liver.

(A) Overlap of rhythmic metabolites between cell autonomous human U2 OS model and mouse liver. Eighteen metabolites are found to be oscillatory in both systems. (B) Metabolite enrichment of the 18 clock regulated metabolites specifically demonstrates the importance of cysteine and methionine metabolism. (C) Expansion of the metabolic pathway involving three metabolites in the methylation pathway, including SAH (S-adenosylhomocysteine), methionine and 5’-methylthioadenosine. Metabolites in blue are also involved but not found to be overlapped between U2 OS and liver, and grey boxes represent putative methylation targets including several known clock-modifying targets such as histones. (D) Analysis of difference between peak phase of metabolites (orange) and transcripts involved in their synthesis (blue) in mouse liver. Open orange circles indicate metabolite phases shifted with one period length. The phase difference (metabolite phase minus transcript phase) is indicated as purple triangles.
To test whether the 18 MCMs were regulated by primary outputs of the clock, we investigated their levels in U2 OS cells after knockdown of BMAL1, CRY1, and CRY2. As expected, all were arrhythmic after BMAL1 knockdown. Overall these MCMs (Figure 5D) follow the expected trend of a shorter period with CRY1 knockdown and lengthened period with CRY2 knockdown. There were exceptions, e.g. rather than changing period, MTA cycling was ablated when either CRY1 or CRY2 were depleted, while SAH cycling and UDP-glucose was ablated only with CRY1 KD. Together, these results argue that interactions of the transcriptional and metabolic clocks influences molecules involved in methylation, oxidative stress, and glycosylation, which have been shown by many others to play a role in epigenetic mechanisms.
In order to further assess the cell autonomous metabolite rhythms, we studied synchronized primary mouse hepatocytes. Due to known dampening of the molecular clock in isolated hepatocytes (Hughes et al, 2009), a limited time course was performed (4 h collection of 24 h). Verification of key clock genes confirmed that expected core clock gene transcripts were cycling (Figure 7A, S5A). Clear temporal variation was noted in a number of metabolites (Figure 7B, S5B), with 45/154 (29%) metabolites showing significant circadian rhythms using the same criteria as the U2 OS cell experiment (BH.Q<0.2 in JTK_Cycle). This is very similar to the total cycling metabolites in U2 OS, and reduced compared to liver which is likely a function of being cell autonomous, dampening rhythms and the relatively low-resolution of temporal sampling.
Figure 7. Primary hepatocytes exhibit global metabolic cycling.

A) Cultured hepatocytes were synchronized with 0.1 μM dexamethasone, and mRNA was collected at the indicated timepoints after synchronization. Expression of Bmal1 (Arntl), Rev-erbβ (NR1D1), and Rev-erbβ (Nr1d2) were determined by qPCR, normalized to expression of 36B4 (Rplp0), as has been described (Gréchez-Cassiau, 2008) previously (Data are averages from two biological replicate experiments). (B) Temporal concentrations of circadian cycling metabolites with lowest FDR values demonstrating metabolite cycling. Complete data are available in Dataset S1. Data are averages of two to three biological replicate experiments with three analytical replicates and the error bars represent standard error of the mean. (C) Comparison of the JTK reported q-values for circadian cycling between liver and hepatocytes for the 18 MCMs reported here. Metabolites with appreciable cycling are annotated. (D) Overlap in number of metabolites reported to cycle between liver, U2 OS and primary hepatocyte cycling datasets. See also Figure S5.
A comparison of the cycling between primary hepatocytes and liver for the set of multi-cycling metabolites reveals that a subset of the MCMs also cycle in hepatocytes (Figure 7C). Five of these (histidine, butyrlcarnitine, UDP-glucose, oxidized glutathione, and taurine) cycle significantly (BH.Q < 0.2), with nicotinamide, and CMP being close to meeting this threshold. As a result, a subsequent comparison was done with a slightly relaxed threshold (BH.Q < 0.3, p<0.05) to the liver and U2 OS data (Figure 7D). Consistent circadian cycling of these seven metabolites in two cell autonomous systems and one in vivo system presents compelling evidence that these metabolites are indeed directly related to the transcriptional clock. It remains to be seen if the remaining MCMs not observed to be cycling in primary hepatocytes have functional significance in maintenance of circadian function.
MCM metabolites are differentially regulated under conditions of circadian challenge
We note 90 metabolites which cycle in livers from in ad lib fed mice. Feeding, and timing of food intake can be viewed as both an input into the (Damiola, 2000) clock as well as a property controlled by the clock. In our experiment, the DD conditions imply that the influence of feeding on the metabolome will be a circadian property in and of itself. In order to better understand the influence of feeding behavior on our results, we compared the liver cycling metabolite to two studies which have used high fat diet (HFD) and/or time-restricted feeding (tRF) as models of circadian manipulation (Complete comparisons are available as Dataset S2).
The study of Abbondante et al. examined oscillatory behavior in animals fed both ad lib and HFD using 4 h sampling over 24 h. A total of 88 metabolites were measured in both the current and Abbondante studies. Using the same FDR (q < 0.05), 26 metabolites cycle in the Abbondante data with normal chow diet compared to 51 in this study. 20 of these metabolites overlap. Thus, our high-resolution time sampling study nearly doubled the number of cycling metabolites.
Additionally, the effects of HF vs control diet on circadian function was examined using a relaxed FDR for the previous study (BH.Q < 0.20). ~24% cycled under both normal chow and HFD and in our study, including four MCM metabolites (UDP-glucose, methionine, proline, and phosphocholine). An additional five metabolites cycled in our study and under normal chow in Abbondante et al, (~6%) including three MCMs (glycine, CMP and glycerophosphocholine). Conversely, 13 metabolites (~15%) cycled in our study and under HFD, including 6 MCMs (SAH, Taurine, 5-methylthioadensine, glutamate, butrylcarnitine, and oxidized glutathione). A further 13 metabolites (~15%) were cycling only in our study including four MCMs (histidine, nicotinamide, arginine, and malate), while 12 and 16 metabolites (~14% and ~18%) were oscillatory under normal chow and HFD, respectively, in Abbondante et al. and not significantly cycling in this study.
We note with interest that the metabolites cycling under HFD in Abbondate et al included methylation metabolites found to be key MCMs in our study (SAH, 5-methylthioadenosine). Our interpretation is that these metabolites are detectable as oscillating in our study, presumably due to high-resolution sampling, and under HFD perturbation this metabolite cycling is amplified. This reasoning is supported by fact that another metabolite from this pathway, SAM also cycles only under HFD in Abbondante, as well as in our study under ad lib feeding.
In order to examine if there are a set of cycling metabolites which are resistant to clock dysfunction, metabolites which cycled under both ad lib and HFD conditions in liver, as well as in the cell autonomous U2 OS system were compared. Ten metabolites met this criteria, including the MCMs UDP-glucose, methionine, and proline, as well as branched chain amino acids (Leucine/isoleucine, valine), alanine, phenylanaline, inosine, and NAD+.
An alternative approach to examine the influence of feeding on circadian function is through time-restricted feeding. Hatori et al (2012) examined the interaction between tRF and feeding regimes (normal chow vs. HFD). Although this study did not examine directly oscillatory behavior, global comparison of 82 metabolites measured in both studies reveals 27 metabolites (33%) including eight MCM’s (taurine, 5-methylthioadenosine, glutamate, glycine, nicotinamide, phosphocholine, glycerophosphocholine, CMP, and malate) are oscillating in this study and altered by tRF. In contrast, 21 metabolites (25%) including seven MCMs (SAH, histidine, methionine, oxidized glutathione, proline, butrylcarnitine and arginine) oscillate in this study but are not tRF-dependent in Hatori et al., while 13 metabolites (~16%) are time-dependent in Hatori et al, but not oscillating with BH.Q<0.05 in this study. The remaining 21 metabolites (26%) are not oscillating or time-dependent in either study.
Circadian transcription, translation, and metabolite acrophases
To better understand the interaction between transcription and metabolism, we looked at the relationship between the peak expression of the liver transcripts (Hughes et al., 2009) and the MCMs synthesized by their protein products (Figure 6D). Recent studies demonstrate a median 6 h delay between transcription and protein levels in the liver (Robles et al., 2014). We hypothesized that there’s an additional delay between protein levels and function, as measured by metabolite concentrations. Consistent with this, phase differences were just over 12 hours (mean, 12.44 h, median 13.75 h) between cycling transcript and metabolites. This relationship, combined with previous proteome analysis, would imply a delay of ~6 h between protein expression and metabolite peaks. Examination of two published proteomics (Mauvoisin, 2014; Robles et al., 2014) datasets revealed available information on twelve unique rate-limiting enzymes in liver with significant cycling (FDR < 0.2), namely Tdo2, Pck1, Lpin1, Lpin2, Tat, Fads2, Alas1, Hmgcs1, Gne, Fkbp4, Crot, and Impdh2. Of these, metabolite information was available for the tyrosine aminotransferase reaction catalyzed by TAT, with the hydroxyphenylpyruvate product. Peaks for TAT protein (CT18), and hydroxyphenylpyruvate from this study (CT 3.5) roughly agreed with our analysis, although in this case the mRNA acrophase was simulataneous with the protein (CT18) highlighting the complexity of these relationships. Intuitively, the delay between protein peak and product peak is perplexing as most enzymes would be expected to act in a much shorter timescale.
Further analysis of other canonical pathways reveals a variety of interactions between cycling transcripts, metabolites and proteins and their rhythmicity. For example, there is no cycling at any level in the conversion of phosphoenolpyruvate to pyruvate and then lactate. On the other hand, cycling in glucose-6-phospate (measured as hexose-6-phosphate here but dominated by the glucose form) is robust, as is mRNA cycling of Pfkl and Fbp1, but not fructose-1,6-bisphophate nor any protein products. Similarly, robust glucose and glucose transporter (Glut2/Slc2a2) mRNA cycling is observed (but no significant rhythmicity in transporter protein or hexokinase mRNA or protein). This suggests that multiple forms of regulation are at played and stoichiometry alone is not sufficient to understand these relationships. A complete analysis would require additional factors such as substrate and co-factor availability, catabolic protein production, protein PTM state and cellular transport and localization. In sum, the overall picture appears to be an orderly transition between peak cycling of transcripts, proteins, and metabolites with ~ 6 h delays between each step.
In conclusion, we present the first description of comprehensive oscillatory metabolism in mammalian systems under true circadian conditions (DD, with high temporal resolution). We find a similar percentage of detected polar metabolites oscillate as transcripts. Further, we describe the first comprehensive analyses of cell-autonomous metabolite rhythms in mammalian cells. Here, we find a much higher % of metabolites cycle than transcripts. Interestingly, in mouse liver, like transcription and translation, we find harmonics at both the 12- and 8-h frequency. We find a significant overlap in the cycling metabolites between mouse liver and human U2 OS cells, with many of the metabolites known to play a role in epigenetic regulation. Genetic perturbation of the U2 OS system shows that this cycling depends on an intact circadian clock. However, following CRY1 knockdown, rather than adopting a global short period analogous to locomotor activity in Cry1−/− mice, metabolites become frequency de-coupled from transcription, with many adopting an 8-h frequency. Mapping the transcription and metabolite profiling, we found a ~ 12 h difference, or double the average difference found between transcription and translation in a set of conserved metabolites cycling across both systems. In sum, these data show the pervasive influence of the clock on metabolism and complex multi-scale interactions between systems.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Aalim M. Weljie (aalim@upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal preparation and liver collection
High-resolution circadian collection was performed by using 6 week old C57Bl/6J male mice were acquired from Jackson Labs, entrained to a 12 h light: 12 h dark lighting schedule for 1 week before being released into constant darkness. After 18 h in constant darkness, three mice were euthanized in the dark every hour, for 48 hours by cervical dislocation. Liver samples were quickly excised and snap-frozen in liquid nitrogen. (Hughes et al., 2009; Zhang et al., 2014). Food and water were supplied ad libitum throughout. All procedures were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Homozygous Cry1 −/− or Cry2−/− mice were obtained from original Cry1 −/− or Cry2−/− lines of partial C57Bl/6 background and were each backcrossed more than 12 times against the C57Bl/6 background and then these were again crossed. The backcrossed homozygous Cry1 −/− or Cry2−/− mice were maintained in 12 h light: 12 h dark with food and water ad libitum. At approximately 8 weeks of age, mice were individually sacrificed by asphyxiation with CO2 at either ZT10 or ZT22. Liver samples were immediately snap frozen. All procedures were approved by the University of North Carolina Institutional Animal Care and Use Committee.
Cell autonomous U2OS cell collection
U2 OS cells (female) were seeded at 150,000 cells per 35-mm dish in DMEM medium containing 10% fetal bovine serum (FBS), 1X L-glutamine (Gibco), without antibiotics. Cells were transfected with 12 pmol of siRNA using Lipofectamine RNAiMax transfection reagent (Invitrogen) following the manufacturer’s instructions. A negative control siRNA (AllStars Negative control siRNA; Qiagen) was used to control for exogenous RNA introduction into the cell at equal molar amounts of BMAL1, CRY1 and CRY2 siRNA in each condition. Cells were plated 4 days prior to synchronization and allowed to grow to confluence. Cells were synchronized with 0.1 μM dexamethasone. The collection began 48 hours post-synchronization for MS studies with siRNA addition (Baggs et al., 2009). During the collection, cell pellets were collected and snap frozen every two hours for 48 hours (Altman et al., 2015).
Primary mouse hepatocytes
Primary hepatocytes (male) were isolated from 8-weeks-old male C57BL/6J mice, ordered directly from Jackson Labs. Animals were housed in groups of five mice/cage in a pathogen-free barrier facility in a 12-hour light-dark cycle with free access to food and water for 2weeks.
Hepatocytes were isolated using modified two-step perfusion method. The portal vein was cannulated and the liver was perfused with Liver Perfusion Media (Invitrogen) followed by Liver Digestion media, containing Kreb-Ringer Bicarbonate (Sigma) supplemented with 20mM HEPES, 500uM CaCl2, Collagenase/Elastase (Worthington) and DNase I (Worthington) were used for isolation. After the perfusion, liver was removed, disrupted to release cells using cell scrapers. The cell suspension was then filtered through 70μm filter and centrifuged at 50g for 5 minutes at 4°C, washed once in KRB buffer and precipitated in 25% Percoll gradient at 120g for 5 minutes at 4°C. Healthy hepatocytes were plated at a concentration of 3X106 and 1X106 on 10 cm and 100 mm collagen coated dishes respectively, in 1XDMEM supplemented with 10% FBS and1% Penicillin Streptomycin (Miller, et al, JCI, 2011).
METHOD DETAILS
Immunoblots
Liver samples were immersed in M-PER Mammalian Protein Extraction Reagent (Thermo Fisher) supplemented with protease inhibitor cocktail (BD Biosciences, or Promega) and phosphatase inhibitors II and III (Sigma). Samples were then lysed with a Tissue Lyser II (Qiagen), further homogenized by passage through a Qiashredder column (Qiagen), and centrifuged at 16,000 x g for 10 min at 4°C. Supernatants were quantified for protein content using the Bio-Rad DC assay kit (Bio-Rad), with BSA serving as a reference (Thermo Fisher). Proteins were resolved by SDS-PAGE using Criterion pre-cast gradient gels (Bio-Rad). Antibodies used were rabbit anti-Cry1 (Abcam), rabbit anti-phospho p70 S6K Thr. 389 (Cell Signaling), rabbit anti-p70 S6K (Cell Signaling), rabbit anti-pS6 Ser. 235/236 (Cell Signaling), mouse anti-S6 (Cell Signaling), rabbit anti-4EBP1 Thr. 37/46 (Cell Signaling), rabbit anti-4EBP1 (Cell Signaling), and mouse anti-α Tubulin (EMD Millipore). Secondary antibodies used were Alexa-Flour 680 goat anti-rabbit IgG (Life Technologies) and Alexa-Flour 790 goat anti-mouse IgG (Life Technologies). Immunoblots were imaged with the Odyssey CLx infrared imaging system (Licor) and uniformly contrasted.
qPCR and Primers
RNA was extracted from the frozen pellets of U2 OS as well as the mouse primary hepatocytes, using the RNeasy Plus Mini Kit (Qiagen) according to manufacturer’s instructions. U2OS cDNA was prepared using 500 ng of RNA and the qScript RT reagent (Quanta Biosciences), and hepatocyte cDNA was prepared using 2 μg of RNA and TaqMan Reverse Transcription Reagents (Life Technologies). U2OS cDNA was diluted 1:10, and hepatocyte cDNA not diluted, and used as template for quantitative real time PCR (qPCR) with specific human or mouse primers. qPCRs were performed with Power SYBR Green PCR Master Mix (Life Technologies), Taqman Universal PCR Mix (Life Technologies), or PerfeCTA Fast mix II Low Rox (Quanta Biosciences), using the StepONE Plus System (Life Technologies) and the Viia7 system (Life Technologies). Relative mRNA expression levels for hepatocytes were normalized to 36B4 (Gréchez-Cassiau, 2008) and to GAPDH for U2 OS cells, and analyzed using comparative delta-delta CT method. All qPCR primers are listed below:
| Gene Name | Sequence or Product Number | Source |
|---|---|---|
| 36B4 (Rplp0) | TTA TAA CCC TGA AGT GCT CGA C, CGC TTG TAC CCA TTG ATG ATG | IDT |
| REV-ERBα (Nr1d1) | Mm00520708_m1 | Taqman Gene Expression Assay |
| REV-ERBβ (Nr1d2) | Mm.PT.51.12747673 | IDT |
| BMAL1 (Arntl) | Mm00500226_m1 | Taqman Gene Expression Assay |
| Cry1 | Mm00514392_m1 | Taqman Gene Expression Assay |
| Cry2 | Mm01331543_g1 | Taqman Gene Expression Assay |
| GAPDH | Endogenous Human control | Taqman Gene Expression Assay |
| ARNTL | Hs00154147_m1 | Taqman Gene Expression Assay |
| CRY1 | Hs00172734_m1 | Taqman Gene Expression Assay |
| CRY2 | Hs00391360_m1 | Taqman Gene Expression Assay |
Metabolite Extraction
U2OS and hepatocyte cell pellets were thawed on ice and metabolites were extracted using a modified Bligh-Dyer method (Bligh and Dyer, 1959; Tambellini et al., 2013). Briefly, a methanol:chloroform (2:1) mixture (300μl) was added to the cell pellets, then vortexed and sonicated for 15 min. Chloroform and water (100μl each) were then added and samples were vortexed. Organic and aqueous layers were separated by centrifugation at 13,300rpm, for 7mins at 4°C. Aqueous layer was dried in a speed vacuum for 4 h until dry and re-dissolved in a mixture of 50:50 acetonitrile:water. The samples were centrifuged at 13300rpm for 5min to remove fine particulates. The supernatant was transferred to a LC-MS certified sample vial for Ultraperformance liquid chromatography with 95:5 water:acetonitrile 20mM ammonium acetate, pH 9 (solvent A) and 100% acetonitrile (Solvent B), on a Waters Acquity UPLC coupled to a Waters TQD mass spectrometer (Waters Corporation, USA). The same procedure was used for liver tissue analysis with 50 mg of liver tissue extracted per animal.
UPLC-MS based data acquisition for targeted polar metabolite profiling
Liquid chromatography conditions and mass spectrometer parameters were optimized and performed by injecting each 5μl sample onto an XBridge BEH 2.5μm 100mm x 2.1mm amide column using an Acquity H-class UPLC system (Waters Corporation, USA). Total chromatographic separation was 22min initial conditions of 15% A and 85% B at 0.15mL/min, ramped to 70%A and 30% B in 5mins, followed by an isocratic hold for 10mins. The column was washed in 98% A and 2% B and re-equilibrated in the starting conditions before the next injection. Mass spectrometry was performed on a QQQ quadrupole instrumentation, (Xevo TQD or TQS-micro, Waters Corporation, USA) operating using ion-switching with capillary voltage of 3000V and 2000V respectively, and a column temperature of 40°C. The desolvation gas flow was set to 900 L/h and the temperature was set to 450°C. The source temperature was set at 150°C. Data was acquired as multiple reaction monitoring’s (MRM) to target specific metabolites along with validation either through standards or mass spectrometry databases. The entire set of duplicate sample injections were bracketed with a test mix of standard metabolites at the beginning and at the end of the run to evaluate instrument performance with respect to sensitivity and reproducible signal. The sample queue was randomized to remove bias (Rhoades, 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Mass spectroscopy data processing and enrichment analysis
Mass spectroscopy data processing was performed using Waters TargetLynx software (version 4.1). Ion counts were exported from TargetLynx and subsequently processed in R (version 3.2) using a custom-designed processing script. Quality control (QC) samples, which consisted of a pooled sample of all samples, were injected at the beginning of the batch for LC column equilibration and every 6 injections during the analysis to account for instrumental drift. For every metabolic feature, a locally-weighted scatterplot smoothing function (LOESS) was fitted to the QC data, which was then used as a normalization factor for the samples as a function of run (Dunn, 2011) order. Additionally, metabolic features which appeared in less than 50% of the QC samples and displayed a relative standard deviation (RSD) greater than 30% were dropped from the final dataset. In total, 250 endogenous polar compounds were targeted, with 179 passing the above criteria. This is similar to non-lipid compounds in previous work (for example ~175 polar compounds in Abbondante et al (2015). Pathway analysis was conducted using Metabolites Enrichmement Set Analysis (Xia, 2010) (MSEA.com) or IMPALA (Kamburov, 2011). Pathway impact or fraction is the number of metabolites as hits in a pathways compared to the total set.
Statistical parameters such as details of replication and error bar meaning are reported in the figure legends.
Transcript to metabolite phase analysis
To study phase differences between circadian metabolites and their circadian transcripts, EC numbers for direct synthesis enzymes were extracted manually from KEGG pathways related to the 18 common metabolites cycling between mouse liver and human U2 OS cells. Cycling data from high-resolution time-series transcriptome (Hughes et al., 2009) from wild type mouse liver were coordinated. Raw cel files (downloading from GEO database) were analyzed with gcrma() function (gcrma package) and further normalized by normalize Quantiles() function (limma package) implemented in R. Time-series expression profiles and metabolite data were separately analyzed by JTK_CYCLE, version 3 (Hughes, 2010) through meta2d() function (only ‘JTK’ is selected for periodic detection, period length in search was set as 24; MetaCycle package). For extracting annotation information (including gene symbol and EC number) for each probe set, the mouse4302.db package was used. Those probe sets with maximum expression value less than 10^1.45 or lacking annotation were filtered out, and BH.Q values (Benjamini, 1995) were re-calculated based on p-values given by JTK_CYCLE for the remaining probe sets. For genes with multiple probe sets, a representative with smallest p-value and largest amplitude (in the case of multiple probe sets with equal smallest p-value) was selected. Metabolites (FDR < 0.05) and transcripts (FDR < 0.05) were linked based on the EC number to calculate phase differences. As we were focused on synthesis genes, we assumed that the peak phase of a metabolite must follow the peak phase of its associated transcripts. If transcript acrophase preceded metabolite acrophase, the phase of transcript was shifted by one 24 h period length. The phase differences between circadian metabolites and circadian transcripts involved in their synthesis were then calculated. R software (R 3.2.3) was used to perform analysis.
DATA AND SOFTWARE AVAILABILITY
The final metabolite dataset is publically available in the MetaboLights database under accession number MTBLS292 in addition to Supplemental Table 1.
Supplementary Material
Rhythmicity measurement parameters and processed mass spectrometry data tables for mouse liver, U2 OS cell and hepatocyte cycling experiments.
Literature comparison of liver cycling metabolites to previously reported mouse liver cycling datasets.
Highlights.
50% of detected metabolites oscillate in mouse liver
18 metabolites cycle in both liver and in the cell autonomous U2 OS system
These oscillations in the U2 OS system are driven by core clock genes
Genetic perturbation of CRY1 decouples transcriptional and metabolite rhythms
Acknowledgments
We thank Nick Lahens for providing U2 OS transcriptomic cycling numbers and Barry Slaff for assistance in collection of circadian cell culture samples, and Alyssa Kriegermeier for assistance in mouse primary hepatocyte collection protocol. This work is supported by the National Institute of Neurological Disorders and Stroke (5R01NS054794-08 to JBH), the Defense Advanced Research Projects Agency (DARPA-D12AP00025, to John Harer, Duke University), a TAPITMAT award to AMW via the National Center for Advancing Translational Sciences (5UL1TR000003, to Garret Fitzgerald), the National Cancer Institute (NCI) of the National Institutes of Health (NIH) K99CA204593 (to BJA) and R01CA057341 (to CVD), The Leukemia and Lymphoma Society LLS 6106-14 (to CVD), the Abramson Family Cancer Research Institute (to CVD), the National Institute for Diabetes and Digestive and Kidneys Diseases (R01DK098656 to JAB), the National Institute on Aging (R01AG043483 to JAB) and by the Penn Genome Frontiers Institute under a HRFF grant with the Pennsylvania Department of Health.
Footnotes
The authors declare no competing financial interests.
Author Contributions
Conceptualization, S.Y.K., J.B.H., A.M.W.; Methodology, S.Y.K., J.G., A.M.W; Formal Analysis, S.Y.K., G.W., J.B.H., R.C.A, A.M.W.; Investigation, S.Y.K., B.J.A, J.G., S.R., F.C, A.V., A.O.O.G., L.J.F., U.E., B.S., A.Sengupta, S.M., S.G., C.P.S., S.C., I.H.K, J.B.H., A.M.W.; Resources, J.B.H., A.M.W., A.Sancar, J.A.B.; Writing –Original Draft, S.Y.K., A.M.W.; Writing - Review and Editing, S.Y.K., C.V.D, J.B.H., A.M.W.; Visualization, S.Y.K., J.B.H., A.M.W.; Supervision, J.B.H., A.M.W.; Funding Acquisition; J.B.H., A.M.W.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbondante S, Eckel-Mahan KL, Nicholas JC, Baldi Pierre, Sassone-Corsi P. Comparative Circadian Metabolomics reveal Differential Effects of Nutritional Challenge in the Serum and Liver. Journl of Biological Chemistry. 2015:20. doi: 10.1074/jbc.M115.681130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht U. Circadian clocks and mood-related behaviors. Handb Exp Pharmacol. 2013:227–239. doi: 10.1007/978-3-642-25950-0_9. [DOI] [PubMed] [Google Scholar]
- Altman BJ, Hsieh AL, Sengupta A, Krishnanaiah SY, Stine ZE, Walton ZE, Gouw AM, Venkataraman A, Li B, Goraksha-Hicks P, et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. 2015;22:1009–1019. doi: 10.1016/j.cmet.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes LC, Levandovski R, Dantas G, Caumo W, Hidalgo MP. Obesity and shift work: chronobiological aspects. Nutr Res Rev. 2010;23:155–168. doi: 10.1017/S0954422410000016. [DOI] [PubMed] [Google Scholar]
- Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:12. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Baggs JE, Price TS, DiTacchio L, Panda S, Fitzgerald GA, Hogenesch JB. Network features of the mammalian circadian clock. PLoS Biol. 2009;7:e52. doi: 10.1371/journal.pbio.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass J. Circadian topology of metabolism. Nature. 2012;491:348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtold DA, Gibbs JE, Loudon AS. Circadian dysfunction in disease. Trends Pharmacol Sci. 2010;31:191–198. doi: 10.1016/j.tips.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science (New York, NY) 2013;339:6. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini YaHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:11. [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Brown SA, Kunz D, Dumas A, Westermark PO, Vanselow K, Tilmann-Wahnschaffe A, Herzel H, Kramer A. Molecular insights into human daily behavior. Proc Natl Acad Sci U S A. 2008;105:1602–1607. doi: 10.1073/pnas.0707772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C, Briggs W, Paschos GK, Fitzgerald GA, Griffin JL. A metabolomic study of adipose tissue in mice with a disruption of the circadian system. Mol Biosyst. 2015;11:1897–1906. doi: 10.1039/c5mb00032g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaix A, Zarrinpar A, Miu P, Panda S. Time-restricted feeding is apreventative and therapeutic intervention against diverse nutritional challenges. CellMetab. 2014;20:991–1005. doi: 10.1016/j.cmet.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, Sabatini DM. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell. 2016;165:12. doi: 10.1016/j.cell.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua ECP, Shui G, Lee ITG, Lau P, Tan LC, Yeo SC, Lam BD, Bulchand S, Summers SA, Puvanendran K, Rozen SG, Wenk MR, Gooley JJ. Extensive diversity in circadian regulation of plasma lipids and evidence for different circadian metabolic phenotypes in humans. Proc Natl Acad Sci USA. 2013;110:14468–14473. doi: 10.1073/pnas.1222647110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretenet G, Le Clech M, Gachon F. Circadian clock-coordinated 12 Hr period rhythmic activation of the IRE1alpha pathway controls lipid metabolism in mouse liver. Cell Metab. 2010;11:47–57. doi: 10.1016/j.cmet.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Dallmann R, Viola AU, Tarokh L, Cajochen C, Brown SA. The human circadian metabolome. Proc Natl Acad Sci USA. 2012;109:2625–2629. doi: 10.1073/pnas.1114410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiola F, Le Minh N, Preitner N, Kornmann Bt, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes & development. 2000;14:12. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SK, Ang JE, Revell VL, Holmes B, Mann A, Robertson FP, Cui N, Middleton B, Ackermann K, Kayser M, Thumser AE, Raynaud FI, Skene DJ. Effect of sleep deprivation on the human metabolome. Proc Natl Acad Sci USA. 2014;111:10761–10766. doi: 10.1073/pnas.1402663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi R, OK, Ishida N. Rhythms of Hepatic Glycogen Synthesis through Transcriptional Activation of Gys2. The Journal of Biological chemistry. 2010;285 doi: 10.1074/jbc.M110.110361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn BW, BD, David B, Paul B, Eva Z, Sue Francis M, Nadine A, Marie B, Joshau DK, Antony H, John NH, Andrew WN, Ian DW, Douglas BK, Royston G The Human Serum Metabolome (HUSERMET) Consortium. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocols. 2011;6:14. doi: 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- Dyar KA, Ciciliot S, Wright LE, Biensø RS, Tagliazucchi GM, Patel VR, Forcato M, Paz MIP, Gudiksen A, Solagna F, Albiero M, Moretti I, Eckel-Mahan KL, Baldi P, Sassone-Corsi P, Rizzuto R, Bicciato S, Pilegaard H, Blaauw B, Schiaffino S. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab. 2014;3:29–41. doi: 10.1016/j.molmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, Sassone-Corsi P. Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci USA. 2012;109:5541–5546. doi: 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamada H, Komatsu R, Shimba S, Okamura H. Rhythmic nucleotide synthesis in the liver: temporal segregation of metabolites. CellReports. 2012;1:341–349. doi: 10.1016/j.celrep.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan K, Sassone-Corsi P. Metabolism and the circadian clock converge. Physiol Rev. 2013;93:107–135. doi: 10.1152/physrev.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, Sassone-Corsi P. Reprogramming of the circadian clock by nutritional challenge. Cell. 2013;155:1464–1478. doi: 10.1016/j.cell.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Lazar MA. Rev-erbalpha and the circadian transcriptional regulation of metabolism. Diabetes Obes Metab. 2015;17(Suppl 1):12–16. doi: 10.1111/dom.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giskeødegård GF, Davies SK, Revell VL, Keun H, Skene DJ. Diurnal rhythms in the human urine metabolome during sleep and total sleep deprivation. Sci Rep. 2015;5:14843. doi: 10.1038/srep14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogna N, Singh VJ, Sheeba V, Dorai K. NMR-based investigation of the Drosophila melanogaster metabolome under the influence of daily cycles of light and temperature. Mol Biosyst. 2015;11:3305–3315. doi: 10.1039/c5mb00386e. [DOI] [PubMed] [Google Scholar]
- Gréchez-Cassiau A, Rayet Béatrice, Guillaumond Fabienne, Teboul Michèle, Delaunay Franck. The Circadian Clock Component BMAL1 Is a Critical Regulator of p21WAF1/CIP1 Expression and Hepatocyte Proliferation. The Journal of Biological Chemistry. 2008;283:8. doi: 10.1074/jbc.M705576200. [DOI] [PubMed] [Google Scholar]
- Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JAJ, Ellisman MH, Panda S. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M, Deharo L, Pulivarthy SR, Gu J, Hayes K, Panda S, Hogenesch JB. High-resolution time course analysis of gene expression from pituitary. Cold Spring Harb Symp Quant Biol. 2007;72:5. doi: 10.1101/sqb.2007.72.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JE, Kornacker K. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms. 2010;25:8. doi: 10.1177/0748730410379711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hong HK, Chong JL, Indacochea AA, Lee SS, Han M, Takahashi JS, Hogenesch JB. Brain-specific rescue of Clock reveals system-driven transcriptional rhythms in peripheral tissue. PLoS Genet. 2012;8:e1002835. doi: 10.1371/journal.pgen.1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, ST Circadian-Rhythm of Liver-Glycogen Metabolism in Rats - Effects of Hypothalamic-Lesions. The American Journal of Physiology. 1980;238:E21. doi: 10.1152/ajpendo.1980.238.1.E21. [DOI] [PubMed] [Google Scholar]
- Kamburov A, Cavill R, Ebbels TM, Herwig R, Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27:2. doi: 10.1093/bioinformatics/btr499. [DOI] [PubMed] [Google Scholar]
- Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Speizer FE, Hennekens CH. Prospective study of shift work and risk of coronary heart disease in women. Circulation. 1995;92:3178–3182. doi: 10.1161/01.cir.92.11.3178. [DOI] [PubMed] [Google Scholar]
- Li Y, Li G, Görling B, Luy B, Du J, Yan J. Integrative analysis of circadian transcriptome and metabolic network reveals the role of de novo purine synthesis in circadian control of cell cycle. PLoS Comp Biol. 2015;11:e1004086. doi: 10.1371/journal.pcbi.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Papagiannakopoulos T, Kinouchi K, Liu Y, Cervantes M, Baldi P, Jacks T, Sassone-Corsi P. Lung Adenocarcinoma Distally Rewires Hepatic Circadian Homeostasis. Cell. 2016;165:896–909. doi: 10.1016/j.cell.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, Roqueta-Rivera M, Deng C, Osborne TF, Mostoslavsky R, Baldi P, Sassone-Corsi P. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. 2014;158:659–672. doi: 10.1016/j.cell.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvoisin D, Wang J, Jouffe C, Martin E, Atger F, Waridel P, Quadroni M, Gachon F, Naef F. Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc Natl Acad Sci U S A. 2014;111:6. doi: 10.1073/pnas.1314066111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest. 2011;121:11. doi: 10.1172/JCI45942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura H, Miyake S, Sumi Y, Yamaguchi S, Yasui A, Muijtjens M, Hoeijmakers JH, van der Horst GT. Photic induction of mPer1 and mPer2 in cry-deficient mice lacking a biological clock. Science. 1999;286:2531–2534. doi: 10.1126/science.286.5449.2531. [DOI] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, Jacks T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016;24:8. doi: 10.1016/j.cmet.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschos GK, Ibrahim S, Song WL, Kunieda T, Grant G, Reyes TM, Bradfield CA, Vaughan CH, Eiden M, Masoodi M, Griffin JL, Wang F, Lawson JA, Fitzgerald GA. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat Med. 2012;18:1768–1777. doi: 10.1038/nm.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes KR. Shift work and age as interactive predictors of body mass index among offshore workers. Scand J Work Environ Health. 2002;28:64–71. doi: 10.5271/sjweh.648. [DOI] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, Takahashi JS, Imai S, Bass J. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:4. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Rhoades SD, Weljie AM. Comprehensive optimization of LC–MS metabolomics methods using design of experiments (COLMeD) Metabolomics. 2016;12:183. doi: 10.1007/s11306-016-1132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38:369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, et al. Quantitative Phosphoproteomics Reveal mTORC1 Activates de Novo Pyrimidine Synthesis. Science (New York, NY) 2013;339:4. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- Robles MS, Cox J, Mann M. In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet. 2014;10:e1004047. doi: 10.1371/journal.pgen.1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesler WJ, KRL Diurnal-Variations in the Activities of the Glycogen Metabolizing Enzymes in Mouse-Liver. International Journal of Biochemistry. 1985;17 doi: 10.1016/0020-711x(85)90089-8. [DOI] [PubMed] [Google Scholar]
- Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifian A, FS, Pasalar P, Gharavi M, Aminian O. Shift work as an oxidative stressor. J Circadian Rhythms. 2005;3:3. doi: 10.1186/1740-3391-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shostak A, Meyer-Kovac J, Oster H. Circadian regulation of lipid mobilization in white adipose tissues. Diabetes. 2013;62:2195–2203. doi: 10.2337/db12-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417:78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- Tambellini NP, Zaremberg V, Turner RJ, Weljie AM. Evaluation of extraction protocols for simultaneous polar and non-polar yeast metabolite analysis using multivariate projection methods. Metabolites. 2013;3:592–605. doi: 10.3390/metabo3030592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss CA, Levy M, Korem T, Dohnalová L, Shapiro H, Jaitin DA, David E, Winter DR, Gury BenAri M, Tatirovsky E, Tuganbaev T, Federici S, Zmora N, Zeevi D, Dori-Bachash M, Pevsner-Fischer M, Kartvelishvily E, Brandis A, Harmelin A, Shibolet O, Halpern Z, Honda K, Amit I, Segal E, Elinav E. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell. 2016;167:1495–1510.e12. doi: 10.1016/j.cell.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Tran M, Yang Z, Liangpunsakul S, Wang L. Metabolomics Analysis Revealed Distinct Cyclic Changes of Metabolites Altered by Chronic Ethanol-Plus-Binge and Shp Deficiency. - PubMed - NCBI. Alcohol Clin Exp Res. 2016;40:2548–2556. doi: 10.1111/acer.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, et al. A transcription factor response element for gene expression during circadian night. Nature. 2002;418:534–539. doi: 10.1038/nature00906. [DOI] [PubMed] [Google Scholar]
- Wu G, Anafi RC, Hughes ME, Kornacker K, Hogenesch JB. MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinformatics. 2016;32:3351–3353. doi: 10.1093/bioinformatics/btw405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wishart DS. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010;38:8. doi: 10.1093/nar/gkq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, Liu X, Atwood A, Huss JW, 3rd, Janes J, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111:16219–16224. doi: 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwighaft Z, Aviram R, Shalev M, Rousso-Noori L, Kraut-Cohen J, Golik M, Brandis A, Reinke H, Aharoni A, Kahana C, et al. Circadian Clock Control by Polyamine Levels through a Mechanism that Declines with Age. Cell Metab. 2015;22:874–885. doi: 10.1016/j.cmet.2015.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rhythmicity measurement parameters and processed mass spectrometry data tables for mouse liver, U2 OS cell and hepatocyte cycling experiments.
Literature comparison of liver cycling metabolites to previously reported mouse liver cycling datasets.