

A rational approach enables the almost complete suppression of nucleation events in Aβ42 aggregation using designed antibodies.
Keywords: Alzheimer’s disease, Protein aggregation, Chemical Kinetics
Abstract
Antibodies targeting Aβ42 are under intense scrutiny because of their therapeutic potential for Alzheimer’s disease. To enable systematic searches, we present an “antibody scanning” strategy for the generation of a panel of antibodies against Aβ42. Each antibody in the panel is rationally designed to target a specific linear epitope, with the selected epitopes scanning the Aβ42 sequence. By screening in vitro the panel to identify the specific microscopic steps in the Aβ42 aggregation process influenced by each antibody, we identify two antibodies that target specifically the primary and the secondary nucleation steps, which are key for the production of Aβ42 oligomers. These two antibodies act, respectively, to delay the onset of aggregation and to block the proliferation of aggregates, and correspondingly reduce the toxicity in a Caenorhabditis elegans model overexpressing Aβ42. These results illustrate how the antibody scanning method described here can be used to readily obtain very small antibody libraries with extensive coverage of the sequences of target proteins.
INTRODUCTION
The aggregation of the 42-residue form of the amyloid-β peptide (Aβ42) into amyloid fibrils is a key molecular process underlying Alzheimer’s disease (AD) (1–5). Therefore inhibition of the aggregation process of Aβ42 has been among the major therapeutic strategies directed against AD (1–10). Nevertheless, compounds designed for this purpose have yet to reach the clinic (2, 11). These failures are, at least in part, due to the complexity of the aggregation behavior of Aβ42, which involves a series of tightly coupled microscopic steps (12, 13). Because of this complexity, as recent studies on the kinetics of aggregation have shown, the suppression of fibril formation can have surprising outcomes, in some cases even increasing, rather than decreasing, the concentration of potentially toxic oligomeric species (6). It is, therefore, of critical importance for the development of effective therapies to design, in a rational manner, molecules capable of acting selectively on the critical steps of formation of the toxic species in the aggregation process (6).
Over the past decade, increasing evidence has implicated prefibrillar oligomeric species, rather than mature amyloid fibrils, as the major agents responsible for cellular toxicity in AD and similar pathologies (14–19). Recently, major advances in understanding the molecular mechanisms underlying the formation by Aβ42 of such toxic species have been made (13). In particular, it has been shown that, once a small but critical concentration of aggregates has formed through the self-assembly of Aβ42 monomers as a result of primary nucleation events, surface-catalyzed secondary nucleation becomes important because the surfaces of existing fibrils can catalyze the generation of new oligomeric species (13). These oligomers can then grow and convert into additional fibrils, thus providing a positive feedback mechanism that results in rapid aggregate proliferation (13). A consequence of this mechanism is that compounds capable of selectively interfering with either the primary nucleation or the surface-catalyzed secondary nucleation of Aβ42 have the potential to suppress the formation of aggregated forms of Aβ42. Molecular chaperones (20, 21) and carefully selected small molecules (22, 23) have been shown to be able to act in this manner.
Here, we extend this strategy to antibodies with an aim of using them against AD by directly affecting different microscopic steps in Aβ42 aggregation. This goal is inspired by the view that antibodies, which can be obtained with well-established methods such as immunization or phage and associated display methods against a wide variety of targets (24–29), can have wide applicability in diagnostics and therapeutics because of their high target selectivity (30–34). In particular, a traditional and generally effective therapeutic approach of using antibodies is active immunotherapy, which consists of boosting our natural immune defense by the administration of harmless versions of the pathogenic agents (35, 36). This approach, which represents one of the most promising strategies against cancer (35, 37), is currently being explored for the treatment of AD, although with difficulties in balancing the immune response while maintaining efficacy (7–9). Here, we adopt a different approach, as we use antibodies as compounds that directly inhibit the aggregation process of Aβ42.
To exploit the exceptional versatility of antibodies in molecular recognition, we apply a recently developed method for the rational design of single-domain antibodies to specifically bind disordered or otherwise solvent-exposed linear epitopes within a target protein (38, 39). These antibodies tend to preferentially bind the target epitope when the protein is in the aggregated rather than in the monomeric conformation (38), probably because of entropic effects due to the preorganization of the epitope in the aggregated state. We thus generate here a small panel of five human single-domain antibodies (Fig. 1) that are rationally designed to bind Aβ42; we call these antibodies DesAbs (“designed antibodies”). Each DesAb in the panel is designed to target one epitope within the peptide, with consecutive epitopes being selected to achieve close to full coverage of the Aβ42 sequence; we refer to this procedure as “antibody scanning.” This procedure is related to the recently proposed epitope mapping method (40) but differs from it because the antibodies that we describe here are designed rationally, rather than being polyclonal antibodies obtained with immunization techniques.
Fig. 1. Schematic representation of the DesAb panel against Aβ42 generated using the antibody scanning method described in this work.
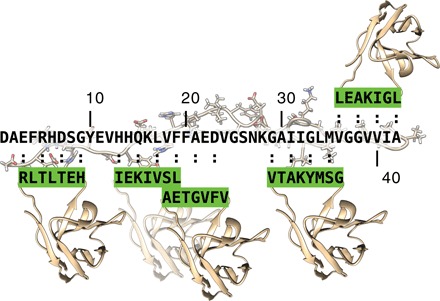
The five target epitopes, which scan the Aβ42 sequence, are shown as green-framed rectangular boxes, whereas the corresponding designed complementary peptides grafted into the CD3 loop of the single-domain human antibody scaffold are highlighted in green.
By combining highly reproducible fluorescence-based aggregation assays with a chemical kinetics framework for protein aggregation analysis (12, 41), we investigate the mechanism of inhibition of Aβ42 aggregation by the five DesAbs in the antibody scanning panel that we produced. This approach does not require previous knowledge of the elusive structures of the toxic species of Aβ42 and could represent a highly sensitive method for the quantitative detection of the effects of potential therapeutic molecules on the microscopic processes that underlie protein aggregation (6, 20, 23). Furthermore, because of the low binding affinity that is often observed in the interactions with disordered proteins such as Aβ42, conventional experimental methods, which have been largely developed in the context of enzyme inhibition, remain challenging for the study of the inhibition of protein aggregation (6). By contrast, the approach that we adopt here is based on chemical kinetics and does not require tight binding between a therapeutic molecule and an aggregation-prone protein in its monomeric conformation (6). Thus, even relatively low (micromolar) affinity binding to the monomeric forms of the target protein can result in large effects on its aggregation behavior (42). These effects can be achieved by a specific binding to aggregates (6) or by a type of binding to monomers that perturbs the populations of their different conformations by decreasing those that are able to nucleate and aggregate (43). Using this strategy, we identify two stable DesAbs of potential therapeutic interest. The first, DesAb18–25, is designed to bind to the central region of Aβ42 (residues 18 to 25: VFFAEDVG), and the second, DesAb29–36, is designed to bind to the C-terminal region of Aβ42 (residues 29 to 36: GAIIGLMV). We find that DesAb18–25 and DesAb29–36 are able to selectively inhibit the primary and the surface-catalyzed secondary nucleation of Aβ42, respectively, and to consistently suppress the Aβ42-mediated toxicity in a Caenorhabditis elegans model.
RESULTS AND DISCUSSION
Generation of an antibody panel against Aβ42 using antibody scanning
In this section, we describe an antibody scanning procedure for the rapid in silico generation of antibody libraries. We applied this procedure to Aβ42 to obtain a pool of antibodies that are capable of binding different epitopes along the sequence of this peptide. From this pool, we then identified those that are capable of selectively inhibiting specific microscopic steps in the Aβ42 aggregation process.
The method developed in this study is designed to generate a small panel of antibodies that carry complementary-determining regions (CDRs) rationally designed in silico to target different linear epitopes that systematically cover the whole sequence of Aβ42. Using this method, we can replace library generation, biopanning, and amplification steps of standard in vitro technologies (29, 44) with a fast computational procedure, focusing the experimental efforts exclusively on functional screening. Specifically, for each target linear epitope along the sequence of Aβ42 (Fig. 1), we perform the following steps: (i) We run the cascade method (38) of designing complementary peptides binding to the target sequence; (ii) we select the most promising peptide by using the scores from the cascade methods itself, which are related to the predicted binding (38), as well as the predicted solubility calculated using the CamSol method (performing these two steps takes only a few minutes on a standard laptop) (45); and (iii) we then graft the selected complementary peptide onto the CDR3 of a human VH (variable region of immunoglobulin heavy chain) domain antibody scaffold, which has previously been shown to be stable and highly tolerant to the replacement of the CDR3 loop (46, 47). The designed peptides are reported in Table 1.
Table 1. List of the different DesAbs used in this work.
| Antibody | Grafted sequence | Target sequence |
| DesAb3–9 | HETLTLR | (3)EFRHDSG(9) |
| DesAb13–19 | LSVIKEI | (13)HHQKLVF(19) |
| DesAb18–25 | VFVGTEA | (18)VFFAEDVG(25) |
| DesAb29–36 | GSMYKATV | (29)GAIIGLMV(36) |
| DesAb36–42 | LGIKAEL | (36)VGGVVIA(42) |
| DesAb-F (38) | FQEAVSG | (70)VVTGVTA(76) (α-synuclein) |
| DesAb15–21 (38) | FKLSVIT | (15)QKLVFFA(21) |
Characterization of the binding of the DesAbs to Aβ42
All the rationally designed DesAb variants were expressed in Escherichia coli and purified, as previously reported (fig. S1) (45). The five different single-domain antibodies are named DesAb3–9, DesAb13–19, DesAb18–25, DesAb29–36, and DesAb36–42, where the subscript identifies the region of the Aβ42 sequence where the antibody is designed to bind (Table 1). Circular dichroism (CD) spectroscopy revealed that all DesAbs have a secondary structure content compatible with the native conformation of a VH domain (Fig. 2A). Furthermore, no differences were detected between the thermal stability of the DesAbs and the original VH scaffold, as reported in the literature (Fig. 2, A and B) (48). These results indicate that the grafting procedure that we used does not produce significant changes in the structure and stability of the original single-domain antibody.
Fig. 2. Structural and functional characterization of the DesAbs.
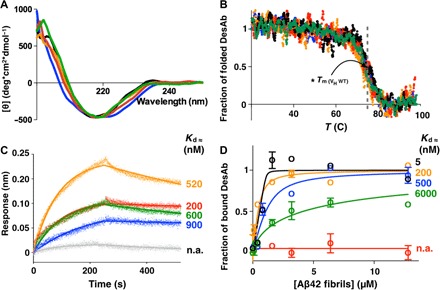
CD spectra (A) and CD thermal denaturation (B) of the DesAbs used in this work. WT, wild type. (B) Denaturation data are reported as fraction of the folded protein (see Materials and Methods). (C) BLI measurements of the binding of the DesAbs to SA sensor chip coated with monomeric biotinylated Aβ42. Each curve was subtracted from a curve of binding of the corresponding DesAb to an uncoated sensor chip; the Kd values of binding to monomeric Aβ42 are reported. Given the proximity of the target peptide of DesAb3–9 to the biosensor surface, the affinity of DesAb3–9 for monomeric Aβ42 was determined with biotin-mediated affinity measurements (fig. S3). n.a., not applicable. (D) Fibril binding experiments of the DesAbs; the Kd values of binding to fibrillar Aβ42 are reported. DesAb3–9, black; DesAb13–19, orange; DesAb18–25, blue; DesAb29–36, green; DesAb36–42, red; DesAb-F (a DesAb that targets α-synuclein), gray. The gray dashed line in (B) indicates the Tm (≈73°C) of the original scaffold as reported in the literature (48).
Biolayer interferometry (BLI) (see Materials and Methods) using streptavidin (SA) biosensor tips coated with N-terminal biotinylated Aβ42 was used to show that DesAb13–19, DesAb18–25, DesAb29–36, and DesAb36–42 bind monomeric Aβ42 with a Kd (dissociation constant) ranging from 200 to 900 nM (Fig. 2C). As a control, we verified that a related DesAb that carries a complementary peptide designed to bind α-synuclein does not bind Aβ42 in the same assay (Fig. 2C, gray curve). In addition, we also show that the Aβ42 DesAbs do not bind α-synuclein (fig. S2). Likewise, in a previous study, we showed that DesAbs designed to bind other amyloidogenic proteins, including DesAb-F (Table 1), do not bind Aβ42, illustrating the specificity of these antibodies (38).
In the case of DesAb3–9, no significant binding signal was detected in this BLI setup, most likely as a result of the close proximity of the target epitope to the surface of the BLI biosensor tip. Therefore, for this DesAb variant, the binding to Aβ42 monomers was studied by means of biotin-mediated affinity measurements (fig. S3). Specifically, we incubated DesAb3–9 in the presence of different concentrations of N-terminal biotinylated Aβ42. The DesAb3–9/Aβ42 complex formed in solution was then removed by incubating the samples in an enzyme-linked immunosorbent assay (ELISA) plate coated with SA. The amount of DesAb3–9 left in solution was quantified by intrinsic fluorescence measurements, and the corresponding fraction of bound DesAb3–9 was plotted as a function of the concentration of Aβ42 monomers in the sample (fig. S3). The affinity calculated in this manner (about 100 nM) was consistent with that of the other DesAbs reported above (in the 200 to 900 nM range).
Furthermore, to verify whether or not the DesAbs interact with their respective target regions of the Aβ42 sequence, representative binding measurements were carried out on DesAb18–25 and DesAb29–36 (fig. S4). BLI measurements of DesAb18–25 and DesAb29–36 were recorded in the presence of the corresponding chemically synthesized target epitopes Aβ18–25 (Ac-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-NH2) and Aβ29–36 (Ac-Gly-Ala-Ile-Ile-Gly-Leu-Met-Val-NH2), respectively (see Materials and Methods). These peptides were the only ones among the target epitopes in the Aβ42 sequence (Fig. 1) that had sufficient solubility under the experimental conditions that we used (see Materials and Methods) for such analysis. Our analysis revealed that both antibodies bind their respective target epitopes, as expected (fig. S4, A and B). As a control of specificity, we performed a similar BLI experiment by swapping the target epitope peptides (that is, we tested the binding of DesAb18–25 with Aβ29–36 and that of DesAb29–36 with Aβ18–25) and, under these conditions, we could not detect significant binding (fig. S4, A and B). In the case of DesAb18–25, we further validated the binding region on Aβ42 by performing a competition assay in the presence of DesAb-Aβ (which we denote here as DesAb15–21), a DesAb previously reported to bind the region Aβ15–21 (38). To do this, we incubated a fluorescently labeled variant of DesAb18–25, which retains a native-like conformation as assessed by CD (fig. S4C), in the presence of an equimolar concentration of N-terminal biotinylated Aβ42 and increasing concentrations of DesAb15–21. The A647-DesAb18–25/N-terminal biotinylated Aβ42 complex was then isolated by means of an ELISA plate coated with SA (see Materials and Methods), and the unbound A647-DesAb18–25 was quantified by fluorescence measurements. As expected, we found that DesAb15–21 is able to compete with DesAb18–25 for the binding to Aβ42 with a median inhibitory concentration (IC50) of 400 nM, which is in the same order of magnitude of the affinity of DesAb18–25 for Aβ.
We then investigated the ability of the DesAbs to bind aggregated species of Aβ42. To do so, we performed biotin-mediated affinity measurements by using fibrils made of N-terminal biotinylated Aβ42 (see Fig. 2D and Materials and Methods). We found that some DesAbs show a preferential binding toward fibrillar species. In particular, the Kd values of binding of DesAb3–9, DesAb13–19, and DesAb18–25 for Aβ42 fibrils are 5, 200, and 500 nM, respectively, 100-, 5-, and 2-fold better than the corresponding values for Aβ42 monomers (Fig. 2D). The other antibodies bind Aβ42 fibrils with an affinity worse than that for the monomers, in agreement with the recent report that the central and the C-terminal regions of Aβ42 are buried within the fibrils (Fig. 2D) (48, 49).
Inhibition by the DesAbs of different microscopic steps in Aβ42 aggregation
The formation of amyloid fibrils in vitro can be monitored using fluorescent dyes, such as thioflavin T (ThT), which specifically interact with the stacked β sheet structure of the amyloid fibrils (see Materials and Methods). The development of a protocol to achieve highly reproducible kinetic data, together with the derivation of an analytical solution to the coupled differential equations that govern amyloid growth (12), has allowed the rates of the microscopic processes that underpin the formation of the oligomeric species of Aβ42 (Fig. 3A) to be extracted from macroscopic measurements of the aggregation kinetics (13). In addition, this type of analysis has enabled the study of the mechanisms of inhibition of protein aggregation by candidate therapeutic molecules (6, 23). This approach consists of measuring ThT fluorescence–based aggregation of Aβ42 under reference conditions and monitoring the changes in the aggregation kinetics upon systematic variations of the concentration of a tested inhibitor. The perturbations in the reaction profiles are then analyzed with a kinetic model to identify the mechanisms of aggregation most affected by the interactions of the tested inhibitor with one or more of the species present in the system.
Fig. 3. The antibody scanning method produces antibodies that affect different microscopic steps in Aβ42 aggregation.
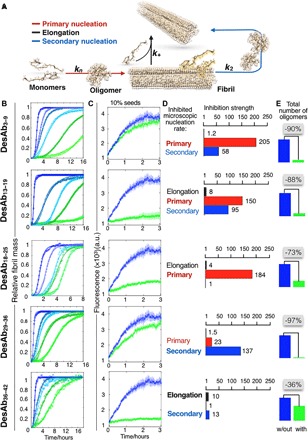
(A) Model of aggregation of Aβ42 showing the primary (red arrow) and the secondary (blue arrow) nucleation of the oligomers and the elongation of the fibrils (black arrow). (B) Solutions containing 2 μM Aβ42 were incubated in the presence of increasing (blue to green) Aβ42 monomer equivalents of the DesAbs (serial dilutions starting from 1 μM DesAb concentration; see fig. S5); each antibody targets a specific epitope within the sequence of Aβ42 (Fig. 1) and inhibits the aggregation of the peptide in a characteristic manner. Continuous lines represent the fits of the data using the integrated rate law for Aβ42 aggregation (see Materials and Methods). (C) Seeded aggregation of Aβ42 in the presence of 10% preformed fibrils with a 0:1 (blue) or 1:1 (green) antibody–to–Aβ42 monomer ratio. a.u., arbitrary units. (D) Bar plot showing the inhibition strength of the DesAbs (which is defined as kAβ42/kAβ42+DesAb) on k+ (black), kn (red), and k2 (blue) rate constants, derived from (B), (C), and fig. S5. The fold change in the presence of the antibodies of each of the rate constants is indicated on the top of the corresponding bar. (E) Relative number of oligomers generated during the aggregation reaction with or without a 1:2 antibody–to–Aβ42 monomer ratio.
By applying this method, we measured by ThT fluorescence the time evolution of the formation of Aβ42 fibrils in the presence of varying concentrations of the different DesAbs. We observed a progressive reduction in the overall rate of aggregation with increasing concentrations of the DesAbs (Fig. 3B), which shows that all DesAbs designed to bind Aβ42 are effective in inhibiting its aggregation, even at substoichiometric concentrations (as low as 1:32 antibody–to–Aβ42 monomer ratios). By contrast, the antibody DesAb-F (Table 1), which showed a strong inhibitory effect on α-synuclein aggregation (38), does not have any detectable effect on the aggregation of Aβ42 (fig. S5). Because DesAb-F differs from the antibodies designed to target Aβ42 discussed here only in the sequence of the complementary peptide grafted in the CDR3 loop, these results indicate that the inhibitory effects observed on the aggregation of Aβ42 specifically originate from the computationally designed peptides (Fig. 3B).
To identify the microscopic processes involved in the aggregation of Aβ42 that are most affected by the interaction with each antibody, we determined the changes in the global parameters k+kn and k+k2 (where k+, kn, and k2 are the elongation, primary, and secondary rate constants, respectively) by fitting the aggregation curves with the integrated kinetic laws described above (fig. S6) (6). In addition, it is particularly important to evaluate the perturbations in the individual microscopic rate constants k+, kn, and k2. This information is relevant in the light of the observation that the failure in clinical trials targeting Aβ aggregation may have resulted from a general and nonspecific inhibition of the aggregation process, potentially leading to the suppression of the formation of high–molecular weight aggregates at the expenses of an increased number of small toxic oligomers (6). To evaluate the changes in the individual microscopic processes, we complemented the kinetic data under unseeded conditions by testing the effect of the DesAbs in an assay that specifically probes the elongation of Aβ42 fibrils (Fig. 3C). For this purpose, the DesAbs were introduced in a reaction mixture containing 2 μM monomeric Aβ42 supplemented with 0.2 μM preformed fibrils. At this high concentration of fibrils, primary and secondary nucleation events are negligible, and only elongation reactions contribute to the increase in the fibrillar mass. We found that, at a 1:1 antibody–to–Aβ42 monomer ratio, DesAb3–9 and DesAb29–36 do not have any significant effect on Aβ42 aggregation kinetics, indicating that they are unable to interfere with the elongation of existing Aβ42 fibrils. Instead, the antibodies DesAb13–19, DesAb18–25, and DesAb36–42 show a strong effect on the seeded aggregation kinetics (Fig. 3C). After measuring the changes in k+ from the seeded experiments, by considering the decreases in k+k2 and k+kn evaluated from unseeded aggregations, we were in the position to determine the decreases in the single microscopic rate constant for each DesAb (see Materials and Methods). We found that, in the presence of the single-domain antibodies raised against the N-terminal region of Aβ42 (DesAb3–9 and DesAb13–19), both primary and secondary nucleation rate constants were changed by up to two orders of magnitude (Fig. 3D). These findings are consistent with a recent report that shows that the manipulation of the N terminus of Aβ42 affects all the microscopic steps in the aggregation process of this peptide (50). However, DesAb18–25, which targets the central region of Aβ42, mainly affects kn, which again decreases by two orders of magnitude, whereas k2 decreases by only about 10-fold (Fig. 3D and Table 2). The two antibodies designed to target C-terminal epitopes (DesAb29–36 and DesAb36–42), by contrast, show a preferential inhibition of k2, which decreases by about two orders of magnitude, whereas kn remains almost unaffected (Fig. 3D and Table 2).
Table 2. Changes in the microscopic rate constants in Aβ42 aggregation in the presence of the different DesAbs used in this work (elongation, k+; primary nucleation, kn; and secondary nucleation, k2).
| Antibody | k+ | knk+ | k2k+ | kn | k2 |
| DesAb3–9 | 1.2 | 205.1 | 68.7 | 173.8 | 58.2 |
| DesAb13–19 | 8.0 | 1210.9 | 760.9 | 150.1 | 94.8 |
| DesAb18–25 | 3.9 | 723.4 | 3.2 | 183.6 | 0.8 |
| DesAb29–36 | 1.5 | 35.1 | 206.2 | 23.4 | 137.4 |
| DesAb36–42 | 9.9 | 7.4 | 127 | 0.8 | 12.9 |
To determine how the reduction of the rate constants of the different microscopic steps affects the formation of Aβ42 oligomers, we estimated the change of the on-pathway oligomeric populations in the presence of the different DesAbs by using the rate constants derived from the kinetic analysis (Fig. 3E). We found that the antibodies with a strong effect on secondary nucleation were the most effective in inhibiting the formation of these oligomers. In particular, DesAb29–36, which predominantly affects secondary nucleation, was the antibody with the strongest effect by decreasing the number of oligomers by 97%. Strikingly, this decrease is larger than that (90%) caused by DesAb13–19, which has a much more pronounced effect on the overall aggregation process reported by the ThT measurements. By contrast, the decrease (36%) caused by DesAb36–42 is much lower than that of DesAb29–36, despite the fact that the overall unseeded aggregation curves look very similar for these two antibodies. These results show how the kinetic analysis used here makes it possible to extract very important—but often hidden—information on on-pathway oligomeric species from macroscopic measurements. Together, these results show that the antibody scanning strategy introduced here is effective for the rapid identification of antibodies that are capable of targeting specific microscopic steps in protein aggregation, without the need for any a priori knowledge about the regions of the peptide or protein that are most important for the self-assembly process.
Rescue by DesAb18–25 and DesAb29–36 of Aβ42-mediated dysfunction in C. elegans
As previously discussed, effective therapeutic strategies against AD that focus on inhibiting Aβ42 aggregation are likely to require the targeting of specific nucleation processes, instead of simply suppressing the formation of high–molecular weight aggregates (6). In the previous section, we have shown that DesAb18–25 and DesAb29–36 have specific effects on the primary and the secondary nucleation rate constants, respectively. In particular, DesAb29–36 almost exclusively affects the secondary nucleation rate, without any detectable effect on the elongation of existing Aβ42 fibrils (Fig. 3, C and D, and Table 2).
To test the effects of the antibodies on the formation of Aβ42 aggregates in vivo, we used a C. elegans model of Aβ42-mediated dysfunction, denoted GMC101, in which human Aβ42 is expressed in body wall muscle cells where it forms aggregates and results in severe age-progressive paralysis (51). Specifically, the effects of the administration of the antibodies observed using this worm model were compared to those observed in control worms (N2, wild type), which do not express Aβ42 (see Materials and Methods). To analyze the effects of the two DesAbs on the toxicity of Aβ42 in worms, we developed a protocol for the transduction of native proteins into living worms based on a commercially available reagent for lipid-mediated transduction of macromolecules into mammalian cells (see Materials and Methods). The effectiveness of the procedure was first tested by administering 20 μM mCherry fluorescent protein encapsulated into the lipid vesicles of the reagent into control worms as suspensions in a MES buffer solution (see Materials and Methods). A confocal microscopy analysis performed after 4 hours of incubation revealed the presence of the specific fluorescence of mCherry (fig. S7), thus indicating that mCherry is delivered effectively into the cells and has retained the native conformation.
We then performed the same protocol using lipid vesicles into which each of the two DesAbs was encapsulated at a concentration of 20 μM and measured the frequency and amplitude of the body bends, which are indicative of the state of the muscle cells and of the overall viability of the worms (Fig. 4). We administered the antibodies at different stages of adulthood to test their effects at different phases of the aggregation process in vivo. The expectation from the in vitro results is that the antibody that inhibits primary nucleation should be most effective in an early administration, whereas that inhibiting secondary nucleation should be most effective in a late administration. Therefore, in one experiment, the DesAbs were administered at days 1 and 3, whereas in another experiment, the DesAbs were administered only at day 6. Phenotypic differences were screened at day 7 in both cases. To correct for secondary effects of the treatments on the fitness of the worms (as, for example, an improvement of motility as a consequence of the metabolism of the lipid molecules of the vesicles), Aβ42 and control worms were treated with empty vesicles and compared with worms treated with the same numbers of antibody-filled vesicles.
Fig. 4. Effects of DesAb18–25 and DesAb29–36 in a C. elegans model of Aβ42-mediated toxicity.
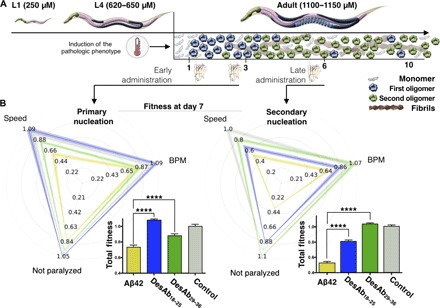
(A) Experimental design for the investigation of the effects of the two selected DesAbs in the C. elegans strain GMC101 (the Aβ42 worm model) compared with strain N2 (the control worm model). The pathological phenotype is induced in the worms by increasing their temperature of incubation from 20° to 24°C, which induces Aβ42 aggregation. A pictorial representation of the populations of monomers (light blue), oligomers formed by primary (blue) and secondary (green) nucleations, and fibrils (maroon) at the different stages (in days) of adulthood of the worms is given to illustrate the aggregation process. (B) Phenotypic fingerprints, which consider speed, body bends per minute (BPM), and fraction not paralyzed or the worms, of Aβ42 worms (C. elegans GMC101; yellow) and control worms (C. elegans N2, WT; gray) treated with empty lipid vesicles and after the administration of DesAb29–36 (green) and DesAb18–25 (blue), screened at day 7 of adulthood. DesAbs were administered starting from a 20 μM concentration (see Materials and Methods) at days 1 and 3 (left) or at day 6 (right). The fingerprints show one representative of three biological replicates that showed similar results. The thickness of the lines represents SEM. The bar plots report the total fitness (see Materials and Methods). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 (relative to untreated worms).
In the presence of both antibodies, the frequency of the body bends, their amplitude, and the viability of Aβ42 worms increase significantly (Fig. 4 and fig. S8; movies S1 to S7 illustrate the effects of the treatment). In particular, the antibody DesAb29–36 that inhibits secondary nucleation shows a substantial effect on all the three measured parameters when administered at day 6 and produces an almost full recovery of the pathological phenotype (Fig. 4). The administration at day 6 of DesAb18–25, which inhibits primary nucleation in vitro, also induces an improvement of the phenotype, but to a significantly lower level than that observed for DesAb29–36. By contrast, the effect of DesAb18–25 is much larger upon early administration at days 1 and 3, compared to late administration, in line with the in vitro findings that a primary nucleation inhibitor loses its efficacy once a sizeable amount of aggregates have formed. As a further control, following the administration at days 1 and 3, we also screened the viability of the worms at day 4 and obtained very similar results to those from the screening at day 7, as discussed above (fig. S9). Notably, no significant effects on the measured phenotypic parameters were observed following the administration of the DesAbs to the control worms (fig. S10) or to a worm model of α-synuclein–mediated toxicity (OW40) (fig. S11A). In addition, no significant effects were detected upon the administration of DesAb-F to the Aβ42 worms (fig. S11B). Together, these results illustrate the efficacy of our strategy. Finally, fluorescence imaging using the amyloid-specific probe NIAD-4 (see Materials and Methods) shows that the treatments using the DesAbs have a direct effect on the amount of Aβ42 aggregates in the worms (fig. S12).
Together, these results show that the administration of both antibodies has a beneficial effect in a C. elegans model of Aβ42-mediated dysfunction. Furthermore, the antibody-specific inhibition of Aβ42 toxicity observed in vivo is consistent with the different effects that the two antibodies have on the microscopic processes in vitro. In particular, DesAb18–25 preferentially affects primary nucleation in vitro, which is the critical process of formation of oligomers at early stages of the aggregation process of Aβ42 (13). In agreement with this observation, we have found that DesAb18–25 is most effective when administered at the onset of the disease when the worms are still relatively young. Similarly, DesAb29–36 is a potent inhibitor of secondary nucleation in vitro and suppresses the effects of Aβ42 aggregation most effectively in vivo when administered at late stages of the worm maturation. This result is in agreement with the in vitro observation that secondary nucleation becomes the predominant mechanism of generation of Aβ42 oligomers once a critical mass of aggregates has been formed (13), which happens later on in the maturation of the worms.
CONCLUSIONS
We have described an antibody scanning strategy for the rapid production of small antibody libraries with full coverage of given target proteins. Because this strategy is based on rational design, the resulting libraries can be of very small size, which makes them particularly amenable to quantitative screening procedures. We have illustrated the efficacy of this approach by generating a pool of antibodies designed to scan the sequence of Aβ42 and then by using kinetic methods to test the ability of these antibodies to inhibit specific microscopic steps in Aβ42 aggregation. This analysis has revealed that all DesAbs have significant effects on Aβ42 aggregation in vitro, and it has allowed us to identify two antibodies that target, respectively, the primary and secondary nucleation of the aggregation process with high selectivity. We have then confirmed that these in vitro results are fully consistent with the effects of the two antibodies in vivo using a C. elegans model of Aβ42-mediated toxicity.
A particularly important aspect of the antibody scanning approach that we have presented is that to design the antibody panel and to identify the most effective antibodies, we did not exploit any a priori knowledge about the structures of the aggregates or about the sequence regions that are most important in determining their formation. The only information that we have used concerned solely the amino acid sequence of Aβ42. Although we have illustrated here the antibody scanning technique by primarily addressing kinetic questions, namely, to identify antibodies that are capable of interfering with specific microscopic steps in the aggregation of Aβ42, in principle it should also be possible to use them to obtain insights into the structural properties of Aβ42 monomers, oligomers, and fibrils. However, these structural studies are complicated by the possibility that the conformational properties of Aβ42 may be affected by the binding of the antibodies. For example, a specific insight that we can already obtain from the present results is that DesAb29–36, which inhibits secondary nucleation, is likely to do so by interacting with secondary oligomers, rather than with the fibril surfaces, because its epitope is not exposed in the two recently reported structures of Aβ42 fibrils (48, 49).
We anticipate that the strategy that we have introduced may find applications for the effective rational identification of a wide range of candidate protein therapeutics against neurodegenerative diseases. More generally, we may also expect that the antibody scanning method will help the functional characterization of proteins by monitoring the changes in their activity when specific regions of their sequences are bound to the corresponding antibodies.
MATERIALS AND METHODS
Design of the antibodies
We summarize here the computational method that we have developed for the identification of complementary peptides that bind to specific linear epitopes in target proteins of interest, which we have grafted onto the CDR loops of domain antibodies. A detailed description of the method is provided by Sormanni et al. (38), together with additional experimental validation. The complementary peptide design procedure consisted of two steps. First, given a target linear epitope, we collected from the Protein Data Bank (PDB) all protein fragments that face in a β strand any subsequence of at least three residues in which the target epitope can be fragmented. Second, complementary peptides predicted to bind the target epitope were built by merging together these fragments using a “cascade method.” In essence, this cascade method started from one of these fragments and extended it to the length of the target epitope by linking it to some of the others. Fragments were linked using three rules: (i) Fragments can be joined together only if found in β strands of the same type (that is, parallel or antiparallel), (ii) all fragments making up a complementary peptide must partly overlap with their neighboring fragments, and (iii) the overlapping regions must be identical both in the sequence and in the backbone hydrogen-bond pattern that is extracted from the β strand where each fragment is found.
Because the identification of the complementary peptides is based on the analysis of amino acid sequences facing each other in β strands in the PDB, the interaction with the target sequence is already shown to be viable in a biological context. In addition, given this design strategy, the resulting complementary peptides are expected to bind the target epitope by enforcing a β strand–like conformation. Therefore, these complementary peptides will be particularly effective in binding to solvent-exposed regions of protein sequences that do not form persistent hydrogen bonds with other parts of the protein molecule, such as in the case of disordered regions (38).
Protein expression and purification
The various complementary peptides were grafted into the CDR3 loop of the DesAb scaffold by means of mutagenic polymerase chain reaction with phosphorylated oligonucleotides (38). The different DesAb constructs were then expressed and purified using pRSET-B vector in E. coli Rosetta-gami 2 (DE3) (Merck Millipore), as previously described (38). Cells were grown for 15 hours at 30°C using Overnight Express Instant TB Medium (Merck Millipore) supplemented with ampicillin (100 μg/ml). Cells were harvested by centrifugation; resuspended in 8 mM Na2HPO4, 15 mM KH2PO4, 137 mM NaCl, and 3 mM KCl (pH 7.3) [phosphate-buffered saline (PBS)] with the addition of one EDTA-Free Complete Protease Inhibitor Cocktail Tablet (Roche) per 500 ml of cell growth; and lysed using sonication. Cell debris was removed using centrifugation at 15,000 rpm (JA-20 rotor, Beckman Coulter). The cleared lysate was loaded onto a Ni2+-NTA Superflow column (Qiagen), previously equilibrated with PBS containing 10 mM imidazole. After washing with PBS containing 40 mM imidazole, the His-tagged DesAbs were eluted with PBS containing 200 mM imidazole and dialyzed extensively against PBS. For all the protein variants used in this study, protein concentration was determined by absorbance measurement at 280 nm using theoretical extinction coefficients calculated with ExPASy ProtParam (52). Aβ42 peptides were expressed in E. coli BL21 (DE3) Gold Strain (Agilent Technologies) and purified, as described previously (13). Aliquots of purified Aβ42 were lyophilized and stored at −80°C.
Circular dichroism
Far-ultraviolet (UV) CD spectra of the DesAbs were recorded using a Jasco J-810 spectropolarimeter equipped with a Peltier holder, using a 0.1-cm-pathlength cuvette. Samples contained 10 μM protein in PBS. The far-UV CD spectra of all DesAbs were recorded from 200 to 250 nm at 20°C, and the spectrum of the buffer was systematically subtracted from the spectra of all DesAbs.
The structural stability of the DesAbs was analyzed by monitoring the CD signal at 207 nm from 20° to 98°C at a rate of 0.5°C min−1. Data points were acquired every 0.1°C with a bandwidth of 1 nm. Analysis of the thermal unfolding curves was performed, assuming a two-state unfolding model.
BLI measurements
The binding between the various DesAbs and monomeric Aβ42 was assayed by means of BLI experiments with a ForteBio Octet RED96 (Pall ForteBio LLC). Specifically, SA biosensor tips were coated with monomeric N-terminal biotinylated Aβ42 (AnaSpec) by incubation in peptide solution (15 μg/ml) for 10 s and then blocked in PBS, 0.1% bovine serum albumin (BSA), and 0.05% Tween 20 for 15 min.
The binding between the immobilized Aβ42 peptide and the DesAbs was monitored in the presence of 5 μM antibody solution in PBS, 0.1% BSA, and 0.02% Tween 20 for 250 s. Then, the dissociation of the DesAbs was monitored in PBS, 0.1% BSA, and 0.05% Tween 20 for 250 s.
The binding of DesAb18–25 and DesAb29–36 to the target peptides (ChinaPeptides) Aβ18–25 (Ac-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-NH2) and Aβ29–36 (Ac-Gly-Ala-Ile-Ile-Gly-Leu-Met-Val-NH2), respectively, was analyzed as follows: Ni-NTA biosensor tips were coated with DesAb18–25 or DesAb29–36 by incubation in an antibody solution (15 μg/ml) for 10 s. DesAb18–25 or DesAb29–36 association and dissociation with 6 and 20 μM peptide, respectively, were monitored in PBS, 1% BSA, and 0.05% Tween 20. As a control of specificity, the binding of DesAb18–25 to Aβ29–36 and that of DesAb29–36 to Aβ18–25 were also tested.
The binding of the different DesAbs to monomeric α-synuclein was analyzed as follows: Ni-NTA biosensor tips were coated with the DesAbs by incubation in an antibody solution (15 μg/ml) for 10 s. The association and dissociation between the immobilized DesAbs and 20 μM α-synuclein were monitored for 300 s in PBS, 1% BSA, and 0.05% Tween 20. In all cases, the binding curves were corrected by subtracting nonspecific binding to the biosensor tips.
Biotin-mediated affinity measurements and fluorescence competition assay
Samples containing 2 μM DesAbs and increasing concentration of monomeric or fibrillar N-terminal biotinylated Aβ42 (0, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, 6.4, and 12.8 μM) were incubated overnight at room temperature. The DesAb/biotinylated Aβ42 complex was then isolated from the solution by incubating the 1:1000 diluted samples in an ELISA plate (Thermo Fisher Scientific), which was previously coated with 50 ng of Alexa 488–streptavidin (Thermo Fisher Scientific) and blocked with 10% (w/v) BSA in PBS (BSA/PBS) overnight at 4°C, following six times washing with PBS. The uniformity of the coating reaction was verified by measuring the fluorescence of Alexa 488 in each well using a CLARIOstar plate reader (BMG Labtech) (fig. S3).
Samples were then removed from the plate, and the DesAb left in the solution was quantified by measuring the maximal intrinsic fluorescence between 300 and 400 nm (≈350 nm) upon excitation at 285 nm. The concentrations of DesAbs and Aβ42 and the excitation and the emission wavelengths used for the analysis were selected to attenuate possible artifacts due to the high sensitivity of tryptophans to small environmental changes in general and to minimize the contribution of the only tyrosine of Aβ42 to the intrinsic fluorescence measurements (fig. S13). The decrease in fluorescence signal was plotted as a function of Aβ42 concentration and analyzed assuming single-site binding using
where ΘL is the fraction of bound ligand, ΔF is the decrease in fluorescence intensity observed at a given concentration of DesAb, ΔFmax is the maximal decrease in fluorescence at saturation, LT and PT are the total ligand and protein concentrations, respectively, and Kd is the apparent dissociation constant of the complex. The fraction of bound ligand was plotted as a function of protein concentration to compare affinities between the different DesAbs.
The fluorescence competition assay between DesAb18–25 and DesAb15–21 [referred to as DesAb-Aβ by Sormanni et al. (38)] was performed as follows. DesAb18–25 was labeled with fluorophore Alexa 647 using the Alexa Fluor 647 Antibody Labeling Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The conformation of the antibody after the labeling reaction was assessed by CD using the same protocol described in the “Circular dichroism” section (fig. S4C). Samples containing 50 nM monomeric N-terminal biotinylated Aβ42 and 50 nM Alexa 647–DesAb18–25 were incubated in the presence of increasing concentrations of DesAb15–21 (0, 100, 200, 400, 800, and 1600 nM) for 2 hours at room temperature. The Alexa 647–DesAb18–25/biotinylated Aβ42 complex was then isolated from the solution by incubating the samples in an ELISA plate (Thermo Fisher Scientific) previously coated with 50 ng per well of Alexa 488–streptavidin (Thermo Fisher Scientific), as described above. After incubation, the samples were transferred in a second plate and the unbound Alexa 647–DesAb18–25 left in the solution was quantified by measuring the fluorescence of Alexa 647 of each well using a CLARIOstar plate reader (BMG Labtech). The same procedure was applied for samples in which biotinylated Aβ42 was not present and the fluorescence value obtained from these samples was assumed to be the fluorescence of 100% unbound Alexa 647–DesAb18–25. The fraction of bound Alexa 647–DesAb18–25 at each concentration of DesAb15–21 was then determined using
where ΘL is the fraction of bound ligand (Alexa 647–DesAb18–25), FBAβ is the fluorescence intensity of Alexa 647–DesAb18–25 of samples containing biotinylated Aβ42, and F−BAβ is the fluorescence intensity of Alexa 647–DesAb18–25 of samples in which biotinylated Aβ42 was not added, at a given concentration of DesAb15–21. The data were then fitted using an inhibition model with variable slope using the program Prism 6 (GraphPad Software).
Aggregation assays
The lyophilized Aβ42 peptide was dissolved in 6 M urea (pH 8.5) and incubated for 30 min at room temperature. This protein solution was then subjected to gel filtration using a Superdex 75 10/300 GL column (GE Healthcare), and the peak corresponding to the monomeric Aβ42 peptide was collected in low-binding test tubes (Corning) on ice (13).
Monomeric Aβ42 peptides were used to prepare solutions at a protein concentration of 2 μM in the presence of increasing amounts of the specified DesAb variant in 20 mM sodium phosphate buffer (pH 8), 200 μM EDTA, and 0.02% NaN3, supplemented with 6 μM ThT (13). Seeded experiments were performed in the presence of 10% preformed fibrils (23) with a 0:1 or 1:1 antibody–to–Aβ42 monomer ratio. Each sample was then pipetted into multiple wells of a 96-well half-area plate of black polystyrene with a clear bottom and polyethylene glycol coating (Corning) (90 μl per well). Plates were sealed to prevent evaporation. Aggregation assays were performed at 37°C under quiescent conditions using a CLARIOstar plate reader (BMG Labtech). The ThT fluorescence was measured through the bottom of the plate every minute with an excitation filter of 440 nm and an emission filter of 480 nm.
Kinetic analysis
The time evolution of the total fibril mass concentration, M(t), in the absence of seeds is described by the following integrated rate law
where the kinetic parameters B±, C±, k, k∞, and are described in detail in the study of Cohen et al. (20) and are functions of the two combinations of the microscopic rate constants k+k2 and knk2, where kn, k+, and k2 are the primary nucleation, elongation, and secondary nucleation rate constants, respectively.
The DesAbs can perturb the aggregation process by inhibiting one or more of the individual microscopic reactions. We can identify the microscopic events that are inhibited by the DesAbs by applying the above equation to describe the macroscopic aggregation profiles shown in Fig. 3 and comparing the set of microscopic rate constants k+k2 and knk2 required to describe the time evolution of the fibril formation in the absence and presence of antibody.
The decrease in the parameter k+ in the presence of DesAbs was calculated from the seeded experiments. On this purpose, we evaluated the rate of the formation of fibrillar aggregates within the first 10% of monomer conversion, r = 2k+P0m, where P0 is the number of seeds introduced in the system and m is the initial monomer concentration. The relative decrease in the apparent rate constant k+ was evaluated by dividing the rate in the presence of DesAbs with the value calculated in their absence. Finally, the decreases in kn and k2 (Table 2) were calculated by dividing the decreases in k+k2 and k+kn (Table 2) obtained under unseeded conditions by the decrease in k+ derived from the seeded aggregation profiles.
The perturbation of the different microscopic reaction rates has markedly different effects on the generation of low–molecular weight oligomeric species. To illustrate this behavior, we calculated the time evolution of the rate of generation of new fibrils (via on-pathway oligomers) from monomers according to the nucleation rate rn(t) given by rn(t) = knm(t)nc + k2M(t)m(t)n2. Because a negligible amount of oligomers was detectable in the system at the end of the reaction, the total number of fibrils present at the end of the process was indicative of the total number of on-pathway oligomers generated during the reaction. This value was calculated by integrating the nucleation rate rn(t) over the reaction.
Strains of C. elegans
The following strains were used in the present work to assess the effect of the antibodies on the toxicity of Aβ42 aggregates:
(1) GMC101; genotype dvIs100 [unc-54p::A-beta-1-42::unc-54 3′-UTR + mtl-2p::GFP]; mtl-2p::GFP constitutively expresses the green fluorescent protein (GFP) in intestinal cells; unc-54p::A-beta-1-42 expresses the human full-length Aβ42 peptide in body wall muscle cells. Shifting L4 or young adult animals from 20° to 25°C promotes Aβ42 aggregation and causes the paralysis of the worms (51).
(2) N2, wild-type C. elegans var Bristol, in this work referred to as control worms or wild-type worms. Generation time is about 3 days. Brood size is about 350 [isolated from mushroom compost near Bristol, England by L. N. Staniland (53)].
(3) OW40 [strain expressing yellow fluorescent protein (YFP)–α-synuclein] genotype zgIs15 [P(unc-54)::α-syn::YFP]IV. In OW40, α-synuclein fused to YFP relocates to inclusions, which are visible as early as day 2 of adulthood and increase in number and size during the aging of the animals (54).
Propagation conditions of C. elegans
C. elegans worms were propagated using standard conditions (53). Briefly, the animals were synchronized by hypochlorite bleaching, hatched overnight in M9 [KH2PO4 (3 g/liter), Na2HPO4 (6 g/liter), NaCl (5 g/liter), and 1 μM MgSO4] buffer, and subsequently cultured at 20°C on nematode growth medium (NGM) [1 mM CaCl2, 1 mM MgSO4, cholesterol (5 μg/ml), 250 μM KH2PO4 (pH 6), Bacto agar (17 g/liter), NaCl (3 g/liter), and casein (7.5 g/liter)] plates seeded with the E. coli strain OP50. Saturated cultures of OP50 were grown by inoculating 50 ml of LB medium [Bacto tryptone (10 g/liter), NaCl (10 g/liter), and Bacto yeast extract (5 g/liter)] with OP50 and incubating the culture for 16 hours at 37°C. NGM plates were seeded with bacteria by adding 350 μl of saturated OP50 to each plate and leaving the plates at 20°C for 2 to 3 days. On day 3 after synchronization, the animals were placed on NGM plates containing 5-fluoro-2′deoxyuridine (FUDR) (75 μM, unless stated otherwise) to inhibit the growth of offspring.
Antibody transduction protocol
About 500 C. elegans worms were incubated overnight in M9 with 20 μM of m-cherry protein (ABE3463, Bioscience Lifesciences) and 40 μM of PULSin (PolyPlus-transfection SA) in a final volume of 1 ml. Motility or imaging procedures were carried out 12 hours after transduction. All experiments were carried out in triplicate, and one experiment that is representative of the three measured is shown.
Automated motility assay on agar plates and imaging of the aggregates
All C. elegans populations were cultured at 20°C and developmentally synchronized from a 4-hour egg lay. At 64 to 72 hours after egg lay (time zero), individuals were shifted to 24°C and transferred to FUDR plates and body movements were assessed over the times indicated. At different ages, specifically at days 1 and 3 [administration protocol 1 (AP1)] or at day 6 (AP2), the animals were washed off the plates and incubated with 20 μM DesAbs overnight in M9 buffer. At day 7, the worms were spread over an OP50 unseeded 9-cm plate, and their movements were recorded at 30 fps (frames per second) using a novel microscopic setup for 30 s or 1 min. Up to 1000 animals were counted in each experiment, unless stated otherwise. Videos were analyzed using a custom-made tracking code. In the case of the AD1, the worms were also screened at day 4 to assess whether the antibodies lose efficacy at day 7 as a consequence of degradation, which may be significant in the case of experiments involving a long period between administration and screening. The total fitness was calculated by summing the mobility, speed, and viability of the worms. Fingerprint and total fitness values were normalized using the values of the control worms.
To visualize the amount of aggregates in the worms, live transgenic worms at day 9 of adulthood were incubated with 1 μM NIAD-4 (0.1% dimethyl sulfoxide in M9 buffer) for 4 hours at room temperature. After staining, animals were allowed to recover on NGM plates for about 24 hours (day 10 of adulthood) to allow destaining via normal metabolism. Stained animals were mounted on 2% agarose pads containing 40 mM NaN3 as anesthetic on glass microscope slides for imaging. Images were captured using a Zeiss Axio Observer D1 fluorescence microscope (Carl Zeiss Microscopy GmbH) with a 20× objective and a 49004 ET-CY3/TRITC filter (Chroma Technology Corp.). Fluorescence intensity was calculated using ImageJ software (National Institutes of Health) and then normalized as the corrected total cell fluorescence. Only the head region was considered because of the high background signal in the guts. All experiments were carried out in triplicate, and the data from one representative experiment are shown. Statistical significance was determined using t tests.
Supplementary Material
Acknowledgments
We thank the biophysical facility and K. Stott at the Department of Biochemistry of the University of Cambridge for the use of the BLI instrument, G. Heller for helpful discussion, and S. Preet and E. Klimont for assistance in protein purification. Funding: This work was supported by the Centre for Misfolding Diseases, University of Cambridge. F.A.A. was supported by a Senior Research Fellowship Award from the Alzheimer’s Society, UK (grant number 317, AS-SF-16-003). Author contributions: F.A.A., P.S., and M.V. designed the research. F.A.A., P.S., M.P., and P.A. performed the research. F.A.A., P.S., M.P., P.A., S.L., T.P.J.K., C.M.D., and M.V. analyzed the data. F.A.A., P.S., M.P., P.A., S.L., T.P.J.K., C.M.D., and M.V. wrote the paper. Competing interests: M.V., F.A.A., and P.S. are authors on a patent held by Cambridge Enterprise LTD related to this work (WO2016091765; published 16 June 2016). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700488/DC1
fig. S1. Purified DesAbs used in this study.
fig. S2. BLI analysis of the interaction of different DesAbs with monomeric α-synuclein.
fig. S3. Biotin-mediated affinity measurement of DesAb3–9 binding to monomeric Aβ42 and setup of the experimental conditions.
fig. S4. DesAb binding specificity assessment and interaction of DesAb18–25 and DesAb29–36 with the respective target peptides.
fig. S5. A DesAb designed to target α-synuclein does not inhibit Aβ42 aggregation.
fig. S6. Effect of the DesAbs on the global parameters k+kn and k+k2 of Aβ42 aggregation.
fig. S7. Transduction of the fluorescent protein mCherry into wild-type worms.
fig. S8. Effects of DesAb18–25 and DesAb29–36 treatments on the C. elegans worms.
fig. S9. Fingerprints of the Aβ42 worms screened at day 4 of adulthood.
fig. S10. Effects of DesAb18–25 and DesAb29–36 treatments on wild-type control worms.
fig. S11. Analysis on the specificity of the treatment with the DesAbs in C. elegans.
fig. S12. Effects of DesAb18–25 and DesAb29–36 treatments on the aggregation of Aβ42 in C. elegans models.
fig. S13. Difference between the spectrum of DesAb18–25 and the background.
movie S1. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with empty vesicles at days 1 and 3 (AP1, early treatment).
movie S2. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with empty vesicles at day 6 (AP2, late treatment).
movie S3. Representative video clip of the control C. elegans worms N2 at day 7 upon treatment with empty vesicles at day 6 (AP2, late treatment).
movie S4. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb18–25 at days 1 and 3 (AP1, early treatment).
movie S5. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb29–36 at days 1 and 3 (AP1, early treatment).
movie S6. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb18–25 at day 6 (AP2, late treatment).
movie S7. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb29–36 at day 6 (AP2, late treatment).
REFERENCES AND NOTES
- 1.Hardy J., Selkoe D. J., The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Aguzzi A., O’Connor T., Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 9, 237–248 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Selkoe D. J., Hardy J., The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sala Frigerio C., De Strooper B., Alzheimer’s disease mechanisms and emerging roads to novel therapeutics. Annu. Rev. Neurosci. 39, 57–79 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Knowles T. P. J., Vendruscolo M., Dobson C. M., The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Arosio P., Vendruscolo M., Dobson C. M., Knowles T. P. J., Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Lemere C. A., Masliah E., Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nat. Rev. Neurol. 6, 108–119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pul R., Dodel R., Stangel M., Antibody-based therapy in Alzheimer’s disease. Expert Opin. Biol. Ther. 11, 343–357 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P., Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 (1999). [DOI] [PubMed] [Google Scholar]
- 10.DeMattos R. B., Bales K. R., Cummins D. J., Dodart J.-C., Paul S. M., Holtzman D. M., Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 8850–8855 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karran E., Hardy J., A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 76, 185–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knowles T. P. J., Waudby C. A., Devlin G. L., Cohen S. I. A., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., Dobson C. M., An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J., Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M., Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G., Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Haass C., Selkoe D. J., Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Cremades N., Cohen S. I. A., Deas E., Abramov A. Y., Chen A. Y., Orte A., Sandal M., Clarke R. W., Dunne P., Aprile F. A., Bertoncini C. W., Wood N. W., Knowles T. P. J., Dobson C. M., Klenerman D., Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149, 1048–1059 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lesné S., Teng Koh M., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H., A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Benilova I., Karran E., De Strooper B., The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 15, 349–357 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Cohen S. I. A., Arosio P., Presto J., Kurudenkandy F. R., Biverstål H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P. J., Linse S., A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arosio P., Michaels T. C. T., Linse S., Månsson C., Emanuelsson C., Presto J., Johansson J., Vendruscolo M., Dobson C. M., Knowles T. P. J., Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Habchi J., Arosio P., Perni M., Costa A. R., Yagi-Utsumi M., Joshi P., Chia S., Cohen S. I. A., Müller M. B. D., Linse S., Nollen E. A. A., Dobson C. M., Knowles T. P. J., Vendruscolo M., An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer’s disease. Sci. Adv. 2, e1501244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Habchi J., Chia S., Limbocker R., Mannini B., Ahn M., Perni M., Hansson O., Arosio P., Kumita J. R., Challa P. K., Cohen S. I. A., Linse S., Dobson C. M., Knowles T. P. J., Vendruscolo M., Systematic development of small molecules to inhibit specific microscopic steps of Abeta42 aggregation in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 114, E200–E208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradbury A. R. M., Sidhu S., Dübel S., McCafferty J., Beyond natural antibodies: The power of in vitro display technologies. Nat. Biotechnol. 29, 245–254 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoogenboom H. R., Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 23, 1105–1116 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Lee C. C., Perchiacca J. M., Tessier P. M., Toward aggregation-resistant antibodies by design. Trends Biotechnol. 31, 612–620 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Miersch S., Sidhu S. S., Synthetic antibodies: Concepts, potential and practical considerations. Methods 57, 486–498 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Sidhu S. S., Phage display in pharmaceutical biotechnology. Curr. Opin. Biotechnol. 11, 610–616 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Winter G., Griffiths A. D., Hawkins R. E., Hoogenboom H. R., Making antibodies by phage display technology. Annu. Rev. Immunol. 12, 433–455 (1994). [DOI] [PubMed] [Google Scholar]
- 30.Carter P. J., Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 317, 1261–1269 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Elvin J. G., Couston R. G., van der Walle C. F., Therapeutic antibodies: Market considerations, disease targets and bioprocessing. Int. J. Pharm. 440, 83–98 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Goodman M., Market watch: Sales of biologics to show robust growth through to 2013. Nat. Rev. Drug Discov. 8, 837 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Leader B., Baca Q. J., Golan D. E., Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 7, 21–39 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Pavlou A. K., Reichert J. M., Recombinant protein therapeutics—Success rates, market trends and values to 2010. Nat. Biotechnol. 22, 1513–1519 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg S. A., Yang J. C., Restifo N. P., Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 10, 909–915 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couzin-Frankel J., Cancer immunotherapy. Science 342, 1432–1433 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Scott A. M., Wolchok J. D., Old L. J., Antibody therapy of cancer. Nat. Rev. Cancer 12, 278–287 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Sormanni P., Aprile F. A., Vendruscolo M., Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 112, 9902–9907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aprile F. A., Sormanni P., Vendruscolo M., A rational design strategy for the selective activity enhancement of a molecular chaperone toward a target substrate. Biochemistry 54, 5103–5112 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Bergh J., Zetterström P., Andersen P. M., Brännström T., Graffmo K. S., Jonsson P. A., Lang L., Danielsson J., Oliveberg M., Marklund S. L., Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc. Natl. Acad. Sci. U.S.A. 112, 4489–4494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohen S. I. A., Vendruscolo M., Dobson C. M., Knowles T. P. J., From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 421, 160–171 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Sevigny J., Chiao P., Bussière T., Weinreb P. H., Williams L., Maier M., Dunstan R., Salloway S., Chen T., Ling Y., O’Gorman J., Qian F., Arastu M., Li M., Chollate S., Brennan M. S., Quintero-Monzon O., Scannevin R. H., Moore Arnold H., Engber T., Rhodes K., Ferrero J., Hang Y., Mikulskis A., Grimm J., Hock C., Nitsch R. M., Sandrock A., The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Heller G. T., Sormanni P., Vendruscolo M., Targeting disordered proteins with small molecules using entropy. Trends Biochem. Sci. 40, 491–496 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Clackson T., Hoogenboom H. R., Griffiths A. D., Winter G., Making antibody fragments using phage display libraries. Nature 352, 624–628 (1991). [DOI] [PubMed] [Google Scholar]
- 45.Sormanni P., Aprile F. A., Vendruscolo M., The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 427, 478–490 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Barthelemy P. A., Raab H., Appleton B. A., Bond C. J., Wu P., Wiesmann C., Sidhu S. S., Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J. Biol. Chem. 283, 3639–3654 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Ladiwala A. R. A., Bhattacharya M., Perchiacca J. M., Cao P., Raleigh D. P., Abedini A., Schmidt A. M., Varkey J., Langen R., Tessier P. M., Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc. Natl. Acad. Sci. U.S.A. 109, 19965–19970 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wälti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Böckmann A., Güntert P., Meier B. H., Riek R., Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colvin M. T., Silvers R., Zhe Ni Q., Can T. V., Sergeyev I., Rosay M., Donovan K. J., Michael B., Wall J., Linse S., Griffin R. G., Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szczepankiewicz O., Linse B., Meisl G., Thulin E., Frohm B., Frigerio C. S., Colvin M. T., Jacavone A. C., Griffin R. G., Knowles T., Walsh D. M., Linse S., N-terminal extensions retard Aβ42 fibril formation but allow cross-seeding and coaggregation with Aβ42. J. Am. Chem. Soc. 137, 14673–14685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McColl G., Roberts B. R., Pukala T. L., Kenche V. B., Roberts C. M., Link C. D., Ryan T. M., Masters C. L., Barnham K. J., Bush A. I., Cherny R. A., Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 7, 57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gill S. C., Von Hippel P. H., Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 (1989). [DOI] [PubMed] [Google Scholar]
- 53.Brenner S., The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Ham T. J., Thijssen K. L., Breitling R., Hofstra R. M. W., Plasterk R. H. A., Nollen E. A. A., C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLOS Genet. 4, e1000027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700488/DC1
fig. S1. Purified DesAbs used in this study.
fig. S2. BLI analysis of the interaction of different DesAbs with monomeric α-synuclein.
fig. S3. Biotin-mediated affinity measurement of DesAb3–9 binding to monomeric Aβ42 and setup of the experimental conditions.
fig. S4. DesAb binding specificity assessment and interaction of DesAb18–25 and DesAb29–36 with the respective target peptides.
fig. S5. A DesAb designed to target α-synuclein does not inhibit Aβ42 aggregation.
fig. S6. Effect of the DesAbs on the global parameters k+kn and k+k2 of Aβ42 aggregation.
fig. S7. Transduction of the fluorescent protein mCherry into wild-type worms.
fig. S8. Effects of DesAb18–25 and DesAb29–36 treatments on the C. elegans worms.
fig. S9. Fingerprints of the Aβ42 worms screened at day 4 of adulthood.
fig. S10. Effects of DesAb18–25 and DesAb29–36 treatments on wild-type control worms.
fig. S11. Analysis on the specificity of the treatment with the DesAbs in C. elegans.
fig. S12. Effects of DesAb18–25 and DesAb29–36 treatments on the aggregation of Aβ42 in C. elegans models.
fig. S13. Difference between the spectrum of DesAb18–25 and the background.
movie S1. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with empty vesicles at days 1 and 3 (AP1, early treatment).
movie S2. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with empty vesicles at day 6 (AP2, late treatment).
movie S3. Representative video clip of the control C. elegans worms N2 at day 7 upon treatment with empty vesicles at day 6 (AP2, late treatment).
movie S4. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb18–25 at days 1 and 3 (AP1, early treatment).
movie S5. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb29–36 at days 1 and 3 (AP1, early treatment).
movie S6. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb18–25 at day 6 (AP2, late treatment).
movie S7. Representative video clip of the Aβ42 C. elegans worms GMC101 at day 7 upon treatment with DesAb29–36 at day 6 (AP2, late treatment).