

Abstract
Studies indicate that elevated interleukin-6 (IL-6) levels engage IL6Rα-gp130 receptor complexes to activate signal transducer and activator of transcription 3 (STAT3) that is hyperactivated in many cancers including head and neck squamous cell carcinoma (HNSCC). Our previous HCS campaign identified several hits that selectively blocked IL-6-induced STAT3 activation. This study describes our investigation of the mechanism(s) of action of three of the four chemical series that progressed to lead activities: a triazolothiadiazine (864669), amino alcohol (856350), and an oxazole-piperazine (4248543). We demonstrated that all three blocked IL-6-induced upregulation of the cyclin D1 and Bcl-XL STAT3 target genes. None of the compounds exhibited direct binding interactions with STAT3 in surface plasmon resonance (SPR) binding assays; neither did they inhibit the recruitment and binding of a phospho-tyrosine-gp130 peptide to STAT3 in a fluorescence polarization assay. Furthermore, they exhibited little or no inhibition in a panel of 83 cancer-associated in vitro kinase profiling assays, including lack of inhibition of IL-6-induced Janus kinase (JAK 1, 2, and 3) activation. Further, 864669 and 4248543 selectively inhibited IL-6-induced STAT3 activation but not that induced by oncostatin M (OSM). The compounds 864669 and 4248543 abrogated IL-6-induced phosphorylation of the gp130 signaling subunit (phospho-gp130Y905) of the IL-6-receptor complex in HNSCC cell lines which generate docking sites for the SH2 domains of STAT3. Our data indicate that 864669 and 4248543 block IL-6-induced STAT activation by interfering with the recruitment, assembly, or activation of the hexamer-activated IL-6/IL-6Rα/gp130 signaling complex that occurs after IL-6 binding to IL-6Rα subunits.
Electronic supplementary material
The online version of this article (doi:10.1007/s12154-017-0169-9) contains supplementary material, which is available to authorized users.
Keywords: STAT3 pathway, Interleukin-6 (IL-6), STAT3 inhibitors, Head and neck cancer
Introduction
Signal transducer and activation of transcription 3 (STAT3) belongs to a family of transcription factors activated by a variety of cytokines and soluble growth factors including the interleukin 6 (IL-6) family cytokines and many others [6]. IL-6, a multifunctional cytokine, exerts its action through interleukin 6 receptor (IL-6R) complexed with gp130 which thereupon dimerizes and initiates intracellular signaling [29], through activated Janus kinases (JAKs) which phosphorylate STAT3.
STAT3 activation has been implicated downstream of IL-6 receptor complex engagement leading to numerous alterations required for tumor growth [11, 47]. Numerous studies have demonstrated that phosphorylation at a single tyrosine residue (Y705) in the transactivation domain of STAT3 contributes to the development, progression, and maintenance of many cancers including head and neck squamous cell carcinoma (HNSCC) [1, 17] and correlated with a shorter overall survival in a cohort of patients [16, 53]. Thus, STAT3 is considered a novel therapeutic target due to its association with malignant transformation. Although different strategies such as phosphopeptide inhibitor (PY*LKTK); peptidomimetic analog; ISS 610 [45, 46]; small-molecule inhibitors such as STA-21, S3I-201, and Stattic [37, 41]; and several oligonucleotide approaches such as antisense RNA or siRNA approaches [9, 22, 27] have been developed to target STAT3, only a few have progressed to in vivo efficacy, pharmacology, or toxicity testing [52]. STAT3 decoy oligonucleotide is another novel strategy that is being used to inhibit the activity of several target transcription factors [39, 52]. A chemically modified cyclic STAT3 decoy resulted in a stable therapeutic compound that retained potency and target specificity and demonstrated anti-tumor activity after systemic delivery [39] with no apparent toxicity [38], suggesting that clinical development of the cyclic STAT3 decoy may yield an effective therapeutic outcome.
We previously reported a high content screening campaign that identified four chemical series that selectively inhibited IL-6-induced pSTAT3 activation over interferon gamma (IFNγ)-induced pSTAT1 activation, inhibited the growth of HNSCC cell lines, and were progressed into chemistry hit-to-lead activities. The present studies were carried out to delineate the mechanism(s) of action of three representatives of these series: the triazolothiadiazines (864669), the amino alcohols (856350), and the oxazole-piperazines (4248543). Our results demonstrate that two of the series, triazolothiadiazine 864669 and oxazole-piperazine 4248543, selectively inhibit IL-6-induced pSTAT3Tyr705 activation in HNSCC cells relative to either oncostatin M (OSM)-induced pSTAT3Tyr705 activation or IFNγ-induced pSTAT1Tyr701 activation. The selective inhibition of IL-6-induced pSTAT3Tyr705 activation appears to involve inhibition of the phosphorylation of a specific tyrosine residue (Tyr 905) of the gp130 subunits in the activated IL-6 receptor complex that prevents the recruitment of latent STAT3 and the subsequent JAK-mediated phosphorylation of STAT3Tyr705 and ensuing downstream events.
Materials and methods
Cell culture
The HNSCC cell line Cal33 was a kind gift from Jean Louis Fischel, Centre Antoine Lacassagne, Nice, France. U266B1 lymphocytes were obtained from the ATTC (ATCC® TIB-196™).
Antibodies and reagents
Anti-pSTAT3Tyr705, anti-pSTAT1Tyr701, STAT3, and STAT1 antibodies were purchased from Cell Signaling Technology Inc. (Danvers, MA). Cyclin D1, Bcl-XL, and gp130 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-pgp130Tyr905, anti-PIAS3, anti-SOCS1, anti-SOCS3, and β-tubulin antibodies were purchased from Abcam Inc. (Cambridge, MA). Anti-phospho-JAK1, anti-phospho-JAK2, anti-phospho-JAK3, JAK1, JAK2, and JAK3 antibodies were purchased from Millipore (Billerica, MA). The goat anti-rabbit IgG (H+L)-horseradish peroxidase conjugate secondary antibody was from Bio-Rad Laboratories (Hercules, CA). Recombinant human interleukin-6 (IL-6), interferon gamma (IFNγ), and oncostatin M (OSM) were purchased from R&D Systems, Inc. (Minneapolis, MN).
Western blotting analysis of STAT3 signaling in HNSCC cells
Cal33 cells were treated with 3, 10, 30, and 100 μM concentrations of the compounds for 2 h and 45 min, followed by stimulation with IL-6 (50 ng/ml, 15 min) and harvested. Western analyses were done to determine pSTAT3Tyr705, STAT3, cyclin D1, Bcl-XL, pJAK1, pJAK2, pJAK3, JAK1, JAK2, and JAK3 protein expression. β-Tubulin was used as a loading control.
Kinome profiling
Kinase assays were conducted using the KinaseProfiler service of EMD Millipore.
Compound handling
864669 [23], 4248543, and 856350 and structurally related analogs were synthesized, purified, structurally verified, and provided by the University of Pittsburgh Chemical Diversity Center [23], or the determination of the 50% inhibition concentrations (IC50), 10-point 2-fold serial dilutions of test compounds in 100% DMSO were performed using a 384-well P30 dispensing head on the EP3 liquid handling platform as described previously [18, 19].
Automated pSTAT3 HCS assay protocols
The automated protocols for the pSTAT3 HCS assay has been described previously [18, 19].
Image acquisition on the ImageXpress ultra confocal automated imaging platform
For the pSTAT3 and pSTAT1 HCS assays, the IXU was set up to acquire two images using a 20× 0.45 NA ELWD objective in each of two fluorescent channels [18, 19].
Image analysis using the translocation-enhanced module
The fluorescent signal of Hoechst 33342-stained nuclei captured in channel 1 (Ch 1) images were used by the translocation-enhanced (TE) image analysis module to define an “inner” nuclear mask and an “outer” cytoplasm mask in channel 2 (Ch 2) [18, 19].
Effect of compounds on pgp130Tyr905 expression in HNSCC cell line by western analyses
Cal33 cells were plated, serum starved, and treated with 3, 10, 30, and 100 μM concentrations of the compounds for 2 h and 45 min followed by stimulation with IFN-γ (50 ng/ml, 15 min). Cells were harvested, and western analyses were done to determine pgp130Tyr905 and gp130 protein expression. β-Tubulin was used as a loading control.
Data processing, visualization, statistical analysis, and curve fitting
HCS data processing for the pSTAT3 activation assays were performed using ActivityBase™ (IDBS, Guildford, UK) and CytoMiner (UPDDI) as described previously [18, 19].
Supplemental methods
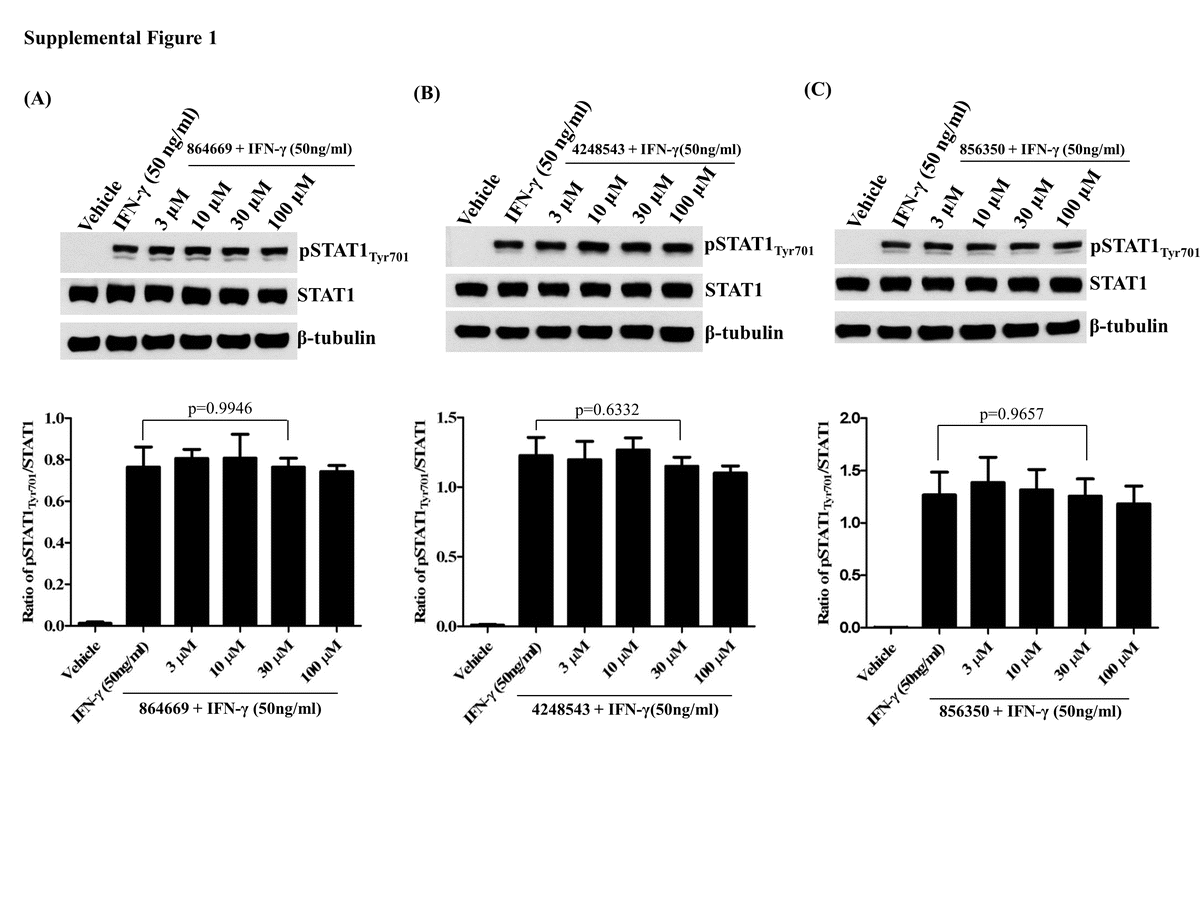
Effect of 864669 and 4248543 on STAT1 signaling in HNSCC cells
Cal33 cells were treated with 3, 10, 30, and 100 μM concentrations of the compounds for 2 h and 45 min followed by stimulation with IFN-γ (50 ng/ml, 15 min). The cells were harvested, and western analyses were done to probe for pSTAT1Tyr701 and STAT1. β-Tubulin was used as a loading control.
IL-6-receptor binding assay in U266 cell plasma membranes
The U266 B-lymphocyte cell line has previously been shown to express a high number of IL-6 receptor sites (11,000) per cell [43], and we developed a 96-well filtration format 125I-IL-6 binding assay using U266-derived plasma membranes isolated as described previously [4]. To prepare plasma membranes, U266B1 lymphocytes were grown, harvested, and centrifuged. Cell pellets were re-suspended in ice-cold buffer A (50 mM Tris-HCl, pH 7.4), mechanically sheared, and centrifuged. The supernatant was centrifuged to pellet the U266 membranes. The membrane pellets were then homogenized in buffer A, and then aliquots were flash frozen in liquid nitrogen. The protein concentration of the plasma membrane preparation was determined using a Pierce BCA Protein Assay Kit.
Binding reactions were performed in round-bottom 96-well plates (Costar) that contained 1% BSA (final), 25 μg of U266 plasma membranes, 100 nM–50 μM test compounds, and 1 nM 125I-IL6. 125I-IL6 binding was allowed to proceed on ice for 2 h with periodic agitation every 10–15 min. Non-specific background control wells contained all the reaction components except U266 plasma membranes.
Effect of 864669 and 4248543 on the negative regulators of STAT3
Cal33 cells were serum starved and treated with 3, 10, 30, and 100 μM concentrations of the compounds for 2 h and 45 min. Cells were stimulated with IL-6 (50 ng/ml, 15 min) and harvested, and cell lysates were analyzed to determine PIAS3, SOCS1, and SOCS3 protein expression by western analyses. β-Tubulin was used as a loading control.
Results
A high-content screening (HCS) campaign of a 95,000-compound subset of the NIH Molecular Library Screening Centers Network (MLSCN) small-molecule library identified four chemical hit series that selectively inhibited IL-6-induced STAT3 pathway activation in head and neck cancer cell lines that were subsequently progressed into hit-to-lead activities: the triazolothiadiazines, an oxazole-piperazine singleton, the amino alcohols, and the 2-guanidinoquinazolines [19, 23, 24]. We describe here our efforts to elucidate the mechanism(s) of action (MOA) of three representative hit compounds from the triazolothiadiazine, oxazole-piperazine, and amino alcohol series, namely, 864669, 4248543, and 856350, respectively.
Structures
Structures of 864669, 4248543, and 856350 are included in Fig. 1.
Fig. 1.

Structures of 864669, 4248543, and 856350
Inhibition of IL-6-induced pSTAT3Tyr705 activation and STAT3-mediated target gene expression
Consistent with the primary HCS assay [19], our western blotting analysis confirmed that all three compounds significantly inhibited IL-6-induced pSTAT3Tyr705 activation (p value ≤0.05) in Cal33 HNSCC cells in a concentration-dependent manner (Fig. 2a–c). For example, pre-exposure to 30 μM of the triazolothiadiazine 864669 inhibited IL-6-induced pSTAT3Tyr705 expression by 85% (p = 0.0036), cyclin D1 expression by 68% (p = 0.0378), and Bcl-XL expression by 40% (p = 0.0464) in Cal33 cells (Fig. 2a). Similarly, pre-treatment with 30 μM of the oxazole-piperazine 4248543 inhibited IL-6-induced pSTAT3Tyr705 expression by 62% (p = 0.0182), cyclin D1 expression by 78% (p = 0.0264), and Bcl-XL expression by 41% (p = 0.0545) in Cal33 cells (Fig. 2b). Treatment with 30 μM of the amino alcohol 856350 inhibited IL-6-induced pSTAT3Tyr705 expression by 62% (p = 0.0528) and cyclin D1 expression by 88% (p = 0.0077) in Cal33 cells (Fig. 2c). Although the 40% decrease in IL-6-induced Bcl-XL expression produced by 30 μM 856350 was not significant (p = 0.4069), the apparent inhibition of Bcl-XL expression exhibited a concentration-dependent trend (Fig. 2c). Importantly, these studies demonstrate that all three compounds inhibit STAT3-mediated transcriptional activation of target genes that promote cell proliferation and survival by inhibiting IL-6-induced STAT3 tyrosine phosphorylation rather than reducing STAT3 protein levels.
Fig. 2.

a 864669, b 4248543, and c 856350 downmodulates IL6-induced upregulation of pSTAT3Tyr705 and decreases STAT3 target gene expression. Cal33 cells were serum starved and treated with increasing concentrations of 864669, 4248543, and 856350, respectively. At the end of 2 h and 45 min, cells were stimulated with IL6 (50 ng/ml, 15 min) and harvested and protein lysates were prepared and ran on a 10% SDS-polyacrylamide gel. A significant decrease in pSTAT3Tyr705, cyclin D1, and Bcl-XL expression was observed in Cal33 cells treated with 864669 (Fig. 2a) and 4248543 (Fig. 2b), and 856350 (Fig. 2c) at 30 μM compared to cells stimulated with IL-6. The only exception was Bcl-XL expression (Fig. 2c) in Cal33 cells treated with 856350 where a dose-dependent decrease was observed although it was not significant. Results are from three separate experiments
Kinase inhibition
Activation of the IL-6 receptor complex by ligand binding triggers Janus kinase (JAK) auto-phosphorylation and activation, phosphorylation of specific intracellular tyrosine residues of the gp130 signaling subunit, recruitment of latent STAT3 via Src homology 2 (SH2) domain interactions, and ultimately JAK-mediated phosphorylation of STAT3Tyr705 [36]. The concentrations used for the kinase assay were the IC50 values determined earlier [18, 19]. Our work demonstrated that the compounds failed to alter the protein expression levels of IL-6-induced pJAK1, pJAK2, and pJAK3 or JAK1, JAK2, and JAK3 (Fig. 3a–c) as determined by western blot. In addition to cytokine receptor-associated JAKs, STAT3 can also be phosphorylated at the Tyr 705 residue by a number of growth factor receptor and SRC family tyrosine kinases [33, 49]. To evaluate whether any of the compounds were kinase inhibitors, we tested them in a KinaseProfiler panel of 83 in vitro kinase assays (EMD Millipore, now Eurofins Pharma Discovery Services UK Limited). At their respective cell-based IC50 concentrations, none of the compounds significantly inhibited any of the 83 kinases, including members of the JAK, SRC, and growth factor receptor tyrosine kinase families (data not shown), suggesting that these compounds are unlikely to exert their effects through kinase inhibition.
Fig. 3.

a 864669, b 4248543, and c 856350 do not inhibit JAKs in Cal33 cells. Cal33 cells were serum starved and treated with increasing concentrations of 864669, 4248543, and 856350, respectively. At the end of 2 h and 45 min, cells were stimulated with IL6 (50 ng/ml,15 min) and harvested and protein lysates were prepared and ran on a 10% SDS-polyacrylamide gel to determine the expression of pJAK1, JAK1, pJAK2, JAK2, pJAK3, and JAK3. β-Tubulin was used as a loading control. 864669 (Fig. 3a), 4248543 (Fig. 3b), and 856350 (Fig. 3c) do not inhibit any of the JAKs at the concentration tested. Results are from three separate experiments
Selective inhibition of IL-6-induced versus OSM-induced STAT3 pathway activation
All IL-6-type cytokines recruit gp130 to their signaling complexes, auto-phosphorylate, and activate associated JAKs and recruit STAT proteins (Fig. 4a) [13, 14]. IL-6 binds to IL-6 receptor alpha subunits (IL-6Rα, gp80) on cells that express this receptor, and this binary complex then recruits a gp130 signaling subunit to form an active IL-6/IL-6Rα/gp130/JAK signaling complex that mediates the phosphorylation of the tyrosine-705 residue of STAT3, resulting in the formation of pSTAT3 dimers that traffic into the nucleus and bind to specific DNA response elements to activate the transcription of target genes (Fig. 4a) [13, 14, 17, 26, 50]. In contrast, oncostatin M (OSM) binds to two distinct heterodimer receptor complexes formed between a gp130 subunit and either the OSM receptor (OSM-R) or the leukemia inhibitory factor receptor (LIF-R) without first binding to an alpha receptor subunit (Fig. 4a) [13, 14]. Nevertheless, activation of each of the three receptor complexes, IL-6/IL-6Rα/gp130/JAK, OSM/OSMR/gp130/JAK, or OSM/LIFR/gp130/JAK, triggers identical downstream signal transduction events with respect to STAT3 pathway activation (Fig. 4a) [13, 14]. The three compounds selectively block STAT3 activation over IFN-γ-induced STAT1 activation, and inhibit the growth of HNSCC cells in vitro [18, 19]. To determine whether they inhibited pSTAT3Tyr705 activation by IL-6 and OSM, we conducted concentration-dependent inhibition (IC50) assays with both cytokines (Fig. 4b–e). Consistent with our published data [19], the triazolothiadiazine 864669 (Fig. 4b) and the oxazole-piperazine 4248543 (Fig. 4c) both inhibited IL-6-induced pSTAT3Tyr705 activation in a concentration-dependent manner. However, neither of these compounds inhibited OSM-induced pSTAT3Tyr705 activation (Fig. 4b, c). In marked contrast, the amino alcohol 856350 (Fig. 4d) and the pan JAK inhibitor Pyridone 6 (Fig. 4e) inhibited pSTAT3Tyr705 activation induced by both IL-6 and OSM with similar potencies. To further validate the differential effects of the three on OSM-induced pSTAT3Tyr705 activation, we performed western blotting analysis similar to those described for Fig. 2, except that 50 ng/ml OSM was used as the activating stimulus (Fig. 5). Neither 864669 (Fig. 5a) nor 4248543 (Fig. 5b) inhibited the OSM-induced upregulation of pSTAT3Tyr705; neither did they alter cyclin D1 or Bcl-XL expression levels. In contrast, 856350 significantly inhibited the OSM-induced upregulation of pSTAT3Tyr705 (p = 0.0486 at 30 μM) and cyclin D1 expression (p = 0.02 at 30 μM), and although the downregulation of Bcl-XL was not statistically significant, there was a clear concentration-dependent effect (Fig. 5c). Collectively, these data indicate that 864669 and 4248543 selectively inhibited IL-6-induced pSTAT3Tyr705 activation induced by IL-6 but not by OSM. In contrast, the amino alcohol 856350 inhibited pSTAT3Tyr705 activation induced by both IL-6 and OSM.
Fig. 4.

a STAT3 activation pathways for interleukin-6 and oncostatin M. In classical IL-6 signal transduction, IL-6 binds to IL-6 receptor alpha subunits (IL-6Rα, gp80) on cells that express this receptor, and this binary complex then recruits a gp130 signaling subunit to form an IL-6/IL-6Rα/gp130 heterotrimer and then two of these interact to form a hexamer-activated IL-6/IL-6Rα/gp130 signaling complex [13, 14, 26]. Homo-dimerization of the IL-6/IL-6Rα/gp130 heterotrimers triggers phosphorylation of the gp130-associated Janus kinase, and subsequent phosphorylation of tyrosine residues on the intracellular cytoplasmic tail of gp130 generates docking sites for the SH2 domains of STAT3 [13, 14, 26, 50]. JAK-mediated phosphorylation of the tyrosine-705 residue of STAT3 recruited to the activated IL-6/IL-6Rα/gp130/JAK complex enables reciprocal interactions between SH2 domains of pSTAT3 monomers to form dimers which then translocate into the nucleus [13, 14, 17, 26, 50]. In the nucleus, pSTAT3 dimers bind to specific DNA response elements in the promoters that activate the transcription of target genes that favor the development, progression, and maintenance of HNSCC and many other tumors [7, 10, 15, 17, 25]. In contrast, oncostatin M (OSM) can bind to two distinct heterodimer receptor complexes without first binding to an alpha receptor subunit and then recruiting gp130 [13, 14]. OSM initiates signal transduction by binding to heterodimer complexes formed between a gp130 subunit and either the OSM (OSMR) or leukemia inhibitory factor (LIFR) signaling receptors [13, 14]. Activation of either the IL-6/IL-6Rα/gp130/JAK, OSM/OSMR/gp130/JAK, or OSM/LIFR/gp130/JAK receptor complex triggers identical downstream signal transduction events with respect to STAT3 recruitment, tyrosine-705 phosphorylation of STAT3, pSTAT3 dimerization, and trafficking to the nucleus and transcriptional regulation of STAT3 target genes. b–e Inhibition of IL-6-induced versus OSM-induced pSTAT3Tyr705 activation by b 864669 and c 4248543 but not d 856350 and e Pyridone 6. Three hundred eighty-four-well plates were seeded with Cal33 HNSCC cells and serum starved for 24 h. The cells were exposed to the indicated concentrations of the selected compounds for 3 h and then treated with either 50 ng/ml of IL-6 or 50 ng/ml OSM for 15, fixed and stained with Hoechst and a mouse monoclonal anti-pSTAT3Tyr705 antibody. Images were acquired on the ImageXpress Ultra confocal HCS platform. To calculate the percent inhibition of IL-6-induced pSTAT3 activation, the mean average inner pSTAT3Tyr705 intensity values of the 0.2% DMSO minimum plate control wells (n = 32) and the 50 ng/ml IL-6 or OSM maximum plate control wells (n = 32) were used to normalize the mean average inner intensity pSTAT3Tyr705 values of the compound-treated wells and to represent 100 and 0% inhibition of IL-6-induced pSTAT3 activation, respectively. The mean ± SD (n = 3) percent inhibition of IL-6-induced pSTAT3 activation (open circle) and the mean ± SD (n = 3) percent inhibition of OSM-induced pSTAT3 activation (filled circle) at the indicated compound concentrations are presented. Representative experimental data from one of numerous independent experiments are shown
Fig. 5.

a 864669 and b 4248543 do not abrogate oncostatin M (OSM)-induced upregulation of pSTAT3 and STAT3 target gene expression; however, c 856350 downmodulates OSM-induced upregulation of pSTAT3 and decreases STAT3 target gene expression. Cal33 cells were serum starved and treated with increasing concentrations of 864669, 4248543, and 856350, respectively. At the end of 2 h and 45 min, cells were stimulated with OSM (50 ng/ml, 15 min) and harvested and protein lysates were ran on a 10% SDS-polyacrylamide gel. 864669 (a) and 4248543 (b) do not inhibit pSTAT1Tyr705, cyclin D1, and Bcl-XL in Cal33 cells stimulated with OSM. Results are from two separate experiments. 856350 (c) causes a significant decrease in pSTAT3Tyr705 (p = 0.0486) and cyclin D1 (p = 0.02) expression levels when compared to cells stimulated with OSM. Bcl-XL expression showed a decreasing trend. Results are from two separate experiments
Blockade of pgp130Tyr905 expression
To further investigate the MOA by which the triazolothiadiazine 864669 and oxazole-piperazine 4248543 selectively inhibit IL-6-induced pSTAT3Tyr705 activation, we developed a western blotting assay to determine if these compounds inhibited the phosphorylation of a specific tyrosine residue (Tyr 905) of the gp130 subunit (Fig. 6). Latent non-phosphorylated STAT3 is recruited to the pgp130Tyr905 subunits of the activated IL-6 receptor complex via SH2 domain interactions, and this ultimately leads to JAK-mediated phosphorylation of STAT3Tyr705. Cal33 cells were treated with 3, 10, 30, or 100 μM of the compounds for 3 h and then stimulated with IL-6 (50 ng/ml, 15 min). Both 864669 and 4248543 inhibited the IL-6-induced upregulation of pgp130Tyr905 expression in a concentration-dependent manner (Fig. 6). At 30 μM, both 864669 (Fig. 6a, p = 0.0388) and 4248543 (Fig. 6b, p = 0.0163) significantly inhibited the IL-6-induced upregulation of pgp130Tyr905 expression. In contrast, neither compound significantly altered the gp130 expression levels of Cal33 cells.
Fig. 6.

a 864669 and b 4248543 downmodulate IL6-induced upregulation of pgp130Tyr905 expression. Cal33 cells were serum starved and treated with increasing concentrations of 864669 and 4248543, respectively. At the end of 2 h and 45 min, cells were stimulated with IL6 (50 ng/ml, 15 min) and harvested and protein lysates were ran on a 10% SDS-polyacrylamide gel. A significant decrease in pgp130Tyr905 (p = 0.0338) expression was observed in Cal33 cells treated with 864669 at 30 μM (a). Similarly, 4248543 at 30 μM showed a significant decrease in pgp130Tyr905 (p = 0.0163) expression (b). Results are from three separate experiments
STAT signaling pathway selectivity, IL-6-receptor binding, and effect of 864669 and 4248543 on the negative regulators of STAT3
Since activated STAT3 is oncogenic and STAT1 has tumor suppressor activities [2], one of the hit selection criteria for the STAT3 pathway HCS campaign was that compounds should exhibit ≥3-fold selectivity for the inhibition of IL-6-induced pSTAT3Tyr705 activation relative to inhibition of IFNγ-induced pSTAT1Tyr701 activation [19]. However, because of the high sequence homology between STAT1 and STAT3 and their propensity to form STAT1-STAT3 heterodimers, we re-examined the effects of 3 h pre-exposure to various concentrations of 864669, 4248543, and 856350 on IFNγ-induced pSTAT1Tyr701 activation and total STAT1 expression levels by western blotting. Consistent with our previous HCS data [19], none inhibited either IFNγ-induced pSTAT1Tyr701 activation or total STAT1 protein expression levels (data not shown). Further, apparent selectivity for the triazolothiadiazine and oxazole-piperazine for inhibiting IL-6-induced versus OSM-induced STAT3 activation prompted us to formulate a hypothesis that these compounds might be interfering with IL-6 binding to IL-6Rα and/or the subsequent recruitment of gp130 and assembly of the activated IL-6/IL-6Rα/gp130/JAK receptor complex. To test whether 864669 and 4248543 antagonized the binding of I125-IL6 to the IL-6 receptor complex, we developed a 96-well filtration format I125-IL6 radio ligand binding assay with plasma membranes isolated from U266 cells. Neither 864669 or any of the 864669 analogs nor 4248543 or any of the 4248543 series analogs competitively inhibited I125-IL-6 binding to U266 plasma membranes at concentrations ≤50 μM (data not shown). These data indicate that the ability of triazolothiadiazine 864669 and oxazole-piperazine 4248543 to selectively inhibit IL-6-induced pSTAT3Tyr705 activation is not mediated through antagonism of IL-6 receptor binding. In addition, 864669 and 4248543 were tested to assess their effect on STAT3 negative regulators such as PIAS3, SOCS1, and SOCS3 expression by western blotting. None inhibited PIAS3, SOCS1, or SOCS3 protein expression levels at the concentrations tested (data not shown).
Discussion
Cumulative evidence strongly implicates IL6 deregulation in a variety of cancers [21], and there is a correlation between increased expression of IL6 and lower patient survival rates [44]. Elevated levels of IL6 have been associated with tumor progression through stimulation of angiogenesis, proliferation, migration, drug resistance, and inhibition of cancer cell apoptosis [12, 31]. In HNSCC, high serum IL-6 levels have been correlated with tumor metastasis [51]. Further, elevated IL-6 expressions have been shown to contribute to erlotinib resistance in HNSCC and thus blockade of the IL-6 signaling pathway may be an effective therapeutic strategy to overcome resistance to erlotinib [42]. High serum IL6 levels have been detected in patients with lymphoma, lung [35], prostate [32, 40], breast [5], and colorectal cancer [48]. These data imply an oncogenic role for IL-6, and indeed, blocking IL-6 signaling is an anti-cancer strategy currently in clinical development. In multiple myeloma, clinical trials with neutralizing anti-IL-6 antibodies demonstrated good anti-tumor efficacy; however, antibody treatment resulted in a reservoir of free IL-6 in the circulation [20]. To circumvent such difficulties, the humanized anti-IL-6 receptor (IL-6R) antibody tocilizumab was developed which prevents binding of IL-6 to IL-6R, and only shows mild systemic elevation of IL-6 [20].
Many studies have indicated a crucial role of STAT3 in the downstream signal transduction of IL-6 [30] and their role in the development of cancer [48]. In cancer cells, the constitutive or elevated activation of STAT3 could also result from defects in the negative regulators of STAT3 [3, 28, 34], excessive stimulation of STAT3, positive feedback loops that maintain constitutive activation of STAT3, and/or somatic mutations that confer STAT3 hyperactivation.
The present study was conducted as part of a hit-to-lead exercise to elucidate the mechanism(s) of action of compounds from the triazolothiadiazine, oxazole-piperazine, and amino alcohol series, namely, 864669, 4248543, and 856350, respectively, as selective inhibitors of IL-6-induced STAT3 pathway activation. The three compounds 864669, 4248543, and 856350, and structurally related analogs from each series (data not shown), inhibited IL-6-induced pSTAT3Tyr705 activation and decreased expression of the STAT3 target genes cyclin D1 and Bcl-XL. Consistent with our high content screening strategy [23], none of the compounds inhibited either IFNγ-induced pSTAT1Tyr701 activation or total STAT1 protein expression. Further, in a KinaseProfiler panel of 83 in vitro kinase assays, none of the compounds significantly inhibited any of the kinases in the panel, including members of the JAK, known to mediate STAT3 signaling. In addition, western blot analyses in Cal33 HNSCC cells demonstrated that 864669, 4248543, and 856350 did not inhibit either the IL-6-induced auto-phosphorylation of pJAK1, pJAK2, and pJAK3 or their protein expression levels, confirming that they do not exert their effects on STAT3 signaling through JAK kinase inhibition. Western blot analysis of the expression of the negative modulators of STAT3 such as SOCS3 or PIAS3 in HNSCC cells indicated that exposure to the compounds had no noticeable effects on the expression of these regulatory proteins. Further, the compounds showed selectivity for IL-6-induced pSTAT3Tyr705 activation versus OSM-induced pSTAT3Tyr705 activation. Further, both 864669 and 4248543 inhibited the IL-6-induced upregulation of pgp130Tyr905 expression in a concentration-dependent manner without altering the gp130 expression levels of Cal33 cells.
Both IL-6 and STAT3 have been implicated in the initiation and progression of tumorigenesis, and direct targeting of STAT3 has long been considered a plausible strategy for cancer therapy. Although a variety of STAT3-targeted approaches have been pursued [8], few have progressed to clinical candidates. The triazolothiadiazine 864669 and oxazole-piperazine 4248543 selectively inhibits IL-6-induced pSTAT3Tyr705 activation in HNSCC cells relative to either OSM-induced pSTAT3Tyr705 activation or IFNγ-induced pSTAT1Tyr701 activation. These two compounds do not alter the protein expression levels of either STAT3 or its negative regulators. Neither do they block ligand binding to the IL-6 receptor complex nor the auto-phosphorylation of the associated JAK kinase. Rather, the selective inhibition of IL-6-induced pSTAT3Tyr705 activation appears to involve inhibition of the phosphorylation of a specific tyrosine residue (Tyr 905) of the gp130 subunits in the activated IL-6 receptor complex that prevents the recruitment of latent STAT3 and the subsequent JAK-mediated phosphorylation of STAT3Tyr705. It is possible that 864669 and 4248543 inhibit IL-6 induced STAT activation by blocking the IL-6-induced recruitment and/or assembly of the IL-6 receptor hexamer complex that occurs after IL-6 binding to IL-6Rα subunits. Further, it will be important to determine whether these selective small-molecule inhibitors of IL-6-induced STAT3 activation exhibit in vivo anti-tumor efficacy.
Electronic supplementary material
{kind=link}
(GIF 89 kb)
{kind=link}
(GIF 58 kb)
{kind=link}
(GIF 43 kb)
Compliance with ethical standards
Funding sources
This project has been funded in part with Federal Funds from the National Cancer Institute, National Institutes of Health, under Contract No. HSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This study was supported by NExT-CBC Project ID #1015, S08-221 Task Order 6 “STAT3 Pathway Inhibitor HCS” (Grandis, PI), NCI Chemical Biology Consortium, Pittsburgh Specialized Application Center (PSAC) (Lazo and Johnston co-PIs), and University of Pittsburgh Chemical Diversity Center (Huryn, PI). The project was also supported in part by funds from the American Cancer Society (Grandis) and a Head and Neck Spore P50 award (Grandis, CA097190).
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12154-017-0169-9) contains supplementary material, which is available to authorized users.
References
- 1.Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18:254–267. doi: 10.1038/cr.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: a matter of balance. JAKSTAT. 2012;1:65–72. doi: 10.4161/jkst.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brantley EC, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 2008;14:4694–4704. doi: 10.1158/1078-0432.CCR-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter JW, et al. Configuring radioligand receptor binding assays for HTS using scintillation proximity assay technology. Methods Mol Biol (Clifton, NJ) 2002;190:31–49. doi: 10.1385/1-59259-180-9:031. [DOI] [PubMed] [Google Scholar]
- 5.Chang Q, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia (New York, NY) 2013;15:848–862. doi: 10.1593/neo.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dziennis S, Alkayed NJ. Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev Neurosci. 2008;19:341–361. doi: 10.1515/REVNEURO.2008.19.4-5.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007;251:199–210. doi: 10.1016/j.canlet.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. STAT inhibitors for cancer therapy. J Hematol Oncol. 2013;6:90. doi: 10.1186/1756-8722-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao L, et al. Down-regulation of signal transducer and activator of transcription 3 expression using vector-based small interfering RNAs suppresses growth of human prostate tumor in vivo. Clin Cancer Res. 2005;11:6333–6341. doi: 10.1158/1078-0432.CCR-05-0148. [DOI] [PubMed] [Google Scholar]
- 10.Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13:5665–5669. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- 11.Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38:904–910. doi: 10.1016/j.ctrv.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/bj20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jing N, Tweardy DJ. Targeting Stat3 in cancer therapy. Anti-Cancer Drugs. 2005;16:601–607. doi: 10.1097/00001813-200507000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Johnson DE. Targeting proliferation and survival pathways in head and neck cancer for therapeutic benefit. Chin J Cancer. 2012;31:319–326. doi: 10.5732/cjc.011.10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv. 2011;11:18–26. doi: 10.1124/mi.11.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston PA, Sen M, Hua Y, Camarco D, Shun TY, Lazo JS, Grandis JR. High-content pSTAT3/1 imaging assays to screen for selective inhibitors of STAT3 pathway activation in head and neck cancer cell lines. Assay Drug Dev Technol. 2014;12:55–79. doi: 10.1089/adt.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnston PA, et al. HCS campaign to identify selective inhibitors of IL-6-induced STAT3 pathway activation in head and neck cancer cell lines. Assay Drug Dev Technol. 2015;13:356–376. doi: 10.1089/adt.2015.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung IH, Choi JH, Chung YY, Lim GL, Park YN, Park SW. Predominant activation of JAK/STAT3 pathway by interleukin-6 is implicated in hepatocarcinogenesis. Neoplasia (New York, NY) 2015;17:586–597. doi: 10.1016/j.neo.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaPorte M, Wang, Z., Colombo, R., Garzan, A., Peshkov, VA., Liang, M., Johnston, PA., Schurdak, ME., Sen, M., Camarco, DP., Hua, Y., Pollock, NI., Lazo, JS., Grandis, JR., Wipf, P., Huryn, DM. (2016) Optimization of pyrazole-containing 1,2,4-triazolo-[3,4-b] thiadiazines, a new class of STAT3 pathway inhibitors Bioorganic & medicinal chemistry letters [DOI] [PMC free article] [PubMed]
- 24.LaPorte MG, et al. 2-Guanidinoquinazolines as new inhibitors of the STAT3 pathway. Bioorg Med Chem Lett. 2014;24:5081–5085. doi: 10.1016/j.bmcl.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leeman RJ, Lui VW, Grandis JR. STAT3 as a therapeutic target in head and neck cancer. Expert Opin Biol Ther. 2006;6:231–241. doi: 10.1517/14712598.6.3.231. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Xiao H, Lin L, Jou D, Kumari V, Lin J, Li C. Drug design targeting protein-protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning: discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 interface. J Med Chem. 2014;57:632–641. doi: 10.1021/jm401144z. [DOI] [PubMed] [Google Scholar]
- 27.Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65:2532–2536. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- 28.Lui VW, et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc Natl Acad Sci U S A. 2014;111:1114–1119. doi: 10.1073/pnas.1319551111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 2012;122:143–159. doi: 10.1042/CS20110340. [DOI] [PubMed] [Google Scholar]
- 30.Mitsuyama K, Matsumoto S, Masuda J, Yamasakii H, Kuwaki K, Takedatsu H, Sata M. Therapeutic strategies for targeting the IL-6/STAT3 cytokine signaling pathway in inflammatory bowel disease. Anticancer Res. 2007;27:3749–3756. [PubMed] [Google Scholar]
- 31.Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer. 2014;110:469–478. doi: 10.1038/bjc.2013.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6) BJU Int. 2014;113:986–992. doi: 10.1111/bju.12452. [DOI] [PubMed] [Google Scholar]
- 33.Park OK, Schaefer TS, Nathans D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci U S A. 1996;93:13704–13708. doi: 10.1073/pnas.93.24.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peyser ND, Freilino M, Wang L, Zeng Y, Li H, Johnson DE, Grandis JR. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene. 2016;35:1163–1169. doi: 10.1038/onc.2015.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schafer ZT, Brugge JS. IL-6 involvement in epithelial cancers. J Clin Invest. 2007;117:3660–3663. doi: 10.1172/JCI34237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaper F, et al. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J. 1998;335(Pt 3):557–565. doi: 10.1042/bj3350557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 38.Sen M, et al. Systemic administration of a cyclic signal transducer and activator of transcription 3 (STAT3) decoy oligonucleotide inhibits tumor growth without inducing toxicological effects. Mol Med. 2014;20:46–56. doi: 10.2119/molmed.2013.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sen M, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58:1008–1015. doi: 10.1016/S0090-4295(01)01405-4. [DOI] [PubMed] [Google Scholar]
- 41.Siddiquee K, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanam A, Love-Homan L, Joseph TS, Espinosa-Cotton M, Simons AL. Upregulated interleukin-6 expression contributes to erlotinib resistance in head and neck squamous cell carcinoma. Mol Oncol. 2015;9:1371–1383. doi: 10.1016/j.molonc.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taga T, Kawanishi Y, Hardy RR, Hirano T, Kishimoto T. Receptors for B cell stimulatory factor 2. Quantitation, specificity, distribution, and regulation of their expression. J Exp Med. 1987;166:967–981. doi: 10.1084/jem.166.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011;3:177–189. doi: 10.2147/CMR.S18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turkson J, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 46.Vultur A, et al. Cell-to-cell adhesion modulates Stat3 activity in normal and breast carcinoma cells. Oncogene. 2004;23:2600–2616. doi: 10.1038/sj.onc.1207378. [DOI] [PubMed] [Google Scholar]
- 47.Waldner MJ, Foersch S, Neurath MF. Interleukin-6—a key regulator of colorectal cancer development. Int J Biol Sci. 2012;8:1248–1253. doi: 10.7150/ijbs.4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang SW, Sun YM. The IL-6/JAK/STAT3 pathway: potential therapeutic strategies in treating colorectal cancer (review) Int J Oncol. 2014;44:1032–1040. doi: 10.3892/ijo.2014.2259. [DOI] [PubMed] [Google Scholar]
- 49.Wang YZ, Wharton W, Garcia R, Kraker A, Jove R, Pledger WJ. Activation of Stat3 preassembled with platelet-derived growth factor beta receptors requires Src kinase activity. Oncogene. 2000;19:2075–2085. doi: 10.1038/sj.onc.1203548. [DOI] [PubMed] [Google Scholar]
- 50.Wilks AF. The JAK kinases: not just another kinase drug discovery target. Semin Cell Dev Biol. 2008;19:319–328. doi: 10.1016/j.semcdb.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 51.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res: MCR. 2011;9:1658–1667. doi: 10.1158/1541-7786.MCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang HF, Lai R. STAT3 in cancer-friend or foe? Cancers (Basel) 2014;6:1408–1440. doi: 10.3390/cancers6031408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(GIF 89 kb)
(GIF 58 kb)
(GIF 43 kb)