

Abstract
Triggering receptor expressed on myeloid cells 2 (TREM2) is a single transmembrane molecule uniquely expressed in microglia. TREM2 mutations are genetically linked to Nasu-Hakola disease and associated with multiple neurodegenerative disorders, including Alzheimer's disease. TREM2 may regulate microglial inflammation and phagocytosis through coupling to the adaptor protein TYRO protein-tyrosine kinase-binding protein (TYROBP). However, there is no functional system for monitoring this protein-protein interaction. We developed a luciferase-based modality for real-time monitoring of TREM2-TYROBP coupling in live cells that utilizes split-luciferase complementation technology based on TREM2 and TYROBP fusion to the C- or N-terminal portion of the Renilla luciferase gene. Transient transfection of human embryonic kidney 293 cells with this reporter vector increased luciferase activity upon stimulation with an anti-TREM2 antibody, which induces their homodimerization. This was confirmed by ELISA-based analysis of the TREM2-TYROBP interaction. Antibody-mediated TREM2 stimulation enhanced spleen tyrosine kinase (SYK) activity and uptake of Staphylococcus aureus in microglial cell line BV-2 in a kinase-dependent manner. Interestingly, the TREM2 T66M mutation significantly enhanced luciferase activity without stimulation, indicating constitutive coupling to TYROBP. Finally, flow cytometry analyses indicated significantly lower surface expression of T66M TREM2 variant than wild type or other TREM2 variants. These results demonstrate that our TREM2 reporter vector is a novel tool for monitoring the TREM2-TYROBP interaction in real time.
Keywords: Alzheimer disease, cell culture, genetic polymorphism, microglia, phagocytosis, plasmid, Alzheimer's disease
Introduction
Triggering receptor expressed on myeloid cells 2 (TREM2)2 is expressed by cells of the myeloid lineage (1), especially microglia (2, 3). TREM2 is mostly found in intracellular stores but can rapidly translocate to the cell surface upon exocytotic stimulation (4). The adaptor protein TYRO protein-tyrosine kinase-binding protein (TYROBP; also known as DAP12) is an intracellular signaling molecule for TREM2 signaling (for reviews, see Refs. 2 and 5). Upon receptor ligation, tyrosine residues within the TYROBP immunoreceptor tyrosine-based activation motif region undergo phosphorylation by Src kinases. This leads to inflammatory signaling and reorganization of the actin cytoskeleton via activation of small guanine nucleotide-binding proteins such as Rac and Cdc42 for endocytic functions (6).
The clinical significance of TREM2 and TYROBP was first highlighted by their mutations in a rare autosomal recessive inflammatory neurodegenerative disorder, Nasu-Hakola disease. Early studies revealed that Nasu-Hakola disease patients carried loss-of-function mutations in TYROBP or TREM2 (7–9). The presence of these mutations results in exaggerated responses to Toll-like receptor activation and elevated levels of proinflammatory cytokines in plasma and brain (10–12). More recent studies revealed that specific variants in TREM2 were associated with late-onset AD, Parkinson's disease, amyotrophic lateral sclerosis (13), and frontotemporal dementia (FTD) (13–19). The TREM2 R47H variant was associated with AD and FTD (15, 16), whereas the T66M variant was associated with FTD or FTD-like syndrome (19), and the S116C variant was found in one case of FTD (19).
It is unclear how these mutations affect the development of neurodegenerative diseases. Recent in vitro data suggest that intracellular TREM2 is processed into a soluble form (sTREM2), and this form may have biological functions (20). Disease-associated TREM2 mutations may result in deficient processing and secretion of sTREM2 that lead to an accumulation of immature TREM2 in the cytoplasm (20). Although its biological function is unknown, sTREM2 is detected in the cerebrospinal fluid (CSF) and plasma of healthy normal patients. The level of sTREM2 in CSF was significantly lower in AD and FTD patients of one study (20), whereas other studies reported it to be elevated (21, 22).
The role of TREM2 in amyloid clearance has been under intense investigation. APP+PS1 mice expressing familial AD-linked variants of amyloid precursor protein (13) and presenilin-1 (PS1) show reduced accumulation of amyloid-β peptide (Aβ) by disruption of TREM2 (23), whereas others report enhanced Aβ clearance in 5XFAD mice lacking TREM2 (24). These apparently contrasting results suggest that TREM2 function can be affected by mutated PS1 and should be investigated in more physiological models.
The disease-linked TREM2 mutations affect phagocytic function of myeloid cells. Phagocytosis of Aβ was impaired due to expression of R47H or T66M TREM2 mutation (20). Patients with disease-associated TREM2 variants may then have a build-up of Aβ and apoptotic debris due to deficient processing of TREM2 and impaired phagocytosis. To investigate TREM2 and its disease-associated mutations, we developed a bioluminescence-based assay that allows us to monitor real-time TREM2 coupling to TYROBP in vitro with a construct that utilizes split-luciferase complementation technology.
Results
TREM2-CLuc-IRES-TYROBP-NLuc construct complements luciferase activity in response to anti-TREM2 antibody
For examination of the real-time protein-protein interaction between TREM2 and TYROBP, we developed a mammalian expression vector (TREM2-CLuc-IRES-TYROBP-NLuc) that utilizes the “bait-prey” split-Renilla luciferase complementation assay (Fig. 1A) (25–27). Renilla luciferase catalyzes the oxidation of coelenterazine, a luciferin, to emit light (28). The amount of light emitted is measured as relative light units (RLU) and can be directly correlated to the magnitude of protein-protein interaction (Fig. 1B). The TREM2-CLuc-IRES-TYROBP-NLuc plasmid was transfected into human embryonic kidney (HEK) 293 cells followed by stimulation with specific ligands. Luciferase activity was measured over 30 min, and area under the curve (AUC) was calculated. HEK293 cells transfected with TREM2-CLuc-IRES-TYROBP-NLuc showed some basal level of luciferase activity that may represent spontaneous TREM2 coupling to TYROBP that occurs independently of ligand, although this was not significantly higher than background (Fig. 2, A and B). As expected, our TREM2-CLuc-TYROBP negative control construct elicited luciferase activity that was also no higher than background (Fig. 2, A and B). It has previously been shown that a TREM2-cross-linked antibody can activate TREM2 (29). Indeed, monoclonal anti-TREM2 antibody stimulation at 10 and 20 μg/ml induced significantly higher luciferase activity up to a 3-fold net increase over time compared with unstimulated transfected cells (Fig. 2, E and F). Treatment of transfected cells with 20 μg/ml antibody did not induce a significantly higher effect compared with 10 μg/ml, suggesting its saturated binding to TREM2. Treatment of transfected cells with control IgG resulted in only a maximum of about a 0.3-fold net increase at 1 μg/ml (supplemental Fig. S1, A and B). Although this difference was statistically significant, there is a greater biological change upon stimulation with anti-TREM2 antibody. Research suggests that TREM2 activation may be increased in response to the presence of Aβ because its expression is increased in amyloid plaque-associated microglia in the APP mouse brain (30). However, there was no significant effect of Aβ42 stimulation (Fig. 2, C and D) or anti-TYROBP antibody (Fig. 2, G and H) even at the highest concentrations. These data demonstrate that our TREM2-CLuc-IRES-TYROBP-NLuc construct is capable of being utilized to measure the TREM2-TYROBP interaction by ligand stimulation.
Figure 1.
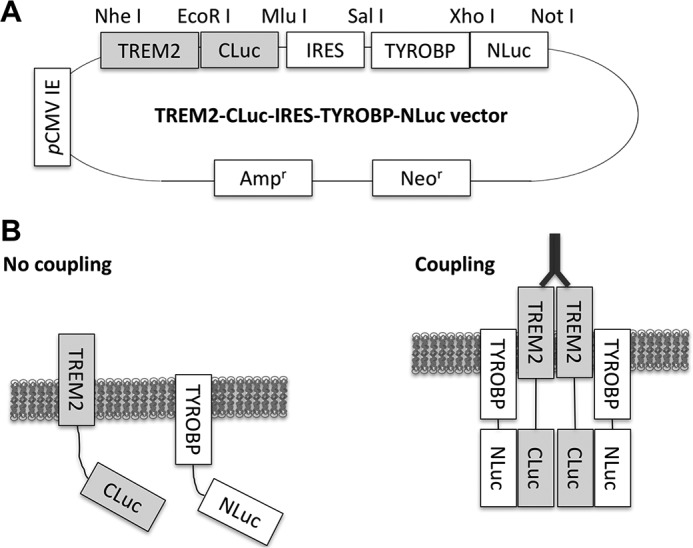
TREM2-CLuc-IRES-TYROBP-NLuc split-Renilla luciferase reporter system. The TREM2-CLuc-IRES-TYROBP-NLuc vector contains CMV immediate early (IE) promoter, the C terminus of the Renilla luciferase gene fused to the cytoplasmic region of TREM2 (TREM2-CLuc), IRES, and the N-terminal region of the Renilla luciferase gene fused to the N-terminal region of TYROBP (TYROBP-NLuc) (A). The unique restriction sites for cloning are depicted. Ampr, ampicillin resistance gene; Neor, neomycin resistance gene. In a resting state, wild-type TREM2 and TYROBP show limited coupling. Upon stimulation with agonistic ligand (such as dimerizing anti-TREM2 antibody), TREM2 interacts with TYROBP, which reconstitutes luciferase activity by coupling of the C- and N-terminal regions of the split-Renilla luciferase gene (B).
Figure 2.

Anti-TREM2 antibody, but not Aβ or anti-TYROBP antibody (Ab), induces dose-dependent increase in the TREM2-TYROBP interaction in HEK293 cells. HEK293 cells were transfected with internal control reporter pRL-TK, TREM2-CLuc-TYROBP negative control, or our TREM2-CLuc-IRES-TYROBP-NLuc plasmid prior to stimulation with ligand. Time-course data (A, C, E, and G) were assessed for luminescence over time by AUC analysis (B, D, F, and H) in RLU for n = 3 for each group. Error bars represent S.D. ααα denotes p < 0.001 versus all other groups. * denotes p < 0.05 versus TREM2/PBS as determined by one-way ANOVA and Tukey's post-test.
Anti-TREM2 antibody induces TREM2 coupling to TYROBP in HEK293 cells
To support our luciferase data, we transfected HEK293 cells with TREM2-CLuc-IRES-TYROBP-NLuc and stimulated with anti-TREM2 antibody followed by quantification of the TREM2-TYROBP interaction by sandwich ELISA (Fig. 3A). For this assay, treatment with the tyrosine phosphatase inhibitor sodium orthovanadate and cross-linker dimethyl 3,3′-dithiobispropionimidate (DTBP) was necessary. This is because TREM2 signaling presumably is dependent on tyrosine phosphorylation of TYROBP as well as the transient nature of their interaction due to the secondary association of TYROBP with SHIP1 (31, 32). Cells stimulated with anti-TREM2 antibody showed a significant increase in TREM2-TYROBP interactions versus unstimulated cells, suggesting that increased stabilization of TREM2 coupling to TYROBP was due to antibody treatment (Fig. 3B).
Figure 3.

Quantification of TREM2·TYROBP complex by “sandwich” ELISA. HEK293 cells were transfected with TREM2-CLuc-IRES-TYROBP-NLuc followed by anti-TREM2 antibody (Ab) stimulation to induce TREM2 coupling to TYROBP and protein cross-linking with DTBP and phosphatase inhibitor sodium pervanadate (Na3O4V) (A). Following cell lysis, protein was incubated in anti-TYROBP-coated wells and labeled for TREM2 with biotinylated anti-TREM2·HRP-conjugated streptavidin complex for quantification of TREM2·TYROBP protein complexes. Average (Ave) OD was quantified by ELISA. Data represent the arithmetic difference between a given treatment condition and the average OD of untransfected controls (n = 4 per group) (B). Significant differences were determined by one-way ANOVA followed by Tukey's post-test. Error bars represent S.D. †† and ††† denote p < 0.01 and p < 0.001, respectively, versus untransfected cells. ### denotes p < 0.001 versus unstimulated cells transfected with TREM2-TYROBP. ** and *** denote p < 0.01 and 0.001, respectively, versus those indicated.
Anti-TREM2 antibody stimulation enhances phagocytosis of Staphylococcus aureus in BV-2 microglial cells
To further investigate whether the TREM2-TYROBP interaction facilitates phagocytic activity, we performed a phagocytosis assay with the BV-2 mouse microglial cell line and S. aureus particles conjugated with Alexa Fluor 594 and fluorescence-activated cell sorting (FACS). BV-2 cells endogenously express TREM2 and TYROBP (33, 34). We confirmed the endogenous distribution of TREM2 by flow cytometry and found that 25.3% of TREM2 is expressed on the cell surface (supplemental Fig. S2). We stimulated BV-2 cells with anti-TREM2 antibody to induce endogenous TREM2-TYROBP coupling. Cells stimulated with anti-TREM2 antibody were labeled with a donkey anti-goat Alexa Fluor 488-conjugated secondary antibody. Gating of cells was done using unstained cells or cells positive for Alexa Fluor 488 or 594 alone (Fig. 4, A–D). The total percentage of phagocytic cells is indicated in each graph (Fig. 4, E–I). The S. aureus Alexa Fluor 594+ population was then separated into phagocytic high (Hi) versus low (Lo) groups (Fig. 4, E–G), and phagocytosis was determined by analyzing the percentage of cells in each group over the total cell population (Fig. 4H). Untreated cells had a significantly lower percentage of phagocytic Hi (3.9%) populations versus phagocytic Lo (11.2%) populations. Treatment of microglia with anti-TREM2 antibody significantly increased both the percentage of phagocytic Hi (by 60%) and Lo populations (by 138.1%) as compared with untreated cells. In control experiments, treatment of BV-2 cells with control IgG did not significantly increase low and high phagocytosis-dependent mechanisms. A greater proportion of cells were in the high-uptake population after treatment with anti-TREM2 antibody as compared with the IgG group, indicating that activation of TREM2 is more responsible for increasing the phagocytic Hi population. These findings are substantiated by the use of TREM2-specific inhibitor to spleen tyrosine kinase (SYK) (Fig. 4H). Control IgG-stimulated phagocytosis was insensitive to SKY inhibition in low-phagocytosis cell populations. TREM2-dependent mechanisms were sensitive to treatment with the SYK inhibitor in both phagocytic Lo (Fig. 4H) and phagocytic Hi populations (Fig. 4H). To explore whether the observed trend toward increased phagocytosis in control IgG-treated BV2 cells is accomplished through Fc receptors, the normal goat F(ab′)2 fragment was also tested; it failed to increase the population of low- or high-phagocytic groups (Fig. 4H). These data indicate that enhancing the TREM2-TYROBP interaction by anti-TREM2 antibody stimulation increases the phagocytic Hi population, which biologically links the split-luciferase activity to phagocytic functions of myeloid cells.
Figure 4.

Anti-TREM2 antibody stimulation enhances phagocytic capability of BV-2 microglial cell line. Cells were gated based on unstained cells (A), anti-TREM2 antibody-treated cells (B), or S. aureus-treated cells (C). Cells that were treated with anti-TREM2 antibody (Ab) and S. aureus particles are shown in D. The phagocytic cell population (percentage is indicated above plot) was further separated into phagocytic Lo versus Hi populations (E–G) (52). In H, relative distributions of the phagocytic cell populations into low versus high indices are represented by FlowJo software and graphical analysis. Significant differences in low or high populations were determined by one-way ANOVA followed by Tukey's post-test. Error bars represent S.D. #, ##, and ### denote p < 0.05, 0.01, and 0.001, respectively, versus those groups indicated.
TREM2 induces activation of SYK
Coupling of TREM2 with TYROBP activates intracellular signaling via recruitment of SYK. To directly demonstrate the intracellular signaling of TREM2 after antibody stimulation, we stimulated the parental BV-2 microglial cell line with anti-TREM2 or nonspecific IgG in the presence or absence of a SYK inhibitor (CAS 622387-85-3) and immunoprecipitated SYK from the cell lysate. The immunoprecipitate was then used in an in vitro SYK activity assay that measured SYK's functional conversion of ATP to ADP (Fig. 5). Treatment with SYK inhibitor alone reveals background kinase activity in the immunoprecipitants, but stimulation of BV-2 cells with anti-TREM2 antibody induced significantly higher activation of SYK in the absence of SYK inhibitor. Control IgG stimulation had no effect on SYK activity. These data further illustrate that anti-TREM2 antibody stimulation can activate specific TREM2 intracellular signaling pathways.
Figure 5.
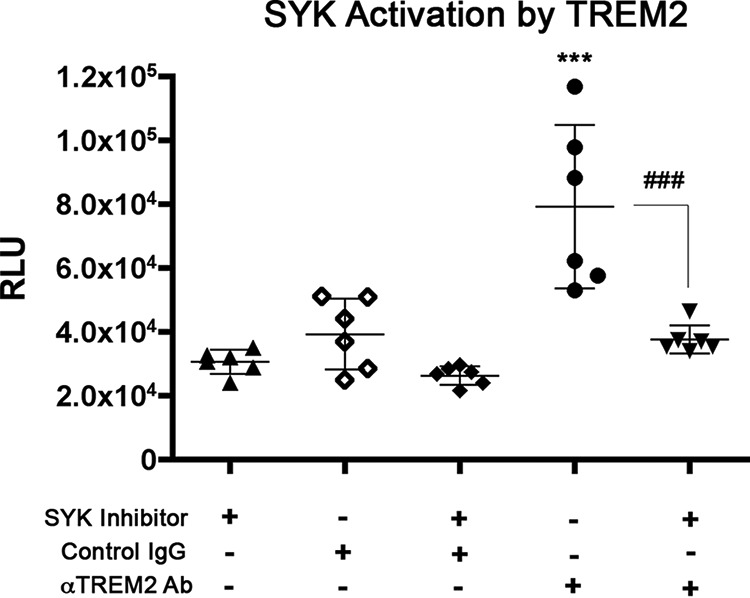
SYK activation by TREM2. Non-transfected BV-2 cells expressing endogenous mouse TREM2 were stimulated with anti-TREM2 antibody (Ab) or control IgG for a period of time. Cells were lysed, and SYK was extracted through immunoprecipitation. Activity was then quantified in an ADP-Glo kinase assay. Significant differences were determined by one-way ANOVA followed by Bonferroni post-test. Error bars represent S.D. *** denotes p < 0.001 versus untransfected cells, and ### denotes p < 0.001 versus TREM2 WT group with antibody stimulation + SYK inhibitor.
T66M mutation of TREM2 induces enhanced basal luciferase activity
We next examined the effect of disease-associated TREM2 mutation of TYROBP coupling in our system. For that purpose, TREM2 variants associated with AD (R47H), Nasu-Hakola disease and FTD (T66M), and a case of FTD (S116C) were inserted into the TREM2-CLuc-IRES-TYROBP-NLuc vector. Immunostaining of permeabilized cells showed mostly similar distribution of TREM2 and TYROBP across all groups, indicative of their intracellular interactions (supplemental Fig. S3A). Transfection of HEK293 cells with wild-type (WT) TREM2 and its variants followed by ELISA for protein levels of TREM2 (supplemental Fig. S3B) and TYROBP (supplemental Fig. S3C) shows no differences in expression efficiency. On average, the ratio of TREM2/TYROBP expression was around 1.0 for each variant, indicating no difference in expression (supplemental Fig. S3D). The effect of anti-TREM2 antibody stimulation on the coupling of TREM2 variants with TYROBP was monitored by real-time luciferase activity (Fig. 6, A, C, and E) and AUC (Fig. 6, B, D, and F). Interestingly, the T66M mutation elicited higher luciferase activity without stimulation and did not show enhanced luciferase activity by anti-TREM2 antibody stimulation, showing the autonomous coupling of the T66M variant with TYROBP in response to anti-TREM2 antibody (Fig. 6, C and D). Unlike WT TREM2, variant TREM2 constructs did not show significant differences in luciferase activity in response to increasing antibody doses. Control experiments demonstrate that the stimulating effect of anti-TREM2 antibody is specific because treatment with control IgG induced no change in any of the TREM2 variants (supplemental Fig. S1, C–H).
Figure 6.

The T66M mutation in TREM2 elicits enhanced TREM2 coupling to TYROBP in HEK293 cells. HEK293 cells were transfected with TREM2-CLuc-IRES-TYROBP-NLuc plasmid prior to stimulation with ligand. Time-course data (A, C, and E) were assessed for AUC (B, D, and F) in RLU for n = 3 for each group. Significant differences were determined by one-way ANOVA followed by Tukey's post-test. Error bars represent S.D. † and ††† denote p < 0.05 and 0.001, respectively, versus unstimulated WT TREM2-transfected cells. ** and *** denote p < 0.01 and 0.001, respectively. Ab, antibody.
T66M TREM2 variant shows reduced cell surface expression of TREM2
We also determined whether there was a difference in cell surface expression compared with WT TREM2. For this purpose, the cell surface TREM2+ populations were assessed by FACS analysis of TREM2-phycoerythrin (PE)-stained HEK293 cells that were untransfected or transfected with WT TREM2 or R47H, S116C, or T66M variant (Fig. 7, A and B). TREM2 is mostly localized in the cytoplasm, which is reflected by the low number of cell surface TREM2+ cells (8.67% in TREM2 WT, 6.37% in R47H, 6.11% in S116C, and 2.77% in T66M group). The number of cell surface TREM2+ cells was less than 50% of other groups. To establish that the differences induced by variant TREM2 constructs were not due to differences in transfection efficiency, the cells were also stained with TREM2-PE after permeabilization, which showed similar numbers of TREM2+ cells (30.3% in TREM2 WT, 28.1% in R47H, 25.6% in S116C, and 25.8% in T66M; Fig. 7, C and D). Mean fluorescence intensity (MFI) of gated TREM2 cells from the roughly 10,000 cells was significantly higher in the TREM2 WT and S116C groups versus untransfected groups, whereas R47H and T66M groups showed significantly lower MFIs of gated TREM2+ cells versus the TREM2 WT group (Fig. 7E). There was no difference in the MFI of TREM2+ cells in permeabilized cells (Fig. 7F), demonstrating similar expression of total TREM2 in transfected cells. Taken together, these data suggest that the T66M mutation induces constitutive coupling with TYROBP as evident with the higher split-luciferase activity, which may prevent its cell surface expression.
Figure 7.

Reduced cell surface expression of TREM2 T66M mutant. A and B, flow cytometry analysis of TREM2 signal (x axis) in untransfected (blue) and transfected cells (red) with TREM2 WT, R47H, S116C, or T66M after gating with forward and side scatter profiles for live cells (not shown) (A). The numbers in A show the percent fraction of TREM2+ cells in transfected cells, and B shows profiles of TREM2+ cells after gating in A. C and D, the same experiment was performed in permeabilized cells for untransfected (blue) and transfected cells (red). The numbers in C show the percent fraction of TREM2+ cells in transfected cells, and D shows profiles of TREM2+ cells after gating in C. Average fluorescence intensity was measured for TREM2+ cells for each group in unpermeabilized (E) and permeabilized conditions (F). Significant differences were determined by one-way ANOVA followed by Bonferroni post-test. Error bars represent S.D. * and ** denote p < 0.05 and 0.01 versus TREM2 WT group. ††† denotes p < 0.001 versus untransfected cells (E and F).
Discussion
TREM2 has both neuropathological and genetic associations with AD. However, how it affects onset or progression of disease is unclear due to inconsistent study results. In primary microglial cells, TREM2 promoted phagocytosis of apoptotic neurons without stimulating cytokine production (35). This suggests that disease-associated mutations in TREM2 may impair phagocytosis of apoptotic neurons and increase the release of proinflammatory cytokines. Microglia surrounding amyloid plaques show elevated expression of TREM2 in AD (3, 36, 37). A recent examination of AD-like transgenic mice and post-mortem human AD brains revealed that these TREM2-expressing microglia tightly surround amyloid fibrils to promote their compaction, and TREM2 haplodeficiency or expression of the R47H polymorphism correlates with a reduction in this plaque compaction and increased axonal dystrophy in the regions surrounding plaques (37). Levels of TREM2 were also significantly elevated in the hippocampi of aged APP+PS1 mice, and overexpression of TREM2 in primary microglia from these mice facilitated phagocytosis of Aβ42 in vitro (38). In support of these findings, a recent study found that APP+PS1 mice deficient in TREM2 had reduced association of macrophages with Aβ plaques and reduced expression of proinflammatory cytokines such as interleukin-6 (IL-6) and IL-1β (23). These mice also had reduced levels of amyloid deposition in the hippocampi compared with APP+PS1/TREM2+/+ controls (23) despite the predicted reduction in phagocytic capability of macrophages with deficient TREM2 expression. These studies suggest that TREM2 is induced by the interaction of microglia with amyloid plaques and potentially facilitates their clearance while reducing the amount of damage done to neighboring neurons. These findings are contradictory to an earlier study showing that 5XFAD mice lacking TREM2 have enhanced accumulation of microglia around amyloid plaques (24). These contradictory findings suggest that TREM2 may function differently in mouse versus human brains and during different disease stages and that both enhancers and inhibitors of TREM2 signaling should be investigated to understand the species-specific difference in TREM2 function.
In this study, we successfully developed a split luciferase-based monitoring system in live cells to allow for real-time analysis of TREM2 coupling to TYROBP using a bioluminescence-based assay. Our TREM2-CLuc-IRES-TYROBP-NLuc construct shows a dose-dependent increase in luciferase activity in response to monoclonal anti-TREM2 antibody stimulation, indicative of TREM2-TYROBP binding. We used this construct to study the effects of AD- and FTD-associated TREM2 mutations on the TREM2-TYROBP interaction. We show that a TREM2-specific monoclonal antibody can stimulate TREM2-TYROBP coupling in a dose-dependent manner. Our data, however, do not show positive stimulation of TREM2-TYROBP coupling by human Aβ42 peptides, which are mainly dimers. This suggests that Aβ stimulation of microglia induces TREM2 expression in the AD brain even though it may not be the ligand of TREM2. A recent study demonstrated that microglia are more efficient at taking up Aβ when it forms a complex with a lipoprotein, such as low-density lipoprotein, compared with Aβ by itself (39). The degree of Aβ uptake is also dependent on the degree of TREM2 expression in microglia (39). However, more detailed examinations are necessary to determine whether the TREM2-Aβ interaction is specific to conformations, such as oligomers, protofibrils, or fibrils.
Microglia show increased phagocytosis of apoptotic neurons when coated with apoE (40), suggesting that TREM2 may function to activate microglial phagocytosis of apoE-coated cells that are undergoing apoptosis. Phosphatidylserine is first detected by milk fat globule-EGF factor 8 (41, 42), which is secreted by microglia and can bind to integrin receptors (αvβ3 or αvβ5) on the cell surface (43). In addition, phosphatidylserine on the apoptotic body is also detected by growth arrest-specific gene 6 and protein S, which stimulate the Tyro3, Axl, and Mertk subfamily of receptor tyrosine kinases for the immune response and phagocytosis (44–46). The interaction of TREM2 with apoptotic bodies may also require a bridging molecule similar to milk fat globule-EGF factor 8, growth arrest-specific gene 6, or protein S, which are not secreted by HEK293 cells. A more physiological model will be necessary to complement the missing molecule for the development of an apoptotic body-stimulated TREM2 reporter system.
We also demonstrated that stimulation of microglial cells with an anti-TREM2 antibody enhances their ability to phagocytose S. aureus, but not yeast-derived zymosan, by BV-2 cells. This is consistent with the previous report showing the TREM2-dependent uptake of bacteria (S. aureus, Escherichia coli, and Francisella tularensis) but not zymosan (47). Although it is still unclear which bacterial molecules are the binding targets of TREM2, carbohydrates and lipopeptides are likely good candidates. This is a convenient alternative measure to assess the effect of TREM2 activation on phagocytosis in the murine microglial cell line. This assay can be utilized in conjunction with TREM2 variant cell lines to further examine how polymorphisms in TREM2 can affect TREM2-TYROBP signaling and phagocytic capability in vitro. The sensitivity of low- and high-phagocytic TREM2-stimulated cell populations to SYK inhibition also demonstrates the activation of SYK-dependent phagocytic mechanisms. Although treatment with control IgG was able to stimulate phagocytosis, it was insensitive to SYK inhibition, demonstrating that not all Fc receptors are SYK-dependent. Potent activation of SYK by TREM2 supports these data and illustrates a TREM2-SYK-dependent mechanism for phagocytosis by microglial cells.
Our split-luciferase system is of great value to further examine the effects of TREM2 polymorphisms in AD as these have the greatest association with risk of AD after the well known genetic risk factor apoE. We demonstrate a significant effect of the T66M variant on constitutive TREM2 coupling to TYROBP. This was unexpected because the homozygous T66M TREM2 mutation was previously identified in Nasu-Hakola disease and predicted to be a loss-of-function mutation (9). Other Nasu-Hakola disease-linked mutations of TREM2 or TYROBP consistently cause loss-of-function mutations (48). Our data show that the T66M mutation is a gain-of-function mutation in terms of its coupling to TYROBP. However, our FACS-based analysis of the cell surface expression TREM2 variants shows that the T66M variant is poorly expressed on the cell surface compared with other TREM2 variants. TREM2 can be expressed on the cell surface by ionomycin treatment-induced Ca2+ influx and exocytotic stimulation (4). We tested ionomycin treatment of HEK293 cells after transient transfection of TREM2, but it was unchanged (data not shown). This T66M mutation of TREM2 may cause constitutive coupling with TREM2, which induces SYK activation and endocytosis of the complex, suggesting that T66M TREM2 is mostly retained intracellularly and does not engage in phagocytic function on the surface. Thus, we speculate that T66M is a gain-of-coupling mutation but consequently a loss-of-phagocytic-function mutation. Our finding on the T66M TREM2 variant also replicates those found in Kleinberger et al. (20) and Song et al. (49) who show reduced cell surface expression of TREM2 carrying the T66M mutation. Dysfunctional processing of T66M TREM2 resulted in an accumulation of immature TREM2 and reduced production of plasma membrane-bound or sTREM2 in cell medium in vitro, which supported their discovery that AD patients show reduced levels of sTREM2 in the CSF (20). However, these findings are contrary to a more recent report that AD patients had significantly higher sTREM2 concentrations in the CSF that correlated with markers of neurodegeneration and glial activation (22). Further study is needed to support these findings. The T66M mutation may result in hyperactive TREM2-TYROBP signaling and impaired processing or a TREM2 that is detrimental to immune homeostasis and phagocytosis, thus increasing the risk for AD.
In summary, we demonstrated that expression of the T66M mutation in HEK293 cells resulted in constitutive TREM2 activation and coupling to TYROBP. Thus, the T66M TREM2-CLuc-IRES-TYROBP-NLuc construct would be a good platform to screen inhibitors of TREM2-TYROBP coupling. Our TREM2-CLuc-IRES-TYROBP-NLuc construct would be more suitable for screening enhancers for TREM2-TYROBP coupling. These reporter constructs and complementing phagocytosis and cell surface expression assays may be useful for identifying ligands that can restore normal TREM2 functioning and coupling to TYROBP in patients with disease-associated polymorphisms in TREM2.
Experimental procedures
Plasmid design for TREM2-CLuc-IRES-TYROBP-NLuc constructs
We chose an internal ribosome entry site (IRES) backbone vector (pIRES; Clontech) under the control of the CMV immediate early promoter to allow for two cloning sites to express both human TREM2 and TYROBP genes as split-luciferase fusion proteins from a single mRNA (Fig. 1A). All restriction sites are unique for each insert. All amplifications by PCR were done using Pfu UltraII Hot start PCR Master Mix (Agilent Technologies, Santa Clara, CA). Human TREM2 (GenBankTM accession number NM_018965.2; 693 bp) and TYROBP cDNA sequences (GenBank accession number BT009851.1; 342 bp) were amplified by PCR from the human brain-derived cDNA library as a template using primer sets as listed in Table 1. The C terminus of the Renilla luciferase gene (CLuc) was amplified by PCR from pDuex-Bait vector, and the N terminus of the Renilla luciferase gene (NLuc) was amplified from pDuex-Prey vector using the primer sequences given in Table 1 (26). All primers were custom synthesized by Invitrogen. The PCR amplicons were purified after gel electrophoresis using the QIAquick Gel Extraction kit (Qiagen, Valencia, CA) and ligated into the TOPO® TA cloning pCR2.1 vector (Invitrogen). After confirmation of the DNA sequences of TREM2, TYROBP, CLuc, and NLuc DNA fragments, the plasmids were digested with restriction enzymes from New England Biolabs (M0202; Ipswich, MA) as indicated in Fig. 1A, purified after gel electrophoresis using QIAquick Gel Extraction kit, and sequentially inserted into the pIRES vector to develop the TREM2-CLuc-IRES-TYROBP-NLuc plasmid. Point mutations for R47H (16, 19), T66M (19), and S116C of human TREM2 (19) were designed and incorporated into the vector by GenScript (Piscataway, NJ). DNA plasmids were purified using the Endo-Free Plasmid Maxi kit for transfection studies (Qiagen).
Table 1.
Primer sequences for PCR amplification
For, forward; Rev, reverse.
| Primer | Sequence |
|---|---|
| TREM2 | |
| For | 5′-AAAGCTAGCATGGAACCTCTCCGGCTGCTC |
| Rev | 3′-TTGAATTCGAGCTCTCTAGACGTGTCTCTCAGCCCTGGCA |
| TYROBP | |
| For | 5′-TTGTCGACATGGGGGGACTTGAACCCTGCA |
| Rev | 3′-TTGCGGCCGCCTCGAGTTTGTAATACGGCCTCTGTGT |
| CLuc | |
| For | 5′-TTGAATTCGGTGGTGGCGGTTCAGGCGGA |
| Rev | 3′-TTACGCGTTAGCAGCACCCGGGGCAGCA |
| NLuc | |
| For | 5′-TTCTCGAGGGTGGTGGCGGTTCAGGCGGAGGTGGCTCT |
| Rev | 3′-TTGCGGCCGCCTACTTAACGAGAGGGATCTCGCGAG |
Cell culture
Murine microglia BV-2 cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen) at 1.5 × 106/well in 6-well plates. HEK293 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 2.2 × 106/well in 10-cm dishes.
Luciferase assays
HEK293 cells were plated at 4 × 104/well for n = 3/group in poly-d-lysine-coated wells in a 96-well plate. Cells were then transfected with either 100 ng of Renilla luciferase reporter vector containing the herpes simplex virus thymidine kinase promoter (pRL-TK, E2241, Promega, Madison, WI) as an internal control reporter or 200 ng of TREM2-CLuc-IRES-TYROBP-NLuc or variant constructs in Opti-MEM reduced serum medium (Invitrogen) utilizing the Lipofectamine 2000 transfection system (Invitrogen) according to the manufacturer's recommendations. For testing TREM2 variants, we included an additional control group with 200 ng of the WT TREM2-TYROBP lacking the N-terminal component of the Renilla luciferase gene (TREM2-CLuc-TYROBP).
Cells were stimulated with 10, 30, or 100 μg/ml Aβ42; 1, 5, 10, or 20 μg/ml anti-human TREM2 antibody (rat monoclonal IgG2b clone 237920, MAB17291, R&D Systems, Minneapolis, MN); 1, 5, 10, or 20 μg/ml control rat IgG2b (eBioscience/Thermo Fisher Scientific, Inc.); or 5, 10, or 20 μg/ml anti-TYROBP antibody (rabbit polyclonal IgG, sc-20783, Santa Cruz Biotechnology, Dallas, TX) 24 h after transfection. Constructs containing point mutations associated with neurological disease were tested in a similar manner with the exclusion of anti-TYROBP antibody and Aβ protein as these showed no effect in testing of our WT TREM2-CLuc-IRES-TYROBP-NLuc construct. Immediately after addition of ligand, the 96-well plate was placed into a CentroXS3 luminometer (Berthold Technologies, Bad Wildbad, Germany). CentroXS3 has the capacity for automated pipetting of multiple substrates and can measure luciferase activity from each individual well. The instrument was programmed to inject 50 μl of 30 μm ViviRen live cell Renilla luciferase substrate (E6491, Promega) into the well and measure luciferase activity immediately after injection at 0.5-s integration before injecting substrate into the next well. The luminescence was measured from each individual well in succession at 0.5-s integration repeatedly over 30 min.
TREM2-TYROBP cross-linking assay
To confirm that luciferase activity measured in our luminescence assays was due to a physical interaction between TREM2 and TYROBP, we performed a cross-linking assay with protein quantification by sandwich ELISA. Fig. 3A illustrates the experimental scheme used for measuring concentration of TREM2·TYROBP protein complexes. HEK293 cells were plated in a 100-mm dish at 70% confluence before transfection with 10 μg of TREM2-CLuc-IRES-TYROBP-NLuc with Lipofectamine 2000. Cells were then stimulated with 20 μg/ml anti-TREM2 antibody (goat polyclonal IgG, AF1828, R&D Systems) for 20 min in DMEM to induce TREM2 coupling to TYROBP. To stabilize the TREM2-TYROBP interaction for measurement by ELISA, we utilized adapted protocols to induce protein cross-linking (31, 50, 51). Briefly, we replaced media with DMEM containing 4 mm DTBP (D2388, Sigma-Aldrich), an amidine protein cross-linker with an 11.9-Å spacer arm, and 10 mm phosphatase inhibitor sodium vanadate with 0.6% hydrogen peroxide (sodium pervanadate; Sigma-Aldrich) for 10 min at 37 °C. Sodium vanadate was necessary to detect the interaction of TREM2 and tyrosine-phosphorylated TYROBP, which would be inactivated by the association of tyrosine phosphatase SHIP1 (31, 32). After incubation, media were aspirated, and cells were washed with PBS before lysing in radioimmune precipitation assay buffer (50 mm Tris hydrochloride, 150 mm sodium chloride, 1 mm EDTA, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, and 1% Nonidet P-40) containing 1% protease inhibitor phenylmethylsulfonyl fluoride and 1 mm sodium pervanadate for 1 h on ice. Tris quenches cross-linking by any remaining DTBP. The lysate was centrifuged at 14,000 rpm for 30 min at 4 °C. Protein was quantified using the BCA Protein Assay kit (Pierce) according to the manufacturer's instructions.
TREM2-TYROBP sandwich ELISA
Tissue culture-grade 96-well plates were coated with 50 μl of anti-TYROBP antibody (rabbit polyclonal IgG, sc-20783) at a concentration of 1 μg/ml in coating buffer (0.2 m sodium bicarbonate, pH 9.4) overnight at 4 °C. Coated wells were washed and then blocked with 10% normal goat serum in PBS for 1 h at room temperature. HEK293 protein lysates from the TREM2-TYROBP cross-linking assay were applied to TYROBP-coated wells at 100 μg in 100 μl of radioimmune precipitation assay buffer and incubated overnight at 4 °C. To label TYROBP protein cross-linked with TREM2, TYROBP-bound protein was labeled with a biotinylated anti-human TREM2 antibody (goat polyclonal IgG, BAF1828, R&D Systems)·HRP-conjugated streptavidin (21126, Life Technologies) complex (0.5 μg/ml each) in 0.1% Triton X-100 in PBS for 1 h at room temperature. Wells were then incubated with 100 μl of a stabilized chromogen, tetramethylbenzidine, for 30 min in the dark at room temperature followed by addition of 100 μl of Stop solution (Life Technologies). Optical density (OD) was read at 450 nm with a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA), and results were quantified by the arithmetic difference between the given treatment and the average OD of untransfected controls for n = 4 per group.
Phagocytosis assay
To determine whether activation of TREM2 enhanced the ability of BV-2 cells to phagocytose, BV-2 microglial cells were plated at 1.0 × 106/well in a 6-well plate. Cells were then serum-starved overnight before the phagocytosis assay. Some cells were pretreated with 14 μm SYK inhibitor (CAS 622387-85-3, EMD Millipore, Billerica, MA) for 30 min to test TREM2-specific inhibition of phagocytosis. Alexa Fluor 594-tagged S. aureus particles (S-23372, Life Technologies) were prepared beforehand by centrifugation at 10,000 × g followed by resuspension in DMEM and 15-min sonication in a water bath sonicator. BV-2 cells were then stimulated with 20 μg/ml anti-human TREM2 antibody (goat polyclonal IgG, BAF1828), goat IgG control, or goat F(ab′)2 fragment control (ChromPure 005-000-006, Jackson ImmunoResearch Laboratories, West Grove, PA) and a 1:5 ratio of cells:S. aureus particles for 30 min. Cells were trypsinized immediately after completion of the assay to generate a single cell suspension and remove non-phagocytosed S. aureus and prepared for analysis of phagocytic cells in technical duplicate by flow cytometry. Blocking of nonspecific Fc-mediated interactions was done with 4 μl of anti-mouse CD16/CD32/100 μl for 20 min on ice. To label cells treated with anti-TREM2 antibody, cells were incubated with 5 μg/ml donkey anti-goat Alexa Fluor 488 secondary antibody (A11055, Invitrogen) for 30 min. Cells not treated with Alexa Fluor 594-tagged S. aureus particles or Alexa Fluor 488 secondary antibody were used to gate cells that were positive for Alexa Fluor 488 or 594 signal. Cells were examined by flow cytometry within 1 h of completing the staining procedure using a BD LSR II instrument with BD FACSDiva 6.2.1 software (BD Biosciences) and analyzed with FlowJo software (FlowJo LLC, Ashland, OR).
SYK activity assay
Parental BV-2 cells were stimulated in suspension with either anti-TREM2 antibody (BAF1828) or nonspecific IgG control (AB-108-C, R&D Systems) at a concentration of 5 μg/ml each in Opti-MEM at 37 °C for 30 min. SYK inhibitor (CAS 622387-85-3) was also administered in tandem at a concentration of 14 μm. Cells were lysed on ice in lysis buffer (25 mm Tris-HCl, pH 7.4, Triton X-100, 150 mm NaCl, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm EDTA, 50 mm NaF, and 1 mm sodium orthovanadate), and cell lysate was extracted and incubated with anti-SYK rabbit polyclonal antibody (sc-573, Santa Cruz Biotechnology) for 3 h. Immunoprecipitation was then conducted with 20 μl of protein A-agarose beads (ab193254, Abcam). Next SYK was eluted from the beads with 0.1 m glycine for 10 min and stabilized with 50 mm Tris. SYK extracts were then assessed for levels of activity using an ADP-Glo assay kit (V6930, Promega) according to the manufacturer's procedure.
Flow cytometry for TREM2/TYROBP expression
HEK293 cells were plated at 0.5 × 106/well in a 6-well plate for n = 1 per construct. Cells were then transfected with 1600 ng of TREM2-CLuc-IRES-TYROBP-NLuc (WT TREM2); TREM2-CLuc-TYROBP (NC TREM2) control plasmid; or R47H, T66M, or S116C variant constructs in the same manner as described previously. 24 h after transfection, cells were trypsinized and prepared for flow cytometry.
Cells were analyzed for cell surface or intracellular expression of TREM2 and TYROBP. For analysis of intracellular TREM2 and TYROBP, cells were fixed in 4% paraformaldehyde followed by permeabilization with 0.5% Tween 20 in PBS containing 1% bovine serum albumin. Cells used for cell surface expression of protein remained unfixed and unpermeabilized to maintain the integrity of the cell membrane. Cells were then incubated with anti-TYROBP antibody (rabbit polyclonal IgG, sc-20783) and PE-conjugated anti-human/mouse TREM2 (rat monoclonal IgG2b clone 237920, FAB17291P) for 1 h. This was followed by 30-min secondary antibody incubation with donkey anti-rabbit conjugated to Alexa Fluor 647 (A-31573, Invitrogen) at 10 μg/ml for fluorescence labeling of TYROBP. At least 10,000 cells were analyzed by flow cytometry using a BD LSR II instrument with BD FACSDiva 6.2.1 software and FlowJo software. Gating was aided by analysis of single positive PE-TREM2-only and TYROBP-Alexa Fluor 647-only cells as well as negative control cells that were treated with neither anti-TREM2 nor anti-TYROBP antibody.
Immunofluorescence
HEK293 cells (1 × 105 cells/well) were plated on poly-d-lysine-coated coverslips in 24-well plates, transfected with 800 ng of TREM2-CLuc-IRES-TYROBP-NLuc (WT TREM2) or R47H, T66M, or S116C variant constructs as described under “Flow cytometry for TREM2/TYROBP expression” and 48 h post-transfection fixed by 4% paraformaldehyde in PBS for 20 min. For the cell surface detection of TREM2, cells were blocked by 5% donkey serum in PBS for 30 min and stained with 1:200 anti-TREM2 rat monoclonal antibody as described above followed by washing and permeabilization of the cells with 0.5% Triton X-100 and blocking with 5% donkey serum prior to staining with 1:1000 anti-TYROBP rabbit polyclonal antibody as described above. For total TREM2 staining, cells were first permeabilized with 0.5% Triton X-100 and blocked with 5% goat serum in PBS for 30 min followed by staining with anti-TREM2 rat and anti-TYROBP rabbit antibodies in a similar manner. After washing, the bound primary antibodies were detected by 1:500 Alexa Fluor 594-conjugated donkey anti-rat and Alexa Fluor 488-conjugated donkey anti-rabbit antibodies (Invitrogen), respectively. The immunostained cells on the coverslips were mounted on Superfrost Plus slides (Fisher Scientific) with Fluoromount mounting medium (Sigma-Aldrich), and images were captured using a Nikon Eclipse TE-2000U with attached charge-coupled device camera with Nikon CFI Plan Fluor 60× oil objective.
TREM2 and TYROBP ELISA
HEK293 cells were transfected with the WT and R47H, T66M, and S116C variants of the TREM2-CLuc-IRES-TYROBP-NLuc plasmid using Lipofectamine 2000. Cells were then lysed in lysis buffer (150 mm NaCl, 1% Triton X-100, 2 mm EDTA, 25 mm Tris-HCl, pH 7.4), and the lysates were analyzed for total protein concentration using a BCA Protein Assay kit and quantification of TREM2 and TYROBP in commercial ELISA kits for human TREM2 (RAB1091, Sigma-Aldrich) and human TYROBP (LS-F13489, LifeSpan Biosciences, Inc., Seattle, WA).
Statistics
All data are presented as mean values ± S.D. For comparison between two groups, data were analyzed by Student's t test. For multiple mean comparisons, data were analyzed by one or two-way ANOVA followed by Tukey or Bonferroni post hoc analysis using statistics software (Prism 4.0, GraphPad Software, Inc., San Diego, CA). Significance is considered any value of p ≤ 0.05.
Author contributions
M. M. V. and K. A. C. designed and performed experiments, developed figures and tables, and wrote the manuscript. S. I. and A. Y.-K. designed and performed experiments and developed figures. G. Y. constructed DNA plasmids. L. K. performed tissue culture for the experiments. T. I. conceptualized and outlined the study, designed the DNA plasmid construction scheme, supervised all the authors, interpreted data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Naohiro Kato at the Louisiana State University for pDuex-Bait and pDuex-Prey vectors, the Flow Cytometry Core Facility at Boston University School of Medicine for expertise and guidance in experiments, and Dr. Hirohide Asai for assisting in gene constructions.
This work was supported by National Institutes of Health Predoctoral Training Grant 5T32 GM008541 (to M. M. V. and K. A. C.) and Grant RF1 AG054199-01 (to T. I.), a Henry I. Russek Award (to M. M. V.), Boston University Undergraduate Research Opportunities Program Awards (to G. Y. and L. K.), the Alzheimer's Association (to T. I.), the BrightFocus Foundation (to T. I.), and the Massachusetts Neuroscience Consortium (to T. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S3.
- TREM2
- triggering receptor expressed on myeloid cells 2
- TYROBP
- TYRO protein-tyrosine kinase-binding protein
- TYRO
- tyrosine
- AD
- Alzheimer's disease
- FTD
- frontotemporal dementia
- sTREM2
- soluble form of TREM2
- CSF
- cerebrospinal fluid
- APP
- amyloid precursor protein
- PS1
- presenilin-1
- Aβ
- amyloid-β peptide
- RLU
- relative light units
- IRES
- internal ribosome entry site
- AUC
- area under the curve
- DTBP
- dimethyl 3,3′-dithiobispropionimidate
- Hi
- high
- Lo
- low
- SYK
- spleen tyrosine kinase
- PE
- phycoerythrin
- MFI
- mean fluorescence intensity
- CLuc
- C terminus of the Renilla luciferase gene
- NLuc
- N terminus of the Renilla luciferase gene
- ANOVA
- analysis of variance.
References
- 1. Colonna M. (2003) TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453 [DOI] [PubMed] [Google Scholar]
- 2. Klesney-Tait J., Turnbull I. R., and Colonna M. (2006) The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 3. Schmid C. D., Sautkulis L. N., Danielson P. E., Cooper J., Hasel K. W., Hilbush B. S., Sutcliffe J. G., and Carson M. J. (2002) Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83, 1309–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sessa G., Podini P., Mariani M., Meroni A., Spreafico R., Sinigaglia F., Colonna M., Panina P., and Meldolesi J. (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 20, 2617–2628 [DOI] [PubMed] [Google Scholar]
- 5. Ivashkiv L. B. (2008) A signal-switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. N. Rev. Immunol. 8, 816–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vicente-Manzanares M., and Sánchez-Madrid F. (2004) Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 4, 110–122 [DOI] [PubMed] [Google Scholar]
- 7. Mizuno N., Uematsu S., and Ono M. (1975) [Pustular psoriasis (Zumbusch) treated with 8-methoxypsoralen phototherapy (author's transl)]. Nihon Hifuka Gakkai Zasshi 85, 587–594 [PubMed] [Google Scholar]
- 8. Kondo T., Takahashi K., Kohara N., Takahashi Y., Hayashi S., Takahashi H., Matsuo H., Yamazaki M., Inoue K., Miyamoto K., and Yamamura T. (2002) Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology 59, 1105–1107 [DOI] [PubMed] [Google Scholar]
- 9. Paloneva J., Manninen T., Christman G., Hovanes K., Mandelin J., Adolfsson R., Bianchin M., Bird T., Miranda R., Salmaggi A., Tranebjaerg L., Konttinen Y., and Peltonen L. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamerman J. A., Jarjoura J. R., Humphrey M. B., Nakamura M. C., Seaman W. E., and Lanier L. L. (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J. Immunol. 177, 2051–2055 [DOI] [PubMed] [Google Scholar]
- 11. Turnbull I. R., Gilfillan S., Cella M., Aoshi T., Miller M., Piccio L., Hernandez M., and Colonna M. (2006) Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524 [DOI] [PubMed] [Google Scholar]
- 12. Hamerman J. A., Tchao N. K., Lowell C. A., and Lanier L. L. (2005) Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat. Immunol. 6, 579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cady J., Koval E. D., Benitez B. A., Zaidman C., Jockel-Balsarotti J., Allred P., Baloh R. H., Ravits J., Simpson E., Appel S. H., Pestronk A., Goate A. M., Miller T. M., Cruchaga C., and Harms M. B. (2014) TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guerreiro R. J., Lohmann E., Brás J. M., Gibbs J. R., Rohrer J. D., Gurunlian N., Dursun B., Bilgic B., Hanagasi H., Gurvit H., Emre M., Singleton A., and Hardy J. (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., et al. (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., Rujescu D., Hampel H., Giegling I., Andreassen O. A., Engedal K., et al. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rayaprolu S., Mullen B., Baker M., Lynch T., Finger E., Seeley W. W., Hatanpaa K. J., Lomen-Hoerth C., Kertesz A., Bigio E. H., Lippa C., Josephs K. A., Knopman D. S., White C. L. 3rd, Caselli R., et al. (2013) TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol. Neurodegener. 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuyvers E., Bettens K., Philtjens S., Van Langenhove T., Gijselinck I., van der Zee J., Engelborghs S., Vandenbulcke M., Van Dongen J., Geerts N., Maes G., Mattheijssens M., Peeters K., Cras P., Vandenberghe R., et al. (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol. Aging 35, 726.e11–726.e19 [DOI] [PubMed] [Google Scholar]
- 19. Borroni B., Ferrari F., Galimberti D., Nacmias B., Barone C., Bagnoli S., Fenoglio C., Piaceri I., Archetti S., Bonvicini C., Gennarelli M., Turla M., Scarpini E., Sorbi S., and Padovani A. (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 35, 934.e7–934.e10 [DOI] [PubMed] [Google Scholar]
- 20. Kleinberger G., Yamanishi Y., Suárez-Calvet M., Czirr E., Lohmann E., Cuyvers E., Struyfs H., Pettkus N., Wenninger-Weinzierl A., Mazaheri F., Tahirovic S., Lleó A., Alcolea D., Fortea J., Willem M., et al. (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra86. [DOI] [PubMed] [Google Scholar]
- 21. Piccio L., Deming Y., Del-Águila J. L., Ghezzi L., Holtzman D. M., Fagan A. M., Fenoglio C., Galimberti D., Borroni B., and Cruchaga C. (2016) Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 131, 925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heslegrave A., Heywood W., Paterson R., Magdalinou N., Svensson J., Johansson P., Öhrfelt A., Blennow K., Hardy J., Schott J., Mills K., and Zetterberg H. (2016) Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol. Neurodegener. 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jay T. R., Miller C. M., Cheng P. J., Graham L. C., Bemiller S., Broihier M. L., Xu G., Margevicius D., Karlo J. C., Sousa G. L., Cotleur A. C., Butovsky O., Bekris L., Staugaitis S. M., Leverenz J. B., et al. (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Cella M., Mallinson K., Ulrich J. D., Young K. L., Robinette M. L., Gilfillan S., Krishnan G. M., Sudhakar S., Zinselmeyer B. H., Holtzman D. M., Cirrito J. R., and Colonna M. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paulmurugan R., and Gambhir S. S. (2003) Monitoring protein-protein interactions using split synthetic Renilla luciferase protein-fragment-assisted complementation. Anal. Chem. 75, 1584–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fujikawa Y., and Kato N. (2007) Split luciferase complementation assay to study protein-protein interactions in Arabidopsis protoplasts. Plant J. 52, 185–195 [DOI] [PubMed] [Google Scholar]
- 27. Deng Q., Wang D., Xiang X., Gao X., Hardwidge P. R., Kaushik R. S., Wolff T., Chakravarty S., and Li F. (2011) Application of a split luciferase complementation assay for the detection of viral protein-protein interactions. J. Virol. Methods 176, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matthews J. C., Hori K., and Cormier M. J. (1977) Purification and properties of Renilla reniformis luciferase. Biochemistry 16, 85–91 [DOI] [PubMed] [Google Scholar]
- 29. Takahashi K., Prinz M., Stagi M., Chechneva O., and Neumann H. (2007) TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 4, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frank S., Burbach G. J., Bonin M., Walter M., Streit W., Bechmann I., and Deller T. (2008) TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447 [DOI] [PubMed] [Google Scholar]
- 31. Bouchon A., Hernández-Munain C., Cella M., and Colonna M. (2001) A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194, 1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peng Q., Malhotra S., Torchia J. A., Kerr W. G., Coggeshall K. M., and Humphrey M. B. (2010) TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal. 3, ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hsieh C. L., Koike M., Spusta S. C., Niemi E. C., Yenari M., Nakamura M. C., and Seaman W. E. (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhong L., Chen X. F., Zhang Z. L., Wang Z., Shi X. Z., Xu K., Zhang Y. W., Xu H., and Bu G. (2015) DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi K., Rochford C. D., and Neumann H. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Melchior B., Garcia A. E., Hsiung B. K., Lo K. M., Doose J. M., Thrash J. C., Stalder A. K., Staufenbiel M., Neumann H., and Carson M. J. (2010) Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro 2, e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan P., Condello C., Keene C. D., Wang Y., Bird T. D., Paul S. M., Luo W., Colonna M., Baddeley D., and Grutzendler J. (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang T., Tan L., Zhu X. C., Zhang Q. Q., Cao L., Tan M. S., Gu L. Z., Wang H. F., Ding Z. Z., Zhang Y. D., and Yu J. T. (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 39, 2949–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yeh F. L., Wang Y., Tom I., Gonzalez L. C., and Sheng M. (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-β by microglia. Neuron 91, 328–340 [DOI] [PubMed] [Google Scholar]
- 40. Atagi Y., Liu C. C., Painter M. M., Chen X. F., Verbeeck C., Zheng H., Li X., Rademakers R., Kang S. S., Xu H., Younkin S., Das P., Fryer J. D., and Bu G. (2015) Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuller A. D., and Van Eldik L. J. (2008) MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J. Neuroimmune Pharmacol. 3, 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Y., Yang X., Guo C., Nie P., Liu Y., and Ma J. (2013) Essential role of MFG-E8 for phagocytic properties of microglial cells. PLoS One 8, e55754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hanayama R., Tanaka M., Miwa K., Shinohara A., Iwamatsu A., and Nagata S. (2002) Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187 [DOI] [PubMed] [Google Scholar]
- 44. Lew E. D., Oh J., Burrola P. G., Lax I., Zagorska A., Traves P. G., Schlessinger J., and Lemke G. (2014) Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife 3, e03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsou W. I., Nguyen K. Q., Calarese D. A., Garforth S. J., Antes A. L., Smirnov S. V., Almo S. C., Birge R. B., and Kotenko S. V. (2014) Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 289, 25750–25763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van der Meer J. H., van der Poll T., and van 't Veer C. (2014) TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood 123, 2460–2469 [DOI] [PubMed] [Google Scholar]
- 47. N'Diaye E. N., Branda C. S., Branda S. S., Nevarez L., Colonna M., Lowell C., Hamerman J. A., and Seaman W. E. (2009) TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J. Cell Biol. 184, 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bianchin M. M., Capella H. M., Chaves D. L., Steindel M., Grisard E. C., Ganev G. G., da Silva Júnior J. P., Neto Evaldo S., Poffo M. A., Walz R., Carlotti Júnior C. G., and Sakamoto A. C. (2004) Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy—PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell. Mol. Neurobiol. 24, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song W., Hooli B., Mullin K., Jin S. C., Cella M., Ulland T. K., Wang Y., Tanzi R. E, and Colonna M. (2017) Alzheimer's disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 13, 381–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rizvi S. M., and Raghavan M. (2010) Mechanisms of function of tapasin, a critical major histocompatibility complex class I assembly factor. Traffic 11, 332–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daws M. R., Lanier L. L., Seaman W. E., and Ryan J. C. (2001) Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 31, 783–791 [DOI] [PubMed] [Google Scholar]
- 52. Iqbal K., Alonso A. C., Gong C. X., Khatoon S., Pei J. J., Wang J. Z., and Grundke-Iqbal I. (1998) Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J. Neural. Transm. Suppl. 53, 169–180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.