

Abstract
Background and Purpose
Pharmacological strategies aimed to facilitate neuronal renewal in the adult brain, by promoting endogenous neurogenesis, constitute promising therapeutic options for pathological or traumatic brain lesions. We have previously shown that non‐tumour‐promoting PKC‐activating compounds (12‐deoxyphorbols) promote adult neural progenitor cell (NPC) proliferation in vitro and in vivo, enhancing the endogenous neurogenic response of the brain to a traumatic injury. Here, we show for the first time that a diterpene with a lathyrane skeleton can also activate PKC and promote NPC proliferation.
Experimental Approach
We isolated four lathyranes from the latex of Euphorbia plants and tested their effect on postnatal NPC proliferation, using neurosphere cultures. The bioactive lathyrane ELAC (3,12‐di‐O‐acetyl‐8‐O‐tigloilingol) was also injected into the ventricles of adult mice to analyse its effect on adult NPC proliferation in vivo.
Key Results
The lathyrane ELAC activated PKC and significantly increased postnatal NPC proliferation in vitro, particularly in synergy with FGF2. In addition ELAC stimulated proliferation of NPC, specifically affecting undifferentiated transit amplifying cells. The proliferative effect of ELAC was reversed by either the classical/novel PKC inhibitor Gö6850 or the classical PKC inhibitor Gö6976, suggesting that NPC proliferation is promoted in response to activation of classical PKCs, particularly PKCß. ELAC slightly increased the proportion of NPC expressing Sox2. The effects of ELAC disappeared upon acetylation of its C7‐hydroxyl group.
Conclusions and Implications
We propose lathyranes like ELAC as new drug candidates to modulate adult neurogenesis through PKC activation. Functional and structural comparisons between ELAC and phorboids are included.
Abbreviations
- ASCL1
achaete scute homologue 1
- BrdU
bromodeoxyuridine
- BSO
buthione sulfoximine
- DCX
doublecortin
- DG
dentate gyrus of the hippocampus
- EGSH
glutathione ethyl ester
- GFAP
glial fibrilary acidic protein
- NPC
neural progenitor cells
- NSC
neural stem cells
- SOX2
(sex determining region Y)‐box 2
- SVZ
subventricular zone
- TAP
transit amplifying progenitor
Introduction
Central nervous system insults, such as stroke or traumatic brain injury, are characterized by an irreversible neuronal loss, for which there is no adequate treatment at present. Neurons are post‐mitotic cells that can only be generated de novo from neural stem cells (NSC) through a process known as neurogenesis. In mammals, adult neurogenesis occurs throughout life fundamentally in two brain areas: the dentate gyrus of the hippocampus (DG) and the subventricular zone (SVZ). NSC within these regions divide asymmetrically to produce undifferentiated multipotent neural progenitor cells (NPC), which, under physiological conditions, primarily commit to the neuronal lineage (Alvarez‐Buylla and Garcia‐Verdugo, 2002; Goldman, 2003; Aimone et al., 2014). Endogenous neurogenesis increases significantly upon brain injury, and newly generated neurons have been found in brain lesions of different aetiologies (Parent et al., 1997; Jin et al., 2001); some of these new neurons arise from neuroblasts generated in distant neurogenic regions after they have migrated to the injury site, while others have been reported to generate locally at the site of injury from astrocytes reprogrammed into neurogenic cells (Magnusson et al., 2014). Nonetheless, the neuroregenerative capacity of the brain under pathological conditions is rather limited (Fallon et al., 2000; Arvidsson et al., 2002; Nakatomi et al., 2002; Buffo et al., 2008; Susarla et al., 2014). Hence, in order to improve neuronal replacement within an injured region of the CNS, it is necessary to find new drugs to promote different aspects of neurogenesis, like NSC activation, NPC proliferation or neuronal differentiation.
Diterpenes constitute a vast group of 20‐carbon‐skeleton compounds derived from geranyl geranyl pyrophosphate, which, by cyclization, generate polycyclic structures (i.e. jatrophanes, lathyranes, daphnanes or tiglianes) that are abundant in the latex of Euphorbia plants (Vasas and Hohmann, 2014). Several tiglianes with a phorboid structure significantly increase NPC proliferation by targeting and activating one or more, still unidentified, isoforms of PKC. These phorboids included the commercially available PMA and the non‐tumour‐promoting phorboid prostratin, in addition to six natural products isolated by us from the plant Euphorbia resinifera. Two of these compounds, which did not show tumour‐promoting activities, prostratin and ER272 (13‐O‐isobutyroyl‐12‐deoxyphorbol), also augment endogenous neurogenesis in response to a traumatic brain injury in vivo (Geribaldi‐Doldán et al., 2016). Interestingly, Sung et al. (2007) have reported that cerebral hypoxia induces NPC proliferation through a PKC‐dependent mechanism, suggesting that the neurogenic response to brain insults may be mediated by PKC activation.
The PKC family of serine/threonine kinases is composed of 10 members, which share a highly conserved catalytic domain linked to a more divergent amino‐terminal regulatory domain. When inactive, PKCs are auto‐inhibited by a pseudosubstrate sequence located in the regulatory domain, which occupies the substrate‐binding pocket preventing catalysis. PKCs are activated when distinct second messengers and/or allosteric modulators bind to the regulatory domain, displacing the pseudosubstrate region from the active site and transiently recruiting PKCs to the plasma membrane, all of which allows for substrate binding and PKC activation (Rosse et al., 2010; Black and Black, 2012). Based on their specific activators, PKCs are subdivided into three subfamilies: (i) the classical or conventional PKCs (cPKCs α, β and γ), activated by Ca2+, DAG and phosphatidylserine (PS); (ii) the novel PKCs (nPKCs δ, ε, θ and η), activated by DAG and PS; and (iii) the atypical PKCs (aPKCs ζ and λ/ι), which require PS and protein–protein interactions (Rosse et al., 2010).
Several PKCs are expressed in neurogenic regions (Minami et al., 2000) and participate in distinct signalling cascades initiated by growth factors (GF), often determining GF specificity (Corbit et al., 2000). Atypical PKCs have been involved in the NSC‐to‐neuron transition during development, and their indirect activation with metformin also increases neurogenesis in the adult brain (Wang et al., 2012). A novel PKC, PKCε, seems to be important for the PMA‐mediated astrocytic differentiation of NPC (Steinhart et al., 2007). It is plausible that additional isoforms of PKC may be implicated in other particular aspects of adult neurogenesis, like NSC self‐renewal, proliferation, survival or neuronal differentiation.
Certain diterpenes with a lathyrane skeleton can mimic prostratin and other PKC activators in their ability to antagonize HIV‐1 latency in Jurkat cells, by a PKC‐dependent mechanism (Avila et al., 2010). These have been reported, based on its structures, as non‐tumour‐promoting agents; thus, our first aim was to test the capacity of four different lathyranes to promote NPC proliferation, in an attempt to identify new molecules useful to improve neuronal replacement through PKC modulation (Blumberg et al., 2008; Geribaldi‐Doldán et al., 2016). We show that one of these lathyranes, which we denominated ELAC, stimulates NPC proliferation in vivo and in vitro by a mechanism involving classical PKC activation and without negatively affecting stemness. The functional interactions between ELAC and the physiologically relevant basic FGF (bFGF/FGF2) and EGF are described. We also demonstrate the singular importance of ELAC's C7‐hydroxyl group for PKC activation. Structural characteristics required for the bioactivity of lathyranes are discussed.
Methods
O‐acetylation of the C7 position in ELAC
A solution of ELAC (30 mg) in pyridine (3 mL) and acetic anhydride (3 mL) was magnetically stirred at room temperature for 48 h. The reaction mixture, diluted in ethyl acetate (50 mL), was treated with HCl 1N (3 × 50 mL), dried on anhydrous sodium sulphate and solvent‐removed at reduced pressure. Resulting crude reaction mixture was purified by HPLC (80:20 hexane : ethyl acetate) to yield 15 mg of Ac‐ELAC (3,7,12‐tri‐O‐acetyl‐8‐O‐tigloylingol), an amorphous solid. [α]D 20 = −25 (c = 0.1 mg·mL−1, CHCl3). IR (KBr) ʋmáx (cm−1): 3440, 1735, 1700, 1645, 1370, 1365. 1H–NMR (400 MHz, CDCl3): δ (ppm) 6.83 (1H, qq, J = 7.5, 1.5, H3´), 5.58 (1H, sa, H5), 5.33 (1H, d, J = 8, H3), 5.11 (1H, d, J = 1.5, H7), 4.86 (1H, dd, J = 11.0, 3.5, H12), 4.61 (1H, dd, J = 11.0, 2.0, H8), 2.93 (1H, dq, J = 7.5, 4.0, H13), 2.78 (1H, dd, J = 15, 9.0, H1α), 2.46 (1H, m, H2), 2.12 (3H, s, C7‐COCH3), 2.11 (3H, d, J = 1.0, CH317), 2.09 (3H, s, C12‐COCH3), 2.07 (3H, s, C3‐COCH3), 1.79 (3H, s, CH35´)*, 1.78 (3H, s, CH34´)*, 1.69 (1H, d, J = 15.0, H1β), 1.31 (1H, dd, J = 10.5, 8.5, H9), 1.1 (1H, m, H11), 1.1 (3H, s, CH319), 1.05 (3H, d, J = 7.5, CH320), 0.96 (3H, d, J = 7.5, CH316), 0.81 (3H, s, CH318). 13C–NMR (100 MHz, CDCl3): δ (ppm) 207.6 (C14, CO), 170.7 (C3‐COCH3), 170.3 (C12‐COCH3), 169.8 (C7‐COCH3), 167.1 (C8‐COCH3), 139.9 (C6), 137.9 (C3´), 128.2 (C2´), 116.8 (C5), 77.3 (C7), 76.3 (C3), 73.4 (C4), 71.2 (C8), 71.1 (C15), 70.8 (C12), 43.0 (C13), 31.4 (C1), 30.9 (C11), 29.4 (C2), 29.2 (CH318), 25.0 (C9), 21.1 (COCH3), 20.9 (COCH3), 20.6 (COCH3), 19.3 (C10), 17.4 (CH317), 16.9 (CH316), 16.1 (CH319), 14.5 (C4´)*, 13.4 (CH320), 11.9 (C5´)* (* interchangeable signals). HRMSESI(+): Observed m/z 597.2688 [M + Na]+ (calculated for C31H42O10Na 597.2676).
Animal subjects
CD1 mice were used throughout this study. Animals were housed under controlled conditions of temperature (21–23°C) and light (LD 12:12) with free access to food (AO4 standard maintenance diet, SAFE, Épinay‐sur‐Orge, France) and water. Care and handling of animals were performed according to the Guidelines of the European Union Council (2010/63/EU), and the Spanish regulations (65/2012 and RD53/2013) for the use of laboratory animals. Animal experiment protocols were approved by the “Dirección General de la Producción Agrícola y Ganadera” of the Andalusian “Consejería de Agricultura Pesca Y Desarrollo Rural”. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
The total number of animals used was as follows: 24 2‐month‐old adult mice and 120 7‐day‐old pups. Adult mice were randomized during the first week after birth by cross fostering, and male mice were used at the age of 2 months.
SVZ cell isolation and culture
NPC were isolated from the SVZ of 7 day postnatal (P7) mice following the same procedure described before (Torroglosa et al., 2007), and were cultured as described elsewhere (Rabaneda et al., 2008). The killing of P7 mice was done by decapitation without anaesthesia, and six pups were used for each independent culture. EGF (20 ng·mL−1, from GIBCO) and bFGF (10 ng·mL−1; from PeproTech, Frankfurt, Germany) were used for culture expansion, but only bFGF was present in most experimental settings, unless otherwise indicated.
Neurosphere assays and secondary neurosphere measurements
To test the effects of our compounds on NPC proliferation, single cells from mechanically disaggregated neurospheres were plated in anti‐adherent 96‐well plates (Corning, NY, USA) at a density of 20 000 cells mL−1 (4000 cells in 200 μL per well), making triplicates for each condition in each experiment. Lathyranes and other pharmacological agents were added at the time of seeding, and neurosphere size and number were measured 72 h later, as previously described (Geribaldi‐Doldán et al., 2016). Experiments were repeated a minimum of five independent times, and each of these five independent experiments was run in triplicate. In terms of total numbers of neurospheres measured, a minimum of 540 neurospheres had their size measured per condition.
Some of these primary neurospheres were mechanically disaggregated again after completion of the 72 h treatments with the different compounds, seeded at an identical density (4000 cells in 200 μL per well) in triplicates and cultured for an extra 72 h in the absence of any added drugs; then, the number of secondary neurospheres formed from pretreated NPC was determined by direct counting (Geribaldi‐Doldán et al., 2016). Quantification of the size and number of neurospheres were blinded (samples were coded and experiments were performed and quantified by different individuals).
Cell culture adhesion, treatment and transfection
Neurosphere cells were centrifuged, resuspended in defined medium with growth factors and seeded. Cell transfection with specific siRNAs was performed 18 h after seeding; for this, cells were changed to antibiotic‐free medium and transfected using Lipofectamine 2000, following the manufacturer's instructions. Lipofectamine was removed 24 h later, ELAC was added and cells were maintained for 48 additional hours before being fixed for Ki67 immunocytochemistry.
PKC kinase assay
Neurospheres were disaggregated, and 20 000 single cells were seeded per well. Treatments (5 μM prostratin, 5 μM ELAC or none) were added for 1 h prior to cell centrifugation (200 g, 5 min) and lysis. Protein content was measured in the lysates using the BCA method (ThermoFisher Scientific, Rockford, IL, USA), and 1.5 μg of crude protein was used per assay. The amount of PKC kinase activity was measured in each sample using the PKC Kinase Activity Assay Kit (Abcam, Cambridge, U.K.; cat. no. ab139437), following the manufacturer's instructions. Positive controls (20–60 ng of purified active PKC supplied by the kit) and blanks (diluent only) were included in each independent determination. Blanks were subtracted from measurements before comparisons were made.
Immunocytochemistry
Ki67 immunostaining was performed as previously described (Geribaldi‐Doldán et al., 2016). Nestin and sex determining region Y‐box 2 (Sox2) immunostaining was performed in intact neurospheres, formed during a 72 h period after which neurospheres were carefully collected and centrifuged for 10 min at 900 × g in a Cytospin 4 Cytocentrifuge (ThermoFisher Scientific, Rockford, IL, USA), allowing gentle deposition of the cell aggregates onto 12‐mm‐diameter round coverslips. Cells were then fixed with 4% paraformaldehyde at RT for 30 min. Then, standard protocols for immunocytochemistry were followed (Geribaldi‐Doldán et al., 2016). The primary antibodies used for this were goat anti‐nestin and rabbit anti‐Sox2 (both at 1:100, from Santa Cruz Biotechnology, Santa Cruz, CA, USA); the secondary antibodies used were donkey anti‐rabbit IgG labelled with AlexaFluor® 488 (1:1000) and donkey anti‐goat IgG labelled with AlexaFluor® 594 (1:5000), from Invitrogen (Carlsbad, CA, USA). Samples were coded before immunostaining, and blinded quantifications of labelled cells were done in five independent experiments; each of these five independent experiments was performed in triplicate. Experiments were performed and quantified by different individuals. Original fluorescence colours have been changed in some of the pictures shown, using the ImageJ software, to enhance the visualization of markers.
Measurement of pyknotic nuclei
Cell death was estimated by counting pyknotic nuclei in the same sample preparations used for immunocytochemistry (see previous paragraph). Nuclei showing chromatin hypercondensation (apoptotic and/or necrotic) were detected by DAPI staining and expressed as a percentage of total nuclei number. Samples were coded and blinded quantifications of pyknotic nuclei were done in five independent experiments; each of these five independent experiments was performed in triplicate. Experiments were performed and quantified by different people.
Cell viability assays
Cells were seeded at a density of 20 000 cells mL−1 and cultured in the absence (none) or presence of specific treatments (ELAC, ELAC + BSO, ELAC + EGSH) and left for 48 h. Then, cultures were disaggregated and mixed with trypan blue (0.04% w.v‐1 in PBS). Viable cells (those excluding the trypan blue dye) were counted using a haemocytometer under an inverted microscope and are expressed as percentage of the total number of cells. Samples were coded and blinded quantifications were done in five independent experiments; each of these five independent experiments was performed in triplicate. Experiments were performed and quantified by different people.
RNA isolation, reverse transcription and real‐time quantitative PCR
Total RNA isolation from neurosphere cultures, reverse transcription and RT‐qPCR to relatively quantify the expression of PKCα, β and γ were performed as previously described (Romero‐Grimaldi et al., 2011), except that 18S rRNA was used as the housekeeping transcript, and a minimum of five independent neurosphere cultures were used for each determination. All PCR reactions within each experiment were run in duplicates. Amplification specificity was confirmed by melting‐curve analysis of the PCR products. The relative expression of each mRNA was calculated as 2−ΔCt, where ΔCt = Ct (target mRNA) − Ct (18S). No signal was detected in non‐template or non‐RT controls.
Primer sequences (5′–3′) and annealing temperatures used were the following: for PKCα, Fw: TGAATCCTCAGTGGAATGAGT, Rw: GGTTGCTTTCTGTCTTCTGAA, 55°C; for PKCβ, Fw: CCCGAAGGAAGCGAGGGCAATGAAG, Rw: AGTTCATCTGTACCCTTCCGCTCTG, 63°C; for PKCγ, Fw: TGAGAGAGTGCGGATGGGCCCC, Rw: GCAGGCGTCCTGGGCTGGCACC, 65°C; for 18S, Fw: CTCAACACGGGAAACCTCAC, Rw: CGCTCCACCAACTAAGAACG, 55°C.
Central administration of ELAC, brain processing and immunohistochemistry
Adult male mice weighting 35–40 g were anaesthetized with an i.p. injection ketamine/xylazine cocktail containing 120 mg·kg−1 of ketamine (Imalgene®, Boehringer Ingelheim, Ingelheim, Germany) and 20 mg·kg−1 of xylacine (Rompun®, Bayer, Leverkusen, Germany) diluted to a final volume of 250 μL with PBS and placed on a stereotaxic frame (Kopf Instruments). To determine depth of anaesthesia we softly pinched the mouse paws handly until no reflexes were observed. A small trepanation was made 0.8 mm lateral to Bregma, where the needle of a 5 μL Hamilton syringe was introduced 2.4 mm below the brain surface. Ultra‐filtered ELAC, diluted in PBS at a concentration of 5 μM (n = 6), or vehicle (n = 6), was injected ipsilaterally into the right lateral ventricles. Single injections of 2 μL per brain were performed through a time‐frame of 10 min; afterwards, the syringe was left for 5 min before removal. After i.c.v. administration, mice were used in two different experimental approaches described below.
A set of control and ELAC‐treated mice (6 + 6) received daily i.p. injections of the thymidine analogue and cell‐division marker bromodeoxyuridine (BrdU, 120 mg·kg−1, diluted in PBS to a final volume of 250 μL per injection) during 3 days, starting the day of the surgical procedure. Then, 72 h after the i.c.v. injections, mice were deeply anaesthetized with i.p. injections of 250 μL of Dolethal® (Vetoquinol, Lure, France), containing a lethal 50 mg dose of pentobarbital, and perfused with 4% paraformaldehyde via the ascending aorta. Brains were removed and sliced in 30 μm serial sections containing the entire SVZ. These mice (control and ELAC‐treated) were used to analyse proliferation within the SVZ.
Another set of mice (six control mice and six ELAC‐treated mice) received i.p. injections of BrdU (120 mg·kg−1, diluted in PBS to a final volume of 250 μL per injection) during 3 days after ELAC administration, starting the day of the surgical procedure. On day 10 after i.c.v injection, mice were deeply anaesthetized with i.p. injections of 250 μL of Dolethal® (Vetoquinol, Lure, France), containing a lethal 50 mg dose of pentobarbital, and perfused with 4% paraformaldehyde via ascending aorta. Brains were removed and olfactory bulb (OB) was sliced in 30 μm serial sections. This set of mice was used to analyse the presence of mature neurons in the OB that had incorporated BrdU.
Sample sizes were chosen based on previous articles in which a similar analysis was performed (Rabaneda et al., 2008; Romero‐Grimaldi et al., 2008; Geribaldi‐Doldán et al., 2016). Mice were coded, and treatment (control or ELAC) was assigned randomly to code numbers and applied. In addition blind quantifications were performed.
Brain processing and immunohistochemical detection of the proliferation marker BrdU, the transient amplifying cell marker achaete scute homologue 1 (ASCL1), the NSC marker Sox2 and the astrocyte and NSC marker glial fibrilary acidic protein (GFAP), the early neuronal differentiation marker doublecortin (DCX) and the mature neuron maker NeuN were performed as previously described (Rabaneda et al., 2008).
Primary antibodies used were mouse monoclonal anti‐BrdU (1:100) from Dako (Hamburg, Germany) or rat monoclonal anti‐BrdU (1:100) from Abcam (Cambridge, UK), mouse monoclonal anti‐ASCL1 (1:100) from BD Pharmingen (San Jose, CA, USA), rabbit monoclonal anti‐SOX2 (1:200) and mouse polyclonal anti‐GFAP both from Cell Signalling (Beverly, MA,USA), goat polyclonal anti‐DCX from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and rabbit monoclonal anti‐NeuN (1:100) from Abcam (Cambridge, UK). Secondary antibodies used were Alexa Fluor 488 donkey anti‐mouse, Alexa Fluor 594 donkey anti‐mouse, Alexa Fluor 405 goat anti‐mouse, Alexa Fluor 594 donkey anti‐rat, Alexa Fluor 488 donkey anti‐rabbit, Alexa Fluor 594 donkey anti‐rabbit and Alexa Fluor 594 donkey anti‐goat (all at 1:1000, from Life Tech).
Quantification of neurogenesis in brain sections
SVZ cells positive for BrdU, DCX, ASCL1, GFAP and NeuN were estimated as previously described (Rabaneda et al., 2008; Geribaldi‐Doldán et al., 2016). Positive cells were counted throughout the entire lateral and laterodorsal walls of the lateral ventricles (where the SVZ NPC are located) in every fifth section; 14–16 sections per brain were analysed under fluorescence microscopy at 20× magnification. A confocal microscope (OLYMPUS FV1000) was used to take images from triple‐labelled brain sections. Mice were coded depending on the treatment and quantification of cells in brain slices was done in a blinded analysis.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analysis was performed using the computer programme IBM SPSS Statistics 22. When more than one treatment group were compared, statistical analyses were performed using one‐way ANOVA followed by a post hoc Bonferroni's test unless otherwise indicated. Student's t‐test was used when only one treatment group was compared with the control. Differences were considered significant at values of P < 0.05. In general, sample size used in statistical analysis was n = 6 for in vivo experiments and n = 5 for in vitro experiments. In the latter, each individual n value was obtained by averaging triplicates within each experiment, except for the qRT‐PCR data in which each individual n value was the average of duplicates.
Reagents
The lathyranes ELAC [3,12‐di‐O‐acetyl‐8‐O‐tigloilingol; CAS number: (58749‐62‐5)], EOF2 [7,8,12‐tri‐O‐acetyl‐3‐O‐(4‐methoxyphenyl)acetylingol; CAS number: (944799‐47‐7)], EOF3 [7,12‐di‐O‐acetyl‐8‐O‐methyl‐3‐O‐phenylacetylingol; CAS number: (944799‐48‐8)] and ELAF12‐2 [(2S,3S,4S,8R,9S,11R,15R)‐8,15‐diacetoxy‐3‐isobutyroxy‐14‐oxolathyra‐(5E),(12E)‐diene; CAS number: (1217313‐10‐4)] were isolated and purified in our laboratory, as described previously (Daoubi et al., 2007; Avila et al., 2010). Briefly, purification by semipreparative HPLC was performed with a Hitachi/Merck L‐6270 apparatus equipped with a differential refractometer detector (RI‐7490). A LiChrospher® Si 60 (10 μm) LiChroCart® (250 × 10 mm) column was used in isolation experiments. Silica gel (Merck) was used for column chromatography. TLC was performed on Merck Kieselgel 60 F254, 0.25 mm thick. Infrared spectra were recorded on a FTIR spectrophotometer and reported as wavenumbers (cm−1). 1H and 13C NMR measurements were obtained on a 400 MHz spectrometer with SiMe4 as the internal reference. Chemical shifts were referenced to CDCl3 (δH 7.25, δC 77.0), and NMR assignments were made by a combination of 1D and 2D techniques. Multiplicities are described using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and br = broad. High‐resolution mass spectrometry was performed with a QTOF mass spectrometer in positive ion ESI mode.
The commercial PKC activators PMA (13‐O‐acetyl‐12‐O‐tetradecanoylphorbol, also known as TPA) and prostratin (13‐O‐acetyl‐12‐deoxyphorbol) were both purchased from Sigma‐Aldrich (St. Louis, MO, USA). The general PKC inhibitor bisindolylmaleimide I (also known as GF109203X, GFX or Gö6850) and the classical PKC inhibitor Gö6976 were from Calbiochem (Millipore, Billerica, MA, USA) and were added to the cells at final concentrations of 0.5 and 0.16 μM respectively. Stock solutions of all PKC interacting compounds were prepared in DMSO and pre‐diluted in culture medium before addition to cell cultures. When cultures were co‐treated with PKC inhibitors and activators, PKC inhibitors were added to the cells 30 min before addition of PKC activators. Specific small interference RNAs (siRNAs) were from Thermo Scientific Dharmacon (Lafayette, CO, www.dharmacon.com).
Other products, unless otherwise indicated, were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
ELAC, a natural product with lathyrane structure, activates PKC and neural progenitor cell proliferation
Four diterpenes with lathyrane skeleton were screened to test whether they could stimulate NPC proliferation through PKC activation. These lathyranes were as follows: ELAC, isolated from Euphorbia lactea; EOF2 and EOF3, from Euphorbia officinarum; and ELAF12‐2, from Euphorbia laurifolia (Figure 1).
Figure 1.
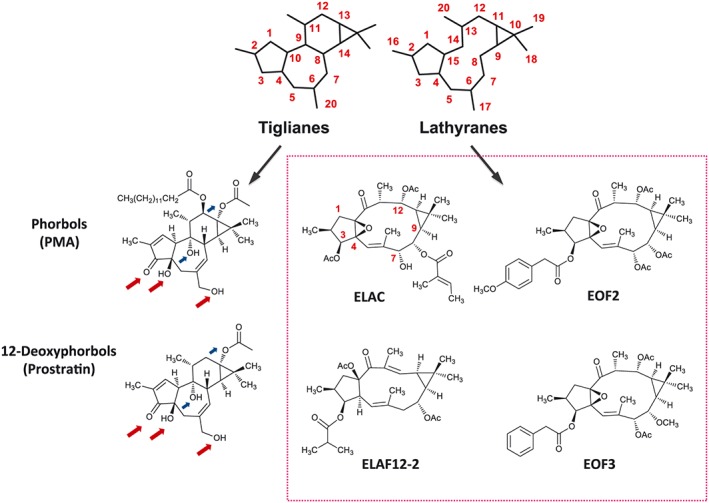
Schematic representations of the chemical skeletons of tiglianes and lathyranes, two classes of polycyclic diterpenes that are abundant in the latex of Euphorbia plants. Tiglianes like phorbols and 12‐deoxyphorbols – being PMA and prostratin the stereotypical members of these types, respectively – are well‐known PKC activators that promote the proliferation of NPC (Geribaldi‐Doldán et al., 2016). Red arrows pinpoint C3, C4 and C20‐bound oxygen residues, identical in PMA and prostratin, thought to be important for PKC recognition and activation. Blue arrows pinpoint other oxygen residues that might also be involved in PKC activation. The tumourigenicity of phorbols like PMA makes them unsuitable for clinical research, while less toxic molecules, such as prostratin, are more propitious pharmacological candidates. In the search for new molecules with clinical value, and given the basic structure similarity between tiglianes and lathyranes, we have tested whether lathyranes could also activate PKC and induce NPC proliferation; the four lathyranes tested are shown inside the box. Abbreviations: Ac, acetyl.
NPC proliferation was analysed using neurosphere culture assays, in which the size and number of newly formed neurospheres is estimated. The rationale for the use of this culture system is as follows: when NPC divide, daughter cells remain gently attached to each other forming spherical cell aggregates known as neurospheres; the size reached by neurospheres is indicative of NPC proliferation and survival, while the number of neurospheres formed after subsequent subcultures is a measure of NPC self‐renewal capacity and survival (Ramirez‐Castillejo et al., 2006; Torroglosa et al., 2007).
We cultured NPC for 72 h in bFGF‐containing medium supplemented with or without the different lathyranes, added at concentrations (1–5 μM) previously proven effective for 12‐deoxyphorbols (Geribaldi‐Doldán et al., 2016). Treatment with ELAC (but not with EOF2, EOF3 or ELAF12‐2) significantly increased neurosphere size (Figure 2A, B), without modifying neurosphere number (Figure 2C). Scaling up ELAC concentration to 10 μM did not result in a further increase on neurosphere size, being 5 μM the most effective concentration.
Figure 2.
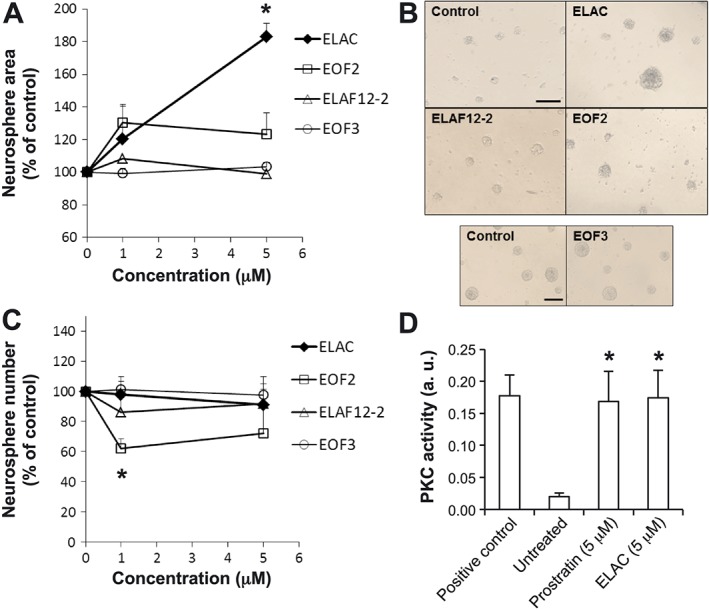
Effect of lathyranes on NPC proliferation in vitro. Proliferation was tested in NPC cultures grown in the presence of bFGF. (A) Graph shows the effect of increasing concentrations of lathyranes (ELAC, EOF2, EOF3 and ELAF12‐2) on neurosphere area after 72 h of culture. A minimum of 540 neurospheres were measured per treatment. (B) Phase‐contrast microphotographs of neurospheres cultured for 72 h in the absence or presence of different lathyranes at a concentration of 5 μM. Scale bar indicates 200 μm. (C) Graph shows the effect of increasing concentrations of lathyranes on the number of neurospheres formed after 72 h of treatment. (D) PKC activity was measured in lysates obtained from cells that had been treated for 1 h with 5 μM prostratin, 5 μM ELAC or none, using a PKC activity assay kit. PKC activation induced by ELAC was similar to that induced by prostratin and was equivalent to the kinase activity observed in the positive control (consisting of 60 ng of purified active PKC). Results are the mean ± SEM of five independent experiments, and each individual experiment was performed in triplicates. *P < 0.05 when compared with control (untreated) cells in a Student's t‐test; a.u.: absorbance units.
The level of PKC activation induced by ELAC in NPC was directly measured using a commercial elisa‐like PKC‐activity assay kit (Figure 2D). Although this kit does not distinguish between classical, novel or atypical PKC activities, results showed that ELAC could activate pan‐PKC activity to an extent comparable with that of the 12‐deoxyphorbol prostratin (Figure 2D).
With the exception of ELAC, no other lathyranes from the ones tested had any significant effect on neurosphere size (Figure 2A, B), although a negative effect on neurosphere number was observed with EOF2 treatment (Figure 2C).
Since oxidative stress may impair neurogenesis in mice (Bokara et al., 2011; Bokara et al., 2016), we decided to analyse whether, in addition to its proliferative effect, ELAC was exerting a cytoprotective effect against oxidative stress on NPC. Thus, oxidative stress was induced in NPC by buthione sulfoximine (BSO) treatment, and cell survival was analysed in the presence and absence of ELAC. Glutathione ethyl ester (EGSH) was used as positive control. We could observe that as expected, BSO induced cell death in NPC cultures; however, ELAC treatment promoted survival in BSO‐treated NPC, and the effect was similar to that of EGSH (Supporting Information Figure S1).
A free C7‐hydroxyl group in ELAC is essential for its bioactivity
Crystallographic analysis performed by Zhang et al. (1995) revealed that three oxygen residues in phorbol esters allow for their interaction with the PKC C1b domain and, therefore, should be important for PKC recognition and activation. These oxygen residues are located in positions C3, C4 and C20 of the tigliane skeleton (see red arrows in Figure 1), which are also conserved in PKC‐activating 12‐deoxyphorbols, like prostratin (Figure 1). Equivalent oxygenated functions in the lathyrane skeleton could be those present at positions C3, C4 and C7 (see ELAC in Figure 1). A free hydroxyl group at C7 seemed relevant for ELAC's activity, since this functional group was either absent or protected as acetate in the inactive lathyrane compounds ELAF12‐2, EOF2 and EOF3.
Previous results with 12‐deoxyphorbols show that acetylation of the C20 hydroxyl group results in decreased potency for PKC activation (Geribaldi‐Doldán et al., 2016). In order to test whether ELAC's C7‐hydroxyl could be understood as a PKC interacting point – in a similar way to C20 hydroxyls of phorbols and 12‐deoxyphorbols – we acetylated the C7‐hydroxyl in ELAC (Figure 3A) by treatment with acetic anhydride in pyridine and tested the ability of the corresponding acetylated compound (Ac‐ELAC) to increase NPC proliferation. We observed that, while treatment with 5 μM ELAC significantly increased neurosphere size (Figure 3B, C), treatment with Ac‐ELAC at an identical concentration had no effect on neurosphere area.
Figure 3.
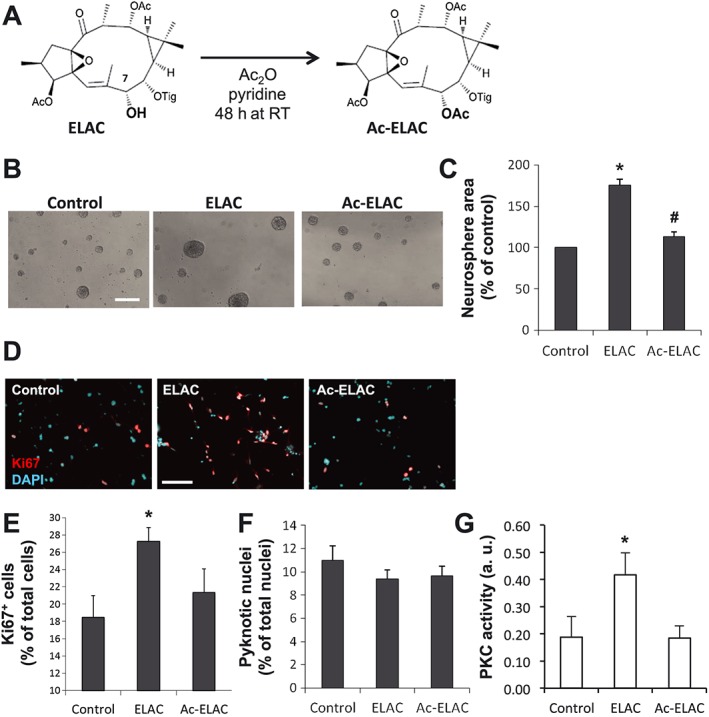
The effect of ELAC on NPC proliferation is lost upon acetylation at the C7‐hydroxyl. (A) Scheme of the acetylation reaction in which ELAC is O‐acetylated at the C7‐hydroxyl group to obtain the product Ac‐ELAC, performed with acetic anhydride (Ac2O) in pyridine medium for 48 h. (B) Phase‐contrast microphotographs of neurospheres cultured for 72 h in the absence or presence of ELAC (5 μM) or Ac‐ELAC (5 μM). Scale bar indicates 200 μm. (C) Graph shows the effect of ELAC (5 μM) or Ac‐ELAC (5 μM) on neurosphere area after 72 h of culture. A minimum of 540 neurospheres was measured per condition. (D) Fluorescence microphotographs of neurosphere‐derived adhered cells grown for 48 h with or without 5 μM ELAC or 5 μM Ac‐ELAC. Cells were immunostained to detect the proliferation marker Ki67 (red), which labels the nuclei of cells that have not yet withdrawn from the cell‐cycle, and total nuclei were counterstained with DAPI (cyan). Scale bar indicates 100 μm. (E) Quantification of Ki67+ cells in culture preparations as those described in (D); Ki67+ cells are expressed as percentage of total cells. (F) Quantification of pyknotic nuclei in culture preparations as those described in (D), expressed as percentage of total nuclei. In all cases, cells were cultured in the presence of bFGF. (G) PKC activity was measured in lysates obtained from cells that had been treated for 1 h with 5 μM ELAC, 5 μM AC‐ELAC, or none, using a PKC activity assay kit. PKC activation induced by ELAC was equivalent to the kinase activity observed in the positive control (consisting of 60 ng of purified active PKC), and no PKC activity was induced by AC‐ELAC. Results are the mean ± SEM of five independent experiments, and each individual experiment was performed in triplicates. *P < 0.05 when compared with control in a Student's t‐test; #P < 0.05 when compared with ELAC in a Student's t‐test. Abbreviations: Ac, acetyl; Tig, tigloyl.
The proliferative effect of ELAC in NPC – and the lack of effect of Ac‐ELAC – was further confirmed by a second technique: the immunocytochemical detection of the proliferation marker Ki67. For this, neurosphere‐derived cells were seeded onto an adherent surface and cultured with or without 5 μM ELAC or 5 μM Ac‐ELAC for 48 h. Then, Ki67 immunostaining revealed that, compared with the control condition, treatment with ELAC significantly increased Ki67+ cells by 152%, an increase that was not observed with Ac‐ELAC (Figure 3D, E). The percentage of pyknotic nuclei was not changed by any of these treatments (Figure 3F). In order to demonstrate that ELAC acetylation impaired its interaction with PKC, total PKC activity in homogenates of NPC treated with either ELAC or AC‐ELAC was tested. The twofold increase in PKC activity induced by ELAC could not be observed in NPC treated with Ac‐ELAC (Figure 3G). Overall, these data indicated that integrity of the C7‐hydroxyl group in ELAC is necessary for NPC proliferation induction.
A classical PKC is responsible for ELAC‐induced NPC proliferation
If ELAC increased NPC proliferation through a mechanism involving PKC activation, this increase should then be prevented by PKC inhibitors (Way et al., 2000). As shown in Figure 4A, B, the increase on neurosphere size observed in the presence of ELAC was fully reverted by concomitant addition of the PKC inhibitor Gö6850 (also known as bisindolylmaleimide I, GF109203X or GFX), which inhibits classical and novel PKCs. This result indicated that the effect of ELAC on NPC proliferation was indeed PKC‐dependent and ruled out atypical PKCs as candidate kinases responsible for this effect. Moreover, addition of Gö6976, an inhibitor specific for calcium‐dependent (classical) PKCs, also abolished the proliferative effect of ELAC on NPC in a concentration‐dependent manner (Figure 4A, B). In contrast, inhibitors against other related kinases like PKG did not change the effect of ELAC on neurosphere area (Figure 4A). None of the tested inhibitors alone had any effect on basal (bFGF‐dependent) neurosphere size (Figure 4A), except for the cPKC inhibitor Gö6976 (0.16 μM), which decreased this basal NPC proliferation to 84% of control. At the indicated concentrations, none of these treatments induced any significant change in cell viability (data not shown). In conclusion, these results indicated that ELAC induced NPC proliferation by activating one or more classical PKC isoforms.
Figure 4.
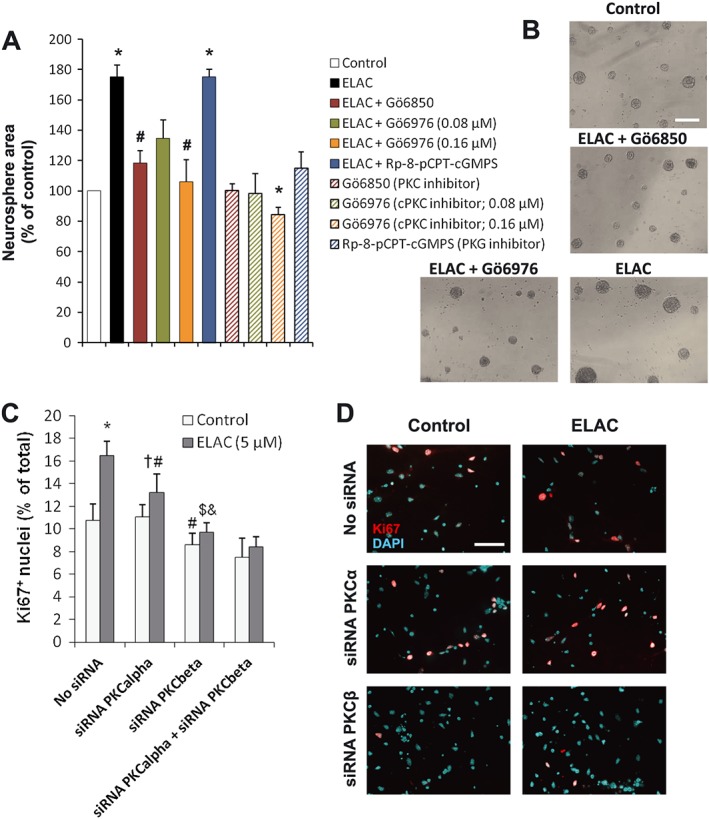
The effect of ELAC on NPC proliferation is mediated by activation of classical PKCs. (A) Proliferation was tested in NPC cultures grown for 72 h in the absence or presence of 5 μM ELAC and/or Gö6850 (0.5 μM), an inhibitor of both classical and novel PKCs; Gö6976 (0.08–0.16 μM), an inhibitor of classical (calcium‐dependent) PKCs; or Rp‐8‐pCPT‐cGMPS (10 μM), an inhibitor of PKG, as a negative control. Results show that the proliferative effect of ELAC in NPC is fully dependent on classical PKC activation. Results are the mean ± SEM of five independent experiments. Each individual experiment was performed with triplicate samples (a minimum of 540 neurospheres were measured per condition). *P < 0.05 when compared with control in a Student's t‐test; #P < 0.05 when compared with ELAC alone in a Student's t‐test. (B) Phase‐contrast microphotographs of neurospheres cultured for 72 h in the indicated conditions, as further described in A (the concentration of Gö6976 shown is 0.16 μM). Scale bar indicates 200 μm. (C) Quantification of Ki67+ cells in culture preparations as those described in D; Ki67+ cells are expressed as percentage of total cells. Results are the mean ± SEM of five independent experiments. Each individual experiment was performed with triplicate samples. * Indicates different from rest of groups in one‐way ANOVA; # different from untransfected control treated with ELAC; †different from untreated PKCα siRNA‐transfected cultures; $ different from ELAC‐treated cultures transfected with PKCα siRNA; and & different from untransfected ELAC‐treated cultures. (D) Fluorescence microphotographs of neurosphere‐derived adhered cells transfected with PKCα or PKCβ siRNA and grown for 48 h with or without 5 μM ELAC. Cells were immunostained to detect the proliferation marker Ki67 (red), which labels the nuclei of cells that have not yet withdrawn from the cell‐cycle, and total nuclei were counterstained with DAPI (cyan). Scale bar indicates 100 μm.
In order to delimit which specific cPKC isoform/s could be triggering NPC proliferation, mRNA expression analyses of all cPKCs were performed in NPC cultures. As shown in Table 1, PKCβ mRNA was consistently the most abundant in all NPC samples tested. PKCα mRNA was also expressed in NPC, although it was less abundant than PKCβ mRNA; in contrast, PKCγ mRNA was only detectable in trace amounts, being around 100 times less abundant than PKCβ mRNA (Table 1). These results suggested that PKCα, PKCβ or both were likely responsible for the proliferative effect of ELAC on NPC.
Table 1.
Real‐time qPCR of classical PKC isoforms in NPC
| PKC β | PKC α | PKC γ | |
|---|---|---|---|
| Average Ct | 21.33 ± 0.44 | 24.08 ± 0.27 | 28.37 ± 0.57 |
| Primers used | Fw:CCCGAAGGAAGCGAGGGCAATGAAG Rw:AGTTCATCTGTACCCTTCCGCTCTG | Fw:TGAATCCTCAGTGGAATGAGT Rw:GGTTGCTTTCTGTCTTCTGAA | Fw:TGAGAGAGTGCGGATGGGCCCC Rw:GCAGGCGTCCTGGGCTGGCACC |
| Amplicon size | 227 | 325 | 104 |
| Annealing temperature | 63 | 55 | 65 |
| Relative abundance | +++ | + | − or −/+ |
Measurement of the mRNA expression of classical PKCs in NPC. Total RNA was isolated from neurosphere cultures and subjected to reverse transcription and real‐time qPCR. The mRNAs for PKCα, β and γ were measured and normalized to the levels of 18S rRNA. The table shows mRNA abundance for each cPKC in relation to the most‐abundant PKCβ mRNA. The table also shows average Ct values (PCR‐cycle number in which the fluorescence of the amplified sequence becomes detectable) for each cPKC mRNA
Consequently, we next tried to identify the classical PKC isoform responsible for the ELAC‐induced proliferative effect. Thus, proliferation studies in which Ki67 was detected were conducted using cell disaggregated from neurospheres and transfected with either siRNA against PKCα, PKCβ or a combination of both. These cells were assayed in the presence and absence of ELAC. Non‐transfected cells were used as control. It was found that PKCα siRNA had no effect on the percentage of Ki67+ cells in untreated cultures; however, the increase of Ki67+ cells induced by ELAC in control cells was partially reduced in cells transfected with PKCα siRNA. Additionally, in the presence of PKCβ siRNA proliferation of untreated NPC cultures was slightly reduced, and no proliferative effect of ELAC was observed in cells transfected with ELAC + PKCβ siRNA. The combination of both PKCα and PKCβ siRNA induced effects similar to those of PKCβ siRNA. These results indicated that blocking PKCβ expression avoided the proliferative effect of ELAC, suggesting that the effect of ELAC on NPC proliferation was mainly mediated by PKCβ activation, although PKCα might also be partially involved (Figure 4C, D).
ELAC induces NPC proliferation in a growth factor‐dependent manner
The proliferative effect of ELAC was comparatively tested in NPC cultures grown in the presence of bFGF alone, EGF alone or a combination of both GF. As shown in Figure 5A, B, the most potent effect of ELAC on NPC proliferation (a 183% increase on neurosphere area) was observed in the presence of bFGF alone; in contrast, ELAC only induced a 148% neurosphere area increment when in the presence of EGF, and no neurosphere area increment at all was induced by ELAC when bFGF and EGF were concomitantly added to NPC cultures (Figure 5A, B). Slightly different results were obtained when PKC activation was triggered by prostratin instead of ELAC, because prostratin showed no preferential synergy whether in combination with bFGF alone or EGF alone and also because prostratin still increased NPC proliferation farther above the combination bFGF + EGF (Figure 5A, B).
Figure 5.
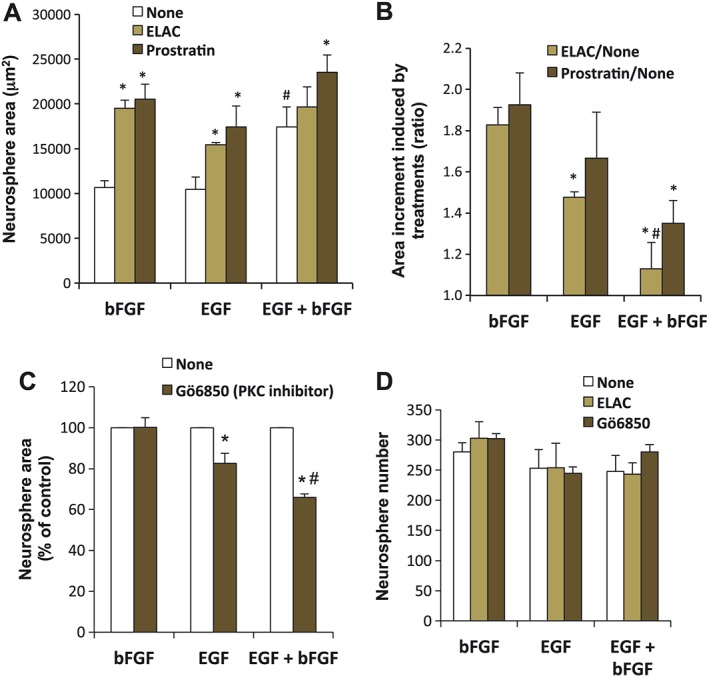
Comparative effect of ELAC and prostratin on EGF‐ and bFGF‐induced NPC proliferation. (A) Proliferation was tested in NPC cultures grown in the presence of bFGF (10 ng·mL−1), EGF (20 ng·mL−1), or a combination of both, with or without addition of 5 μM ELAC or 5 μM prostratin. Graph shows the effect of these treatments on neurosphere area after 72 h of culture. *P < 0.05 when compared with their respective control; #P < 0.05 when compared with either EGF or bFGF alone. (B) The data from (A) are presented here in a different manner, showing neurosphere area increments induced by ELAC or prostratin (ratio treatment/none) whether in the presence of bFGF, EGF or a combination of both; *P < 0.05 when compared with the same treatment ratio in bFGF‐stimulated cultures; #P < 0.05 when compared with the same treatment ratio in EGF‐stimulated cultures. (C) Proliferation was tested in NPC cultures grown in the presence of bFGF, EGF or a combination of both, with or without addition of the PKC inhibitor Gö6850 (0.5 μM). Graph shows the effect of these treatments on neurosphere area after 72 h of culture. *P < 0.05 when compared with their respective control; #P < 0.05 when compared with EGF + Gö6850. (D) Graph shows the effect of the indicated treatments on neurosphere number after 72 h of culture. Results are the mean ± SEM. Each individual experiment was performed with triplicate samples (the size of a minimum of 540 neurospheres was measured per condition).
We tested whether the different interaction of ELAC with EGF and/or bFGF was related to a distinct capacity of these GF, to intrinsically activate PKC by themselves. In agreement with this hypothesis, PKC inhibition did not significantly modify the basal proliferative effect of bFGF (Figure 5C), but it partially reduced the proliferative effect of EGF; furthermore, this reduction in proliferation induced by PKC inhibition was even more pronounced in the case of the combination EGF + bFGF (Figure 5C). Thus, an inverse correlation existed between the effect of ELAC on NPC proliferation and the intrinsic capacity of the GF present to activate PKC. Finally, none of the treatments described induced any significant change in the number of neurospheres formed (Figure 5D).
ELAC stimulates NPC proliferation without affecting the proportion of Sox2+ cells
To rule out that treatment with ELAC was affecting the NPC capacity to continue producing neural progenitors, neurospheres formed after 72 h in the presence of ELAC, Ac‐ELAC or none were processed for immunocytochemistry to detect the nuclear expression of Sox2 and the cytoskeletal marker nestin, an intermediate‐filament protein expressed by undifferentiated multipotent neural progenitors (Figure 6A, C). Treatment with ELAC produced a small, but significant, increase on the percentage of Sox2+ cells (Figure 6C, D), while the percentage of nestin+ progenitors was not affected. As expected, Ac‐ELAC had no effect on Sox2 or nestin expression.
Figure 6.
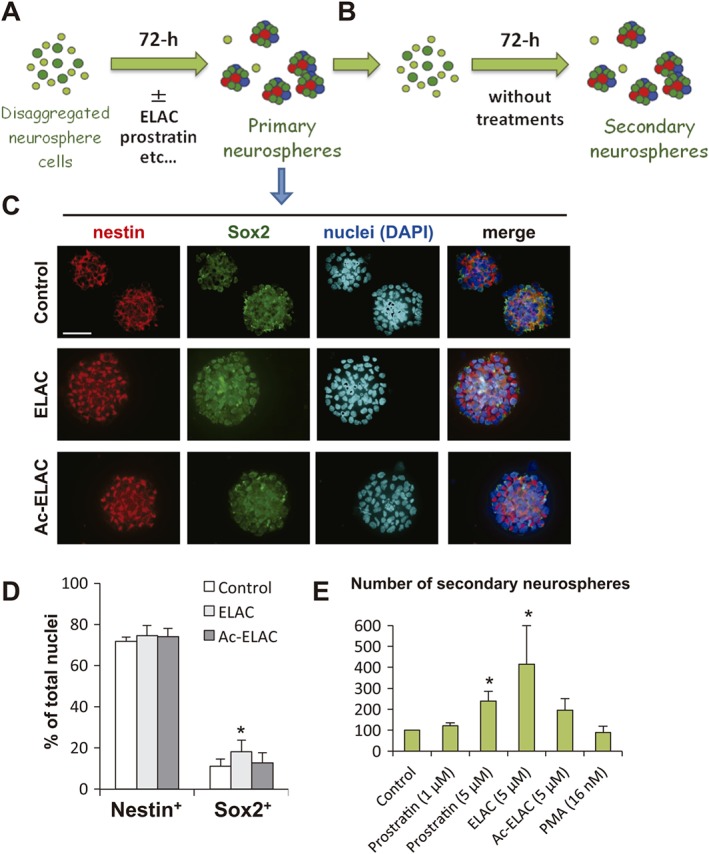
Effect of the lathyrane ELAC on stemness and self‐renewal capacity. (A) Scheme of the procedure followed to test the effect of different compounds on the expression of the transcription factor Sox2 (an NSC marker) and nestin (an intermediate neural progenitor marker) in neurospheres formed during 72 h in the presence of these compounds (primary neurospheres); see Methods for details. (B) Primary neurospheres grown for 72 h in the presence of different compounds were disaggregated, and single cells were cultured for another 72 h in the absence of any treatment, in order to determine the capacity of these cells to generate new secondary neurospheres after treatment withdrawal. (C) Examples of fluorescence images of primary neurospheres formed after treatment with ELAC (5 μM), Ac‐ELAC (5 μM) or none (control), immunostained for nestin (red) and Sox2 (green), as indicated; nuclei are counterstained with DAPI (cyan or blue). Scale bar indicates 50 μm. (D) Quantification of the percentage of neurosphere cells expressing nestin and Sox2 after 72 h of treatment with ELAC (5 μM), Ac‐ELAC (5 μM) or none (control). (E) Number of secondary neurospheres formed from disaggregated single cells that had previously been treated for 72 h with the indicated compounds, as explained in (A and B). Results are the mean ± SEM of five independent experiments. Each individual experiment was performed in triplicates; *P < 0.05 when compared with control in a Student's t‐test performed for paired samples.
The number of secondary neurospheres formed from cells that had previously been treated for 72 h with PKC activators was also determined. Disaggregated cells from ELAC‐, Ac‐ELAC‐, prostratin‐ or PMA‐treated cultures were reseeded at a density of 20 000 cells mL−1 in fresh control medium and cultured for another 72 h (see scheme in Figure 6A, B). The number of newly formed secondary neurospheres was then counted. Pretreatment with 5 μM prostratin, as previously reported (Geribaldi‐Doldán et al., 2016), increased the number of secondary neurospheres formed and so did pretreatment with 5 μM ELAC. No effect of PMA or Ac‐ELAC on secondary‐neurosphere number was observed (Figure 6D).
In vivo administration of ELAC increases NPC proliferation in the adult SVZ
The effect of the lathyrane diterpene ELAC on NPC proliferation was studied in vivo, by injecting the drug into the adult‐mouse lateral ventricle and studying its effect on SVZ neurogenesis. Single i.c.v. doses of ELAC (5 μM, 2 μL), or vehicle, were unilaterally administered, and proliferating cells were concurrently labelled by BrdU intraperitoneal injections for 3 days, after which brains were fixed and subjected to immunohistochemical detection of BrdU. In vehicle‐injected mice, a significant but modest increase on BrdU+ cells was observed in the ipsilateral SVZs when compared with the corresponding contralateral sides in a paired t‐test, indicating that the injection procedure itself mildly induced NPC proliferation (Figure 7A, B), probably due to the small lesion caused by the procedure. Remarkably, treatment with ELAC significantly and robustly increased the total number of BrdU+ cells present in the ipsilateral SVZs, which was ~140% larger than that observed in vehicle‐injected ipsilateral SVZs and ~165% larger than that observed in contralateral SVZs.
Figure 7.
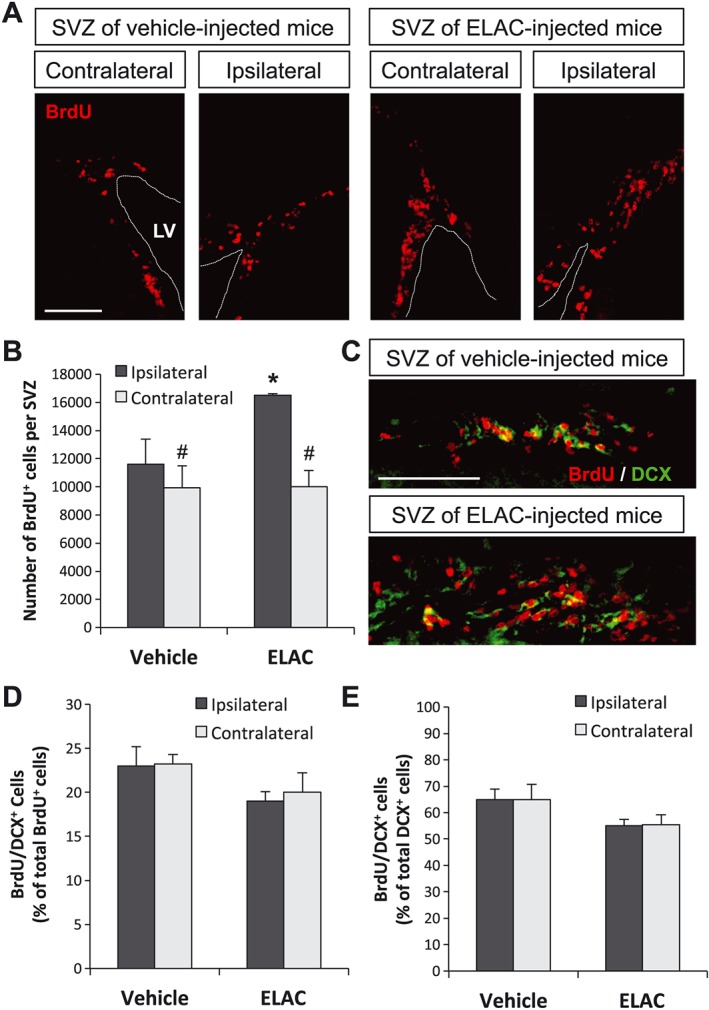
Effect of in vivo administration of ELAC on SVZ neurogenesis. Adult mice were injected in the right lateral ventricles with ELAC (5 μM, 2 μL; n = 6) or vehicle (n = 6) and then daily injected with the cell‐division marker BrdU (120 mg·kg−1) for 3 days. (A) Fluorescence microscopy images of brain coronal sections showing the ipsilateral and contralateral SVZs of mice treated with ELAC or vehicle; sections were processed for immunohistochemical detection of BrdU+ nuclei (red). (B) Quantification of BrdU+ cells within the SVZ of mice that had received i.c.v. injections of ELAC or vehicle. (C) Fluorescence microscopy images showing a close‐up of the laterodorsal corner of ELAC‐ or vehicle‐injected (ipsilateral) SVZs; BrdU (red) and the early neuronal marker DCX (green) were detected by immunohistochemistry. (D) Quantification of the percentage of BrdU+ cells that co‐express the early neuronal marker DCX. (E) Quantification of the percentage of DCX+ cells that co‐express BrdU. Results are expressed as the mean ± SEM, n = 6 animals per condition. *P < 0.05 when comparing ELAC‐ with vehicle‐injected SVZs in a one‐way ANOVA test; #P < 0.05 when comparing ipsilateral and contralateral SVZs within the same animal group in a Student's t‐test for paired samples. Abbreviations: LV, lateral ventricle. Scale bar: 100 μm.
In order to understand the cellular targets of ELAC and to identify whether ELAC was exerting its effects on NSCs, transit amplifying progenitors (TAPs) or neuroblasts, we studied the co‐localization of BrdU with different markers. We used BrdU/GFAP/Sox2 to detect proliferation of NSCs, BrdU/ASCL1 to detect proliferation of TAPs and BrdU/DCX to detect proliferation of neuroblasts. We observed that in mice treated with ELAC, the percentage of BrdU+/DCX+ cells found in either the ipsilateral or contralateral SVZ was similar (Figure 7C–E). In contrast, in mice treated with ELAC, a significant increase in the percentage of BrdU+/ASCL1+ cells was observed in both the ipsilateral and contralateral SVZ (Figure 8A, B), indicating that ELAC is exerting its effects on these cells. Regarding the proliferation of NSCs, we observed that in the ipsilateral SVZ of control mice, the number of proliferative stem cells (BrdU+/Sox2+/GFAP+) was reduced. This reduction was not statistically significant in the presence of ELAC (Figure 8A, C) suggesting that ELAC to some extent might induce the proliferation of NSCs in the injured SVZ.
Figure 8.
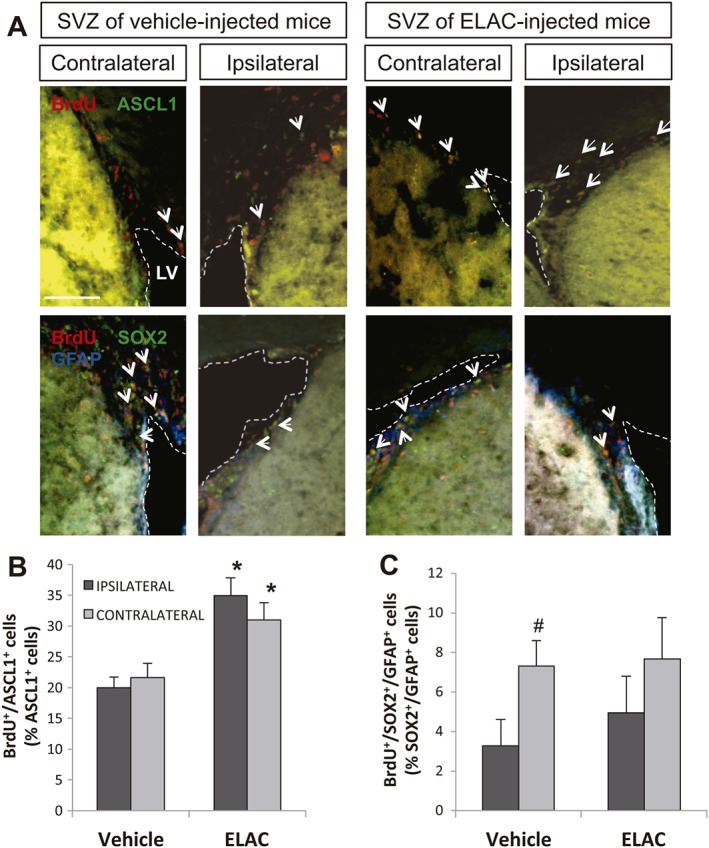
Effect of in vivo administration of ELAC on SVZ proliferating cells. Adult mice were injected in the right lateral ventricles with ELAC (5 μM, 2 μL; n = 6) or vehicle (n = 6) and then daily injected with the cell‐division marker BrdU (120 mg·kg−1) for 3 days. (A) Upper panels show fluorescence microscopy images of brain coronal sections showing the ipsilateral and contralateral SVZs of mice treated with ELAC or vehicle; sections were processed for immunohistochemical detection of BrdU+ nuclei (red) and ASCL1 (green). Lower panels show fluorescence microscopy images of brain coronal sections showing the ipsilateral and contralateral SVZs of mice treated with ELAC or vehicle; sections were processed for immunohistochemical detection of BrdU+ nuclei (red), Sox2 (green) and GFAP (blue). (B) Quantification of BrdU+/ASCL1+ cells within the SVZ of mice that had received i.c.v. injections of ELAC or vehicle. (C) Quantification of BrdU+/Sox2+/GFAP+ cells within the SVZ of mice that had received i.c.v. injections of ELAC or vehicle. *P < 0.05 when comparing ELAC‐ with vehicle‐injected SVZs in a one‐way ANOVA test; #P < 0.05 when comparing ipsilateral and contralateral SVZs within the same animal group in a Student's t‐test for paired samples. Abbreviations: LV, lateral ventricle. Scale bar: 100 μm.
We next sought to determine whether proliferating cells in the presence of ELAC were progressing towards a neuronal lineage. In order to test for this, mice received i.c.v. injections of either vehicle or ELAC and were left for 10 days. BrdU was administered to these mice during 3 days after ELAC administration, starting the day of the surgical procedure. On the 10 day post injections, OBs from these mice were analysed for the presence of BrdU+/NeuN+ cells. We observed an increase in the amount of NeuN+ cells that had incorporated BrdU in mice treated with ELAC, in both the ipsilateral and contralateral SVZ, although this tendency was not statistically significant (Figure 9A, B). This suggested that ELAC might be promoting the generation of new neurons in vivo.
Figure 9.
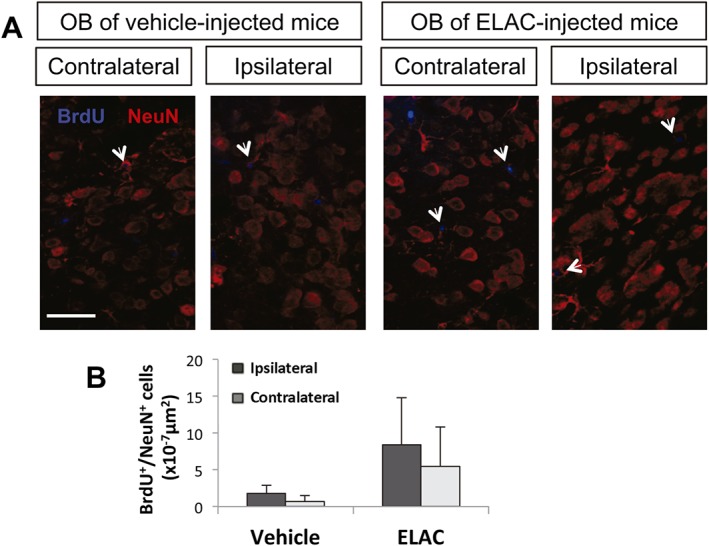
Effect of in vivo administration of ELAC on newly formed OB neurons. Adult mice were injected in the right lateral ventricles with ELAC (5 μM, 2 μL; n = 6) or vehicle (n = 6) and then injected with the cell‐division marker BrdU (120 mg·kg−1) during 3 days after treatment administration starting on the day of the surgical procedure and were killed on day 10 after surgery. (A) Fluorescence microscopy images of brain coronal sections showing the ipsilateral and contralateral OBs of mice treated with ELAC or vehicle; sections were processed for immunohistochemical detection of BrdU+ nuclei (red) and NeuN (blue). (B) Quantification of BrdU+/NeuN+ cells within the SVZ of mice that had received i.c.v. injections of ELAC or vehicle.
Discussion
Stem cell therapy may provide novel therapeutic approaches for neuronal replacement in the CNS. However, insufficient generation of endogenous – or exogenous – neural progenitors may limit its applications. One way to promote the proliferation of adult, postnatal or embryonic NPC is by pharmacological activation of PKC (Lai and Feng, 2004; Morishita et al., 2007; Geribaldi‐Doldán et al., 2016). Non‐tumorigenic PKC activators for medical research include 12‐deoxyphorbols (Das and Rahman, 2014; Hezareh, 2005; Sanchez‐Duffhues et al., 2011; Szallasi and Blumberg, 1991) like prostratin (Figure 1) or ER272, which increase NPC proliferation in the two main neurogenic regions of the adult mouse brain, the SVZ and the DG (Geribaldi‐Doldán et al., 2016).
We investigated herein whether macrocyclic diterpenes with lathyrane skeleton, structurally similar to 12‐deoxyphorbols (Figure 1), could also activate PKC and promote NPC proliferation, in an attempt to identify additional harmless molecules for neurological research – notably, lathyranes have specifically been reported as anti‐tumorigenic in the literature (Pusztai et al., 2007). We screened four different lathyranes isolated from Euphorbia plants, from which ELAC strongly stimulated NPC proliferation in vitro at concentrations identical to those reported to work best for prostratin in a comparable assay (Geribaldi‐Doldán et al., 2016). ELAC and prostratin also increased PKC activity to similar extents in elisa assays.
The lack of activity of the other three lathyranes tested (EOF2, EOF3 and ELAF12‐2) led us to the hypothesis that the C7‐hydroxyl group present only in ELAC could be a structural requirement for PKC recognition and activation. Crystal structure of PKCδ's C1B domain in complex with phorbol 13‐acetate has revealed that C3‐, C4‐ and C20‐oxygen residues in the phorbol establish important hydrogen bonds with the protein (Zhang et al., 1995); additional oxygenated groups implicated in the activation of classical and novel PKCs (blue arrows in Figure 1) include those at C9 (Wender et al., 1998) or C13 (Hritz et al., 2004), which are present in prostratin but only partially represented in ELAC. Thus, to shed some light on the nature of ELAC‐PKC interaction, we conducted a structural comparison of prostratin with ELAC based on skeleton overlapping and equivalent functional group matching (Krauter et al., 1996). A conformation for ELAC compatible with the n.O.e effects observed in 1H–NMR was used (Figure 10A). Superposition to prostratin was done based on the spatial coincidence of oxygenated groups capable of hydrogen‐bond interactions (Figure 10B), revealing a correspondence between C3‐, C4‐, C9‐, C13‐ and C20‐oxygens in prostratin with C3‐, C4‐, C14‐, C12‐ and C7‐oxygens in ELAC. The importance of ELAC's C7‐hydroxyl for PKC binding was tested by preparing a C7‐acetylated derivative (Ac‐ELAC) and analysing its bioactivity. Only ELAC – but not Ac‐ELAC – increased NPC proliferation, in terms of both neurosphere area and Ki67 expression. These results could not be attributed to a toxic effect, since neither ELAC nor Ac‐ELAC induced any amount of cell death. Thus, a free C7‐hydroxyl group seems essential for the bioactivity of ELAC. This is consistent with the lack of activity shown by ELAF12‐2, devoid of oxygenated functions at C4, C7 and C12 (Figure 1) and EOF2/EOF3, whose C7 positions are acetylated.
Figure 10.
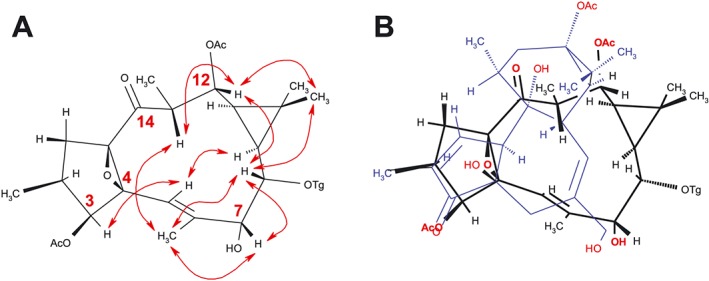
Structural comparisons of ELAC with prostratin. (A) Conformation of ELAC, consistent with selected nuclear Overhauser enhancements (red arrows) observed by NOESY2D NMR experiments. (B) The conformation of prostratin was superimposed on that of ELAC, based on matching of functional groups that might be involved in hydrogen‐bond interactions with PKC. Prostratin is shown in blue, and oxygenated functional groups are in red. Abbreviations: Ac, acetyl; Tg, tigloyl.
Previous results demonstrate that prostratin acts in synergy with the neurogenic‐niche factor bFGF to increase postnatal NPC proliferation by activating a DAG‐dependent PKC, that is, a classical or novel PKC, which in turn uses both ERK1/2 and PI3K as downstream targets. Prostratin alone is enough to significantly induce cyclin D1 expression in NPC cultures, but when combined with bFGF, it boosts the expression of both cyclin D1 and cyclin E (Geribaldi‐Doldán et al., 2016). The lathyrane ELAC, as well as phorboids, may activate more than one PKC isoenzyme at a time, but each PKC is likely to have specific cellular functions (Rosse et al., 2010) so that a small subset of PKCs, or even a single isoform, may be responsible for enhancing NPC proliferation when activated. Our experiments revealed that the effects of ELAC on NPC proliferation were mediated by the activation of PKCβ and that PKCα was partially involved. ELAC‐induced NPC proliferation was completely reverted not only by the classical/novel PKC inhibitor Gö6850 but also by the classical PKC inhibitor Gö6976, selective for calcium‐dependent cPKCs. Measurements of mRNA expression for the different cPKC isoforms in cultured NPC revealed that PKCβ was consistently the most abundant cPKC in all NPC cultures tested, followed by PKCα. The mRNA for PKCγ was virtually undetectable, in agreement with previous reports showing that PKCγ expression in the CNS is restricted to post‐mitotic neurons (Saito and Shirai, 2002). Thus, PKCα and/or PKCβ were tested for their implication in the proliferative effect of ELAC. We have demonstrated by inhibiting PKCβ expression that activation of this isoform is necessary for ELAC to induce its proliferative effect and that PKCα activation participates as well in triggering this effect. Although the majority of scientific research on PKC has focused, over the past decades, on the role of PKC in cancer, and it is often difficult to dissect physiological functions of specific PKC isoforms from those reported in the context of aberrant protein overexpression, several PKCs, including PKCα and β, have been consistently reported to induce proliferation in various cell types, including glioma and neuroblastoma cells (Altman et al., 1992; Finkenzeller et al., 1992; Brodie et al., 1998; Svensson et al., 2000), although PKCβI and βII isotypes may have opposing effects on proliferation (Yamamoto et al., 1998). PKCα and β can both activate ERK1/2 via Raf1 in fibroblasts (Schönwasser et al., 1998). Interestingly, PKCα (but not PKCβ) activates the cyclin D1 promoter and, to a minor extent, the cyclin E promoter in fibroblasts, inducing faster growth rates (Soh and Weinstein, 2003). In summary, ELAC‐induced NPC proliferation is due to pharmacological activation of PKCβ and to some extent PKCα activation.
Postnatal NPC proliferation is highly dependent on ERK1/2 and Akt signalling pathways, activated by GF like EGF or bFGF, which induce a phospho‐ERK1/2‐driven up‐regulation of cyclins D1 and E, and a phospho‐Akt‐dependent degradation of cyclin‐dependent kinase inhibitors, like p27 (Sung et al., 2007; Torroglosa et al., 2007; Rabaneda et al., 2008; Qiu et al., 2009; Geribaldi‐Doldán et al., 2016). While directly activating ERK1/2 through Ras, some GF can as well activate ERK1/2 further by an indirect, Ras‐independent, mechanism, mediated through a PLCγ‐PKC‐Raf1 signalling pathway (Schönwasser et al., 1998; Mason et al., 2006; Black and Black, 2012; Kinehara et al., 2013). We show in here that EGF increased postnatal NPC proliferation by a mechanism that in part involved PKC activation, while bFGF‐induced proliferation did not strictly depend on net PKC activity, in accordance with previous reports (Pende et al., 1997; Morishita et al., 2007). However, bFGF did enhance EGF‐mediated activation of PKC when both GF were added together (Figure 5C); thus, cooperative PKC activation induced by EGF + bFGF could mediate the strong proliferative response of NPC to this GF combination (Reynolds and Weiss, 1992; Belenguer et al., 2016). Interestingly, the lathyrane ELAC and the phorboid prostratin were able to substitute for either GF in combinatory treatments, still obtaining maximum increases on neurosphere size (Figure 5A). These results suggest that PKC activators might be useful to palliate deficient or delayed GF signalling after CNS damage.
Local GF administration has proven useful to increase neuronal replacement in different animal models of adult brain damage (Fallon et al., 2000; Teramoto et al., 2003), but small proteins like GF are difficult to deliver into the brain through non‐invasive routes. Thus, the use of small molecules that could easily reach brain parenchyma to mimic or synergize with GF action should be advantageous. Nevertheless, pharmacological promotion of excessive proliferation may result in NSC/NPC pool exhaustion (Ramirez‐Castillejo et al., 2006; Lopez‐Toledano and Shelanski, 2007), so candidate molecules should be tested for their capacity to preserve their capacity to continue producing neural progenitors. Nuclear expression of the transcription factor Sox2, a marker for multipotential NSCs (Ellis et al., 2004), has been reported indispensable for maintaining self‐renewal capacity in neurogenic niches (Favaro et al., 2009). In the context of NPC cultures, a loss of self‐renewal capacity would result in decreased expression of Sox2 and decreased neurosphere numbers as subcultures go on (Ramirez‐Castillejo et al., 2006; Lopez‐Toledano and Shelanski, 2007). We analysed Sox2 expression in intact neurospheres obtained after treatment with ELAC and observed that this compound slightly, but significantly, augmented the proportion of Sox2+ NPC in culture and also increased the number of secondary neurospheres formed following drug withdrawal and cell dispersion, while the inactive Ac‐ELAC had no effect on either measurement. Therefore, ELAC not only augmented the absolute number of multipotent progenitors by its effect on proliferation – the high proportion of nestin+ multipotent precursors observed in control neurospheres (~75%) was not changed by ELAC treatment – but it also mildly increased the proportion of cells that retained Sox2.
The lathyrane ELAC also promoted adult NPC proliferation in vivo. A single i.c.v. injection of ELAC significantly increased the number of BrdU+ cells in the adult SVZ, further beyond the small increase observed in vehicle‐injected mice due to the injection procedure (Figure 7). In addition, our in vivo studies have allowed us to identify the cellular target of ELAC, demonstrating that ELAC mainly promoted proliferation of TAPs, and at a smaller extent, it might be facilitating the proliferation of NSCs (Figure 8). Nonetheless, proliferation of neuroblasts in the SVZ did not significantly change with the treatment (Figure 7C–E), but a tendency for ELAC to promote the generation of mature neurons from NPCs in the OB was observed that did not reach statistical significance (Figure 9).
In conclusion, we showed for the first time that ELAC, a lathyrane diterpene, stimulates NPC proliferation in vivo and in vitro through PKCβ activation. This highlights ELAC as a potential agent in the development of drugs to treat disorders associated with a reduction in neurogenesis.
Author contributions
M.M.‐C. and C.C. conceived the work, did the experimental design, data acquisition and analysis, discussion of results, article preparation and writing. N.G.‐D., M.C.‐V. and J.D.‐R. performed the experimental design, data acquisition and analysis and discussion of results. E.F.‐G. did the data acquisition and discussion of results. F.G.‐B., P.H.‐F., A.D.‐A. and C.V. carried out the data acquisition and analysis and discussion of results. A.J.M.‐S. and R.H.‐G. did the experimental design, data analysis, discussion of results, manuscript preparation and writing. M.D. performed the data acquisition.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Protective effect of ELAC on cell death induced by oxidative stress. Cells isolated from neurospheres were seeded and cultured under floating conditions for 48 h in the presence and absence of BSO (100 μM), ELAC (5 μM), ELAC + BSO (5 μM and 100 μM, respectively) and EGSH + BSO (5 mM and 100 μM, respectively) and allowed to form neurospheres. Then cells were disaggregated and the percentage of death and surviving cells were measured by trypan blue exclusion.
Acknowledgements
This work was supported by the Spanish Consejería de Innovación, Ciencia y Empleo, Junta de Andalucía (grant numbers P10CTS6639 and P07‐FQM‐02925) and the Ministerio de Economía y Competitividad (grant number BFU2015‐68652‐R and MINECO/FEDER).
Murillo‐Carretero, M. , Geribaldi‐Doldán, N. , Flores‐Giubi, E. , García‐Bernal, F. , Navarro‐Quiroz, E. A. , Carrasco, M. , Macías‐Sánchez, A. J. , Herrero‐Foncubierta, P. , Delgado‐Ariza, A. , Verástegui, C. , Domínguez‐Riscart, J. , Daoubi, M. , Hernández‐Galán, R. , and Castro, C. (2017) ELAC (3,12‐di‐O‐acetyl‐8‐O‐tigloilingol), a plant‐derived lathyrane diterpene, induces subventricular zone neural progenitor cell proliferation through PKCβ activation. British Journal of Pharmacology, 174: 2373–2392. doi: 10.1111/bph.13846.
References
- Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH (2014). Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev : 991–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman A, Mally MI, Isakov N (1992). Phorbol ester synergizes with Ca2+ ionophore in activation of protein kinase C (PKC)alpha and PKC beta isoenzymes in human T cells and in induction of related cellular functions. Immunology 76: 465–471. [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Buylla A, Garcia‐Verdugo JM (2002). Neurogenesis in adult subventricular zone. J Neurosci 22: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8: 963–970. [DOI] [PubMed] [Google Scholar]
- Avila L, Perez M, Sanchez‐Duffhues G, Hernandez‐Galan R, Munoz E, Cabezas F et al. (2010). Effects of diterpenes from latex of Euphorbia lactea and Euphorbia laurifolia on human immunodeficiency virus type 1 reactivation. Phytochemistry 71: 243–248. [DOI] [PubMed] [Google Scholar]
- Belenguer G, Domingo‐Muelas A, Ferron SR, Morante‐Redolat JM, Farinas I (2016). Isolation, culture and analysis of adult subependymal neural stem cells. Differentiation 91: 28–41. [DOI] [PubMed] [Google Scholar]
- Black AR, Black JD (2012). Protein kinase C signaling and cell cycle regulation. Front Immunol 3: 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg P, Kedei N, Lewin N, Yang D, Czifra G, Pu Y et al. (2008). Wealth of opportunity – the C1 domain as a target for drug development. Curr Drug Targets 9: 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokara KK, Kwon KH, Nho Y, Lee WT, Park KA, Lee JE (2011). Retroviral expression of arginine decarboxylase attenuates oxidative burden in mouse cortical neural stem cells. Stem Cells Dev 20: 527–537. [DOI] [PubMed] [Google Scholar]
- Bokara KK, Kim JH, Kim JY, Lee JE (2016). Transfection of arginine decarboxylase gene increases the neuronal differentiation of neural progenitor cells. Stem Cell Res 17: 256–265. [DOI] [PubMed] [Google Scholar]
- Brodie C, Kuperstein I, Acs P, Blumberg PM (1998). Differential role of specific PKC isoforms in the proliferation of glial cells and the expression of the astrocytic markers GFAP and glutamine synthetase. Brain Res Mol Brain Res 56: 108–117. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP et al. (2008). Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A 105: 3581–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Soh JW, Yoshida K, Eves EM, Weinstein IB, Rosner MR (2000). Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol Cell Biol 20: 5392–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoubi M, Marquez N, Mazoir N, Benharref A, Hernandez‐Galan R, Munoz E et al. (2007). Isolation of new phenylacetylingol derivatives that reactivate HIV‐1 latency and a novel spirotriterpenoid from Euphorbia officinarum latex. Bioorg Med Chem 15: 4577–4584. [DOI] [PubMed] [Google Scholar]
- Das J, Rahman GM (2014). C1 domains: structure and ligand‐binding properties. Chem Rev 114: 12108–12131. [DOI] [PubMed] [Google Scholar]
- Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S et al. (2004). SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci 26: 148–165. [DOI] [PubMed] [Google Scholar]
- Fallon J, Reid S, Kinyamu R, Opole I, Opole R, Baratta J et al. (2000). In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci U S A 97: 14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro R, Valotta M, Ferri AL, Latorre E, Mariani J, Giachino C et al. (2009). Hippocampal development and neural stem cell maintenance require Sox2‐dependent regulation of Shh. Nat Neurosci 12: 1248–1256. [DOI] [PubMed] [Google Scholar]
- Finkenzeller G, Marme D, Hug H (1992). Inducible overexpression of human protein kinase C alpha in NIH 3T3 fibroblasts results in growth abnormalities. Cell Signal 4: 163–177. [DOI] [PubMed] [Google Scholar]
- Geribaldi‐Doldán N, Flores‐Giubi E, Murillo‐Carretero M, García‐Bernal F, Carrasco M, Macías‐Sánchez AJ et al. (2016). 12‐Deoxyphorbols promote adult neurogenesis by inducing neural progenitor cell proliferation via PKC activation. Int J Neuropsychopharmacol 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S (2003). Glia as neural progenitor cells. Trends Neurosci 26: 590–596. [DOI] [PubMed] [Google Scholar]
- Hezareh M (2005). Prostratin as a new therapeutic agent targeting HIV viral reservoirs. Drug News Perspect 18: 496–500. [DOI] [PubMed] [Google Scholar]
- Hritz J, Ulicny J, Laaksonen A, Jancura D, Miskovsky P (2004). Molecular interaction model for the C1B domain of protein kinase C‐gamma in the complex with its activator phorbol‐12‐myristate‐13‐acetate in water solution and lipid bilayer. J Med Chem 47: 6547–6555. [DOI] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP et al. (2001). Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci U S A 98: 4710–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinehara M, Kawamura S, Tateyama D, Suga M, Matsumura H et al. (2013). Protein kinase C regulates human pluripotent stem cell self‐renewal. PLoS One 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauter G, Von der Lieth CW, Schmidt R, Hecker E (1996). Structure/activity relationships of polyfunctional diterpenes of the tigliane type. A pharmacophore model for protein‐kinase‐C activators based on structure/activity studies and molecular modeling of the tumor promoters 12‐O‐tetradecanoylphorbol 13‐acetate and 3‐O‐tetradecanoylingenol. Eur J Biochem / FEBS 242: 417–427. [DOI] [PubMed] [Google Scholar]
- Lai C, Feng L (2004). Neuregulin induces proliferation of neural progenitor cells via PLC/PKC pathway. Biochem Biophys Res Commun 319: 603–611. [DOI] [PubMed] [Google Scholar]
- Lopez‐Toledano MA, Shelanski ML (2007). Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind). J Alzheimers Dis 12: 229–240. [DOI] [PubMed] [Google Scholar]
- Magnusson JP, Goritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O et al. (2014). A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science (New York, NY) 346: 237–241. [DOI] [PubMed] [Google Scholar]
- Mason JM, Morrison DJ, Basson MA, Licht JD (2006). Sprouty proteins: multifaceted negative‐feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 16: 45–54. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami H, Owada Y, Suzuki R, Handa Y, Kondo H (2000). Localization of mRNAs for novel, atypical as well as conventional protein kinase C (PKC) isoforms in the brain of developing and mature rats. J Mol Neurosci 15: 121–135. [DOI] [PubMed] [Google Scholar]
- Morishita R, Ueda H, Ito H, Takasaki J, Nagata K, Asano T (2007). Involvement of Gq/11 in both integrin signal‐dependent and ‐independent pathways regulating endothelin‐induced neural progenitor proliferation. Neurosci Res 59: 205–214. [DOI] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N et al. (2002). Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110: 429–441. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17: 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M, Fisher TL, Simpson PB, Russell JT, Blenis J, Gallo V (1997). Neurotransmitter‐ and growth factor‐induced cAMP response element binding protein phosphorylation in glial cell progenitors: role of calcium ions, protein kinase C, and mitogen‐activated protein kinase/ribosomal S6 kinase pathway. [DOI] [PMC free article] [PubMed]
- Pusztai R, Ferreira MJ, Duarte N, Engi H, Molnar J (2007). Macrocyclic lathyrane diterpenes as antitumor promoters. Anticancer Res 27: 201–205. [PubMed] [Google Scholar]
- Qiu J, Takagi Y, Harada J, Topalkara K, Wang Y, Sims JR et al. (2009). p27kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem Cells 27: 920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaneda LG, Carrasco M, Lopez‐Toledano MA, Murillo‐Carretero M, Ruiz FA, Estrada C et al. (2008). Homocysteine inhibits proliferation of neuronal precursors in the mouse adult brain by impairing the basic fibroblast growth factor signaling cascade and reducing extracellular regulated kinase 1/2‐dependent cyclin E expression. FASEB J 22: 3823–3835. [DOI] [PubMed] [Google Scholar]
- Ramirez‐Castillejo C, Sanchez‐Sanchez F, Andreu‐Agullo C, Ferron SR, Aroca‐Aguilar JD, Sanchez P et al. (2006). Pigment epithelium‐derived factor is a niche signal for neural stem cell renewal. Nat Neurosci 9: 331–339. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (New York, NY) 255: 1707–1710. [DOI] [PubMed] [Google Scholar]
- Romero‐Grimaldi C, Moreno‐Lopez B, Estrada C (2008). Age‐dependent effect of nitric oxide on subventricular zone and olfactory bulb neural precursor proliferation. J Comp Neurol 506: 339–346. [DOI] [PubMed] [Google Scholar]
- Romero‐Grimaldi C, Murillo‐Carretero M, Lopez‐Toledano MA, Carrasco M, Castro C, Estrada C (2011). ADAM‐17/tumor necrosis factor‐alpha‐converting enzyme inhibits neurogenesis and promotes gliogenesis from neural stem cells. Stem Cells 29: 1628–1639. [DOI] [PubMed] [Google Scholar]
- Rosse C, Linch M, Kermorgant S, Cameron AJM, Boeckeler K et al. (2010). PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol 11: 103–112. [DOI] [PubMed] [Google Scholar]
- Saito N, Shirai Y (2002). Protein kinase C gamma (PKC gamma): function of neuron specific isotype. J Biochem 132: 683–687. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Duffhues G, Vo MQ, Perez M, Calzado MA, Moreno S, Appendino G et al. (2011). Activation of latent HIV‐1 expression by protein kinase C agonists. A novel therapeutic approach to eradicate HIV‐1 reservoirs. Curr Drug Targets 12: 348–356. [DOI] [PubMed] [Google Scholar]
- Schönwasser DC, Marais RM, Marshall CJ, Parker PJ (1998). Activation of the mitogen‐activated protein kinase/extracellular signal‐regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol 18: 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh JW, Weinstein IB (2003). Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem 278: 34709–34716. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhart R, Kazimirsky G, Okhrimenko H, Ben‐Hur T, Brodie C (2007). PKCepsilon induces astrocytic differentiation of multipotential neural precursor cells. Glia 55: 224–232. [DOI] [PubMed] [Google Scholar]
- Sung SM, Jung DS, Kwon CH, Park JY, Kang SK et al. (2007). Hypoxia/reoxygenation stimulates proliferation through PKC‐dependent activation of ERK and Akt in mouse neural progenitor cells. Neurochem Res 32: 1932–1939. [DOI] [PubMed] [Google Scholar]
- Susarla B, Villapol S, Yi JH, Geller H, Symes A (2014). Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice. ASN Neuro 6: e00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson K, Zeidman R, Troller U, Schultz A, Larsson C (2000). Protein kinase C beta1 is implicated in the regulation of neuroblastoma cell growth and proliferation. Cell Growth Differ 11: 641–648. [PubMed] [Google Scholar]
- Szallasi Z, Blumberg PM (1991). Prostratin, a nonpromoting phorbol ester, inhibits induction by phorbol 12‐myristate 13‐acetate of ornithine decarboxylase, edema, and hyperplasia in Cd‐1 mouse skin. Cancer Res 51: 5355–5360. [PubMed] [Google Scholar]
- Teramoto T, Qiu J, Plumier JC, Moskowitz MA (2003). EGF amplifies the replacement of parvalbumin‐expressing striatal interneurons after ischemia. J Clin Invest 111: 1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroglosa A, Murillo‐Carretero M, Romero‐Grimaldi C, Matarredona ER, Campos‐Caro A, Estrada C (2007). Nitric oxide decreases subventricular zone stem cell proliferation by inhibition of epidermal growth factor receptor and phosphoinositide‐3‐kinase/Akt pathway. Stem Cells (Dayton, Ohio) 25: 88–97. [DOI] [PubMed] [Google Scholar]
- Vasas A, Hohmann J (2014). Euphorbia diterpenes: isolation, structure, biological activity, and synthesis (2008–2012). Chem Rev 114: 8579–8612. [DOI] [PubMed] [Google Scholar]
- Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L et al. (2012). Metformin activates an atypical PKC‐CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 11: 23–35. [DOI] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL (2000). Identification of PKC‐isoform‐specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21: 181–187. [DOI] [PubMed] [Google Scholar]
- Wender PA, DeBrabander J, Harran PG, Jimenez JM, Koehler MF, Lippa B et al. (1998). The design, computer modeling, solution structure, and biological evaluation of synthetic analogs of bryostatin 1. Proc Natl Acad Sci U S A 95: 6624–6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Acevedo‐Duncan M, Chalfant CE, Patel NA, Watson JE, Cooper DR (1998). The roles of protein kinase C beta I and beta II in vascular smooth muscle cell proliferation. Exp Cell Res 240: 349–358. [DOI] [PubMed] [Google Scholar]
- Zhang G, Kazanietz MG, Blumberg PM, Hurley JH (1995). Crystal structure of the cys2 activator‐binding domain of protein kinase C delta in complex with phorbol ester. Cell 81: 917–924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Protective effect of ELAC on cell death induced by oxidative stress. Cells isolated from neurospheres were seeded and cultured under floating conditions for 48 h in the presence and absence of BSO (100 μM), ELAC (5 μM), ELAC + BSO (5 μM and 100 μM, respectively) and EGSH + BSO (5 mM and 100 μM, respectively) and allowed to form neurospheres. Then cells were disaggregated and the percentage of death and surviving cells were measured by trypan blue exclusion.