

Abstract
Status epilepticus (SE) is a common neurological emergency that results from the failure of the mechanisms responsible for seizure termination or the initiation of mechanisms that lead to abnormally prolonged seizures. Although the failure of inhibitory mechanisms during SE is well understood, the seizure-initiating mechanisms are poorly understood. We tested whether hippocampal α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated transmission was enhanced during SE and assessed the underlying molecular mechanism. In animals in self-sustaining limbic SE the amplitudes of the miniature, spontaneous, and AMPA-evoked excitatory currents recorded from the CA1 pyramidal neurons were larger than those recorded in the controls. The evoked EPSCs rectified inwardly. In these animals, the surface expression of GluA1 subunit-containing AMPARs was increased in the CA1 pyramidal neurons. The phosphorylation of the GluA1 subunit on S831 and S845 residues was reduced in animals in SE. In contrast, the GluA1 subunit surface expression and AMPAR-mediated neurotransmission of dentate granule cells (DGCs) was not altered. Treating animals in SE with the NMDAR antagonist MK-801 or with diazaepam blocked the increased surface expression of the GluA1 subunits. NMDAR blockade also prevented the dephosphorylation of the S845 residue but not that of S831. Targeting NMDARs and AMPARs may provide novel strategies to treat benzodiazepine-refractory SE.
Keywords: Status epilepticus, AMPA receptor, mEPSCs, GluA1 subunit phosphorylation, MK-801, benzodiazepines
Introduction
Most seizures are brief and end spontaneously, but some trigger neuronal plasticity such that they transform into a self-perpetuating state. This is referred to as status epilepticus (SE), which is a common neurological emergency that affects 10–41 per 100,000 individuals. The overall mortality associated with SE approaches 20%, and in those who survive, it can cause neuronal injury and death (Betjemann & Lowenstein, 2015;DeLorenzo et al., 1996). SE is initiated in part by repeated or prolonged seizures reducing γ-aminobutyric acid type-A receptor (GABAR)-mediated inhibition, and benzodiazepines, which modulate GABA-A receptors, are the most effective treatment for SE (Alldredge et al., 2001;Silbergleit et al., 2012;Treiman et al., 1998;Chamberlain et al., 2014;Glauser et al., 2016). However, benzodiazepines lose their efficacy as continuous seizure activity occurs, as determined by EEG findings, and SE becomes self-sustaining (Goodkin et al., 2008;Kapur & MacDonald, 1997;Naylor et al., 2005;Terunuma et al., 2008). Studies have revealed that GABAR-mediated inhibitory synaptic transmission is reduced in the hippocampi of animals in SE, due in part to the internalization of synaptic GABARs (Goodkin et al., 2008;Kapur & Coulter, 1995;Kapur & MacDonald, 1997;Naylor et al., 2005;Terunuma et al., 2008).
There is growing evidence that AMPAR plasticity may also play a role in maintaining SE in that, the internalization of the GluA2 subunit is accelerated during SE (Rajasekaran et al., 2012). Furthermore, the benzodiazepine-refractory SE in experimental animals can be terminated by attenuating the fast excitatory transmission mediated by AMPAR (Fritsch et al., 2010; Pitk+ñnen et al., 2007;Rajasekaran et al., 2012; Hanada et al., 2014). However, it is unknown whether AMPAR-mediated transmission is enhanced during SE and the mechanisms that cause this enhancement are unclear.
In the hippocampus, AMPARs are dimers of GluA1/GluA2 and GluA2/GluA3 subunits (Wenthold et al., 1996). The GluA1 subunits play a dominant role in the activity-dependent recruitment of AMPARs at the synapses (Henley & Wilkinson, 2016). Here, we demonstrate the enhanced AMPAR-mediated excitatory transmission of CA1 pyramidal neurons accompanied by the increased surface expression and dephosphorylation of the GluA1 subunit of AMPARs. AMPAR plasticity can be blocked by treatment with agents that dampen the neuronal activity.
Materials and Methods
All studies were performed in accordance with protocols approved by the Animal Care and Use Committee of the University of Virginia. All chemicals were obtained from Sigma (St. Louis, MO, USA) unless otherwise stated. Adult male Sprague-Dawley rats (200–250 g) were used.
Electrode implantation and induction of status epilepticus
Bipolar insulated stainless steel electrodes were stereotaxically implanted over the hippocampus and cortex in adult male rats as described previously (Rajasekaran et al., 2012). The animals were given methyl scopolamine (1 mg/kg, intraperitoneal, ip) 30 min prior to the induction of SE. SE was induced by continuous hippocampal stimulation (CHS) (Lothman et al., 1989) or by the administration of pilocarpine (320 mg/kg, ip) (Rajasekaran et al., 2012). For the studies on SE duration, animals were monitored continuously by video-EEG. For biochemical and electrophysiological studies, the animals were visually monitored, and the time of first grade 5 (Racine score) seizure, which corresponded to the onset of continuous electrographic activity, was noted (Racine, 1972). Continuous seizure activity marks the development of benzodiazepine resistance and the establishment of benzodiazepine refractory SE or established SE (ESE) (Treiman, 2007).
In some of the experiments, animals were treated with MK-801 (2 mg/kg, ip) alone or in combination with diazepam (10 mg/kg, ip) at 10 min after the onset of ESE
Hippocampal slice preparation
For biochemical studies the animals were decapitated under halothane anesthesia 60 min after the first stage 5 behavioral seizure was identified using the Racine scale criteria (Racine, 1972), whereas for electrophysiological studies, the animals were sacrificed 40 min after the first tonic-clonic seizure. The brains were removed and immersed in ice-cold oxygenated dissection buffer (4°C, 95% O2, and 5% CO2) containing (in mM) 65.5 NaCl, 2 KCl, 5 MgSO4, 1.1 KH2PO4, 1 CaCl2, 10 dextrose, and 113 sucrose (300 mOsm), and horizontal hippocampal slices (300 μm) were prepared with a vibratome (VT1200S; Leica, Wetzlar, Germany).
Electrophysiology
Following slicing, the slices were placed in an interface chamber containing oxygenated artificial cerebrospinal fluid (aCSF) at room temperature (25°C) and allowed to equilibrate for 20 min. The aCSF contained (in mM) 124 NaCl, 4 KCl, 1 MgCl2, 25.7 NaHCO3, 1.1 KH2PO4, 10 dextrose, and 2.5 CaCl2 (300 mOsm). AMPA (2.5 μM)-evoked whole-cell currents were recorded from the pyramidal neurons in the hippocampal slices in the ESE and control animals (Rajasekaran et al., 2012). The slices were perfused with oxygenated aCSF containing DL-AP5 (50 μM) and picrotoxin (50 μM) to block the NMDA and GABAA receptors, respectively, at a rate of 2–3 ml/min. The patch electrode was filled with an internal solution containing (in mM) 115 cesium methane sulfonate, 20 CsCl, 10 KCl, 10 HEPES, 0.5 sodium-EGTA, 2.5 MgCl2, 5 Mg-ATP and 5 lidocaine, pH 7.3, 285 mOsm. The neurons were voltage-clamped at −65 mV. Action potential-independent miniature excitatory post-synaptic currents (mEPSCs) were recorded from CA1 pyramidal neurons (supplementary figure 1) and DGCs, spontaneous EPSCs (sEPSCs) were also recorded from CA1 pyramidal neurons. Synaptic currents were analyzed using the MiniAnalysis software as described previously (Sun & Kapur, 2012). The threshold for detecting synaptic currents was set at 5 times the root mean square noise.
Evoked excitatory postsynaptic currents (eEPSCs) were recorded from CA1 pyramidal cells in response to the electrical stimulation of Schaffer collateral axons by a bipolar tungsten microelectrode (FHC, Bowdoin, ME, USA) placed in stratum radiatum.
The plasticity of AMPARs during SE is rapid and transient. Hence, all recordings were completed within 2 hr of slicing. To avoid issues associated with nesting of the data, only one cell per animal was recorded in most instances (Aarts et al., 2014).
Biotinylation and BS3 cross-linking assay
GluA1 subunit surface expression was determined using a biotinylation assay (Goodkin et al., 2008) or a BS3 cross-linking assay (Grosshans et al., 2002). Four to six hippocampal slices from one hemisphere were incubated in ice-cold (2–4°C) artificial CSF (aCSF) containing BS3 (1 mg/ml) at 4°C for 40 min with constant shaking, and 4–6 hippocampal slices from the other hemisphere were simultaneously incubated in ice-cold aCSF alone. Following incubation, the slices were washed twice with ice-cold aCSF containing 10 mM Tris-Cl pH 7.4 to stop the cross-linking reaction and remove the remaining BS3 reagent. The regions of interest were microdissected, and the tissue was lysed in RIPA lysis buffer as described before and was resolved by SDS-PAGE (Goodkin et al., 2008;Rajasekaran et al., 2012). A phosphatase inhibitor cocktail (Thermo Fisher Scientific) was included in the lysis buffer when pGluA1 subunit expression was studied. The antibodies used in this study include anti-GluA1 subunit antibody (MAB2263, Millipore, 1:1000 dilution), anti-pGluA1 S831 (04–823 Millipore, 1:1000), anti-pGluA1 S845 (04–1073 Millipore, 1:1000).and anti-β-actin antibody (A2228, Sigma Aldrich, 1:5000). Horseradish peroxidase conjugated anti-mouse or anti-rabbit secondary antibodies (Biorad, 1:5000 dilution) were used. The Western blot signal was detected using a chemiluminescent reagent (Perkin Elmer), and the blots were exposed on a Chemidoc Touch imaging system (BioRad). The signal intensity was quantified by densitometric scanning, and the expression of the protein of interest was normalized to that of β-actin in each sample.
Power spectrum analysis
The power analysis of EEG was performed as previously described (Raol et al., 2009). The EEG data were digitized at 400 Hz. Power calculations were performed and graphically displayed using the Scilab software. For spectral analysis, the EEGs were filtered using 1-Hz high-pass, 70-Hz low-pass and 60-Hz notch filters and binned in 5-sec intervals, and the power within each period was determined.
Statistical analysis
The data are expressed as the mean ± SEM. Statistical analysis was performed in the GraphPad Prism software using the t-test or Wilcoxon matched-pairs signed-rank test, or ANOVA with post hoc Dunn’s multiple comparison test or Kruskal-Wallis test followed by Dunn’s multiple comparison test and a p value less than 0.05 was considered significant.
Results
Increased AMPAR-mediated synaptic transmission of CA1 pyramidal neurons of animals in SE
To determine whether AMPAR-mediated synaptic transmission was enhanced during SE, hippocampal slices were prepared from rats that had undergone 40 minutes of pilocarpine-induced SE and were allowed to recover for 20 minutes. The miniature AMPAR-mediated EPSCs were recorded from CA1 pyramidal neurons of SE animals (Fig. 1A). The mEPSCs recorded from the CA1 pyramidal neurons of SE animals were larger than those recorded from control animals (Fig. 1B, 22.8 ± 1.1 pA, N= 11 neurons from 9 SE animals vs 18.5 ± 0.7 pA, N= 13 neurons from 7 naïve animals, p<0.05, t-test). However, the frequency (0.59 ± 0.12 Hz vs 0.47 ± 0.094 Hz), rise time (0.61 ± 0.04 ms vs 0.61 ± 0.07 ms), and decay (4.99 ± 0.52 ms, vs 5.4 ± 0.66 ms) of mEPSCs did not change (p > 0.05). The spontaneous EPSCs recorded from CA1 pyramidal neurons of SE animals were also larger (Fig. 1B), but, their frequency was unchanged (1.56 ± 0.46 Hz vs 0.85 ± 0.34 Hz, p > 0.05).
Figure 1. AMPAR-mediated neurotransmission of CA1 pyramidal neurons was enhanced in animals in SE.

SE was induced in adult male rats by a high dose of pilocarpine (350 mg/kg). Brain slices were prepared 40 minutes after the first behavioral seizure and were allowed to recover for 20 min. (A) The averaged action potential-independent synaptic currents (mEPSCs) recorded from CA1 pyramidal neurons of an animal in SE (red) and a naïve animal (black). (B) Mean of the median mEPSC and sEPSC amplitude. The values represent the mean ± SEM (N= 11 neurons from 9 SE animals and 13 neurons from 7 naïve animals for mEPSCs, * p < 0.05, t-test and N= 9 neurons of 7 SE animals and 9 neurons of 6 animals for sEPSCs,* p < 0.05. t-test). (C) Rectification properties of evoked EPSCs recorded from CA1 neurons of control (black) and SE (red) animals. The neurons were held at different voltages (-70 mv to +40 mV) and EPSCs were evoked (eEPSCs) by electrical stimulation of Schaffer collaterals. The eEPSCs recorded from the CA1 neuron of a representative control (black) and SE (red) animal are shown in (a). The amplitude of currents recorded in the same cells displayed in (a) plotted against the holding potentials are shown in (b). (D) Representative whole-cell AMPA-evoked current recorded from CA1 pyramidal neurons of SE (60 min after the first stage 5 behavioral seizure) and naïve animals. AMPA (2.5 μM, black bar) was bath applied after recording a baseline holding current.
Increased neuronal activity incorporates GluA1 subunit-containing AMPARs into synapses, which is likely to increase the proportion of GluA1 homomers and reduce the GluA1/GluA2 hetero dimers at the synapse. Because GluA2-lacking AMPARs produce rectifying channels, we expected an increased rectification of AMPAR currents in SE animals (Rajasekaran et al., 2012). The neurons were held at different voltages (−70 mv to +40 mV). The EPSCs evoked by the electrical stimulation of Schaffer collaterals (eEPSCs) were non-rectifying in control animals but were inwardly rectifying in SE animals (Fig. 1C). The rectification index (RI), which is a ratio of eEPSC amplitude recorded at −40 mV to +40 mV, was smaller in SE animals (0.35 ± 0.079, N= 8 cells/7 animals) than RI in control animals (0.96 ± 0.065, N= 8 cells/5 animals, p < 0.0001).
GluA1-containing AMPARs are expressed in both the extra-synaptic membrane and the synapses and can be activated by exogenously applying AMPA. The bath application of AMPA (2.5 μM) evoked a large current from CA1 pyramidal neurons of both control and SE animals (Fig. 1D, 20.6 ± 3.8 pA/pF, N= 7 neurons 6 animals vs 12.5 ± 1.5 pA/pF, N= 10 neurons 6 animals, p < 0.05).
DGCs inhibit the spread of seizure activity into the hippocampus proper (Heinemann et al., 1992;Lothman et al., 1992) and are resistant to SE-induced cell death (Grooms et al., 2000;Fujikawa, 1996a). There was no evidence of the enhancement of AMPAR-mediated neurotransmission of DGCs of animals in pilocarpine-induced SE. Neither the frequency (0.34 ± 0.06 Hz, N= 6 neurons from 4 animals vs 0.59 ± 0.1 Hz, N=6 neurons from 4 animals, p > 0.05) nor the amplitude of the mEPSCs (11.31 ± 0.5 pA vs 12.6 ± 0.6 pA) recorded from the DGCs of SE animals were altered. However, these events had a faster 10–90% rise-time (1.26 ± 0.05, p < 0.05) and a more rapid decay (8.3 ± 0.4 vs 11.6 ± 0.7, p < 0.05).
Since the mEPSCs recorded from DGCs of control animals were smaller than those of CA1 neurons of control animals, we determined whether the basal AMPAR expression was different in these two regions. Indeed the expression of GluA1 subunit normalized to that of βactin in DGCs was lower than that in the CA1 neurons. (0.88 ± 0.07 vs 1.305 ± 0.06, n=4, p<0.05).
Increased surface expression of GluA1 subunits in animals in SE
We next tested whether SE induces plasticity of GluA1 subunit-containing AMPARs. Therefore, we determined whether the surface expression of GluA1 subunits during SE was also increased in rats in pilocarpine-induced SE (Fig. 2A). Surface proteins were isolated 60 min after the first stage 5 behavioral seizure, which corresponds to the onset of continuous electrographic activity. The surface expression of the GluA1 subunit in the CA1 region of SE rats was higher than that of controls (161 ± 23% of that in controls, N= 5).
Figure 2. Surface expression of the GluA1 subunit of AMPARs was elevated in animals in SE.

SE was induced by high dose pilocarpine or continuous hippocampal stimulation (CHS) and biochemical assays were performed 60 min following the first stage 5 behavioral seizure. (A) The surface expression of the GluA1 subunit of AMPARs was higher in the CA1 region microdissected from rats in pilocarpine-induced SE compared to that in naïve animals (panel surface). Total GluA1 subunit protein, determined in 1/10th fraction of the lysate used to pull down biotin-bound surface proteins (total input), was similar in SE and control animals. (B) A ratio of surface-expressed GluA1 subunits to the total input was higher in rats in pilocarpine SE (N= 5, * p<0.05 t-test). The expression of β-actin was used as a loading control. (C) The expression of the GluA1 subunit in the intracellular proteins isolated from the CA1 region of control and pilocarpine SE animals, determined using a BS3 cross-linking assay. (D) Mean ratio of intracellular to total protein expression in control and pilocarpine SE animals (N= 7 each, ** p < 0.005, t-test). (E) The total and intracellular expression of the GluA1 subunit in the CA1 region of control and CHS SE animals. (F) The ratio of intracellular to total proteins in control and CHS-SE animals (N= 7 animals each, * p < 0.05, t-test).
An alternate way to measure the surface and intracellular fractions of proteins is to cross-link all surface proteins, eliminate them from detection, and measure the intracellular fraction (Grosshans et al., 2002). We used BS3 (Bis (sulfosuccinimidyl) suberate), which is an amine-to-amine cross linker, to cross-link surface proteins. The BS3 assay revealed reduced expression of the GluA1 subunit in the intracellular fraction of the CA1 region of SE animals. The intracellular expression of the GluA1 subunit was decreased in the CA1 region of 60 min SE hippocampi compared to controls, and the total expression of the GluA1 subunit was similar (Fig. 2C, D). Because the total GluA1 subunit expression was unaltered during SE, this assay confirmed the increased surface expression of the GluA1 subunit in the CA1 region of SE animals (51 ± 10% of that in control, N= 7). The increased GluA1 subunit surface expression in the CA1 region of SE animals was further confirmed in the electrical stimulation model (CHS) (Lothman et al., 1989). The intracellular expression of the GluA1 subunit in the CA1 region of CHS-SE animals was also less than that in controls (Fig. 2E, F, 56 ± 13% of that in controls, N= 7). These studies demonstrated that AMPAR-mediated transmission is enhanced during continuous seizure activity of self-sustaining SE.
We also determined whether GluA1 subunit surface expression increased in the DG region of pilocarpine-induced SE animals. The intracellular to total expression ratio in SE animals was 0.20 ± 0.06, similar to that in controls (0.29 ± 0.03, N= 5 each, p > 0.05, t-test). Thus, consistent with the electrophysiology experiments, the intracellular expression of the GluA1 subunit in proteins isolated from the DG subfield was similar to that in naïve animals (104 ± 9% of controls, n=5, p>0.05).
GluA1 subunits were dephosphorylated during SE
Phosphorylation/dephosphorylation of conserved serine/threonine residues of GluA1 subunits of AMPARs regulate their surface membrane trafficking (Song & Huganir, 2002). We determined whether the phosphorylation residues S831 and S845 in the GluA1 subunit were altered during SE. Proteins were isolated from the CA1 region of animals in pilocarpine-induced SE and blotted with pS831 and pS845 antibodies. The signal for pS831 and pS845 GluA1 in SE animals was weaker than that in naïve animals (Fig. 3A). The normalized expression of the pS831 GluA1 subunit expression in SE animals was 66 ± 8% of that in controls (n=6, p<0.05) and that of pS845 GluA1 was 58 ± 10% (n=6, p<0.05) (Fig. 3B).
Figure 3. Phosphorylation of S831 and S845 residues of the GluA1 subunit was reduced in animals in SE.
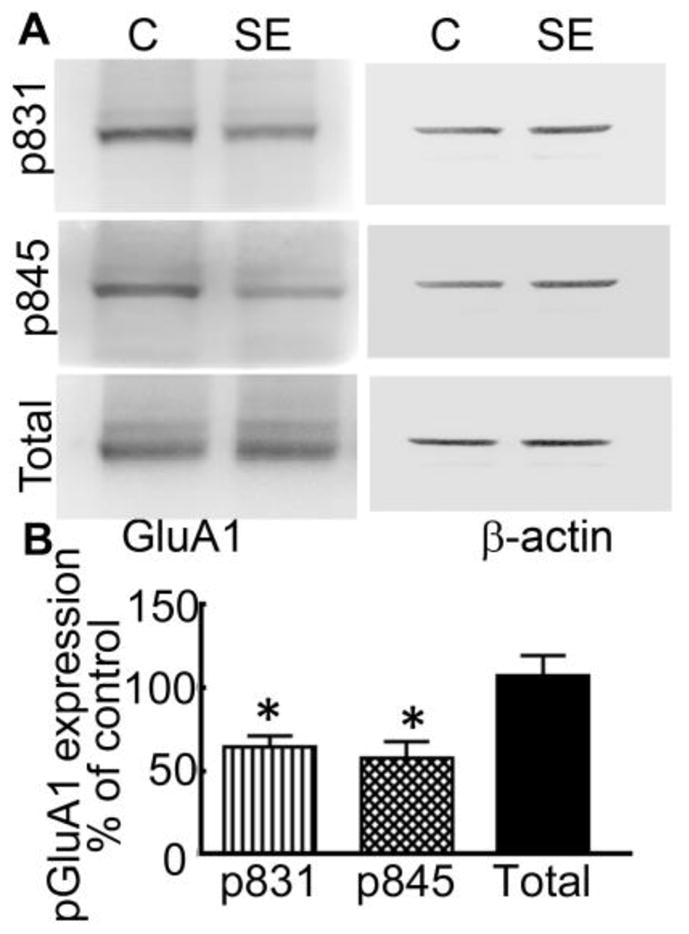
(A) The expression of the GluA1 subunit phosphorylated on residues S831 and S845 in proteins isolated from the CA1 area of SE (40 min after the first stage 5 behavioral seizures induced by pilocarpine) and naïve animals. The expression of β-actin was determined in each gel for normalization. The expression of the GluA1 subunit was also determined in each sample. (B) The expression of pGluA1 and total GluA1 subunit was normalized to that of β-actin and presented relative to that in naïve animals (N= 5, * p<0.005, Wilcoxon matched-pairs signed-rank test).
MK-801 treatment prevented the AMPAR plasticity during SE
NMDAR activation regulates the surface membrane insertion of GluA1 subunit-containing AMPARs during LTP (Derkach et al., 2007;Malinow & Malenka, 2002). NMDARs are activated during SE (Naylor et al., 2013) and may play a role in the increased surface expression of GluA1 subunit. To block NMDARs, animals were treated with MK-801 (2 mg/kg), a non-competitive NMDAR antagonist that acts via the phencyclidine (PCP) site (Foster & Wong, 1987), 10 min after the first stage 5 behavioral seizure occurred. The surface expression of the GluA1 subunit determined using a biotinylation assay in MK-801-treated SE animals was similar to that in controls (Fig. 4A and B, 120 ± 24% of that in controls, N= 5, p > 0.05). A BS3 assay also revealed that MK-801 treatment prevented an increase in surface expression of GluA1 subunits during SE (Fig. 4C). The ratio of intracellular to total expression of GluA1 subunits in MK-801-treated animals was also similar to that in controls (Fig. 4D, 0.47 ± 0.17 vs. 0.34 ± 0.09, N= 6, p > 0.05). Treatment of animals with MK-801 alone without SE for 50 min did not alter the intracellular expression of GluA1 subunits (0.51 ± 0.1, n=6, p>0.05).
Figure 4. Blocking of NMDAR activation during ESE prevented the increased surface expression of the GluA1 subunit.

MK-801 (2 mg/kg) was administered at 10 min after the first stage 5 behavioral seizure induced by pilocarpine. Control and MK-801-treated or untreated SE animals were processed simultaneously. (A) The surface expression of the GluA1 subunit in the microdissected CA1 region of MK-801-treated or untreated SE and naïve animals. (B) The surface expression of the GluA1 subunit normalized to the input (N=5 each, * p < 0.05, ANOVA with Dunn’s post hoc multiple comparison test). (C) The intracellular expression levels of the GluA1 subunit in the control and MK-801-treated SE animals were comparable. The intracellular GluA1 subunit expression in an untreated 60 min-ESE animal, shown for comparison, was lower. (D) Intracellular to total expression ratio of the GluA1 subunit in control and SE animals treated or untreated with MK-801 (N= 6 each, * p < 0.05, ANOVA with Dunn’s post hoc multiple comparison test). (E) The expression of GluA1 subunit phosphorylated on residues S831 and S845. The expression of β-actin is shown as loading control. (F). The GluA1 pS831 and GluA1 p845 expression in naïve and MK-801-treated SE animals (N= 5 each). The expression of GluA1 phosphorylated on S831 and S845 in animals in SE that did not receive any treatment is shown for comparison (N=5), **p<0.005, and *** p<0.001, Kruskal-Wallis test followed by Dunn’s multiple comparison test.
Next, we determined whether MK-801 treatment also prevented the dephosphorylation of the GluA1 subunit. The expression of GluA1 pS845 in MK-801-treated SE and naïve animals was similar (Fig. 4E, F). However, the expression of GluA1 pS831 was significantly lower than in naïve animal (Fig. 4E, F).
Combination of MK-801 and diazepam terminates ESE
The blockade of NMDARs prevented GluA1 subunit upregulation. We determined whether MK-801 combined with benzodiazepine treatment after the start of continuous seizure activity would terminate SE. Animals were treated with MK-801 (2 mg/kg) or diazepam (10 mg/kg, ip) either alone or in combination 10 min after the seizure activity became continuous (Fig. 5A–C). The EEG power in the 1–60 Hz frequency range remained high for 120 min in the animals treated with MK-801 alone or diazepam alone (Fig. 5A and B). However, the combination of diazepam and MK-801 rapidly decreased the EEG power (Fig. 5C). Visual analysis of the EEG traces was performed to determine the point of SE termination, which was defined by the slowing of spike wave discharges to below 2 Hz and the development of arrhythmic patterns without the recurrence of seizures for 60 min. SE continued unabated in the MK-801-treated animals (377 ± 89 min, N= 5, Fig. 5D) and in the diazepam-treated animals (367 ± 113 min, N= 5, Fig. 5D). However, a combination of MK-801 and diazepam rapidly terminated the SE (28 ± 16 min, N= 6, p < 0.05 ANOVA with post hoc Dunn’s multiple comparison test, Fig. 5D).
Figure 5. The combination of diazepam and MK-801 terminated benzodiazepine-refractory SE.

The EEG power spectrum during the first 120 min of ESE in the representative animals treated with (A) diazepam alone (10 mg/kg), (B) MK-801 alone (2 mg/kg) or (C) a combination of diazepam and MK-801 at 10 min ESE triggered by pilocarpine. In the spectrum, increasing power is represented by the change in color from blue to red. The low power at 60 Hz is due to the application of a notch filter to reduce the 60-cycle artifact. Conventional EEG traces from the same animal at baseline and at 10, 60, and 120 min ESE are shown below the power display. The combination of MK-801 and diazepam led to the rapid termination of seizures, after which the EEG returned to the baseline level, whereas seizures continued despite the treatments when these drugs were administered alone. (D) Mean ± SEM of seizure duration from the onset of continuous seizure activity (N=5 each for diazepam or MK-801 alone and N=6 for the diazepam and MK-801 combination treatment, * p<0.05, ANOVA with Dunn’s post hoc multiple comparison test).
A combination of MK-801 and diazepam terminated SE, we determined whether treatment with diazepam alone also affected the GluA1 subunit surface expression. Animals in pilocarpine-induced SE were treated with diazepam (10 mg/kg) 10 min after the first stage 5 behavioral seizure and diazepam treatment also prevented the increase in surface expression of GluA1 subunit (intracellular/total expression ratio 0.46 ± 0.1 vs 0.41 ± 0.5, n=5, p.>0.05).
Discussion
The major findings of this study are 1) during self-sustaining SE, AMPAR-mediated EPSCs recorded from CA1 pyramidal neurons were potentiated, and the surface expression of the GluA1 subunits was increased; 2) GluA1 subunits were dephosphorylated in animals in SE; and 3) SE-induced increase in surface expression of GluA1 subunit was suppressed by diazepam and MK-801. This and previous studies suggest that SE is sustained by two distinct mechanisms, reduction of GABA-A receptor-mediated inhibition and enhanced AMPAR- and NMDAR-mediated transmission (Goodkin et al., 2008;Naylor et al., 2005;Naylor et al., 2013;Rajasekaran et al., 2012;Terunuma et al., 2008). Once seizures have become refractory to benzodiazepines, a combination of drugs targeted at GABARs and glutamatergic transmission is necessary to terminate seizures (Martin & Kapur, 2008;Rice & DeLorenzo, 1999).
This study revealed enhancement of AMPAR-mediated synaptic transmission and increased surface expression of GluA1 subunit-containing AMPARs on CA1 pyramidal neurons during SE. Trafficking of the GluA1 subunit also plays a role in other forms of activity-dependent plasticity such as long-term potentiation (LTP), homeostatic plasticity, open eye potentiation during the formation of ocular dominance columns, and hypoxia-induced seizures (Rakhade et al., 2008). The AMPAR-mediated neurotransmission of CA1 pyramidal neurons was augmented. The enhancement of glutamatergic transmission could contribute to the cell loss and circuit reorganization that follows SE. CA1 pyramidal neurons are more susceptible to SE-induced cell death than DGCs (Pollard et al., 1994;Fujikawa, 1995;Fujikawa, 1996;Todorovic et al., 2012;Sankar et al., 1998;Morrison et al., 1996;Grooms et al., 2000) (also(Sloviter et al., 1996)) and are commonly injured during prolonged limbic SE (Chen et al., 2007). This injury is believed to be excitotoxic (Fujikawa et al., 2000); however, there is limited direct evidence of enhanced glutamatergic transmission during SE.
The current study suggests that the enhanced synaptic AMPAR-mediated transmission is in part due to increased surface expression of the GluA1 subunit, which may be a substitute for the GluA2 subunit, rendering the receptor calcium-permeable (He et al., 2009). These calcium-permeable AMPARs may in turn contribute to neuronal injury (Liu & Zukin, 2007). Increased AMPAR-mediated neurotransmission of CA1 pyramidal neurons could sustain SE by increasing neuronal synchronization and could thus facilitate the spread of seizures. CA1 neurons provide output of the hippocampal tri-synaptic circuit, and they project to the subiculum and parasubiculum, septum, mediodorsal thalamus, and prefrontal cortex (Lothman et al., 1991). Metabolic and EEG seizure circuit mapping studies have demonstrated activation of these regions during prolonged SE (Lothman et al., 1991;VanLandingham & Lothman, 1991). Engagement of these circuits in seizure activity may thus trigger plasticity within these interconnected networks, further spreading and sustaining seizure activity. The amplitude of mEPSCs recorded from DGCs of SE animals was unchanged, but the kinetics of rise and decay of the currents were altered. Furthermore, the whole-cell AMPA-evoked currents of CA1 pyramidal neurons of SE animals also appeared to desensitize faster than the current evoked in neurons of control animals. Current study suggests increased surface expression of the GluA1 subunit and a previous study noted decreased surface expression of GluA2 subunit during SE. This likely changed the proportion of GluA1/GluA2 subunits in AMPARs on CA1 pyramidal neurons during SE. Changes in subunit composition alter desensitization rates. In addition to the subunit composition, other factors regulate AMPAR desensitization including flip/flop variation, and Q/R editing (Mosbacher et al., 1994;Wright & Vissel, 2012;Sommer et al., 1990). Currently it is not known whether the AMPARs expressed on CA1 pyramidal neurons and DGCs of SE animals differ from those of controls with respect to these properties; which may contribute to the observed differences in the properties of mEPSCs.
S831 and S845 are the major phosphorylation sites in the GluA1 subunit; S831 is phosphorylated by CaMKII and PKC whereas S845 is phosphorylated by PKA (Roche et al., 1996;Song & Huganir, 2002). The phosphorylation/dephosphorylation of these residues regulate the surface membrane trafficking of AMPARs during LTP and long-term depression (Song & Huganir, 2002). LTP is associated with increased phosphorylation of GluA1 subunits, although it may not be absolutely necessary for the induction of LTP (Hayashi et al., 2000)(but also (Lee et al., 2000)). Furthermore, phosphorylation of these residues of GluA1 subunit was also increased after one or more hours following hypoxia-induced seizures in neonatal animals (Rakhade et al., 2008). In contrast, we found that the dephosphorylation of the GluA1 subunit, particularly that of S845, was associated with increased surface expression of AMPARs during SE. The differences in the findings of the current study and that by Rakhade and colleagues could be due to the time of tissue harvesting, while the animals were seizing at the time tissue was harvested in our study, the animals were in recovery following hypoxia-induced seizures in the former study. In addition, age of the animals, the type of ictogenic stimuli used may also contribute to the observed differences. The activity of CaMKII, PKC, and PKA, which phosphorylate GluA1 subunits is also reduced or unaltered in animals in SE (Bronstein et al., 1988;Jope et al., 1992;Kochan et al., 2000;Perlin et al., 1992;Singleton et al., 2005;Terunuma et al., 2008) (but also see (Bracey et al., 2009). In contrast, the activity of protein phosphatases is increased and could underlie the observed dephosphorylation of GluA1 subunits (Kurz et al., 2001). The synaptic expression of the GluA1 subunit is unaltered in double phosphomutant mice (GluA1 S831A and S845A)(Lee et al., 2003). Thus, phosphorylation-independent mechanisms may regulate the observed plasticity of the GluA1 subunit.
MK-801 and diazepam treatment blocked the increase in surface membrane expression of GluA1 subunits, which suggests that increased neuronal activity triggered this plasticity. Although these agents target distinct neurotransmitter receptors, they may share common cellular mechanisms. Calcium signaling regulates AMPAR trafficking (Anggono & Huganir, 2012) and both diazepam and MK-801 could affect it. GABAR potentiating agents including diazepam can inhibit the activity of voltage-gated calcium channels (Earl & Tietz, 2011; Taft and DeLorenzo 1984). MK-801 treatment would also block calcium influx via NMDARs (Hardingham & Bading, 2003). Whether diazepam treatment affects NMDAR function is not known; however, it could hamper membrane depolarization and the removal of magnesium block that is necessary for NMDAR activation.
The increased glutamatergic transmission of CA1 pyramidal neurons could contribute to cell death and seizure spread. Since seizures continue unabated in diazepam- and MK-801-treated animals despite the effective abolishment of the increased AMPAR expression, the role of this plasticity in sustaining seizures appears to be uncertain. Multiple mechanisms could sustain seizure activity, and diazepam and MK-801 treatments reduce the spike amplitude (Walton & Treiman, 1991), which indicates that the seizure characteristics are altered. Agents that target GABARs and NMDARs rapidly lose their efficacy to terminate seizures after the onset of SE. In contrast, drugs such as GYKI52466 and perampanel that target AMPARs remain efficacious even when administered 30 min after the onset of SE (Hanada et al., 2014), which suggests that AMPAR function is necessary to sustain seizures. Additional studies using mice lacking GluA1 subunit expression in CA1 pyramidal neurons or GluA1 double phospho-deficient mice may provide additional information regarding the role of GluA1 subunit plasticity in sustaining seizures.
The neuroprotective effects of NMDAR blockade during SE are well documented (Fujikawa et al., 1994;Fujikawa, 1995;Prasad et al., 2002). These could be mediated in part by preventing the augmentation of AMPARs. MK-801 treatment blocked the increase in surface expression of GluA1 subunit and prevented the dephosphorylation of S845 residue. Although seizures continued unabated in these animals, an alteration of SE could not be ruled out. This NMDAR regulation of GluA1 subunit plasticity could be better studied in vitro. Indeed, the surface membrane insertion of GluA1 subunit-containing AMPARs during the chemically induced LTP of Schaffer collateral-CA1 synapses is regulated by NMDAR activation (Lu et al., 2001;Man et al., 2003). Blockade of NMDARs during SE synergistically with a benzodiazepine controlled the seizures. A prior study demonstrated the efficacy of a ketamine and diazepam combination therapy in terminating benzodiazepine-refractory SE (Martin & Kapur, 2008). Rapid termination of seizures is a key goal of treatment of SE. Benzodiazepines are the drug of choice for the initial treatment of SE based on several randomized controlled clinical trials (Glauser et al., 2016). These clinical trials revealed that benzodiazepines, such as lorazepam or midazolam, terminate generalized convulsive SE in 55–70% of patients, which suggests that 30–45% of patients are refractory to benzodiazepines. Patients who fail to respond to benzodiazepines are currently treated with second line agents, such as fosphenytoin, levitiracetam, phenobarbital or valproic acid. Patients who fail second-line agents are often treated with anesthetic agents, which are associated with complications (Hocker et al., 2014). This study suggests that ketamine could be used clinically to terminate benzodiazepine refractory SE. Recent clinical studies and individual case reports have also suggested that ketamine can terminate benzodiazepine-refractory SE (Gaspard et al., 2013;Rosati et al., 2012;Mewasingh et al., 2003;Fang & Wang, 2015;Synowiec et al., 2013;Basha et al., 2015). One of the mechanisms that contribute to the efficacy of ketamine may include the prevention of AMPAR plasticity.
Perampanel, which targets AMPARs, is a recently FDA-approved drug for the treatment of seizures. A retrospective study has found that it is effective in patients with refractory nonconvulsive or simple partial SE (Redecker et al., 2015). In contrast, in another study perampanel terminated refractory or super refractory SE only in a small fraction of patients (Rohracher et al., 2015). However, perampanel treatment was started after a significant delay following the failure of several anti-convulsant drugs to stop seizures in that study. A timely administration of perampanel may increase its efficacy.
Ongoing seizure activity appears to be necessary to maintain the increased surface expression of GluA1 subunit-containing AMPARs. The potentiation of EPSCs demonstrated in the current study dissipates over time after termination of SE. The finding of increased mEPSC amplitude shown here is different than findings of our prior study, in which the amplitude of mEPSCs recorded from CA1 neurons of SE animals was unchanged (Rajasekaran et al., 2013). However, slices are prepared after animals are placed under deep anesthesia, which terminates SE. In Rajasekaran et al study, animals were sacrificed 60 minutes after the first grade 5 seizure and the slices were maintained at 32°C for another 30–80 minutes. We reasoned that the GluA1-containing AMPARs may be moving out of synapses in the time interval between sacrifice and recordings. We then modified the protocol to sacrifice the animals at 40 minutes, maintained slices at room temperature for 20 min and recorded only one cell per animal, within 2 hrs of slice preparation (recording at 32°C). The shorter incubation at room temperature preserved the potentiation of synaptic currents. This suggests that the enhancement of EPSC is short lived in slice preparation. In conclusion this study demonstrated increased AMPAR-mediated neurotransmission of CA1 pyramidal neurons during SE. Drugs that target NMDARs and AMPARs may provide novel strategies to treat refractory SE.
Supplementary Material
Recording from a representative CA1 pyramidal neuron of naïve animal illustrating action-potential independent synaptic currents (mEPSCs).
Highlights.
Increased AMPAR-mediated synaptic neurotransmission of CA1 neurons of SE animals
Increased surface expression of GluA1 subunit on CA1 neurons of animals in SE
Dephosphorylation of S831 and S845 residues during SE
MK-801 and diazepam treatment during SE prevents increased GluA1 subunit surface expression
MK-801 prevents dephosphorylation of GluA1 S845
Acknowledgments
This study was supported by NIH grants RO1 NS 040337 and RO1 NS 044370, (JK), and an Epilepsy Foundation postdoctoral fellowship (KR). Some parts of the results are published in abstract form.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts E, Verhage M, Veenvliet JV, Dolan CV, van der Sluis S. A solution to dependency: using multilevel analysis to accommodate nested data. Nat Neurosci. 2014;17:491–496. doi: 10.1038/nn.3648. [DOI] [PubMed] [Google Scholar]
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001;345:631–637. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Current Opinion in Neurobiology. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha MM, Alqallaf A, Shah AK. Drug-induced EEG pattern predicts effectiveness of ketamine in treating refractory status epilepticus. Epilepsia. 2015;56:e44–e48. doi: 10.1111/epi.12947. [DOI] [PubMed] [Google Scholar]
- Betjemann JP, Lowenstein DH. Status epilepticus in adults. Lancet Neurol. 2015;14:615–624. doi: 10.1016/S1474-4422(15)00042-3. [DOI] [PubMed] [Google Scholar]
- Bracey JM, Kurz JE, Low B, Churn SB. Prolonged seizure activity leads to increased Protein Kinase A activation in the rat pilocarpine model of status epilepticus. Brain Research. 2009;1283:167–176. doi: 10.1016/j.brainres.2009.05.066. [DOI] [PubMed] [Google Scholar]
- Bronstein J, Farber D, Wasterlain C. Decreased calmodulin kinase activity after status epilepticus. Neurochem Res. 1988;13:83–86. doi: 10.1007/BF00971859. [DOI] [PubMed] [Google Scholar]
- Chamberlain JM, Okada P, Holsti M, Mahajan P, Brown KM, Vance C, et al. Lorazepam vs diazepam for pediatric status epilepticus: a randomized clinical trial. JAMA. 2014;311:1652–1660. doi: 10.1001/jama.2014.2625. [DOI] [PubMed] [Google Scholar]
- Chen JW, Naylor DE, Wasterlain CG. Advances in the pathophysiology of status epilepticus. Acta Neurol Scand. 2007;115:7–15. doi: 10.1111/j.1600-0404.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46:1029–1035. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Earl DE, Tietz EI. Inhibition of recombinant L-type voltage-gated calcium channels by positive allosteric modulators of GABAA receptors. J Pharmacol Exp Ther. 2011;337:301–311. doi: 10.1124/jpet.110.178244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Wang X. Ketamine for the treatment of refractory status epilepticus. Seizure. 2015;30:14–20. doi: 10.1016/j.seizure.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Foster AC, Wong EH. The novel anticonvulsant MK-801 binds to the activated state of the NMDA receptor in rat brain. Br J Pharmacol. 1987;91:403–409. doi: 10.1111/j.1476-5381.1987.tb10295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch B, Stott JJ, Joelle Donofrio J, Rogawski MA. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia. 2010;51:108–117. doi: 10.1111/j.1528-1167.2009.02205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Res. 1996;725:11–22. doi: 10.1016/0006-8993(96)00203-x. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Daniels AH, Kim JS. The competitive NMDA receptor antagonist CGP 40116 protects against status epilepticus-induced neuronal damage. Epilepsy Res. 1994;17:207–219. doi: 10.1016/0920-1211(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Seizure-induced neuronal necrosis: implications for programmed cell death mechanisms. Epilepsia. 2000;41(Suppl 6):S9–13. doi: 10.1111/j.1528-1157.2000.tb01549.x. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia. 1995;36:186–195. doi: 10.1111/j.1528-1157.1995.tb00979.x. [DOI] [PubMed] [Google Scholar]
- Gaspard N, Foreman B, Judd LM, Brenton JN, Nathan BR, McCoy BM, et al. Intravenous ketamine for the treatment of refractory status epilepticus: A retrospective multicenter study. Epilepsia. 2013;54:1498–1503. doi: 10.1111/epi.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence-based guideline: Treatment of convulsive status epilepticus in children and adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016;16:48–61. doi: 10.5698/1535-7597-16.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grooms SY, Opitz T, Bennett MVL, Zukin RS. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proceedings of the National Academy of Sciences. 2000;97:3631–3636. doi: 10.1073/pnas.050586497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. Analysis of glutamate receptor surface expression in acute hippocampal slices. Sci STKE. 2002;2002:l8. doi: 10.1126/stke.2002.137.pl8. [DOI] [PubMed] [Google Scholar]
- Hanada T, Ido K, Kosasa T. Effect of perampanel, a novel AMPA antagonist, on benzodiazepine-resistant status epilepticus in a lithium-pilocarpine rat model. Pharmacol Res Perspect. 2014a;2:e00063. doi: 10.1002/prp2.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Ido K, Kosasa T. Effect of perampanel, a novel AMPA antagonist, on benzodiazepine-resistant status epilepticus in a lithium-pilocarpine rat model. Pharmacology Research & Perspectives. 2014b;2:e00063–e00. doi: 10.1002/prp2.63. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends in Neurosciences. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Henley JM, Wilkinson KA. Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci. 2016;17:337–350. doi: 10.1038/nrn.2016.37. [DOI] [PubMed] [Google Scholar]
- Hocker S, Tatum WO, LaRoche S, Freeman WD. Refractory and super-refractory status epilepticus--an update. Curr Neurol Neurosci Rep. 2014;14:452. doi: 10.1007/s11910-014-0452-x. [DOI] [PubMed] [Google Scholar]
- Jope RS, Kolasa K, Song L, Ormandy GC. Seizures selectively impair agonist-stimulated phosphoinositide hydrolysis without affecting protein kinase C activity in rat brain. Neurotoxicology. 1992;13:389–400. [PubMed] [Google Scholar]
- Kapur J, Coulter DA. Experimental status epilepticus alters GABAA receptor function in CA1 pyramidal neurons. Ann Neurol. 1995;38:893–900. doi: 10.1002/ana.410380609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochan LD, Churn SB, Omojokun O, Rice A, DeLorenzo RJ. Status epilepticus results in an NMDA receptor-dependent inhibition of Ca2+/calmodulin-dependent kinase II activity in the rat. Neuroscience. 2000;95:735–743. doi: 10.1016/s0306-4522(99)00462-5. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Sheets D, Parsons JT, Rana A, DeLorenzo RJ, Churn SB. A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus. J Neurochem. 2001;78:304–315. doi: 10.1046/j.1471-4159.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends in Neurosciences. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Bekenstein JW, Perlin JB. Self-sustaining limbic status epilepticus induced by ‘continuous’ hippocampal stimulation: electrographic and behavioral characteristics. Epilepsy Res. 1989;3:107–119. doi: 10.1016/0920-1211(89)90038-7. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, III, Stringer JL. Functional anatomy of hippocampal seizures. Prog Neurobiol. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- Lu WY, Man HY, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29(1):243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, et al. Activation of PI3-Kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38(4):611–624. doi: 10.1016/s0896-6273(03)00228-9. Ref Type: Abstract. [DOI] [PubMed] [Google Scholar]
- Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 2008;49:248–255. doi: 10.1111/j.1528-1167.2007.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewasingh LD, Sekhara T, Aeby A, Christiaens FJ, Dan B. Oral ketamine in paediatric non-convulsive status epilepticus. Seizure. 2003;12:483–489. doi: 10.1016/s1059-1311(03)00028-1. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. The Journal of Neuroscience. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, Wasterlain CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiology of Disease. 2013;54:225–238. doi: 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlin JB, Churn SB, Lothman EW, DeLorenzo RJ. Loss of type II calcium/calmodulin-dependent kinase activity correlates with stages of development of electrographic seizures in status epilepticus in rat. Epilepsy Res. 1992;11:111–118. doi: 10.1016/0920-1211(92)90045-u. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Mathiesen C, Rønn LC, Møller, Nissinen J. Effect of novel AMPA antagonist, NS1209, on status epilepticus: An experimental study in rat. Epilepsy Research. 2007;74:45–54. doi: 10.1016/j.eplepsyres.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, et al. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Prasad A, Williamson JM, Bertram EH. Phenobarbital and MK-801, but not phenytoin, improve the long-term outcome of status epilepticus. Ann Neurol. 2002;51:175–181. doi: 10.1002/ana.10085. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rajasekaran K, Joshi S, Kozhemyakin M, Todorovic MS, Kowalski S, Balint C, et al. Receptor trafficking hypothesis revisited: Plasticity of AMPA receptors during established status epilepticus. Epilepsia. 2013;54:14–16. doi: 10.1111/epi.12266. [DOI] [PubMed] [Google Scholar]
- Rajasekaran K, Todorovic M, Kapur J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann Neurol. 2012;72:91–102. doi: 10.1002/ana.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Zhou C, Aujla PK, Fishman R, Sucher NJ, Jensen FE. Early Alterations of AMPA Receptors Mediate Synaptic Potentiation Induced by Neonatal Seizures. The Journal of Neuroscience. 2008;28:7979–7990. doi: 10.1523/JNEUROSCI.1734-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raol YH, Lapides DA, Keating JG, Brooks-Kayal AR, Cooper EC. A KCNQ channel opener for experimental neonatal seizures and status epilepticus. Ann Neurol. 2009;65:326–336. doi: 10.1002/ana.21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redecker J, Wittstock M, Benecke R, Rösche J. Efficacy of perampanel in refractory nonconvulsive status epilepticus and simple partial status epilepticus. Epilepsy & Behavior. 2015;45:176–179. doi: 10.1016/j.yebeh.2015.01.036. [DOI] [PubMed] [Google Scholar]
- Rice AC, DeLorenzo RJ. NMDA receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience. 1999;93:117–123. doi: 10.1016/s0306-4522(99)00132-3. [DOI] [PubMed] [Google Scholar]
- Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rohracher A, Höfler J, Kalss G, Leitinger M, Kuchukhidze G, Deak I, et al. Perampanel in patients with refractory and super-refractory status epilepticus in a neurological intensive care unit. Epilepsy & Behavior. 2015;49:354–358. doi: 10.1016/j.yebeh.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Rosati A, L’Erario M, Ilvento L, Cecchi C, Pisano T, Mirabile L, et al. Efficacy and safety of ketamine in refractory status epilepticus in children. Neurology. 2012;79:2355–2358. doi: 10.1212/WNL.0b013e318278b685. [DOI] [PubMed] [Google Scholar]
- Sankar R, Shin DH, Liu H, Mazarati A, Pereira de Vasconcelos A, Wasterlain CG. Patterns of Status Epilepticus-Induced Neuronal Injury during Development and Long-Term Consequences. The Journal of Neuroscience. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med. 2012;366:591–600. doi: 10.1056/NEJMoa1107494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MW, Holbert WH, II, Ryan ML, Lee AT, Kurz JE, Churn SB. Age dependence of pilocarpine-induced status epilepticus and inhibition of CaM kinase II activity in the rat. Developmental Brain Research. 2005;156:67–77. doi: 10.1016/j.devbrainres.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dean E, Sollas AL, Goodman JH. Apoptosis and necrosis induced in different hippocampal neuron populations by repetitive perforant path stimulation in the rat. J Comp Neurol. 1996;366:516–533. doi: 10.1002/(SICI)1096-9861(19960311)366:3<516::AID-CNE10>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Sommer B, Keinanen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, et al. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Sun J, Kapur J. M-type potassium channels modulate Schaffer collateral-CA1 glutamatergic synaptic transmission. The Journal of Physiology. 2012;590:3953–3964. doi: 10.1113/jphysiol.2012.235820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synowiec AS, Singh DS, Yenugadhati V, Valeriano JP, Schramke CJ, Kelly KM. Ketamine use in the treatment of refractory status epilepticus. Epilepsy Research. 2013;105:183–188. doi: 10.1016/j.eplepsyres.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Taft WC, DeLorenzo RJ. Micromolar-affinity benzodiazepine receptors regulate voltage-sensitive calcium channels in nerve terminal preparations. Proceedings of the National Academy of Sciences. 1984;81:3118–3122. doi: 10.1073/pnas.81.10.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma M, Xu J, Vithlani M, Sieghart W, Kittler J, Pangalos M, et al. Deficits in phosphorylation of GABAA receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J Neurosci. 2008;28:376–384. doi: 10.1523/JNEUROSCI.4346-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic MS, Cowan ML, Balint CA, Sun C, Kapur J. Characterization of status epilepticus induced by two organophosphates in rats. Epilepsy Research. 2012;101:268–276. doi: 10.1016/j.eplepsyres.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–798. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- Treiman DM. Treatment of Convulsive Status Epilepticus. In: Ramsay RE, editor. International Review of Neurobiology The Neurobiology of Epilepsy and Aging. Academic Press; 2007. pp. 273–285. [DOI] [PubMed] [Google Scholar]
- VanLandingham KE, Lothman EW. Self-sustaining limbic status epilepticus. I. Acute and chronic cerebral metabolic studies: limbic hypermetabolism and neocortical hypometabolism. Neurology. 1991;41:1942–1949. doi: 10.1212/wnl.41.12.1942. [DOI] [PubMed] [Google Scholar]
- Walton NY, Treiman DM. Motor and electroencephalographic response of refractory experimental status epilepticus in rats to treatment with MK-801, diazepam, or MK-801 plus diazepam. Brain Res. 1991;553:97–104. doi: 10.1016/0006-8993(91)90235-n. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A, Vissel B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci. 2012;5:34. doi: 10.3389/fnmol.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Recording from a representative CA1 pyramidal neuron of naïve animal illustrating action-potential independent synaptic currents (mEPSCs).