

Abstract
Background
Perturbation of commensal intestinal microbiota has been associated with several autoimmune diseases. Mice deficient in interleukin-1 receptor antagonist (Il1rn −/− mice) spontaneously develop autoimmune arthritis and are susceptible to other autoimmune diseases such as psoriasis, diabetes, and encephalomyelitis; however, the mechanisms of increased susceptibility to these autoimmune phenotypes are poorly understood. We investigated the role of interleukin-1 receptor antagonist (IL-1Ra) in regulation of commensal intestinal microbiota, and assessed the involvement of microbiota subsets and innate and adaptive mucosal immune responses that underlie the development of spontaneous arthritis in Il1rn −/− mice.
Results
Using high-throughput 16S rRNA gene sequencing, we show that IL-1Ra critically maintains the diversity and regulates the composition of intestinal microbiota in mice. IL-1Ra deficiency reduced the intestinal microbial diversity and richness, and caused specific taxonomic alterations characterized by overrepresented Helicobacter and underrepresented Ruminococcus and Prevotella. Notably, the aberrant intestinal microbiota in IL1rn −/− mice specifically potentiated IL-17 production by intestinal lamina propria (LP) lymphocytes and skewed the LP T cell balance in favor of T helper 17 (Th17) cells, an effect transferable to WT mice by fecal microbiota. Importantly, LP Th17 cell expansion and the development of spontaneous autoimmune arthritis in IL1rn −/− mice were attenuated under germ-free condition. Selective antibiotic treatment revealed that tobramycin-induced alterations of commensal intestinal microbiota, i.e., reduced Helicobacter, Flexispira, Clostridium, and Dehalobacterium, suppressed arthritis in IL1rn −/− mice. The arthritis phenotype in IL1rn −/− mice was previously shown to depend on Toll-like receptor 4 (TLR4). Using the ablation of both IL-1Ra and TLR4, we here show that the aberrations in the IL1rn −/− microbiota are partly TLR4-dependent. We further identify a role for TLR4 activation in the intestinal lamina propria production of IL-17 and cytokines involved in Th17 differentiation preceding the onset of arthritis.
Conclusions
These findings identify a critical role for IL1Ra in maintaining the natural diversity and composition of intestinal microbiota, and suggest a role for TLR4 in mucosal Th17 cell induction associated with the development of autoimmune disease in mice.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0278-2) contains supplementary material, which is available to authorized users.
Keywords: Microbiota, Autoimmune arthritis, T helper 17 cells, Toll-like receptors, IL-1 receptor antagonist
Background
Interleukin-1 (IL-1) plays a central role in inflammation and immunity [1]. Activation of IL-1 receptor is physiologically controlled by its structural homologue and natural inhibitor, the IL-1 receptor antagonist (IL-1Ra), encoded by the IL1rn gene [2]. IL1rn knockout (IL1rn −/−) mice are susceptible to a variety of autoimmune diseases including arthritis, psoriasis, diabetes, and encephalomyelitis [3–7]. This indicates a critical role for IL-1Ra in protection against autoimmunity; however, the mechanisms are poorly understood.
We questioned the role of IL-1Ra in regulation of the intestinal microbiota and the involvement of mucosal immune response as an underlying mechanism for the spontaneous autoimmune arthritis in IL1rn −/− mice, which is dependent on T cells and IL-17 [4, 8]. Several studies have associated commensal microbiota with autoimmune disease in mouse models of rheumatoid arthritis (RA), diabetes, and multiple sclerosis [9–13]. Importantly, the diversity and the composition of commensal intestinal microbiota are altered in patients with psoriatic and RA compared with healthy individuals [14–18]. One of the most prominent effects of microbiota is to define the balance between the proinflammatory CD4+ T helper 1 (Th1) and Th17 cells and protective regulatory T (Treg) cells, both at mucosal surfaces and systemically [19–21]. In this context, specific subsets of intestinal microbiota, such as the vancomycin-sensitive segmented filamentous bacteria (SFB), robustly induce differentiation of Th17 cells in small intestine LP (SI-LP) [22, 23]. Th17 cells are considered to play a pathogenic role in a subset of patients with RA by producing proinflammatory mediators, such as IL-17, and inducing osteoclastogenesis [24–28]. Interestingly, SFB colonization has been shown to exacerbate arthritis in K/BxN mice, an autoimmune model of arthritis arising from T cell auto-reactivity to the glycolytic enzyme glucose-6-phosphate isomerase [13, 29]. However, given that SFB were not found in human adults [30, 31], it is important to investigate the involvement of other indigenous microbiota in arthritis.
We previously described that arthritis in IL1rn −/− mice is diminished under germ-free (GF) condition [12]. We also showed that IL1rn −/− arthritis is dependent on the activation of Toll-like receptor 4 (TLR4), which affected systemic Th17 cell differentiation [12]. Here, we characterized the intestinal microbiota present in autoimmune-prone IL1rn −/− mice to clarify the nature of the microbiota that trigger arthritis and the underlying mucosal immune pathways. We also examined the role of TLR4 in the intestinal mucosal immune responses associated with arthritis.
Using high-throughput 16S ribosomal RNA (rRNA) gene sequencing of fecal microbiota, we demonstrate a critical role for IL-1Ra in maintaining the natural diversity and composition of commensal intestinal microbiota. We show that the aberrant IL1rn −/− microbiota increases intestinal Th17 cell differentiation, a phenotype that is transferable to wild-type (WT) mice by the microbiota. We also provide evidence that tobramycin-sensitive indigenous commensal intestinal bacteria contribute to arthritis in IL1rn −/− mice and identify a significant role for TLR4 in mucosal induction of IL-1β and IL-17 prior to the onset of arthritis.
Results
IL-1Ra maintains the biodiversity and richness of commensal intestinal microbiota
To identify intestinal microbiota associated with arthritis, we sequenced fecal bacterial 16S rRNA genes of IL1rn −/− and age- and gender-matched WT control mice. Fecal microbiota were analyzed as an unselected representation of the overall microbial communities in the intestines. Considering differential roles of TLR2 and TLR4 in IL1rn −/− arthritis [12], we sequenced samples of IL1rn −/− Tlr2 −/− and IL1rn −/− Tlr4 −/− mice in parallel. The average sequencing depth and total numbers of reads and operational taxonomic units (OTU) per experimental group as well as the hierarchical weighed UniFrac cluster analysis at the genus level are shown in Additional file 1: Table S1 and Additional file 1: Figure S1.
Principal coordinates analysis (PCoA) based on an unweighted UniFrac analysis of intestinal microbiota showed that IL1rn −/− microbiota is profoundly different from the WT microbiota (Fig. 1a). WT and Il1rn −/− mice formed clear, separate clusters regardless of the cage or litter of origin (Fig. 1a). Strikingly, microbial composition of IL1rn −/− and IL1rn −/− Tlr2 −/− mice were indistinguishable, while IL1rn −/− Tlr4 −/− mice formed another distinct cluster (Fig. 1a). To assess the effects of familial transmission and lineage origin versus the effect of the genotype (WT or IL1rn −/−), we compared the UniFrac distances within a litter with UniFrac distances across litters of the same genotype as well as the opposite genotype, similar to the study by Ubeda et al. This analysis showed that the effect of the lineage origin and litter was limited in our experimental setting, because, as long as the genotype remained the same, the UniFrac distances across different litters were very similar to the UniFrac distances within the litters (Additional file 1: Figure S2A). This was true for both WT and Il1rn −/− groups. Importantly, the UniFrac distances were significantly higher when mice from different genotypes were compared, indicating a higher level of dissimilarity (Additional file 1: Figure S2B). Therefore, the effect of the genotype (Il1rn-deficiency) on the overall microbiota composition was significantly higher than any litter and cage effect. In addition, IL1rn −/− and IL1rn −/− Tlr2 −/− mice showed significantly reduced number of OTUs and loss of microbial diversity based on the Shannon index, the rarefaction curves of phylogenetic distance (PD) whole tree, and the diversity index bootstrapped for the number of retrieved sequences (Fig. 1b–e). IL-1Ra deficiency also resulted in loss of species richness estimated by Chao index (Fig. 1f). These effects were fully or partially restored in IL1rn −/− Tlr4 −/− mice (Fig. 1b–f). Altogether, these data strongly suggest that IL-1Ra plays a critical role in maintaining the intestinal microbial diversity, and that the loss of diversity in IL1rn −/− mice partially depends on TLR4.
Fig. 1.
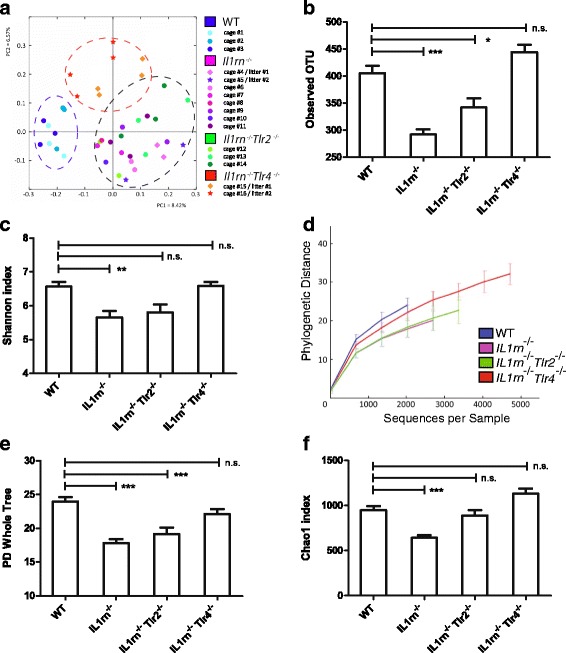
IL-1Ra deficiency skews intestinal microbial composition and reduces its diversity in a TLR4-dependent manner. a Principal coordinates analysis (PCoA) based on an unweighted UniFrac analysis of the intestinal microbial composition where samples of mice from different cages and litters are highlighted with different colors. The position and distance of data points indicates the degree of similarity in terms of both presence and relative abundance of bacterial taxonomies. b Number of observed operational taxonomic units (OTUs). c Shannon index of microbial diversity. d Alpha diversity rarefaction curves of phylogenetic distance (PD) whole tree. e PD whole tree, bootstrapped for 2000 reads per sample, averaged of four trials. f Chao index are shown. Data (mean + SEM) represent 16S rRNA gene 454-pyrosequencing analysis of intestinal microbiota of WT (n = 9), IL1rn −/− (n = 15), IL1rn −/− Tlr2 −/− (n = 8), and IL1rn −/− Tlr4 −/− (n = 8) mice. n.s. not significant,*P ≤ 0.05 and ***P ≤ 0.001, by Mann-Whitney U test. See also Additional file 1: Figure S1 and Additional file 1: Table S1
Specific taxonomic alterations characterize the dysregulated microbiota of autoimmune-prone IL1rn−/− mice
The phylogenetic tree in Fig. 2 summarizes the observed alterations in relative abundances of microbial taxa. Compared with WT microbiota, we found a highly significant overrepresentation of the genus Helicobacter (P = 0.004, Bonferroni corrected), and a significant underrepresentation of the genus Prevotella (P = 0.008, Bonferroni corrected) (Fig. 2 and Additional file 1: Table S2). In addition, IL1rn −/− intestinal microbial composition was characterized by expansion of Butyricimonas, Rikenella, and Streptococcus by 10, 3.7, and 2.4 folds (P = 0.0048, P = 0.0022, and P = 0.0032, respectively, Bonferroni uncorrected), along with a decrease in Parasutterella, Xylanibacter, Ruminococcus, and Barnesiella by 10, 6.9, 2.7, and 1.4 folds (P = 0.040, P = 0.0004, P = 0.0099, and P = 0.0005, respectively, Bonferroni uncorrected), respectively (Fig. 2 and Additional file 1: Table S2). Notably, we were unable to identify any OTUs in our dataset that could be classified as SFB (family Clostridiaceae, genus Candidatus Arthromitus). Moreover, none of the 27 present OTUs assigned to the family Clostridiaceae aligned with the known SFB 16S gene sequences in The Ribosomal Database Project [30]. However, SFB were detectable by qPCR in fecal samples of all WT mice and most of the Il1rn −/− mice (Additional file 1: Table S3). Although WT mice tended to have slightly more SFB, the level of SFB colonization was not significantly different between the groups (Additional file 1: Table S3).
Fig. 2.
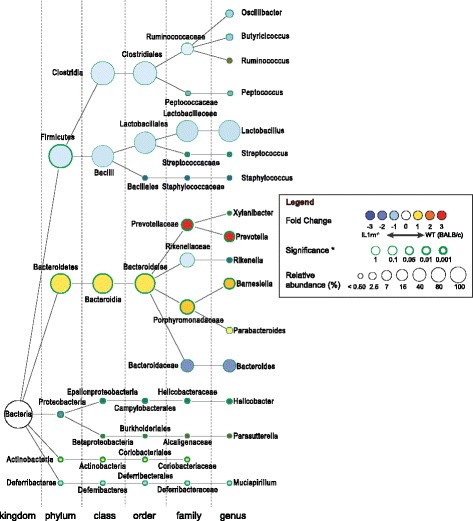
IL-1 receptor antagonist controls relative abundance of specific intestinal microbial taxa. Phylogenetic tree created by Cytoscape software showing specific changes in intestinal microbial community at different taxonomic levels induced by IL-1Ra deficiency. Nodes represent taxa, and the size of each node represents its relative abundance. The color red indicates a decrease and blue represents an increase of relative abundance in IL1rn −/− compared with WT mice. The thickness of the green border indicates the degree of the statistical significance by Mann-Whitney U test. See also Additional file 1: Table S2
Altogether, these data suggest that multiple yet specific microbial taxa are regulated by the physiologic expression of IL-1Ra. Therefore, a complex set of aberrant microbiota may affect the (mucosal) immune response and contribute to the autoimmune disease in IL1rn −/− mice.
IL1rn−/− intestinal microbiota potentiate IL-17 production by intestinal lamina propria lymphocytes
To assess the effect of IL-1Ra deficiency on the mucosal T cell response, we cultured enzymatically isolated lamina propria lymphocytes (LPL) ex vivo in the presence of PMA and ionomycin. The production of the Th1 signature cytokine IFNγ was low and not altered by the IL-1Ra deficiency (Fig. 3a, gating strategy shown in Additional file 1: Figure S3); however, we observed a marked increase in the production of IL-17 by IL1rn −/− LPLs compared with WT LPLs (Fig. 3b). Flow cytometry analysis of lamina propria cells of WT and IL1rn −/− mice verified a significant, clear increase of IL-17-producing TCRβ+ CD4+ cells in IL1rn −/− mice, while TCRβ− cells in LP produced similar amounts of IL-17 in WT and IL1rn −/− mice (Additional file 1: Figure S4). This suggests that Th17 cells, not γδ T cells, are the source of increased IL-17 production in LP of IL1rn −/− mice. Production of IL-4, IL-6, and TNFα was not affected (Fig. 3c and data not shown). Interestingly, the production of IL-17 but not IFNγ by lymphocytes in joint-draining lymph nodes (dLN) was significantly increased in IL1rn −/− mice compared with WT mice (Fig. 3d, e). This was paralleled by a concomitant decrease in the production of the Th2-related cytokine IL-4 in IL1rn −/− mice (Fig. 3f).
Fig. 3.
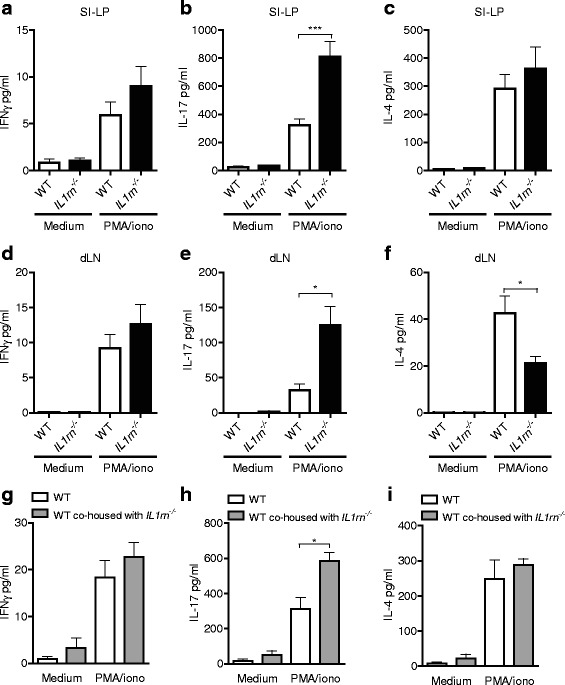
Il1rn −/− intestinal microbiota potentiate IL-17 production in intestinal lamina propria and joint-draining lymph nodes. a–i Production of prototypic Th1, Th17, and Th2 cell cytokines (IFNγ, IL-17A, and IL-4, respectively) by SI-LP (a–c and g–i) and draining lymph node (dLN) lymphocytes (d–f). Cells were isolated from WT and IL1rn −/− mice (a–f) or WT mice transplanted with IL1rn −/− feces and co-housed with IL1rn −/− mice for 10 days (g–i). Cells were stimulated ex vivo with PMA and ionomycin in duplicates for 5 h, and cytokines were measured by Luminex assay. Data represent mean + SEM of a representative experiment with n = 5 (a–c) and n = 3 (d–i) mice per group, each stimulated in duplicate. n.s. not significant, *P ≤ 0.05 and ***P ≤ 0.001, by Mann-Whitney U test. See also Additional file 1: Figure S2
To identify a potential causative relationship between the aberrant microbiota and enhanced mucosal IL-17 production, we transferred IL1rn −/− microbiota to WT mice by oral gavage followed by immediate co-housing of the two mouse strains for up to 6 weeks. Transfer of IL1rn −/− microbiota clearly potentiated IL-17 production by SI-LP T cells in WT recipients as early as 10 days post fecal transfer and co-housing, without affecting IFNγ and IL-4 (Fig. 3g–i). This indicated that IL1rn −/− intestinal microbiota causes a shift in the LP T cell balance in favor of Th17 cells. However, this was not sufficient for the development of arthritis in WT animals during the 6-week follow-up period. This suggests that additional (genetic) susceptibility of the host, as in IL1rn −/− mice, is required for the development of arthritis. Furthermore, co-housing with WT mice did not affect the development of arthritis in IL1rn −/− mice (not shown).
Potentiated Th17 response and spontaneous arthritis in IL1rn−/− mice highly depend on the presence of commensal microbiota
To determine whether the increase in intestinal Th17 cells and spontaneous arthritis in IL1rn −/− mice depends on commensal microbiota, we established GF IL1rn −/− mice. Flow cytometry analysis of LPLs showed that germ-free condition had no significant effect on the percentage of Th1 cells while reducing the numbers of Th1 cells in SI-LP (Fig. 4a, b). In contrast, both the percentage and the number of SI-LP Th17 cells were substantially reduced in GF compared with conventional (CV) mice (Fig. 4c, d). This strongly suggests that the skewed intestinal T cell balance in IL1rn −/− mice is largely microbiota dependent.
Fig. 4.
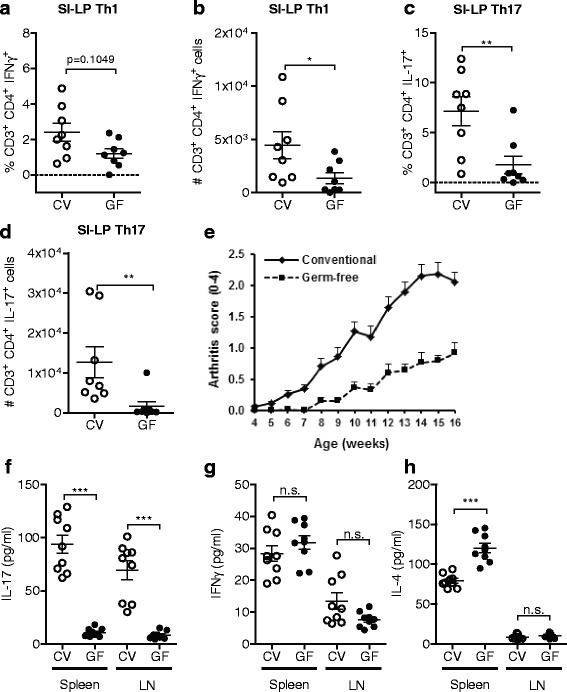
Commensal microbiota drive potentiated Th17 response and spontaneous arthritis in IL1rn −/− mice. a–d Frequency and numbers of IFNγ-producing (a, b) and IL-17-producing (c, d) CD3+CD4+ SI-LP cells. Data are pooled from three independent experiments. e Arthritis severity scores of conventional (CV, n = 12) and germ-free (GF, n = 11) IL1rn −/− mice of a representative experiment. Scale 0–2 for each hind paw. Mean + SEM is shown. f–h Production of IFNγ, IL-17, and IL-4 upon ex vivo stimulation of the spleen and lymph node cells from CV and GF mice with PMA and ionomycin for 6 h, as measured by Luminex assay. n = 3 mice per group of each stimulated in triplicate. n.s. not significant, **P ≤ 0.01 and ***P ≤ 0.001, by Mann-Whitney U test. See Additional file 1: Figure S3 for gating strategy
In agreement with IL-17-dependence of IL1rn −/− arthritis [4, 8] and in line with our previous observations [12], GF IL1rn −/− mice showed a clear sustained protection from arthritis with on average 3 weeks delay in disease onset (Fig. 4e). In addition, transfer of conventional IL1rn −/− microbiota to GF IL1rn −/− mice re-induced arthritis and resulted in a severe disease comparable to that in conventional IL1rn −/− mice (Additional file 1: Figure S5). Therefore, IL1rn −/− commensal microbiota, although not sufficient to induce arthritis in a WT host, are critical for the full development of arthritis in IL1rn −/− mice. Consistently, we also observed a robust reduction of IL-17, but not IFNγ, production in the spleen and most notably in the joint-adjacent lymph nodes of GF mice (Fig. 4f, g). These effects were accompanied by a significant reciprocal increase in Th2-related cytokines IL-4 and IL-10 as well as IL-2 in the spleens of GF mice (Fig. 4h and Additional file 1: Figure S6). These data support modulation of extra-intestinal immune response by intestinal microbiota during arthritis.
Tobramycin-induced alteration of intestinal microbiota suppresses arthritis in IL1rn−/− mice
The lack of microbiota in GF mice is not limited to the intestines. To determine whether intestinal microbiota serve as a relevant trigger for arthritis, we first depleted intestinal microbiota in conventionally housed mice using a cocktail of metronidazole, neomycin, and ampicillin. Treatment of 5-week-old IL1rn −/− mice for only 1 week suppressed arthritis over a sustained period, i.e., 6 weeks after ceasing antibiotics (Fig. 5a). This indicated that abrogation of arthritis in GF mice (Fig. 4c) is not due to an immature immune system and, more importantly, can be reproduced by the sole eradication of intestinal microbiota. Interestingly, colonization of the antibiotic-treated mice with SFB as model organisms inducing SI-LP Th17 cells was sufficient to fully restore arthritis (Fig. 5a).
Fig. 5.
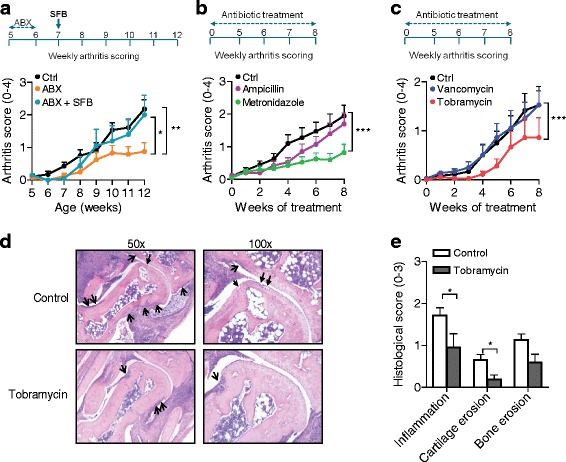
Commensal intestinal anaerobic tobramycin-sensitive microbiota promote arthritis in IL1rn −/− mice. a Arthritis severity scores (0–2 per paw) of IL1rn −/− mice treated with a cocktail of metronidazole, neomycin, and ampicillin (ABX) for 7 days (week 5 to 6), followed by re-colonization with SFB (ABX + SFB) 1 week after ending ABX treatment (at week 7). b, c Arthritis severity scores of IL1rn −/− mice either untreated (Ctrl) or treated with the mentioned antibiotic for 8 weeks. Data show mean + SEM of n = 5–7 (a) and n = 10 (b, c) mice per group. *P ≤ 0.05, ***P ≤ 0.001, by repeated measures ANOVA with Bonferroni correction for multiple testing. d Representative images of ankle joint sections of control and tobramycin-treated mice stained with hematoxylin and eosin illustrating decreased synovial inflammation, cartilage destruction (closed arrows), and bone erosion (open arrows). Original magnification ×50 (left panels) and ×100 (right panels). e Histopathologic scores (mean + SEM) of synovial inflammation, cartilage destruction, and bone erosion in control and tobramycin-treated IL1rn −/− mice. n = 9 per group. *P ≤ 0.05, by Mann-Whitney U test
To determine which subset of IL1rn −/− microbiota triggers arthritis, we first compared the effects of treatment with ampicillin, broadly targeting aerobic bacteria, and metronidazole, broadly targeting anaerobic bacteria. To our surprise, only metronidazole showed efficacy in reducing arthritis severity (Fig. 5b). This suggests involvement of anaerobic bacteria in the progression of arthritis. We next compared the effects of more selective antibiotics tobramycin and vancomycin, the latter of which has been reported to eradicate SFB and inhibit SFB-induced lamina propria Th17 cells and arthritis [13, 22, 32]. These experiments revealed that although SFB were able to exacerbate arthritis in IL1rn −/− mice (Fig. 5a), only tobramycin but not vancomycin significantly diminished arthritis (Fig. 5c). To understand the changes in the microbiota induced by tobramycin treatment, we compared 16S rRNA gene sequences of fecal microbiota at the end-point of tobramycin treatment with the baseline microbiota. Among taxa with >0.1% relative abundance, tobramycin treatment resulted in a near-complete elimination of the genera Helicobacter and Flexispira (both belonging to the family Helicobacteraceae). In addition, a strong and highly significant reduction in the genera Clostridium and Dehalobacterium was observed (Additional file 1: Figure S7 and Table S4). Other changes in the microbiota did not reach the statistical significance after Bonferroni correction for multiple testing. Therefore, tobramycin-induced alterations in these indigenous IL1rn −/− microbiota taxa resulted in suppression of arthritis. This was confirmed by histological examination of arthritic joints which showed a significant reduction of synovial inflammation as well as cartilage destruction and a non-significant reduction in bone erosion upon treatment with tobramycin (Fig. 5d, e).
Aberrations of the intestinal microbiota and LP IL-17 production in IL1rn−/− mice partly depend on TLR4
TLR4 plays a major role in recognition of Gram-negative bacteria [33]. We previously showed that IL1rn −/− Tlr4 −/− mice have a marked and sustained reduction of arthritis [12]. Therefore, we assessed whether TLR4 plays a role in alterations of the intestinal microbiota and the induction of LP Th17 cells. A detailed analysis of the intestinal microbiota showed that in addition to the TLR4-dependent loss of microbial diversity in IL-1Ra-deficient mice (Fig. 1b–f), alterations in Ruminococcus, Streptococcus, and Xylanibacter were partially dependent on TLR4 and were restored in IL1rn −/− Tlr4 −/− mice (Fig. 6a). Abundance of Prevotella was also restored to a statistically significant, yet minor extent (Fig. 6b). In total, 11 out of 44 taxa significantly altered in IL1rn −/− mice were normalized toward the WT levels in IL1rn −/− Tlr4 −/− mice (Additional file 1: Table S2).
Fig. 6.
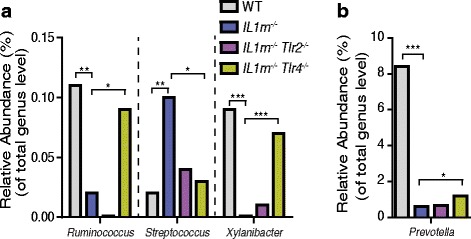
Alteration of specific intestinal microbiota in IL1rn −/− mice is partly TLR4 dependent. a, b Relative abundance of Ruminococcus, Streptococcus, Xylanibacter, and Prevotella in wild-type (WT) (n = 9), IL1rn −/− (n = 15), IL1rn −/− Tlr2 −/− (n = 8), and IL1rn −/− Tlr4 −/− (n = 8) mice. Data represent relative abundances of these genera obtained by 16S rRNA gene sequencing of the fecal microbiota
We next examined the role of TLR4 in the mucosal T cell response in IL1rn −/− mice. Th17 cells require transforming growth factor-β and IL-6, plus IL-1β in mouse, for initial differentiation, and IL-23 for their functional maturation and pathogenic function [24, 34]. To determine the role of TLR4 in response to IL1rn −/− intestinal microbial antigens, we cultured SI-LP mononuclear cells from IL1rn −/− and IL1rn −/− Tlr4 −/− mice ex vivo with autoclaved IL1rn −/− intestinal microbiota. SI-LP mononuclear cells from IL1rn −/− Tlr4 −/− mice produced significantly less IL-1β (Fig. 7a, P = 0.0042). Furthermore, the induction of IL-23 and IL-6 by IL1rn −/− fecal microbiota was partly TLR4-dependent (Fig. 7b, c, P = 0.0014 and P = 0.009, respectively). Reduced cytokine production in IL1rn −/− Tlr4 −/− mice was not due to an altered composition of mononuclear cells in the SI-LP, because the percentage and abundance of CD11c+ MHCII+ DCs as well as distinct subsets of CD103+ CD11b+, CD103+ CD11b−, and CD11b+ CD103− phagocytes were similar between IL1rn −/− and IL1rn −/− Tlr4 −/− mice (data not shown). Importantly, stimulation of IL1rn −/− LP mononuclear cells with IL1rn −/− and IL1rn −/− Tlr4 −/− fecal microbial antigens induced similar concentrations of IL-1β, IL-23, and IL-6 (Additional file 1: Figure S8). This suggests that the altered composition of microbiota in IL1rn −/− Tlr4 −/− mice as such is not responsible for the lower production of these cytokines. These observations imply a significant role for TLR4 in intestinal production of the cytokines involved in LP Th17 differentiation in IL1rn −/− mice.
Fig. 7.
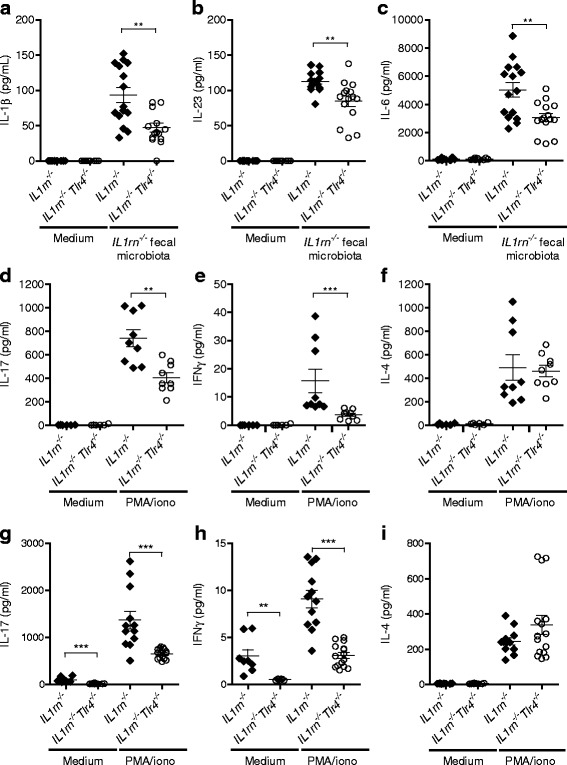
A significant role for TLR4 in intestinal production of cytokines involved in LP Th17 differentiation. a–c Production of IL-1β, IL-23, and IL-6 by SI-LP mononuclear cells of IL1rn −/− and IL1rn −/− Tlr4 −/− mice cultured in the presence of autoclaved IL1rn −/− complete fecal microbial antigens for 24 h. d–f Cytokine production by SI-LP lymphocytes of separately housed IL1rn −/− and IL1rn −/− Tlr4 −/− mice ex vivo stimulated with PMA and ionomycin for 5 h. g–i Cytokine production by SI-LP lymphocytes of IL1rn −/− and IL1rn −/− Tlr4 −/− mice co-housed for 10 days. Cells were stimulated as in d–f. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, by Mann-Whitney U test
When cultured ex vivo with PMA and ionomycin, SI-LP cells from IL1rn −/− Tlr4 −/− mice produced significantly less IL-17 compared with cells from IL1rn −/− Tlr4 +/+ mice before the onset of arthritis (P = 0.0028; Fig. 7d). The amount of IFNγ produced in this culture was about 50 folds lower than IL-17 and was significantly reduced in IL1rn −/− Tlr4 −/− mice as well (Fig. 7e). However, IL-4 levels remained unaffected (Fig. 7f). LP T cells from IL1rn −/− Tlr4 −/− mice still produced significantly less IL-17 and IFNγ when these mice were co-housed with IL1rn −/− (Tlr4 +/+) mice to transfer the microbiota (Fig. 7g–i). Therefore, reduced LP IL-17 production in IL1rn −/− Tlr4 −/− mice is a result of the difference in host TLR4 expression rather than altered microbiota in IL1rn −/− Tlr4 −/− versus IL1rn −/− mice. Stimulation of LP mononuclear cells of these co-housed mice with fecal microbial antigens confirmed that cells from IL1rn −/− Tlr4 −/− mice produce lower amounts of IL-1β, IL-23, and IL-6 regardless of stimulation with IL1rn −/− or IL1rn −/− Tlr4 −/− microbiota (Additional file 1: Figure S9). These data suggest that TLR4 activation contributes to intestinal LP production of IFNγ and most notably IL-17, and these effects precede the onset of arthritis.
Importantly, production of IL-17 but not IFNγ in lymph nodes draining the inflamed joints was diminished in IL1rn −/− Tlr4 −/− mice compared with IL1rn −/− mice (Additional file 1: Figure S10). This is in agreement with our previous study showing that TLR4 induces systemic and local IL-17 production and promotes arthritis in IL1rn −/− mice [12]. Together, these observations suggest an essential role for TLR4 in the induction of intestinal LP IL-17 production associated with extra-intestinal IL-17 levels and the development of arthritis in IL1rn −/− mice.
Discussion
The intestinal microbiome has emerged as a key determinant of health and disease. Although advanced sequencing techniques have enabled microbiome profiling in rheumatic patients, study of the underlying mucosal responses and the functional impact on arthritis is limited in human subjects due to the requirement of invasive techniques. The animal studies presented here demonstrate aberrations in intestinal microbiota in mice developing spontaneous autoimmune arthritis, introduce commensal tobramycin-sensitive microbiota as potential triggers for arthritis, and suggest a role for TLR4 activation in mucosal induction of inflammatory pathways including Th17 induction associated with arthritis.
Our study identifies loss of microbial diversity and specific taxonomic alterations in the microbiota of autoimmune-prone IL1rn −/− mice. Importantly, loss of intestinal microbial diversity and richness also coincides with human autoimmune diseases such as diabetes and rheumatoid and psoriatic arthritis [14, 16, 35]. Among the microbiota increased in IL1rn −/− mice, Streptococcus species are known inducers of chronic TLR-mediated arthritis in animal models when injected intra-articularly [36, 37]. Furthermore, a commensal Helicobacter (H. hepaticus) has been shown to induce IL-23 and mediate T cell-dependent gut inflammation in immunocompromised mice [38]. The decreased Barnesiella in IL1rn −/− mice is consistent with a previous study associating the abundance of Barnesiella with resistance to arthritis in HLA-DRB1*0402 mice [39]. A specific species of Prevotella, P. copri, is overrepresented in patients with new-onset RA [14] and was recently shown to increase colonic Th17 cells and promote arthritis in SKG mice after co-exposure to the fungal component zymosan [18]. On the other hand, P. histicola has been reported to suppress collagen-induced arthritis in transgenic mice expressing RA-susceptibility gene HLA-DQ8 [40]. Therefore, the immunomodulatory effects of the gut microbiota, including Prevotella, are species- and sometimes even strain-dependent. Due to inherently limited resolution of 16S rRNA gene sequencing, our data on the abundance of Prevotella is limited to the genus level and the exact Prevotella species altered in IL1rn −/− mice remain unclear. Overall, it is tempting to speculate that complex alterations in several taxa determine the net mucosal response to affect arthritis. It should also be noted that fecal bacterial community structures do not fully mirror the site-specific luminal or mucosa-associated microbiota profiles and were used in this study as a proxy of the gut microbiota of the Il1rn −/− mice.
Our previous studies showed that IL1rn −/− Tlr2 −/− mice develop a more severe arthritis compared with IL1rn −/− mice [12]. Given that additional TLR2 deficiency did not affect the microbiota of IL1rn −/− mice to a major extent (Fig. 1), we speculate that severe arthritis in IL1rn −/− Tlr2 −/− mice is due to the altered host immune response, specifically reduced function of Treg cells [12], rather than alteration in the microbiome. However, the data regarding lack of a major influence of TLR2 deficiency on Il1rn −/− microbiota should be interpreted with caution due to the absence of littermate Il1rn −/− Tlr2 +/+ mice in our studies.
IL1rn −/− mice had specific expansion of intestinal Th17 cells. The pathogenic relevance of IL-17 in the development of arthritis in IL1rn −/− mice has been demonstrated before, since both IL-17 gene deficiency and treatment with neutralizing anti-IL-17 antibodies inhibit arthritis [4, 8]. A previous study showed that γδ T cells rather than Th17 cells represent most IL-17-producing T cells in the inflamed joints of IL1rn −/− mice [41]. While γδ17 and Th17 cells may have complementary pathogenic roles in the development of IL1rn −/− arthritis, our data suggest that IL-17-producing cells located in lamina propria and induced by IL1rn −/− intestinal microbiota are TCRβ-expressing CD4+ Th17 cells (Fig. 4 and Additional file 1: Figure S4).
The expansion of LP Th17 cells in IL1rn −/− mice was caused by the dysregulated microbiota as confirmed by fecal transfer experiments (Fig. 3g–i). A critical pathogenic link to the spontaneous arthritis was revealed by our germ-free and antibiotic treatment studies (Figs. 4 and 5). Other previous studies which demonstrated the involvement of the gut microbiota in exacerbation of autoimmune arthritis found SFB as the responsible microorganisms. One study showed a role for vancomycin-sensitive microbiota including SFB in the induction of IL-17- and autoantibody-driven arthritis in K/BxN mice [13], and another showed that SFB can lower the activation threshold of self-reactive T cells and promote the differentiation of arthritogenic Th1 cells in a T cell transfer model of arthritis [42]. Our data are the first to demonstrate that although SFB colonization exacerbates arthritis, among the dysregulated indigenous microbiota present in the IL1rn −/− mice, those sensitive to tobramycin, i.e., Helicobacter, Flexispira, Clostridium and Dehalobacterium, are potential candidates to promote arthritis in a genetically susceptible host. This is relevant for human disease, given that SFB were not found in genome-wide sequences of 263 gut metagenomes of human adults [30, 31].
Our data also provide the first evidence for the involvement of TLR4 in defining the intestinal mucosal T cell phenotype. TLR4 activation of LP mononuclear cells by IL1rn −/− microbiota-induced IL-1β, IL-23 and IL-6 (Fig. 7a–c). Microbiota-induced IL-1β is critical for the development of steady-state Th17 cells in the gut [43]. IL-1 also synergizes with IL-6 and IL-23 to regulate early differentiation of Th17 cells and maintain cytokine expression in effector Th17 cells [44]. It was recently shown that infectious triggers such as influenza lung infection and colitis trigger an IL-1β-induced Th17 differentiation and promote arthritis induced by KRN transgenic T cells [45]. Interestingly, a subset of human CD14+ CD163low lamina propria cells expressing both macrophage and DC markers has been found to express TLR4, produce IL-1β and IL-6 upon TLR4 stimulation, and induce Th17 differentiation [46]. However, the specific subset of LP phagocytes that orchestrates the phase-dependent TLR4-mediated mucosal response to microbiota in our studies remains to be determined.
Several studies have shown that TLR4 deficiency and systemic inhibition of TLR4 using specific antagonists or neutralizing antibodies can suppress experimental arthritis [12, 47–49]. Importantly, TLR4 is believed to be hyper-responsive in both blood monocyte-derived DCs and CD14+ synovial fluid macrophages of RA patients compared with healthy controls [50, 51]. Pathways associated with TLR signaling are upregulated in synovial fluid macrophages of patients with RA. A proinflammatory role for TLR4 during arthritis has previously been widely attributed to TLR4 activation by endogenous damage-associated molecular patterns present in the joint rather than microbial agonists [12, 49, 52, 53]. Our observations suggest that TLR4-mediated modulation of the mucosal immune response in intestinal LP may be another function involving TLR4 in arthritis.
Conclusions
Our study reveals a crucial role for IL-1Ra in regulation of the diversity and the composition of intestinal microbiota and a balanced T cell response in the intestinal LP. We show that the aberrant microbiota in IL1rn −/− mice have the capacity to enhance LP Th17 cells which are associated with arthritis, likely via TLR4-induced production of IL-1β, IL-6, and IL-23. Although IL1rn −/− intestinal microbiota do not cause arthritis in a normal (WT) host, these microbiota, in particular tobramycin-sensitive bacteria, contribute to the development of arthritis in IL1rn −/− mice. Our data suggest that the interplay between IL-1Ra, intestinal microbiota, TLR4, and mucosal T cells may serve as a potential predisposing or initiating event in the context of autoimmune disease and provide opportunities to control RA.
Methods
Mice
IL1rn −/− mice on a BALB/c background were kindly provided by Dr. M. Nicklin (Sheffield, UK) [54]. WT BALB/c mice were purchased from Harlan, UK. Mice were co-housed in filter-top non-individually ventilated (non-IVC) cages in the same room in our animal facility for at least 8 weeks prior to feces collection for pyrosequencing. IL1rn −/− Tlr4 −/− mice and their IL1rn −/− Tlr +/+ littermates were generated as described before [12] and used for microbiota sequencing. IL1rn −/− Tlr2 −/− mice were compared to non-littermate IL1rn −/− mice in this study.
Microbiota sequencing and data analysis
Fecal bacterial DNA from 15-week-old mice was isolated using phenol-, chloroform-, isoamyl alcohol-based extraction (Sigma). Sequencing was performed by DNAVision (Charleroi, Belgium) on a Roche 454 GS-FLX System using 16S rRNA gene bar-coded primers targeting the V5-V6 conserved DNA regions (forward primer 784F: 5′-AGGATTAGATACCCTGGTA-3′, reverse primer 1061R: 5′-CRRCACGAGCTGACGAC-3′) [55]. For gene sequence analysis, a customized workflow based on Quantitative Insights Into Microbial Ecology (QIIME version 1.2) was adopted (http://qiime.org/) [56]. Settings recommended in QIIME 1.2 tutorial were applied. Additionally, reads were filtered for chimeric sequences using Chimera Slayer as described before [57]. OTU clustering was performed with settings as recommended by QIIME [58] using an identity threshold of 97%. The Ribosomal Database Project classifier version 2.2 was used for taxonomic classification [59]. Hierarchical clustering of samples was performed using UPGMA with weighted UniFrac as a distance measure as implemented in QIIME 1.2. For statistical analysis and generation of figures, a custom QIIME implemented R-package, SciPy [60] (www.Scipy.org), Graphpad Prism version 5.0, and Microsoft® Office Excel® 2007 were adopted. Presence of SFB was assessed by real-time quantitative PCR (qPCR) on fecal DNA using SFB-specific primers as described before [61]. The delta Ct (cycle threshold) value was calculated for SFB-specific rRNA gene relative to the total (conserved) bacterial 16S rRNA genes amplified using universal bacterial primers to correct for the total bacterial DNA input. Data are presented as delta Ct (ΔCt) and relative SFB expression calculated as 2−ΔCt × 10,000 (Additional file 1: Table S3).
Microbiota transfer and co-housing
IL1rn −/− microbiota were transferred to WT mice by oral gavage of 200 μl of a homogenized IL1rn −/− fecal suspension prepared in sterile PBS. Immediately hereafter, the gavaged WT mice were co-housed with IL1rn −/− mice in the same individually ventilated cage for a period of 6 weeks to ensure sustained microbiota transfer by coprophagy. The control WT mice were gavaged with their own fecal suspension and housed separate from IL1rn −/− fecal-transplanted mice in IVC cages. To verify microbiota of conventional IL1rn −/− mice can trigger arthritis, GF IL1rn −/− mice received either 200 μl of sterile water or 200 μl fecal suspensions of conventional IL1rn −/− mice and were monitored for the development of arthritis for 8 weeks. In some studies, IL1rn −/− mice were co-housed with IL1rn −/− Tlr4 −/− mice for 10 days before analysis of LP T cells.
Antibiotic treatments and reconstitution with SFB
Intestinal microbiota were depleted using a cocktail of metronidazole (Acros Organics), neomycin trisulfate (Sigma), and ampicillin sodium salt (Sigma) (all 1 g/l) provided in drinking water for 1 week. Indicated groups received 200 μl fecal suspensions of SFB-monocolonized mice by oral gavage 1 week after ceasing antibiotics. For single antibiotic treatments, ampicillin sodium salt (1 g/l), metronidazole (1 g/l), vancomycin hydrochloride (0.5 g/l, Fisher Scientific), or tobramycin sulfate (1 g/l, Centrafarm) was added to drinking water for 8 weeks and refreshed once a week. Sucrose (6 g/l) was added to drinking water of all groups including controls during treatments.
Isolation of lamina propria cells
Lamina propria mononuclear cells were isolated from the small intestine and colon after removing mesenteric fat and Peyer’s patches, followed by incubation with 33 mM EDTA on ice for 30 min to remove epithelial cells, and subsequent digestion with 1 mg/ml collagenase-D (Roche) and 10 μg/ml DNAse I (Sigma) at 37 °C for three cycles of 15 min. LP lymphocytes were then harvested at the interphase of a 40:80% percoll gradient (Sigma), washed thoroughly, and used in culture as described below.
Cell cultures and cytokine measurements
SI-LP mononuclear cells (4 × 105 cells/well), LN cells (2 × 105 cells/well), and splenocytes (1 × 105 cells/well) were cultured in round-bottom 96-well plates in the presence of PMA (50 ng/ml; Sigma) and ionomycin (1 μg/ml; Sigma) for 5 or 6 h, as indicated in the figure legends. SI-LP cells were also cultured for 24 h in the presence of IL1rn −/− or IL1rn −/− Tlr4 −/− complete microbial antigens (1:200 v/v ratio) prepared by autoclaving the IL1rn −/− fecal pellets dissolved in PBS, and then centrifuging the suspension at 2000 rpm for 5 min. Cytokine levels in culture supernatants were measured by Luminex using the mouse cytokine/chemokine magnetic bead kit (Milliplex and Bio-Rad).
Flow cytometry
For intracellular cytokine staining, SI-LP cells were incubated with PMA (50 ng/ml; Sigma), ionomycin (1 μg/ml; Sigma), and Brefeldin A (1 μl/ml; BD Biosciences) at 37 °C for 4 h. Cells were stained with fixable viability dye Efluor780 (eBioscience), anti-TCRβ-FITC (Biolegend) or anti-CD3-PE (BD pharmigen), and anti-CD4-APC (Biolegend), then fixed and permeabilized using fixation/permeabilization buffer (eBioscience), and stained with anti-IL-17-FITC (Biolegend), IL-17-PECy7 (Biolegend), or anti-IFNγ-FITC (BD pharmingen) in permeabilization buffer (eBioscience).
Assessment of arthritis
Severity of arthritis was scored using a previously standardized arbitrary scoring system on a 0–2 scale per paw [12]. Arthritis developed only in ankle joints (maximum score of 4). For histology, total ankle joints were isolated and fixed in 4% formaldehyde for 4 days, thereafter decalcified in 5% formic acid and embedded in paraffin. Tissue sections of 7 μm were stained using hematoxylin and eosin to study synovial inflammation, cartilage destruction, and bone erosion. Each parameter was scored on a scale from 0 to 3 in a blinded manner.
Statistics
Group measures are expressed as mean + SEM. Statistical significance was tested using an unpaired two-tailed Mann-Whitney U test to compare two and ANOVA to compare more groups, with Bonferroni correction for multiple testing when applicable (GraphPad Prism 5.0). Arthritis scores were compared using repeated measures ANOVA with Bonferroni correction. Significance is indicated on figures as follows: n.s. (not significant), *P < 0.05, **P < 0.01, ***P < 0.001.
Acknowledgements
We are grateful to Birgitte Walgreen, Liduine van den Bersselaar, Monique Helsen, and Sija Jacobs-Landman for their invaluable support in performing the experiments. We are also grateful to Dr. Janneke Samsom (Erasmus University Medical Center, Rotterdam, The Netherlands) for the histologic assessment of the intestinal tissues.
Funding
This study was performed within the framework of TI Pharma project number D1-101 and was funded by the Netherlands Organization for Scientific Research (VENI grant 916.12.039), the Dutch Arthritis Foundation (AFS 14-1-291), the European BTCure grant 115142-2, and Arthritis National Research Foundation (ANRF 15-A0-00-004310).
Availability of data and materials
All 16S rRNA gene sequencing reads data are publicly available at the European Nucleotide Archive (ENA) database (http://www.ebi.ac.uk/ena) under study accession number PRJEB7447 (or secondary accession number ERP007176).
Authors’ contributions
R.R., T.H.A.E., and S.A. performed the experiments, analyzed the data, and wrote the manuscript. J.B., H.W., J.M., J.U.S., J.K., and S.A.F.T.H. contributed to the sequencing data analysis and interpretation. J.G., P.M.K., M.I.K., and W.B.B. contributed to the data interpretation and scientific discussions. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All animal studies were approved by the institutional review board (Animal Experimentation Committee of Radboud University Medical Center) and were conducted in accordance with the institutional guidelines. The approval numbers for this study were RU-DEC-2012-041, RU-DEC-2013-042, RU-DEC-2013-173, and RU-DEC-2012-305.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Hierarchical clustering of wild-type and IL1rn −/− mice based on intestinal microbiota. Figure S2. The impact of lineage origin versus IL1rn-deficiency on the overall fecal microbiota composition. Figure S3. Gating strategy. Figure S4. Frequencies and numbers of IL-17-producing cells among TCRβ+ and TCRβ− T cell populations with and without CD4 expression. Figure S5. Colonization of germ-free (GF) IL1rn −/− mice with fecal microbiota of conventional IL1rn −/− mice increases the severity of arthritis. Figure S6. Increased expression of Th2/Treg cytokines in the spleens but not in the popliteal lymph nodes (LN) of germ-free IL1rn −/− mice. Figure S7. Effects of 8-week oral tobramycin treatment on microbiota of IL1rn −/− mice assessed by 16S gene sequencing of fecal bacterial DNA. Figure S8. IL1rn −/− and IL1rn −/− Tlr4 −/− microbiota induce similar cytokine response in lamina propria mononuclear cells. Figure S9. Lamina propria mononuclear cells of IL1rn −/− Tlr4 −/− mice co-housed with IL1rn −/− mice produce less Th17-inducing cytokines. Figure S10. Decreased IL-17 production in draining lymph nodes of TLR4 deficient mice. Table 1. The average and total number of (assigned) reads and operational taxonomic units (OTU) per experimental group. Table 2. TLR4 deficiency normalizes specific aberrations in Il1rn −/− intestinal microbiome toward WT level. Table 3. Assessment of the presence of SFB expression in WT, IL1rn −/−, IL1rn −/− Tlr2 −/−, and IL1rn −/− Tlr4 −/− mice. Table 4. Alterations in fecal microbiota by oral tobramycin. (DOCX 1237 kb)
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0278-2) contains supplementary material, which is available to authorized users.
Contributor Information
Rebecca Rogier, Email: Rebecca.Rogier@radboudumc.nl.
Thomas H. A. Ederveen, Email: Tom.Ederveen@radboudumc.nl
Jos Boekhorst, Email: Jos.Boekhorst@nizo.nl.
Harm Wopereis, Email: Harm.Wopereis@wur.nl.
Jose U. Scher, Email: Jose.Scher@nyumc.org
Julia Manasson, Email: Julia.Manasson@med.nyu.edu.
Sanne J. C. M. Frambach, Email: Sanne.Frambach@radboudumc.nl
Jan Knol, Email: Jan.Knol@wur.nl.
Johan Garssen, Email: J.Garssen@uu.nl.
Peter M. van der Kraan, Email: Peter.vanderKraan@radboudumc.nl
Marije I. Koenders, Email: Marije.Koenders@radboudumc.nl
Wim B. van den Berg, Email: Wim.vandenBerg@radboudumc.nl
Sacha A. F. T. van Hijum, Email: Sacha.vanHijum@radboudumc.nl
Shahla Abdollahi-Roodsaz, Phone: 212-598-6417, Email: Shahla.Abdollahi-Roodsaz@radboudumc.nl, Email: S.Abdollahi@nyumc.org.
References
- 1.Joosten LA, Netea MG, Dinarello CA. Interleukin-1beta in innate inflammation, autophagy and immunity. Semin Immunol. 2013;25:416–424. doi: 10.1016/j.smim.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Carter DB, Deibel MR, Jr, Dunn CJ, Tomich CS, Laborde AL, Slightom JL, Berger AE, Bienkowski MJ, Sun FF, McEwan RN, et al. Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein. Nature. 1990;344:633–638. doi: 10.1038/344633a0. [DOI] [PubMed] [Google Scholar]
- 3.Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–320. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A. 2003;100:5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shepherd J, Little MC, Nicklin MJ. Psoriasis-like cutaneous inflammation in mice lacking interleukin-1 receptor antagonist. J Invest Dermatol. 2004;122:665–669. doi: 10.1111/j.0022-202X.2004.22305.x. [DOI] [PubMed] [Google Scholar]
- 6.Matsuki T, Horai R, Sudo K, Iwakura Y. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J Exp Med. 2003;198:877–888. doi: 10.1084/jem.20030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol. 2006;18:399–407. doi: 10.1093/intimm/dxh379. [DOI] [PubMed] [Google Scholar]
- 8.Koenders MI, Devesa I, Marijnissen RJ, Abdollahi-Roodsaz S, Boots AM, Walgreen B, di Padova FE, Nicklin MJ, Joosten LA, van den Berg WB. Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheum. 2008;58:3461–3470. doi: 10.1002/art.23957. [DOI] [PubMed] [Google Scholar]
- 9.Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, Kasper LH. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183:6041–6050. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- 10.Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- 11.Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2011;108:11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, van den Berg WB. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205–216. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21:895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 16.Scher JU, Ubeda C, Artacho A, Attur M, Isaac S, Reddy SM, Marmon S, Neimann A, Brusca S, Patel T, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67:128–139. doi: 10.1002/art.38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, Taneja V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016;8:43. doi: 10.1186/s13073-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, Hirota K, Matsushita M, Furuta Y, Narazaki M, et al. Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheumatol. 2016;68:2646-61. [DOI] [PubMed]
- 19.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 21.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 24.van den Berg WB, McInnes IB. Th17 cells and IL-17 a—focus on immunopathogenesis and immunotherapeutics. Semin Arthritis Rheum. 2013;43:158–170. doi: 10.1016/j.semarthrit.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol. 2015;11:415–429. doi: 10.1038/nrrheum.2015.53. [DOI] [PubMed] [Google Scholar]
- 26.Genovese MC, Greenwald M, Cho CS, Berman A, Jin L, Cameron GS, Benichou O, Xie L, Braun D, Berclaz PY, Banerjee S. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014;66:1693–1704. doi: 10.1002/art.38617. [DOI] [PubMed] [Google Scholar]
- 27.Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Aelion JA, Lee SH, Codding CE, Kellner H, Ikawa T, et al. One-year efficacy and safety results of secukinumab in patients with rheumatoid arthritis: phase II, dose-finding, double-blind, randomized, placebo-controlled study. J Rheumatol. 2014;41:414–421. doi: 10.3899/jrheum.130637. [DOI] [PubMed] [Google Scholar]
- 28.Burmester GR, Durez P, Shestakova G, Genovese MC, Schulze-Koops H, Li Y, Wang YA, Lewitzky S, Koroleva I, Berneis AA, et al. Association of HLA-DRB1 alleles with clinical responses to the anti-interleukin-17A monoclonal antibody secukinumab in active rheumatoid arthritis. Rheumatology (Oxford) 2016;55:49–55. doi: 10.1093/rheumatology/kev258. [DOI] [PubMed] [Google Scholar]
- 29.Block KE, Zheng Z, Dent AL, Kee BL, Huang H. Gut microbiota regulates K/BxN autoimmune arthritis through follicular helper T but not Th17 cells. J Immunol. 2016;196:1550–1557. doi: 10.4049/jimmunol.1501904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sczesnak A, Segata N, Qin X, Gevers D, Petrosino JF, Huttenhower C, Littman DR, Ivanov II. The genome of th17 cell-inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe. 2011;10:260–272. doi: 10.1016/j.chom.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 34.Zuniga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev. 2013;252:78–88. doi: 10.1111/imr.12036. [DOI] [PubMed] [Google Scholar]
- 35.Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, Drew JC, Ilonen J, Knip M, Hyoty H, et al. Toward defining the autoimmune microbiome for type 1 diabetes. Isme j. 2011;5:82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joosten LA, Abdollahi-Roodsaz S, Heuvelmans-Jacobs M, Helsen MM, van den Bersselaar LA, Oppers-Walgreen B, Koenders MI, van den Berg WB. T cell dependence of chronic destructive murine arthritis induced by repeated local activation of Toll-like receptor-driven pathways: crucial role of both interleukin-1beta and interleukin-17. Arthritis Rheum. 2008;58:98–108. doi: 10.1002/art.23152. [DOI] [PubMed] [Google Scholar]
- 37.Abdollahi-Roodsaz S, Joosten LA, Helsen MM, Walgreen B, van Lent PL, van den Bersselaar LA, Koenders MI, van den Berg WB. Shift from toll-like receptor 2 (TLR-2) toward TLR-4 dependency in the erosive stage of chronic streptococcal cell wall arthritis coincident with TLR-4-mediated interleukin-17 production. Arthritis Rheum. 2008;58:3753–3764. doi: 10.1002/art.24127. [DOI] [PubMed] [Google Scholar]
- 38.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez A, Luckey D, Yeoman CJ, Marietta EV, Berg Miller ME, Murray JA, White BA, Taneja V. Loss of sex and age driven differences in the gut microbiome characterize arthritis-susceptible 0401 mice but not arthritis-resistant 0402 mice. PLoS One. 2012;7:e36095. doi: 10.1371/journal.pone.0036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luckey, Marietta D, Luthra E, Harvinder S, Patel R, Murray J, Mangalam A. Abstracts of the American College of Rheumatology/Association of Rheumatology Health Professionals Annual Scientific Meeting. 2012. Commensal gut-derived bacteria as therapy for systemic autoimmune disease. [Google Scholar]
- 41.Akitsu A, Ishigame H, Kakuta S, Chung SH, Ikeda S, Shimizu K, Kubo S, Liu Y, Umemura M, Matsuzaki G, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat Commun. 2015;6:7464. doi: 10.1038/ncomms8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chappert P, Bouladoux N, Naik S, Schwartz RH. Specific gut commensal flora locally alters T cell tuning to endogenous ligands. Immunity. 2013;38:1198–1210. doi: 10.1016/j.immuni.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med. 2012;209:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chevalier N, Tan JK, Mason LJ, Robert R, McKenzie CI, Lim F, Wong CH, Macia L, Thorburn AN, Russ BE, et al. Avenues to autoimmune arthritis triggered by diverse remote inflammatory challenges. J Autoimmun. 2016;73:120–129. doi: 10.1016/j.jaut.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 46.Ogino T, Nishimura J, Barman S, Kayama H, Uematsu S, Okuzaki D, Osawa H, Haraguchi N, Uemura M, Hata T, et al. Increased Th17-inducing activity of CD14+ CD163 low myeloid cells in intestinal lamina propria of patients with Crohn's disease. Gastroenterology. 2013;145:1380–1391.e1381. doi: 10.1053/j.gastro.2013.08.049. [DOI] [PubMed] [Google Scholar]
- 47.Komai-Koma M, Li D, Wang E, Vaughan D, Xu D. Anti-Toll-like receptor 2 and 4 antibodies suppress inflammatory response in mice. Immunology. 2014;143:354–362. doi: 10.1111/imm.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van den Brand BT, Abdollahi-Roodsaz S, Bennink MB, Bussink J, Arntz OJ, van den Berg WB, van de Loo FA. Toll-like receptor 4 in bone marrow-derived cells as well as tissue-resident cells participate in aggravating autoimmune destructive arthritis. Ann Rheum Dis. 2013;72:1407–1415. doi: 10.1136/annrheumdis-2012-202467. [DOI] [PubMed] [Google Scholar]
- 49.Pierer M, Wagner U, Rossol M, Ibrahim S. Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PLoS One. 2011;6:e23539. doi: 10.1371/journal.pone.0023539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007;56:2192–2201. doi: 10.1002/art.22707. [DOI] [PubMed] [Google Scholar]
- 51.Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, van den Hoogen FH, van den Berg WB, Radstake TR. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52:2313–2322. doi: 10.1002/art.21278. [DOI] [PubMed] [Google Scholar]
- 52.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 53.Abdollahi-Roodsaz S, van de Loo FA, van den Berg WB. Trapped in a vicious loop: Toll-like receptors sustain the spontaneous cytokine production by rheumatoid synovium. Arthritis Res Ther. 2011;13:105. doi: 10.1186/ar3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191:303–312. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.New default parameters for uclust OTU pickers. [https://qiime.wordpress.com/2010/12/17/new-defaultparameters-for-uclust-otu-pickers/]. Accessed 25 May 2016.
- 59.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.SciPy open-source software [http://www.scipy.org/]. Accessed 25 May 2016.
- 61.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All 16S rRNA gene sequencing reads data are publicly available at the European Nucleotide Archive (ENA) database (http://www.ebi.ac.uk/ena) under study accession number PRJEB7447 (or secondary accession number ERP007176).