

Abstract
Understanding HIV-1 transmission dynamics is relevant to both screening and intervention strategies of HIV-1 infection. Commonly, HIV-1 transmission chains are determined based on sequence similarity assessed either directly from a sequence alignment or by inferring a phylogenetic tree. This review is aimed at both nonexperts interested in understanding and interpreting studies of HIV-1 transmission, and experts interested in finding the most appropriate cluster definition for a specific dataset and research question. We start by introducing the concepts and methodologies of how HIV-1 transmission clusters usually have been defined. We then present the results of a systematic review of 105 HIV-1 molecular epidemiology studies summarizing the most common methods and definitions in the literature. Finally, we offer our perspectives on how HIV-1 transmission clusters can be defined and provide some guidance based on examples from real life datasets.
Keywords: HIV-1, molecular epidemiology, phylogeny, transmission clusters
Introduction
The classification and clustering of biological organisms and entities has been fundamental to understanding their origins, relationships and evolution [1,2]. Mechanisms of reproductive isolation prevent animals and plants of different species from producing fertile offspring, thereby maintaining species integrity over time, such that biological diversity typically falls into discrete categories or clusters [3]. Reproductive isolation mechanisms are absent or less distinct for viruses. Combined with high mutation rate and ability to adapt swiftly to environmental changes, the genetic diversity of many viruses, such as the HIV-1, exists in much more of a continuum. This makes the definition of discrete clusters at the inter-host and intra-host level particularly challenging [4,5]. However, the rapid evolution leaves measurable footprints in viral genomes that can be associated with transmission dynamics and epidemiology. An HIV-1 transmission cluster can be described as a set of HIV-1 sequences that are aggregated in a nonrandom manner linked to their epidemiology. Over the last two decades, evolutionary theory and sequence analysis have contributed significantly to our understanding of HIV-1 epidemiology, for example by providing information about the time and geographical location of HIV-1 origins [6,7]. Detailed analyses of viral sequences can provide useful information about HIV-1 epidemics by identifying transmission linkages and by elucidating differences in transmission within and between populations [6,8]. Well characterized transmission chains have been compared with sequence-based phylogenies and are often in close agreement [9–14].
Phylogenetic analysis has been used successfully to identify and dissect HIV-1 transmission clusters. When combined with detailed epidemiological and clinical data, the results of such analyses can be of public health relevance, for example by identifying how virus lineages are restricted to, or mix among, different demographic and behavioural subgroups [15–21]. Typically, each HIV-1-infected individual under study is sampled once and represented by a single HIV-1 sequence obtained by bulk Sanger sequencing. The sequences are used to construct a bifurcating evolutionary tree (a phylogeny) in which each virus sequence (taxon) is positioned at a tree tip. Pairs of tree branches share a node that represents the most recent common ancestor (MRCA) of the taxa that have descended from that node. Individuals that share a MRCA are usually considered to be epidemiologically linked, that is to represent a transmission cluster. The lengths of the tree branches usually represent the genetic relatedness between the different ancestors and their descendants. Genetic distances can be linked to time by assuming a so-called molecular clock model [22]. Early molecular clock models assumed that all phylogeny branches evolved at the same rate. However, a constant evolutionary rate is often unrealistic and alternative more flexible molecular clock models have been developed [23–26]. In essence, phylogenetic inference relies on an alignment of genetic sequences, an underlying substitution model to model the process of evolutionary sequence change, an approach or algorithm for inferring the tree, and some measure of statistical support for the relationships given in the tree.
The selection of an appropriate definition for a ‘transmission cluster’, that is a shared MRCA, is complex and needs to take into account both the research question and characteristics of the sequence data, such as the selected genomic region, sequence length, the range of sample collection dates, the distribution of sampling locations, the mode of transmission, the diversity of the genetic variants sampled (within and between HIV-1 subtypes and circulating recombinant forms), the number of sequences, the proportion of the population under study that is sampled, and the degree to which sampling is representative [27]. Hence, it is not surprising that there is no clear consensus on how transmission clusters should be defined. Nevertheless, there is a need for a common strategy among researchers for determining appropriate cluster definitions for typical datasets and research questions. A common rationale would contribute to a better understanding of the HIV-1 pandemic by increasing the comparability between studies [28,29].
Genetic distance, tree building algorithms and node support
Pairwise genetic distances can be either calculated directly from the sequences (the so-called p-distance, or Hamming distance, which equals the observed number of nucleotide differences between two sequences) or computed using a nucleotide substitution model (the expected genetic distance, or so-called d-distance). If genetic distances are computed as the sum of the branch length between two tips in a tree, then they are known as patristic distances [30]. Simple p-distances do not employ a substitution model that describes the evolutionary process and therefore do not account for multiple changes or back mutations at the same site. Consequently, they typically underestimate the true genetic distance between two sequences [31].
Pairwise genetic distances within a transmission cluster of more than two sequences can be summarized in different ways, for example by using the mean, median or the maximum pairwise distance [29,32,33]. Another approach is to associate a sequence to a specific cluster if the distance from that sequence to any other sequence in that cluster is lower than a threshold value – irrespective of the distances to other sequences in the cluster [18,34]. There are advantages and disadvantages to the different approaches, for example the maximum genetic distance has been suggested to be less sensitive to cluster size than cluster definitions relying on mean genetic distances in which one or a few ‘unlinked’ sequences may be erratically included in large clusters because they have minimal influence on the mean distance [35]. Moreover, maximum genetic distance approaches are fast to compute and has been suggested to correlate with time of the MRCA of clusters in molecular clock phylogenies [33].
Both maximum-likelihood and Bayesian tree building approaches use probability models to evaluate the relative plausibility of different phylogenetic topologies, whereas the neighbour-joining approach uses a deterministic tree-building algorithm that generates only a single phylogenetic topology (Table 1 and described in detail in [31]). Traditionally, statistical node support for the relationships in a phylogenetic tree has been evaluated by a statistical technique called bootstrapping [36]. During phylogenetic bootstrapping, site positions in the original alignment are randomly resampled with replacement to produce a set of pseudo-replicate alignments. The tree building approach is then applied to each of these alignments. Clusters of related taxa that are present in a low percentage of the bootstrap trees are weakly supported and vice versa. However, the exact interpretation of bootstrap values is difficult. Higher values are of course better, but what is a reasonable cut-off? It has been suggested that bootstrap values of more than 70% indicate strong support for a group, based on the conclusion that bootstrap supports are conservative measurements [37]. Two other types of statistical tests, that are substantially faster than the bootstrap approach, are the approximate likelihood-ratio test and the zero-branch length test [38–40]. In essence, these test whether each branch in a tree is significantly greater than zero or not (i.e. if the branch exist), and cut-off probabilities of more than 0.9 have been suggested to be conservative and correspond relatively well to bootstrap values more than 70% [39–41].
Table 1.
Components of HIV-1 transmission cluster definitions based on phylogenetic node support.
| Phylogenetic tree reconstruction (examples of commonly used methods) |
| Neighbour-joining |
| Maximum-likelihood |
| Bayesian |
| Substitution model (examples of commonly used substitution models) |
| Jukes–Cantor (JC) |
| Tamura–Nei (TN) |
| General time reversible (GTR) |
| Node support tests (examples of commonly used tests) |
| Bootstrap test |
| Approximate likelihood-ratio test (aLRT) |
| Zero-branch length test |
Instead of relying on one ‘best tree’ or a set of bootstrap pseudo-replicates, Bayesian phylogenetic approaches use Markov chain Monte Carlo sampling to infer a full posterior probability distribution of plausible trees, which should contain all the different tree topologies that are well supported by the data. This set of trees can be used to produce a consensus tree [called a maximum clade credibility (MCC) tree] in which each branch and cluster has an associated probability. In an MCC tree, this probability is the proportion of trees in the posterior probability distribution in which the cluster of interest exists. Bayesian posterior probabilities have been suggested to be a generally less biased predictor of phylogenetic accuracy than bootstrapping [42].
Systematic literature review
We systematically reviewed the scientific literature of HIV-1 molecular epidemiology with the aim to explore current definitions of HIV-1 transmission clusters. Preliminary explorative analyses showed that both the majority of available HIV-1 sequences in Genbank (77%), and HIV-1 transmission network studies (80%) were published after 2005. We therefore limited our review to HIV-1 specific literature published between 2005 and 2016.
A literature search of the PubMed database (http://www.ncbi.nlm.nih.gov/pubmed) was undertaken on 11 April 2016 using the following search and mesh terms: [‘2005’(PDAT): ‘2016’(PDAT)] & (hiv OR ‘human immunodeficiency virus’) & transmission & (cluster∗ OR network∗) & (molecular OR phylogenetic). Previous reviews and opinions were excluded from this review as our aim was to explore cluster definitions used in primary research studies (Fig. 1). Strictly methodological, simulation and case studies were excluded for similar reasons. Non-English articles were excluded for simplicity (in total 18 articles). Two researchers (A.H. and J.E.) independently assessed the eligibility of articles from the literature search. The articles were manually screened, first by title, then by abstract, to assess relevance based on our eligibility criteria. Any discordance between the two reviewers in the list of shortlisted publications was flagged, and discussions held until a consensus on eligibility was reached. Shortlisted articles were imported into EndNote X7 (Thomas Reuters, Philadelphia, Pennsylvania, USA) for further management, and duplicate articles were deleted. After screening, 105 articles remained for full text review [12,17,18,20,33,43–142]. The articles were stratified into three main categories based on the approach that was used to define transmission clusters (Tables 2 and 3).
Fig. 1.
Flow chart showing results from the literature search and inclusion of articles considered in the review.
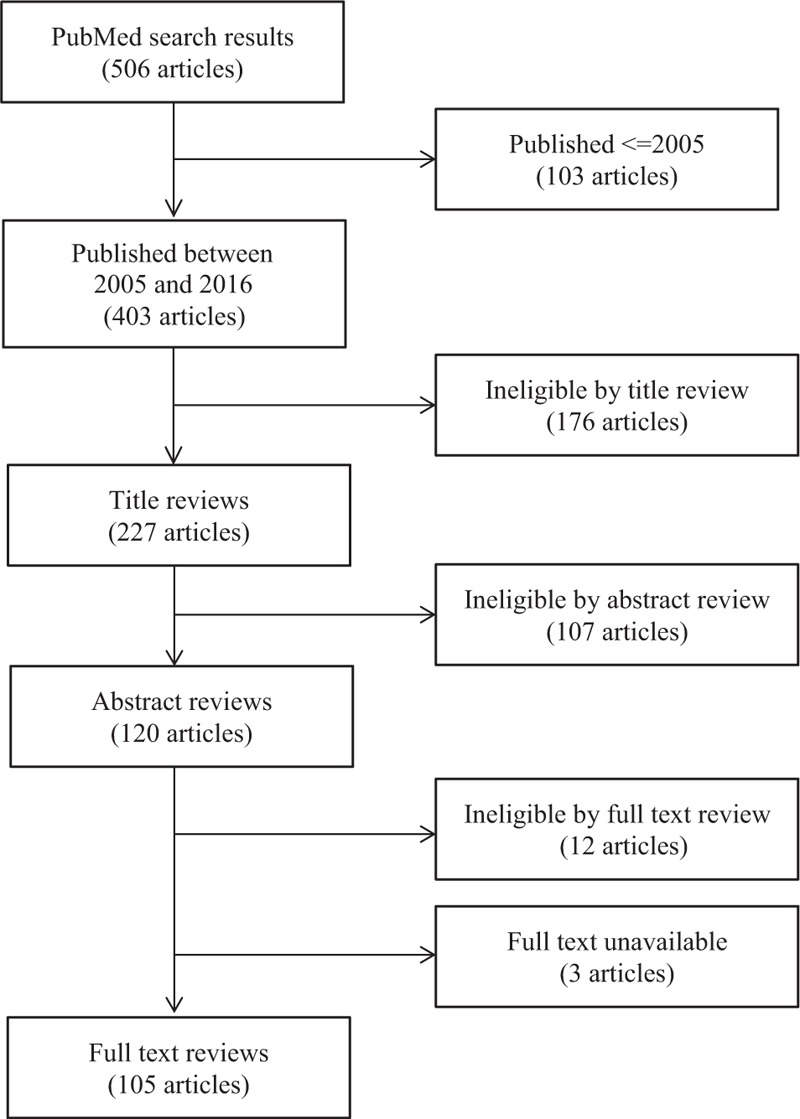
We employed the PubMed search engine (http://www.ncbi.nlm.nih.gov/pubmed) for the literature search strategy. Previous reviews and opinions, strictly methodological articles, simulation work, case studies and non-English papers were excluded from the final full text review.
Table 2.
Three main types of cluster definition.
| Pure phylogenetic transmission cluster definitions based solely on phylogenetic node support |
| Pure distance-based transmission cluster definitions based solely on pairwise genetic distances |
| Combined transmission cluster definitions based on both phylogenetic node support and pairwise genetic distances |
Table 3.
Results of the systematic review of 105 articles employing different strategies to define HIV-1 transmission clusters.
| Categories of cluster definition | Number of articles | Mediana number of sequences (IQRb) | Median study period in years (IQRb) | Median sequence length (IQRb) | Most analysed genetic region (proportion of articles)c | Tree building model used (proportion of articles)c,d | Substitution model used (proportion of articles)c,e | Branch support approach (proportion of articles)c,d | Median cut-off for determining clusters (IQRb) |
| Phylogenetic | 53 | 219 (96–562) | 6 (2–12) | 1100 (895–1497) | pol (88%) | ML (60%) | GTR (73%) | Bootstrap (71%) | 90% (75–90%) |
| Distance-based | 7 | 2747 (179–40950) | 11 (6–16) | 900 (500–1800) | pol (100%) | NA | TN (100%) | NA | 0.015 Substitutions/site (0.014–0.019) |
| Distance-based & phylogenetic | 45 | 534 (131–1413) | 7 (2–12) | 1150 (915–1308) | pol (98%) | ML (76%) | GTR (74%) | Bootstrap (86%) | 90% (90–98%), 0.015 Substitutions/site (0.015–0.038) |
aPairwise comparisons using the Mann–Whitney U test: phylogenetic vs. distance-based, P = 0.036; phylogenetic vs. distance-based & phylogenetic, P = 0.030; distance-based vs. distance-based & phylogenetic, P = 0.13.
bInterquartile range.
cThe most commonly used methodology are presented with the proportion of articles in which this methodology was used.
dML, maximum likelihood; NA, not applicable.
eGTR, general time reversible; TN, Tamura–Nei.
The most common approach was to use a pure phylogenetic definition (50% of the articles), followed by the combined approach (43%) and the pure distance-based approach (7%). No clear difference in study aims was found among the three approaches. Among the seven articles that relied purely on a distance-based cluster definition, the most common approach was to use the Tamura–Nei substitution model, whereas the general time reversible (GTR) model was most popular in phylogenetic-based and combined cluster definitions (Table 3) [143]. Some studies employed more than one tree building methodology. However, the most common was maximum-likelihood (used in 67% of the 98 studies that used a pure or combined phylogenetic cluster definition), followed by neighbour-joining (46%) and Bayesian tree building methodology (28%). Bootstrapping was the most commonly used statistics for branch support with the most common cut-off being 0.9. Studies defining HIV-1 transmission clusters by distance-based methodologies most often used a threshold of 0.015 substitutions/site (Table 3).
Analysis of publication year suggested that the interest in performing cluster analyses of HIV-1 sequence data increased through time, with 17 articles published between 2005 and 2010 compared with 88 articles published between 2011 and 2016. There was a tendency towards increased popularity of the combined approach during the latter period, compared with the pure phylogenetic approach that was previously more popular (Fig. 2). Analysis of future publications will be required to determine whether this is a random fluctuation or a true shift in the most popular approach. The median number of analysed sequences was greatest in articles employing pure distance-based cluster definitions (2747 sequences) and lowest in articles employing pure phylogenetic cluster definitions (219 sequences, Table 3).
Fig. 2.
Number of articles stratified by strategy of HIV-1 transmission cluster determination in relation to availability and number of analysed sequences during the study period.
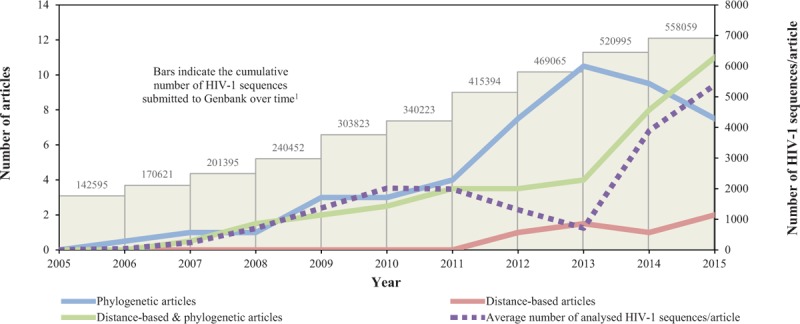
The articles were reviewed for strategy of HIV-1 transmission cluster determination. The lines represent moving averages (per 2 year average) over the study period. The articles were stratified into three categories based on cluster definitions (solid lines): phylogenetic (blue), distance-based (red) and distance-based & phylogenetic (green). The average number of analysed HIV-1 sequences per study is indicated by a dashed purple line. 1The cumulative number of HIV-1 sequences submitted to Genbank is indicated by bars (data collected from the Los Alamos HIV Sequence Database, https://www.hiv.lanl.gov/content/sequence /HIV/mainpage.html).
The average number of analysed HIV-1 sequences per study increased from 41 to 5389 sequences between 2005 and 2015 (Fig. 2). Phylogenetic analysis can be associated with high computational burden, in particular for large sequence datasets. It is possible that the increasing number of available HIV-1 sequences have favoured the generally faster and less parameter-rich distance-based methods in analysis of large sequence datasets (i.e. datasets with several thousands of sequences). Most articles (98 of 105) focused on sequences from a single country, and half of those represented studies in European countries. Only 8% of the studies focused on the African countries. Considering that approximately 70% of all HIV-1 infected individuals live in Africa, this highlights the need of additional studies from the African continent [144]. This is further emphasized by the fact that HIV-1 epidemics in MSM populations in Africa have been recognized only in the last 10 years [145].
All 105 articles based their analyses on sequences produced by Sanger sequencing. The most commonly analysed HIV-1 genetic region was the polymerase gene (pol), and the sequence length was generally around 1000 nucleotides (Table 3). This is likely because pol is used for routine testing of antiretroviral resistance and is the most common HIV-1 genetic sequence that is available in public databases. Although it has been argued that pol has some limitations in giving high enough phylogenetic resolution, it has been used extensively in studies of HIV-1 molecular epidemiology and has been reported to contain sufficient information for analyses of HIV-1 transmission [10,32,132].
Taken together, the literature review showed that the most common phylogenetic methodology was a maximum likelihood approach that uses a substitution model GTR with transmission clusters defined by tree nodes with bootstrap support values of more than 90%.
Real-life examples
Analyses of datasets with high coverage [when a large fraction (>30%) of the total number of infected individuals in a population is represented in the dataset] and longitudinal sampling over extensive periods of time (>10 years) – sometimes comprising sequences from more than one country – can be challenging. It is therefore important to carefully consider the main study aim before determining the cluster definition [28].
To explore and exemplify the effects of different genetic distance thresholds, we performed a comparative analysis of two previously described transmission clusters with different topologies and sequence sampling durations [41]. Both clusters contain HIV-1 pol sequences that have diverged more than 4.5% (0.045 substitutions per site, Fig. 3). Consider, for example, the relatively long and statistically well supported branch that divides the Danish MSM cluster in two (highlighted by an arrow in Fig. 3a). The length of this branch could be due to: (1) no transmissions of this viral strain for a few years, or (2) unsampled transmissions of this viral strain. However, in this particular example, 57% of all newly detected HIV-1 infections in Denmark were sequenced during the study period. Hence, it is perhaps unlikely that the 41 more terminally located Danish sequences would not stem from the same epidemiological introduction as the 14 more basally placed Danish sequences. In addition, the branch ancestral to this cluster was both well supported and relatively long, further supporting this scenario [41]. Moreover, a large number of non-Danish reference sequences selected by genetic similarity (from both Genbank and a large nonpublic sequence database representing surveillance programmes in most European countries) were analysed together with the Danish sequences to maximize the chances of picking up non-Danish links in the original report of this transmission cluster [41,146].
Fig. 3.
Comparison between phylogenetic cluster definitions employing a branch support criteria with or without different distance thresholds.
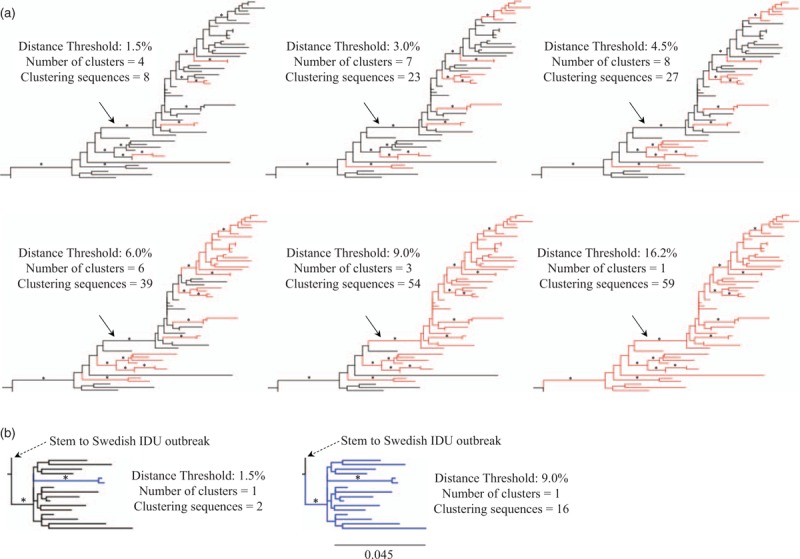
Statistically supported branches are indicated with an asterisk (estimates ≥0.90, as determined by approximate likelihood ratio test with the Shimodaira–Hasegawa-like procedure) [40]. The scale bar indicates the genetic distance in substitutions per site and applies to both panels. Branches highlighted in colour represent sequences found in clusters according to respective cluster definition (as detailed for each tree in the figure). All examples of clusters are defined by a threshold of maximum pairwise distance in substitutions per site between any sequence pair in a cluster. Clusters (a) and (b) were cut out of larger phylogenies analysed in an extensive multinational HIV-1 transmission study with an overall coverage of more than 50% of newly HIV-1 infected individuals in the studied region [41]. (a) Transmission cluster of HIV-1 subtype B infected Danish MSM. The 16.2% distance threshold is the level in which all sequences will be included in the cluster. The relatively long and statistically supported branch dividing the Danish MSM cluster in two parts discussed in the main text are indicated by an arrow. (b) Transmission cluster of HIV-1 CRF01_AE infected Finnish intravenous drug users.
Another example that illustrates the effects of a relatively small distance-based threshold is presented in Fig. 3b. This cluster represents a set of late presenters from a well characterized Finnish HIV-1 outbreak among intravenous drug users (IDUs) in 1998–1999 and is linked to a large Swedish IDU outbreak that occurred 2005–2007 (as established by epidemiological data and discussed in previous publications) [41,108,147]. The longer terminal branches (i.e. higher diversity) observed in this Finnish cluster reflects the fact that these individuals were infected by HIV-1 several years before being sampled (it is likely that the majority of patients were infected during the outbreak in 1998–1999, but the sampling period of the Finnish dataset in this study started first 2003). The Finnish cluster would be reduced to a small cluster of two sequences if the most commonly used distance-based threshold of 1.5% was employed (Table 3 and Fig. 3b). Thus, too small distance-based thresholds may reduce large and long-lived transmission clusters to multiple smaller subclusters. The same principle applies to the Danish MSM cluster discussed above, in which a lower genetic distance threshold would result in several smaller independent, more recent, and potentially active clusters/transmission pairs, instead of the larger cluster as identified by a higher genetic threshold (Fig. 3a). These examples highlight the continuous evolution of HIV-1, which results in an increasing divergence from a founder strain (e.g. the MRCA of a transmission cluster).
Moreover, if the main aim of a study is to determine the number of active transmission clusters, it may be important to also consider nongenetic epidemiological information (e.g. known date of infection or Recent Infection Testing Algorithms) and the social context of the population(s) under study [148,149]. In contrast, a higher distance threshold results in only one or a few larger transmission clusters, implying long-standing and continuous HIV-1 transmission problems in this population. If the aim is to identify both long-lasting and active transmission chains, a stepwise procedure may be useful. A higher threshold could be applied in the first step to identify long-lasting clusters; in a second step, these clusters could then be stratified into active and nonactive clusters based on the existence of subclusters as defined by a lower threshold. This could be particularly useful in datasets that cover a large proportion of the infected population. An alternative sequence-based approach that might reduce the risk of including transmission clusters with important missing links is one that determine clusters using the maximum length of internal branches, instead of mean, median or maximum pairwise genetic distances.
Tools for identification of HIV-1 transmission clusters
Several tools and software have been developed for the identification of transmission clusters from HIV-1 sequence data [29,35,150–154]. Two popular and freely available tools are PhyloPart (https://sourceforge.net/projects/phylopart/) and Cluster Picker (http://hiv.bio.ed.ac.uk/software.html) [29,35]. Both rely on a predetermined phylogenetic tree and allow the user to determine thresholds for either genetic distance, phylogenetic branch support or both. The main difference is that PhyloPart uses a distance threshold that is a user-specified percentile of median pairwise distances, whereas Cluster Picker employs a user-specified maximum pairwise distance threshold. In contrast to PhyloPart and Cluster Picker, the recently developed ‘Gap Procedure’ does not depend on a phylogenetic tree [150]. Instead, pairwise distances are estimated directly from the sequence alignment and sorted by size to identify relatively larger ‘gaps’ between subsets or aggregations of similar distance estimates. By this procedure, the authors argue that there is no requirement for a user-defined and potentially poorly justified a-priori threshold to identify clusters.
Jacka et al.[154] recently used sampling collection dates to infer molecular clock phylogenies and then defined clusters based on lineages existing at a particular point in time (the analysis can be done by the freely available software ClusterByTime). This definition is clearly related to distance-based cluster definitions, because under a strict clock the genetic distance is linearly proportional to time. If rates of evolution vary among lineages, one could argue that time is superior to genetic distance since the same schedule of transmission events would result in clusters with significantly different levels of genetic distance. Further developments also allow for the addition of discrete and continuous traits linked to viral sequences and infections. For example, phylogeographic and Markov jump models can be used to infer the directionality and number of transitions between different traits (e.g. geographic locations or transmission groups) [6,155–157]. Finally, more complex cluster definitions based on simultaneous analysis of epidemiological information and viral sequence data have also been proposed to improve the reconstruction of accurate HIV-1 transmission networks [151,152].
Future research directions
When we assessed studies that have analysed sequence datasets covering a relatively large proportion of the infected population at national or regional scales, it became clear that there is no common strategy to define transmission clusters [12,33,41,45,77,78,131,136,158]. The increasing number of available HIV-1 sequences will make it increasingly difficult to infer phylogenetic trees to determine transmission clusters. A future challenge will therefore be to estimate the level of sequence coverage (i.e. the fraction of the total number of infected individuals in a population) at which the current methods of determining branch support becomes impractical or even uninformative.
Recent developments in sequencing strategies have not only resulted in an increased number of sequences, but also in a wide variety in the quality and accuracy of viral sequences submitted to public databases. Next-generation sequencing (NGS) is superior to Sanger sequencing in detecting low-level variants, but some NGS methodologies suffer from relatively higher error rates and one of the major challenges has been to distinguish technical and analytical errors from true viral diversity [159]. Eshleman et al.[160] analysed HIV-1 sequences from eight index-partner pairs with unlinked HIV-1 sequences (as previously determined by analysis of bulk Sanger sequences) and reported that one of the eight couples in fact was linked when the virus populations were reanalysed by NGS. This indicates that although the correspondence between Sanger and NGS sequences generally seems high, there may be occasions in which bulk Sanger sequences will not adequately represent the entire virus population within an individual. In our literature review, none of the studies used NGS sequences to study HIV-1 transmission dynamics in a geographic region or country. However, with the increasing number of NGS sequences generated in recent years, there will be a need to study both the impact of analysing Sanger versus NGS sequences on a larger population-based scale and the effects of combining both types of sequences in the same analysis of transmission clusters.
The HIV-1 evolutionary dynamics and population genetic forces differ substantially between intra-host and inter-host levels, and by transmission route. Another topic that needs further investigation is therefore how the inclusion of several sequences per patient (either longitudinally collected or multiple clonal sequences from one time-point) impacts the identification of transmission clusters in large sequence datasets [161]. Similarly, further studies are needed on the effects of mixing sequences from individuals infected through different transmission routes. An outbreak among IDUs can for example look very different compared with a transmission cluster with sequences predominantly from MSM or heterosexuals [41,162]. It has been suggested that such differences may be linked to rapid HIV-1 transmissions and lack of transmission bottlenecks in IDU outbreaks [162].
Conclusion and selecting an appropriate cluster definition
HIV-1 infected patients are connected by transmission history and HIV-1 populations accumulate genetic distance over time. Therefore, the genetic distances in a transmission cluster will depend on how long ago it was established. The most suitable definition of an HIV-1 transmission cluster will depend on the hypothesis being tested and the composition of the HIV-1 sequence dataset under study. Consequently, no single method or cut-off will suit all research purposes. However, an approach that combines a genetic distance threshold with a phylogenetic branch support seems to fit most hypotheses and datasets. Moreover, and as exemplified in Fig. 3, loosely set genetic threshold (e.g. larger than the commonly used thresholds of 1.5 or 4.5%) allows inclusion of clusters that span longer time periods. This seems appropriate for datasets with high-sequence coverage of populations followed over long-time periods if the main aim is to understand the long-standing transmission dynamics. A higher threshold will, however, increase the likelihood of including transmission clusters with missing links (i.e. unsampled sequences), and a stricter genetic threshold or a molecular clock analysis may be more appropriate when the aim is to determine recent and epidemiologically active transmission clusters (recently formed clusters have a higher likelihood of still being active).
Studies of viral transmission based on sequence data can provide critical information that would be difficult to obtain through traditional epidemiological methodology and will likely be an increasingly important component in population-based surveillance of infectious diseases. Further developments of accessible and flexible software will be important in future analyses of the increasing number of publicly available HIV-1 sequences and to compare results between studies.
Acknowledgements
A.H. and J.E. performed the literature review. J.E. outlined the review and wrote the article. J.A. and J.E. designed the cluster analysis. All authors read and provided substantial input to the concepts presented in the review. J.E. is supported by the Swedish Research Council (350-2012-6628; 2016-01417) and the Swedish Society for Medical Research. The Kenya Medical Research Institute/Wellcome Trust Research Programme (KWTRP) at the Centre for Geographical Medicine Research-Kilifi is supported by core funding from the Wellcome Trust (#077092). A.H. and E.S. are supported in part by the International AIDS Vaccine Initiative (IAVI), which receives generous support of the American people through the United States Agency for International Development (USAID). A.H. was also supported by funding from the African Research Excellence Fund (AREF, grant # MRF-157-0002-F-HASSA). This work was also supported through the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative [grant # DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [grant # 107752/Z/15/Z] and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of USAID, or the United States Government, AAS, NEPAD Agency, Wellcome Trust or the UK government. This report was published with permission from the Kenya Medical Research Institute (KEMRI).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Linnaeus C. Systema naturæ per regna tria naturæ, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. 10th ed.Stockholm: Laurentius Salvius; 1738. [Google Scholar]
- 2.Darwin C. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. London: John Murray; 1859. [PMC free article] [PubMed] [Google Scholar]
- 3.Barton N, Bengtsson BO. The barrier to genetic exchange between hybridising populations. Heredity (Edinb) 1986; 57 (Pt 3):357–376. [DOI] [PubMed] [Google Scholar]
- 4.Wain-Hobson S. The fastest genome evolution ever described: HIV variation in situ. Curr Opin Genet Dev 1993; 3:878–883. [DOI] [PubMed] [Google Scholar]
- 5.Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 2009; 10:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faria NR, Rambaut A, Suchard MA, Baele G, Bedford T, Ward MJ, et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014; 346:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worobey M, Gemmel M, Teuwen DE, Haselkorn T, Kunstman K, Bunce M, et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 2008; 455:661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esbjornsson J, Mild M, Audelin A, Fonager J, Skar H, Bruun Jorgensen L, et al. HIV-1 transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic countries. Virus Evol 2016; 2:vew010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leitner T, Escanilla D, Franzen C, Uhlen M, Albert J. Accurate reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proc Natl Acad Sci U S A 1996; 93:10864–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemey P, Derdelinckx I, Rambaut A, Van Laethem K, Dumont S, Vermeulen S, et al. Molecular footprint of drug-selective pressure in a human immunodeficiency virus transmission chain. J Virol 2005; 79:11981–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paraskevis D, Magiorkinis E, Magiorkinis G, Kiosses VG, Lemey P, Vandamme AM, et al. Phylogenetic reconstruction of a known HIV-1 CRF04_cpx transmission network using maximum likelihood and Bayesian methods. J Mol Evol 2004; 59:709–717. [DOI] [PubMed] [Google Scholar]
- 12.Bruhn CA, Audelin AM, Helleberg M, Bjorn-Mortensen K, Obel N, Gerstoft J, et al. The origin and emergence of an HIV-1 epidemic: from introduction to endemicity. AIDS 2014; 28:1031–1040. [DOI] [PubMed] [Google Scholar]
- 13.Romero-Severson EO, Bulla I, Leitner T. Phylogenetically resolving epidemiologic linkage. Proc Natl Acad Sci U S A 2016; 113:2690–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romero-Severson E, Skar H, Bulla I, Albert J, Leitner T. Timing and order of transmission events is not directly reflected in a pathogen phylogeny. Mol Biol Evol 2014; 31:2472–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grabowski MK, Lessler J, Redd AD, Kagaayi J, Laeyendecker O, Ndyanabo A, et al. The role of viral introductions in sustaining community-based HIV epidemics in rural Uganda: evidence from spatial clustering, phylogenetics, and egocentric transmission models. PLoS Med 2014; 11:e1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frost SD, Pillay D. Understanding drivers of phylogenetic clustering in molecular epidemiological studies of HIV. J Infect Dis 2015; 211:856–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poon AF, Joy JB, Woods CK, Shurgold S, Colley G, Brumme CJ, et al. The impact of clinical, demographic and risk factors on rates of HIV transmission: a population-based phylogenetic analysis in British Columbia, Canada. J Infect Dis 2015; 211:926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldous JL, Pond SK, Poon A, Jain S, Qin H, Kahn JS, et al. Characterizing HIV transmission networks across the United States. Clin Infect Dis 2012; 55:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brenner B, Wainberg MA, Roger M. Phylogenetic inferences on HIV-1 transmission: implications for the design of prevention and treatment interventions. AIDS 2013; 27:1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wertheim JO, Leigh Brown AJ, Hepler NL, Mehta SR, Richman DD, Smith DM, et al. The global transmission network of HIV-1. J Infect Dis 2014; 209:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher M, Pao D, Brown AE, Sudarshi D, Gill ON, Cane P, et al. Determinants of HIV-1 transmission in men who have sex with men: a combined clinical, epidemiological and phylogenetic approach. AIDS 2010; 24:1739–1747. [DOI] [PubMed] [Google Scholar]
- 22.Zuckerkandl E, Pauling L. Molecular disease, evolution, and genic heterogeneity. New York: Academic Press; 1962. [Google Scholar]
- 23.Britten RJ. Rates of DNA sequence evolution differ between taxonomic groups. Science 1986; 231:1393–1398. [DOI] [PubMed] [Google Scholar]
- 24.Yoder AD, Yang Z. Estimation of primate speciation dates using local molecular clocks. Mol Biol Evol 2000; 17:1081–1090. [DOI] [PubMed] [Google Scholar]
- 25.Rambaut A, Bromham L. Estimating divergence dates from molecular sequences. Mol Biol Evol 1998; 15:442–448. [DOI] [PubMed] [Google Scholar]
- 26.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol 2006; 4:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenner BG, Wainberg MA. Future of phylogeny in HIV prevention. J Acquir Immune Defic Syndr 2013; 63 Suppl 2:S248–S254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novitsky V, Moyo S, Lei Q, DeGruttola V, Essex M. Impact of sampling density on the extent of HIV clustering. AIDS Res Hum Retroviruses 2014; 30:1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prosperi MC, Ciccozzi M, Fanti I, Saladini F, Pecorari M, Borghi V, et al. A novel methodology for large-scale phylogeny partition. Nat Commun 2011; 2:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamming RW. Error detecting and error correcting codes. Bell Syst Tech J 1950; 29:147–160. [Google Scholar]
- 31.Lemey P, Salemi M, Vandamme AM. The phylogenetic handbook – a practical approach to phylogenetic analysis and hypothesis testing. 2nd ed.Cambridge: Cambridge University Press; 2009. [Google Scholar]
- 32.Hue S, Clewley JP, Cane PA, Pillay D. HIV-1 pol gene variation is sufficient for reconstruction of transmissions in the era of antiretroviral therapy. AIDS 2004; 18:719–728. [DOI] [PubMed] [Google Scholar]
- 33.Leigh Brown AJ, Lycett SJ, Weinert L, Hughes GJ, Fearnhill E, Dunn DT, et al. Transmission network parameters estimated from HIV sequences for a nationwide epidemic. J Infect Dis 2011; 204:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heimer R, Barbour R, Shaboltas AV, Hoffman IF, Kozlov AP. Spatial distribution of HIV prevalence and incidence among injection drugs users in St Petersburg: implications for HIV transmission. AIDS 2008; 22:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ragonnet-Cronin M, Hodcroft E, Hue S, Fearnhill E, Delpech V, Brown AJ, et al. Automated analysis of phylogenetic clusters. BMC Bioinformatics 2013; 14:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Efron B, Halloran E, Holmes S. Bootstrap confidence levels for phylogenetic trees. Proc Natl Acad Sci U S A 1996; 93:13429–13434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zharkikh A, Li WH. Statistical properties of bootstrap estimation of phylogenetic variability from nucleotide sequences. I. Four taxa with a molecular clock. Mol Biol Evol 1992; 9:1119–1147. [DOI] [PubMed] [Google Scholar]
- 38.Swofford DL. PAUP∗. Phylogenetic analysis using parsimony (∗and other methods). Version 4. Sunderland, Massachusetts: Sinauer Associates; 2003. [Google Scholar]
- 39.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 2006; 55:539–552. [DOI] [PubMed] [Google Scholar]
- 40.Anisimova M, Gil M, Dufayard JF, Dessimoz C, Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol 2011; 60:685–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esbjörnsson J, Mild M, Audelin A, Fonager J, Skar H, Bruun Jørgensen L, et al. HIV-1 transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic countries. Virus Evol 2016; 2:vew010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alfaro ME, Zoller S, Lutzoni F. Bayes or bootstrap? A simulation study comparing the performance of Bayesian Markov chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol Biol Evol 2003; 20:255–266. [DOI] [PubMed] [Google Scholar]
- 43.Ahumada-Ruiz S, Flores-Figueroa D, Toala-Gonzalez I, Thomson MM. Analysis of HIV-1 pol sequences from Panama: identification of phylogenetic clusters within subtype B and detection of antiretroviral drug resistance mutations. Infect Genet Evol 2009; 9:933–940. [DOI] [PubMed] [Google Scholar]
- 44.Antoniadou ZA, Kousiappa I, Skoura L, Pilalas D, Metallidis S, Nicolaidis P, et al. Short communication: molecular epidemiology of HIV type 1 infection in northern Greece (2009–2010): evidence of a transmission cluster of HIV type 1 subtype A1 drug-resistant strains among men who have sex with men. AIDS Res Hum Retroviruses 2014; 30:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Avila D, Keiser O, Egger M, Kouyos R, Boni J, Yerly S, et al. Social meets molecular: combining phylogenetic and latent class analyses to understand HIV-1 transmission in Switzerland. Am J Epidemiol 2014; 179:1514–1525. [DOI] [PubMed] [Google Scholar]
- 46.Balode D, Skar H, Mild M, Kolupajeva T, Ferdats A, Rozentale B, et al. Phylogenetic analysis of the Latvian HIV-1 epidemic. AIDS Res Hum Retroviruses 2012; 28:928–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartolo I, Zakovic S, Martin F, Palladino C, Carvalho P, Camacho R, et al. HIV-1 diversity, transmission dynamics and primary drug resistance in Angola. PLoS One 2014; 9:e113626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bello G, Afonso JM, Morgado MG. Phylodynamics of HIV-1 subtype F1 in Angola, Brazil and Romania. Infect Genet Evol 2012; 12:1079–1086. [DOI] [PubMed] [Google Scholar]
- 49.Bezemer D, Faria NR, Hassan A, Hamers RL, Mutua G, Anzala O, et al. HIV Type 1 transmission networks among men having sex with men and heterosexuals in Kenya. AIDS Res Hum Retroviruses 2014; 30:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brand D, Moreau A, Cazein F, Lot F, Pillonel J, Brunet S, et al. Characteristics of patients recently infected with HIV-1 non-B subtypes in France: a nested study within the mandatory notification system for new HIV diagnoses. J Clin Microbiol 2014; 52:4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brenner BG, Roger M, Routy JP, Moisi D, Ntemgwa M, Matte C, et al. High rates of forward transmission events after acute/early HIV-1 infection. J Infect Dis 2007; 195:951–959. [DOI] [PubMed] [Google Scholar]
- 52.Callegaro A, Svicher V, Alteri C, Lo Presti A, Valenti D, Goglio A, et al. Epidemiological network analysis in HIV-1 B infected patients diagnosed in Italy between 2000 and 2008. Infect Genet Evol 2011; 11:624–632. [DOI] [PubMed] [Google Scholar]
- 53.Castley AS, Gaudieri S, James I, Gizzarelli LS, Guelfi G, John M, et al. Longitudinal trends in Western Australian HIV-1 sequence diversity and viral transmission networks and their influence on clinical parameters: 2000–2014. AIDS Res Hum Retroviruses 2016; 32:211–219. [DOI] [PubMed] [Google Scholar]
- 54.Chalmet K, Staelens D, Blot S, Dinakis S, Pelgrom J, Plum J, et al. Epidemiological study of phylogenetic transmission clusters in a local HIV-1 epidemic reveals distinct differences between subtype B and non-B infections. BMC Infect Dis 2010; 10:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan PA, Kazi S, Rana A, Blazar I, Dejong CC, Mayer KH, et al. Short communication: new HIV infections at Southern New England academic institutions: implications for prevention. AIDS Res Hum Retroviruses 2013; 29:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan PA, Reitsma MB, DeLong A, Boucek B, Nunn A, Salemi M, et al. Phylogenetic and geospatial evaluation of HIV-1 subtype diversity at the largest HIV center in Rhode Island. Infect Genet Evol 2014; 28:358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan PA, Tashima K, Cartwright CP, Gillani FS, Mintz O, Zeller K, et al. Short communication: transmitted drug resistance and molecular epidemiology in antiretroviral naive HIV type 1-infected patients in Rhode Island. AIDS Res Hum Retroviruses 2011; 27:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang SY, Sheng WH, Lee CN, Sun HY, Kao CL, Chang SF, et al. Molecular epidemiology of HIV type 1 subtypes in Taiwan: outbreak of HIV type 1 CRF07_BC infection in intravenous drug users. AIDS Res Hum Retroviruses 2006; 22:1055–1066. [DOI] [PubMed] [Google Scholar]
- 59.Chen M, Ma Y, Su Y, Yang L, Zhang R, Yang C, et al. HIV-1 genetic characteristics and transmitted drug resistance among men who have sex with men in Kunming, China. PLoS One 2014; 9:e87033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen S, Cai W, He J, Vidal N, Lai C, Guo W, et al. Molecular epidemiology of human immunodeficiency virus type 1 in Guangdong province of southern China. PLoS One 2012; 7:e48747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chin BS, Chaillon A, Mehta SR, Wertheim JO, Kim G, Shin HS, et al. Molecular epidemiology identifies HIV transmission networks associated with younger age and heterosexual exposure among Korean individuals. J Med Virol 2016; 88:1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chin BS, Shin HS, Kim G, Wagner GA, Gianella S, Smith DM. Short communication: increase of HIV-1 K103N transmitted drug resistance and its association with efavirenz use in South Korea. AIDS Res Hum Retroviruses 2015; 31:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ciccozzi M, Madeddu G, Lo Presti A, Cella E, Giovanetti M, Budroni C, et al. HIV type 1 origin and transmission dynamics among different risk groups in Sardinia: molecular epidemiology within the close boundaries of an Italian island. AIDS Res Hum Retroviruses 2013; 29:404–410. [DOI] [PubMed] [Google Scholar]
- 64.Dauwe K, Mortier V, Schauvliege M, Van Den Heuvel A, Fransen K, Servais JY, et al. Characteristics and spread to the native population of HIV-1 non-B subtypes in two European countries with high migration rate. BMC Infect Dis 2015; 15:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davaalkham J, Unenchimeg P, Baigalmaa C, Erdenetuya G, Nyamkhuu D, Shiino T, et al. Identification of a current hot spot of HIV type 1 transmission in Mongolia by molecular epidemiological analysis. AIDS Res Hum Retroviruses 2011; 27:1073–1080. [DOI] [PubMed] [Google Scholar]
- 66.Dennis AM, Hue S, Pasquale D, Napravnik S, Sebastian J, Miller WC, et al. HIV transmission patterns among immigrant Latinos illuminated by the integration of phylogenetic and migration data. AIDS Res Hum Retroviruses 2015; 31:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drescher SM, von Wyl V, Yang WL, Boni J, Yerly S, Shah C, et al. Treatment-naive individuals are the major source of transmitted HIV-1 drug resistance in men who have sex with men in the Swiss HIV cohort study. Clin Infect Dis 2014; 58:285–294. [DOI] [PubMed] [Google Scholar]
- 68.Esbjornsson J, Mild M, Mansson F, Norrgren H, Medstrand P. HIV-1 molecular epidemiology in Guinea-Bissau, West Africa: origin, demography and migrations. PLoS One 2011; 6:e17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fabeni L, Alteri C, Orchi N, Gori C, Bertoli A, Forbici F, et al. Recent transmission clustering of HIV-1 C and CRF17_BF strains characterized by NNRTI-related mutations among newly diagnosed men in central Italy. PLoS One 2015; 10:e0135325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frange P, Meyer L, Deveau C, Tran L, Goujard C, Ghosn J, et al. Recent HIV-1 infection contributes to the viral diffusion over the French territory with a recent increasing frequency. PLoS One 2012; 7:e31695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frentz D, Wensing AM, Albert J, Paraskevis D, Abecasis AB, Hamouda O, et al. Limited cross-border infections in patients newly diagnosed with HIV in Europe. Retrovirology 2013; 10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grgic I, Lepej SZ, Lunar MM, Poljak M, Vince A, Vrakela IB, et al. The prevalence of transmitted drug resistance in newly diagnosed HIV-infected individuals in Croatia: the role of transmission clusters of men who have sex with men carrying the T215S surveillance drug resistance mutation. AIDS Res Hum Retroviruses 2013; 29:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guimaraes ML, Marques BC, Bertoni N, Teixeira SL, Morgado MG, Bastos FI. Assessing the HIV-1 epidemic in Brazilian drug users: a molecular epidemiology approach. PLoS One 2015; 10:e0141372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hofstra LM, Nijhuis M, Pingen M, Mudrikova T, Riezebos-Brilman A, Simoons-Smit AM, et al. Evolution and viral characteristics of a long-term circulating resistant HIV-1 strain in a cluster of treatment-naive patients. J Antimicrob Chemother 2013; 68:1246–1250. [DOI] [PubMed] [Google Scholar]
- 75.Hughes GJ, Fearnhill E, Dunn D, Lycett SJ, Rambaut A, Leigh Brown AJ, et al. Molecular phylodynamics of the heterosexual HIV epidemic in the United Kingdom. PLoS Pathog 2009; 5:e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kapaata A, Lyagoba F, Ssemwanga D, Magambo B, Nanyonjo M, Levin J, et al. HIV-1 subtype distribution trends and evidence of transmission clusters among incident cases in a rural clinical cohort in southwest Uganda, 2004–2010. AIDS Res Hum Retroviruses 2013; 29:520–527. [DOI] [PubMed] [Google Scholar]
- 77.Karlsson A, Bjorkman P, Bratt G, Ekvall H, Gisslen M, Sonnerborg A, et al. Low prevalence of transmitted drug resistance in patients newly diagnosed with HIV-1 infection in Sweden 2003–2010. PLoS One 2012; 7:e33484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kouyos RD, von Wyl V, Yerly S, Boni J, Taffe P, Shah C, et al. Molecular epidemiology reveals long-term changes in HIV type 1 subtype B transmission in Switzerland. J Infect Dis 2010; 201:1488–1497. [DOI] [PubMed] [Google Scholar]
- 79.Kramer MA, Cornelissen M, Paraskevis D, Prins M, Coutinho RA, van Sighem AI, et al. HIV transmission patterns among The Netherlands, Suriname, and The Netherlands Antilles: a molecular epidemiological study. AIDS Res Hum Retroviruses 2011; 27:123–130. [DOI] [PubMed] [Google Scholar]
- 80.Lai A, Bozzi G, Franzetti M, Binda F, Simonetti FR, Micheli V, et al. Phylogenetic analysis provides evidence of interactions between Italian heterosexual and South American homosexual males as the main source of national HIV-1 subtype C epidemics. J Med Virol 2014; 86:729–736. [DOI] [PubMed] [Google Scholar]
- 81.Lai A, Simonetti FR, Zehender G, De Luca A, Micheli V, Meraviglia P, et al. HIV-1 subtype F1 epidemiological networks among Italian heterosexual males are associated with introduction events from South America. PLoS One 2012; 7:e42223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lawyer G, Schulter E, Kaiser R, Reuter S, Oette M, Lengauer T. Endogenous or exogenous spreading of HIV-1 in Nordrhein-Westfalen, Germany, investigated by phylodynamic analysis of the RESINA Study cohort. Med Microbiol Immunol 2012; 201:259–269. [DOI] [PubMed] [Google Scholar]
- 83.Lepej SZ, Vrakela IB, Poljak M, Bozicevic I, Begovac J. Phylogenetic analysis of HIV sequences obtained in a respondent-driven sampling study of men who have sex with men. AIDS Res Hum Retroviruses 2009; 25:1335–1338. [DOI] [PubMed] [Google Scholar]
- 84.Lewis F, Hughes GJ, Rambaut A, Pozniak A, Leigh Brown AJ. Episodic sexual transmission of HIV revealed by molecular phylodynamics. PLoS Med 2008; 5:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li L, Chen L, Liang S, Liu W, Li T, Liu Y, et al. Subtype CRF01_AE dominate the sexually transmitted human immunodeficiency virus type 1 epidemic in Guangxi, China. J Med Virol 2013; 85:388–395. [DOI] [PubMed] [Google Scholar]
- 86.Ng KT, Ong LY, Lim SH, Takebe Y, Kamarulzaman A, Tee KK. Evolutionary history of HIV-1 subtype B and CRF01_AE transmission clusters among men who have sex with men (MSM) in Kuala Lumpur, Malaysia. PLoS One 2013; 8:e67286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li L, Wei D, Hsu WL, Li T, Gui T, Wood C, et al. CRF07_BC Strain Dominates the HIV-1 Epidemic in Injection Drug Users in Liangshan Prefecture of Sichuan, China. AIDS Res Hum Retroviruses 2015; 31:479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li X, Xue Y, Cheng H, Lin Y, Zhou L, Ning Z, et al. HIV-1 genetic diversity and its impact on baseline CD4+T cells and viral loads among recently infected men who have sex with men in Shanghai, China. PLoS One 2015; 10:e0129559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X, Zang X, Ning C, Feng Y, Xie C, He X, et al. Molecular epidemiology of HIV-1 in Jilin province, Northeastern China: emergence of a new CRF07_BC transmission cluster and inter subtype recombinants. PLoS One 2014; 9:e110738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lunar MM, Vandamme AM, Tomazic J, Karner P, Vovko TD, Pecavar B, et al. Bridging epidemiology with population genetics in a low incidence MSM-driven HIV-1 subtype B epidemic in Central Europe. BMC Infect Dis 2015; 15:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mbisa JL, Hue S, Buckton AJ, Myers RE, Duiculescu D, Ene L, et al. Phylodynamic and phylogeographic patterns of the HIV type 1 subtype F1 parenteral epidemic in Romania. AIDS Res Hum Retroviruses 2012; 28:1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mehta SR, Wertheim JO, Brouwer KC, Wagner KD, Chaillon A, Strathdee S, et al. HIV transmission networks in the San Diego–Tijuana border region. EBioMedicine 2015; 2:1456–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Metzner KJ, Scherrer AU, Preiswerk B, Joos B, von Wyl V, Leemann C, et al. Origin of minority drug-resistant HIV-1 variants in primary HIV-1 infection. J Infect Dis 2013; 208:1102–1112. [DOI] [PubMed] [Google Scholar]
- 94.Niculescu I, Paraschiv S, Paraskevis D, Abagiu A, Batan I, Banica L, et al. Recent HIV-1 outbreak among intravenous drug users in Romania: evidence for cocirculation of CRF14_BG and subtype F1 strains. AIDS Res Hum Retroviruses 2015; 31:488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Novitsky V, Bussmann H, Logan A, Moyo S, van Widenfelt E, Okui L, et al. Phylogenetic relatedness of circulating HIV-1 C variants in Mochudi, Botswana. PLoS One 2013; 8:e80589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paraschiv S, Otelea D, Batan I, Baicus C, Magiorkinis G, Paraskevis D. Molecular typing of the recently expanding subtype B HIV-1 epidemic in Romania: evidence for local spread among MSMs in Bucharest area. Infect Genet Evol 2012; 12:1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paraskevis D, Kostaki E, Beloukas A, Canizares A, Aguilera A, Rodriguez J, et al. Molecular characterization of HIV-1 infection in Northwest Spain (2009–2013): investigation of the subtype F outbreak. Infect Genet Evol 2015; 30:96–101. [DOI] [PubMed] [Google Scholar]
- 98.Parczewski M, Leszczyszyn-Pynka M, Witak-Jedra M, Maciejewska K, Rymer W, Szymczak A, et al. Transmitted HIV drug resistance in antiretroviral-treatment-naive patients from Poland differs by transmission category and subtype. J Antimicrob Chemother 2015; 70:233–242. [DOI] [PubMed] [Google Scholar]
- 99.Patino-Galindo JA, Thomson MM, Perez-Alvarez L, Delgado E, Cuevas MT, Fernandez-Garcia A, et al. Transmission dynamics of HIV-1 subtype B in the Basque Country, Spain. Infect Genet Evol 2016; 40:91–97. [DOI] [PubMed] [Google Scholar]
- 100.Peng X, Wu H, Peng X, Jin C, Wu N. Heterogeneous evolution of HIV-1 CRF01_AE in men who have sex with men (MSM) and other populations in China. PLoS One 2015; 10:e0143699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pilon R, Leonard L, Kim J, Vallee D, De Rubeis E, Jolly AM, et al. Transmission patterns of HIV and hepatitis C virus among networks of people who inject drugs. PLoS One 2011; 6:e22245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pineda-Pena AC, Schrooten Y, Vinken L, Ferreira F, Li G, Trovao NS, et al. Trends and predictors of transmitted drug resistance (TDR) and clusters with TDR in a local Belgian HIV-1 epidemic. PLoS One 2014; 9:e101738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prellwitz IM, Alves BM, Ikeda ML, Kuhleis D, Picon PD, Jarczewski CA, et al. HIV behind bars: human immunodeficiency virus cluster analysis and drug resistance in a reference correctional unit from southern Brazil. PLoS One 2013; 8:e69033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Robineau O, Frange P, Barin F, Cazein F, Girard PM, Chaix ML, et al. Combining the estimated date of HIV infection with a phylogenetic cluster study to better understand HIV spread: application in a Paris neighbourhood. PLoS One 2015; 10:e0135367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schulter E, Oette M, Balduin M, Reuter S, Rockstroh J, Fatkenheuer G, et al. HIV prevalence and route of transmission in Turkish immigrants living in North-Rhine Westphalia, Germany. Med Microbiol Immunol 2011; 200:219–223. [DOI] [PubMed] [Google Scholar]
- 106.Shiino T, Hattori J, Yokomaku Y, Iwatani Y, Sugiura W. Phylodynamic analysis reveals CRF01_AE dissemination between Japan and neighboring Asian countries and the role of intravenous drug use in transmission. PLoS One 2014; 9:e102633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Siljic M, Salemovic D, Jevtovic D, Pesic-Pavlovic I, Zerjav S, Nikolic V, et al. Molecular typing of the local HIV-1 epidemic in Serbia. Infect Genet Evol 2013; 19:378–385. [DOI] [PubMed] [Google Scholar]
- 108.Skar H, Axelsson M, Berggren I, Thalme A, Gyllensten K, Liitsola K, et al. Dynamics of two separate but linked HIV-1 CRF01_AE outbreaks among injection drug users in Stockholm, Sweden, and Helsinki, Finland. J Virol 2011; 85:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Skoura L, Metallidis S, Buckton AJ, Mbisa JL, Pilalas D, Papadimitriou E, et al. Molecular and epidemiological characterization of HIV-1 infection networks involving transmitted drug resistance mutations in Northern Greece. J Antimicrob Chemother 2011; 66:2831–2837. [DOI] [PubMed] [Google Scholar]
- 110.Takebe Y, Naito Y, Raghwani J, Fearnhill E, Sano T, Kusagawa S, et al. Intercontinental dispersal of HIV-1 subtype B associated with transmission among men who have sex with men in Japan. J Virol 2014; 88:9864–9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tamalet C, Ravaux I, Moreau J, Bregigeon S, Tourres C, Richet H, et al. Emergence of clusters of CRF02_AG and B human immunodeficiency viral strains among men having sex with men exhibiting HIV primary infection in Southeastern France. J Med Virol 2015; 87:1327–1333. [DOI] [PubMed] [Google Scholar]
- 112.Temereanca A, Ene L, Mehta S, Manolescu L, Duiculescu D, Ruta S. Transmitted HIV drug resistance in treatment-naive Romanian patients. J Med Virol 2013; 85:1139–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Turner D, Amit S, Chalom S, Penn O, Pupko T, Katchman E, et al. Emergence of an HIV-1 cluster harbouring the major protease L90M mutation among treatment-naive patients in Tel Aviv, Israel. HIV Med 2012; 13:202–206. [DOI] [PubMed] [Google Scholar]
- 114.Wang X, Wu Y, Mao L, Xia W, Zhang W, Dai L, et al. Targeting HIV prevention based on molecular epidemiology among deeply sampled subnetworks of men who have sex with men. Clin Infect Dis 2015; 61:1462–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vega Y, Delgado E, Fernandez-Garcia A, Cuevas MT, Thomson MM, Montero V, et al. Epidemiological surveillance of HIV-1 transmitted drug resistance in Spain in 2004–2012: relevance of transmission clusters in the propagation of resistance mutations. PLoS One 2015; 10:e0125699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wilkinson E, Engelbrecht S, de Oliveira T. Detection of transmission clusters of HIV-1 subtype C over a 21-year period in Cape Town, South Africa. PLoS One 2014; 9:e109296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.von Wyl V, Kouyos RD, Yerly S, Boni J, Shah C, Burgisser P, et al. The role of migration and domestic transmission in the spread of HIV-1 non-B subtypes in Switzerland. J Infect Dis 2011; 204:1095–1103. [DOI] [PubMed] [Google Scholar]
- 118.Ambrosioni J, Junier T, Delhumeau C, Calmy A, Hirschel B, Zdobnov E, et al. Impact of highly active antiretroviral therapy on the molecular epidemiology of newly diagnosed HIV infections. AIDS 2012; 26:2079–2086. [DOI] [PubMed] [Google Scholar]
- 119.Audelin AM, Cowan SA, Obel N, Nielsen C, Jorgensen LB, Gerstoft J. Phylogenetics of the Danish HIV epidemic: the role of very late presenters in sustaining the epidemic. J Acquir Immune Defic Syndr 2013; 62:102–108. [DOI] [PubMed] [Google Scholar]
- 120.Bezemer D, van Sighem A, Lukashov VV, van der Hoek L, Back N, Schuurman R, et al. Transmission networks of HIV-1 among men having sex with men in the Netherlands. AIDS 2010; 24:271–282. [DOI] [PubMed] [Google Scholar]
- 121.Brenner BG, Roger M, Moisi DD, Oliveira M, Hardy I, Turgel R, et al. Transmission networks of drug resistance acquired in primary/early stage HIV infection. AIDS 2008; 22:2509–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chan PA, Hogan JW, Huang A, DeLong A, Salemi M, Mayer KH, et al. Phylogenetic investigation of a statewide HIV-1 epidemic reveals ongoing and active transmission networks among men who have sex with men. J Acquir Immune Defic Syndr 2015; 70:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee SS, Tam DK, Tan Y, Mak WL, Wong KH, Chen JH, et al. An exploratory study on the social and genotypic clustering of HIV infection in men having sex with men. AIDS 2009; 23:1755–1764. [DOI] [PubMed] [Google Scholar]
- 124.Cuevas MT, Munoz-Nieto M, Thomson MM, Delgado E, Iribarren JA, Cilla G, et al. HIV-1 transmission cluster with T215D revertant mutation among newly diagnosed patients from the Basque Country, Spain. J Acquir Immune Defic Syndr 2009; 51:99–103. [DOI] [PubMed] [Google Scholar]
- 125.de Silva TI, van Tienen C, Onyango C, Jabang A, Vincent T, Loeff MF, et al. Population dynamics of HIV-2 in rural West Africa: comparison with HIV-1 and ongoing transmission at the heart of the epidemic. AIDS 2013; 27:125–134. [DOI] [PubMed] [Google Scholar]
- 126.Deng W, Fu P, Bao L, Vidal N, He Q, Qin C, et al. Molecular epidemiological tracing of HIV-1 outbreaks in Hainan island of southern China. AIDS 2009; 23:977–985. [DOI] [PubMed] [Google Scholar]
- 127.Dennis AM, Hue S, Hurt CB, Napravnik S, Sebastian J, Pillay D, et al. Phylogenetic insights into regional HIV transmission. AIDS 2012; 26:1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dennis AM, Murillo W, de Maria Hernandez F, Guardado ME, Nieto AI, Lorenzana de Rivera I, et al. Social network-based recruitment successfully reveals HIV-1 transmission networks among high-risk individuals in El Salvador. J Acquir Immune Defic Syndr 2013; 63:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Eyer-Silva WA, Morgado MG. Autochthonous horizontal transmission of a CRF02_AG strain revealed by a human immunodeficiency virus type 1 diversity survey in a small city in inner state of Rio de Janeiro, Southeast Brazil. Mem Inst Oswaldo Cruz 2007; 102:809–815. [DOI] [PubMed] [Google Scholar]
- 130.Feng Y, He X, Hsi JH, Li F, Li X, Wang Q, et al. The rapidly expanding CRF01_AE epidemic in China is driven by multiple lineages of HIV-1 viruses introduced in the 1990s. AIDS 2013; 27:1793–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hue S, Brown AE, Ragonnet-Cronin M, Lycett SJ, Dunn DT, Fearnhill E, et al. Phylogenetic analyses reveal HIV-1 infections between men misclassified as heterosexual transmissions. AIDS 2014; 28:1967–1975. [DOI] [PubMed] [Google Scholar]
- 132.Kaye M, Chibo D, Birch C. Phylogenetic investigation of transmission pathways of drug-resistant HIV-1 utilizing pol sequences derived from resistance genotyping. J Acquir Immune Defic Syndr 2008; 49:9–16. [DOI] [PubMed] [Google Scholar]
- 133.Li Z, Liao L, Feng Y, Zhang J, Yan J, He C, et al. Trends of HIV subtypes and phylogenetic dynamics among young men who have sex with men in China, 2009–2014. Sci Rep 2015; 5:16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lubelchek RJ, Hoehnen SC, Hotton AL, Kincaid SL, Barker DE, French AL. Transmission clustering among newly diagnosed HIV patients in Chicago, 2008 to 2011: using phylogenetics to expand knowledge of regional HIV transmission patterns. J Acquir Immune Defic Syndr 2015; 68:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lukashov VV, Jurriaans S, Bakker M, Berkhout B. Transmission of risk-group specific HIV-1 strains among Dutch drug users for more than 20 years and their replacement by nonspecific strains after switching to low-harm drug practices. J Acquir Immune Defic Syndr 2013; 62:234–238. [DOI] [PubMed] [Google Scholar]
- 136.Mourad R, Chevennet F, Dunn DT, Fearnhill E, Delpech V, Asboe D, et al. A phylotype-based analysis highlights the role of drug-naive HIV-positive individuals in the transmission of antiretroviral resistance in the UK. AIDS 2015; 29:1917–1925. [DOI] [PubMed] [Google Scholar]
- 137.Oster AM, Wertheim JO, Hernandez AL, Ocfemia MC, Saduvala N, Hall HI. Using molecular HIV surveillance data to understand transmission between subpopulations in the United States. J Acquir Immune Defic Syndr 2015; 70:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Panichsillapakit T, Smith DM, Wertheim JO, Richman DD, Little SJ, Mehta SR. Prevalence of transmitted HIV drug resistance among recently infected persons in San Diego, CA 1996–2013. J Acquir Immune Defic Syndr 2016; 71:228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rieder P, Joos B, von Wyl V, Kuster H, Grube C, Leemann C, et al. HIV-1 transmission after cessation of early antiretroviral therapy among men having sex with men. AIDS 2010; 24:1177–1183. [DOI] [PubMed] [Google Scholar]
- 140.Truong HM, Pipkin S, O’Keefe KJ, Louie B, Liegler T, McFarland W, et al. Brief report: recent infection, sexually transmitted infections, and transmission clusters frequently observed among persons newly diagnosed with HIV in San Francisco. J Acquir Immune Defic Syndr 2015; 69:606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yebra G, Ragonnet-Cronin M, Ssemwanga D, Parry CM, Logue CH, Cane PA, et al. Analysis of the history and spread of HIV-1 in Uganda using phylodynamics. J Gen Virol 2015; 96:1890–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li L, Han N, Lu J, Li T, Zhong X, Wu H, et al. Genetic characterization and transmitted drug resistance of the HIV type 1 epidemic in men who have sex with men in Beijing, China. AIDS Res Hum Retroviruses 2013; 29:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 1993; 10:512–526. [DOI] [PubMed] [Google Scholar]
- 144.UNAIDS, UNAIDS. AIDS by the numbers. 2016; http://www.unaids.org/en/resources/documents/2016/AIDS-by-the-numbers [Google Scholar]
- 145.Smith AD, Tapsoba P, Peshu N, Sanders EJ, Jaffe HW. Men who have sex with men and HIV/AIDS in sub-Saharan Africa. Lancet 2009; 374:416–422. [DOI] [PubMed] [Google Scholar]
- 146.SPREAD programme. Transmission of drug-resistant HIV-1 in Europe remains limited to single classes. AIDS 2008; 22:625–635. [DOI] [PubMed] [Google Scholar]
- 147.Liitsola K, Ristola M, Holmstrom P, Salminen M, Brummer-Korvenkontio H, Simola S, et al. An outbreak of the circulating recombinant form AECM240 HIV-1 in the Finnish injection drug user population. AIDS 2000; 14:2613–2615. [DOI] [PubMed] [Google Scholar]
- 148.Murphy G, Parry JV. Assays for the detection of recent infections with human immunodeficiency virus type 1. Euro Surveill 2008; 13:18966. [PubMed] [Google Scholar]
- 149.Mastro TD, Kim AA, Hallett T, Rehle T, Welte A, Laeyendecker O, et al. Estimating HIV incidence in populations using tests for recent infection: issues, challenges and the way forward. J HIV AIDS Surveill Epidemiol 2010; 2:1–14. [PMC free article] [PubMed] [Google Scholar]
- 150.Vrbik I, Stephens DA, Roger M, Brenner BG. The gap procedure: for the identification of phylogenetic clusters in HIV-1 sequence data. BMC Bioinformatics 2015; 16:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Villandre L, Stephens DA, Labbe A, Gunthard HF, Kouyos R, Stadler T, et al. Assessment of overlap of phylogenetic transmission clusters and communities in simple sexual contact networks: applications to HIV-1. PLoS One 2016; 11:e0148459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zarrabi N, Prosperi M, Belleman RG, Colafigli M, De Luca A, Sloot PM. Combining epidemiological and genetic networks signifies the importance of early treatment in HIV-1 transmission. PLoS One 2012; 7:e46156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leigh Brown AJ. HIV-TRACE. http://test.datamonkey.org/hivtrace. [Google Scholar]
- 154.Jacka B, Applegate T, Poon AF, Raghwani J, Harrigan PR, DeBeck K, et al. Transmission of hepatitis C virus infection among younger and older people who inject drugs in Vancouver, Canada. J Hepatol 2016; 64:1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Minin VN, Suchard MA. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol 2008; 56:391–412. [DOI] [PubMed] [Google Scholar]
- 156.Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol 2010; 27:1877–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol 2009; 5:e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Neogi U, Haggblom A, Santacatterina M, Bratt G, Gisslen M, Albert J, et al. Temporal trends in the Swedish HIV-1 epidemic: increase in non-B subtypes and recombinant forms over three decades. PLoS One 2014; 9:e99390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moorthie S, Mattocks CJ, Wright CF. Review of massively parallel DNA sequencing technologies. Hugo J 2011; 5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Eshleman SH, Hudelson SE, Redd AD, Wang L, Debes R, Chen YQ, et al. Analysis of genetic linkage of HIV from couples enrolled in the HIV Prevention Trials Network 052 trial. J Infect Dis 2011; 204:1918–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lemey P, Rambaut A, Pybus OG. HIV evolutionary dynamics within and among hosts. AIDS Rev 2006; 8:125–140. [PubMed] [Google Scholar]
- 162.Maljkovic Berry I, Ribeiro R, Kothari M, Athreya G, Daniels M, Lee HY, et al. Unequal evolutionary rates in the human immunodeficiency virus type 1 (HIV-1) pandemic: the evolutionary rate of HIV-1 slows down when the epidemic rate increases. J Virol 2007; 81:10625–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]