

Abstract
We identified a glycoprotein hormone β-subunit (OGH, also called GPB5) that, as a heterodimer with the α-subunit GPA2, serves as a second ligand for the thyroid-stimulating hormone receptor. Mice in which the OGH gene is deleted (OGH-/-) are indistinguishable from WT littermates in body weight, response to high-fat diet, metabolic parameters, body composition, and insulin tolerance. Mice engineered to transgenically globally overexpress OGH (OGH-TG) develop ≈2-fold elevations in their basal thyroid levels and weigh slightly less than WT littermates despite increased food intake because of an increase in their metabolic rates. Moreover, when OGH-TG mice are challenged with a high-fat diet, they gain significantly less weight and body fat than their WT littermates. The OGH-TG mice also have reduced blood glucose, insulin, cholesterol, and triglycerides. In contrast to other approaches in which the thyroid axis is activated, OGH-TG mice exhibit only minor changes in heart rate and blood pressure. Our findings suggest that constitutive low-level activation of the thyroid axis (via OGH or other means) may provide a beneficial therapeutic approach for combating diet-induced obesity.
Keywords: cholesterol, metabolic rate, thyroid
Thyroid-stimulating hormone (TSH) and the TSH receptor (TSHR) are key proteins in the control of thyroid function. TSH synthesis and release is stimulated by hypothalamic TSH-releasing hormone (TRH) and is down-regulated (inhibited) by thyroid hormone in a classic endocrine negative-feedback loop. The specificity inherent in TSH resides in its unique β-subunit that heterodimerizes with a common α-subunit, which it shares with the other glycoprotein hormones [i.e., follicle-stimulating hormone (FSH), luteinizing hormone, and chorionic gonadotropin]. The primary physiological actions of TSH on the thyroid are stimulation of the synthesis and release of 3,5,3′,5′-tetraiodo-l-thyronine (T4) and 3,5,3′-triiodo-l-thyronine (T3) (together termed thyroid hormone) and promotion of thyroid growth; for example, it has been shown that thyroid development is arrested in mice with targeted disruption of the common α-subunit (1).
Clinically, thyroid hormone is used primarily as a replacement therapy for the patients with hypothyroidism, and it has been considered as a possible therapy for weight reduction (because of its ability to increase metabolism and energy expenditures), for lowering cholesterol, and even to build bone (in osteoporosis). One of the major hurdles in this approach has been the cardiovascular toxicity observed after administration of the endogenous ligands T4 and T3 or nonselective thyroid hormone agonists. The two major subtypes of the thyroid hormone receptors that mediate these responses, TRα and TRβ, are the products of different genes and are also differentially processed to each yield two isoforms. It has been argued that modulation of heart rate and rhythm is mediated predominantly by activation of TRα1 (2–4), and as a result, recent pharmaceutical research efforts have focused on developing specific TRβ1 agonists (5–8).
During a bioinformatic analysis of the human genome sequence, we identified a human gene encoding a homologue of the glycoprotein hormone β-subunits. Because the receptor and function of the predicted protein were not known, we named it OGH (orphan glycoprotein hormone), and we initiated a study of its function by creating genetically modified mice. Specifically, we generated OGH knockout/lacZ knock-in mice (OGH-/-), as well as mice that globally overexpress OGH (OGH-TG). During the construction and analysis of our mouse genetic models, Hsu et al. (9) described a gene, identical to OGH, termed GPB5. They also identified a human homologue, called GPA2, of the common glycoprotein hormone α-subunit. Subsequently, it was shown that GPB5 and GPA2 heterodimerize and that the heterodimer activates the TSHR, and the term thyrostimulin was coined to describe the GPB5/GPA2 heterodimer (10). Thus, the OGH-/- and OGH-TG mice have allowed us to investigate the physiological role of this active β-subunit in normal thyroid physiology, study its effects on various aspects of thyroid activity (TSH, T3/T4, and thyroid histology), and identify key hormonal and metabolic perturbations that translate into important effects in regulation of body weight.
Experimental Procedures
Genetic Manipulations and Animal Procedures. To globally overexpress OGH, the mouse OGH cDNA coding sequence was “knocked-in” to the Rosa26 locus (11) (Fig. 1 A and B). We also generated CJ7 ES cells in which the coding region of the OGH gene was precisely deleted (from initiation to termination codon) and replaced with a lacZ reporter and neomycin selectable marker (Fig. 1B) by using velocigene technology (12). Heterozygous N2 mice (backcrossed to C57BL/6J) were bred to homozygosity, and correct targeting was reconfirmed by Southern blotting (Fig. 1E). All procedures were conducted in compliance with protocols approved by the Regeneron Institutional Animal Care and Use Committee. Animals had free access to either standard chow (5020, Purina) or high-fat diet (HFD; 45% fat; 93075, Harlan Teklad, Madison, WI), as specified.
Fig. 1.

Design and verification of targeted alleles. (A) OGH-TG. (Upper) OGH-TG. (Lower) WT Rosa26. The arrow-labeled OGH indicates adenovirus splice acceptor, mouse OGH cDNA coding sequence, and rabbit β-globin polyA. The arrow-labeled PGK-neo indicates mouse PGK promoter, Tn5 neo, and gene mouse PGK polyA. Restriction sites and probe used for Southern blotting are shown. (B) OGH knockout/lacZ knock-in. (Upper) Endogenous mouse OGH gene. (Lower) Targeted allele. Restriction sites, predicted band size, and probe used for Southern blotting are shown. (C) Southern blot analysis of tail DNA from WT and transgenic (OGH-TG) animals confirming correct targeting. (D) TaqMan analysis of total RNA from three OGH-TG mice and three WT littermates (one male and two females, for each group). H, heart; K, kidney; L, liver. (E) Southern blot analysis of tail DNA from WT, heterozygous, (OGH+/-), and homozygous knockout (OGH-/-) animals.
lacZ and Histological Analyses. Whole-mount lacZ analysis was conducted as described (13). For histomorphometric analysis, skulls were cleared of surrounding tissue and stained with alcian blue and alizarin red. Digital photographs were taken of the dorsal aspect of the skull, and measurements were made by using the public-domain image-analysis program image (available at: http://rsb.info.nih.gov/nih-image). Measurements of the lengths of the nasal, frontal, and parietal bones were performed along the sagittal suture. The width of the parietal bone was measured along a line perpendicular to the midsagittal plane and extending from the sagittal suture to the lateral crest of the parietal bone.
Serum Measurements. Basal serum samples were taken between 10 a.m. and 12 p.m. after an overnight fast and analyzed for glucose, triglycerides, cholesterol, and total T3 by using the 1650 blood-chemistry analyzer (Bayer, Tarrytown, NY). Insulin levels were analyzed by using LincoPlex (Linco Research, St. Charles, MO). TSH and total T4 levels were analyzed by radioimmunoassay (A. F. Parlow, Research and Education Institute, University of Calfornia, Los Angeles).
Indirect Calorimetry, Food Intake, and Body Composition. Metabolic parameters and body composition were assessed as described (14).
Cardiovascular Telemetry. Blood pressure and heart rate measurements were recorded in unrestrained, conscious 12-week-old male mice by implanted transmitters (PA-C20, Datasciences, Minneapolis). According to the manufacturer's instructions, the transmitter device was implanted for carotid artery access to aortic blood flow. After recovery, data were sampled in 10-s intervals every 5 min for 3 days and analyzed by using dataquest software (Data Sciences International, Arden Hills, MN). Data collected from each animal were condensed to single mean values for light- and dark-cycle heart rate, diastolic and systolic blood pressure, with SEM reflecting variability between study animals.
Statistical Analysis. Data are expressed as mean ± SEM. Comparison of means was carried out by using a t test or ANOVA, where appropriate, with the program statview (SAS Institute, Cary, NC). When a significant F ratio was obtained, post hoc analysis was conducted between groups by using a multiple comparison procedure with Bonferroni–Dunn correction of means (ANOVA). P < 0.05 was considered statistically significant.
Results and Discussion
Identification of the OGH Gene. We first identified the second exon of human OGH genomic sequence by homology searches (tblastn) against follicle-stimulating hormone (FSH) of high-throughput genomic sequences (htgs) available within division of GenBank. OGH is encoded within two exons in both humans and mice (as are homologous genes for FSH, luteinizing hormone, and TSH), and upon resequencing, analysis of sequence upstream of the second exon revealed a potential first exon with an appropriate signal sequence. Mouse bacterial artificial chromosomes were identified by hybridization to the human OGH exon2 sequences and sequencing showed a high degree of conservation with the human gene. The human sequence that was identified as OGH was later published as GPB5 (9). Identical sequences for the human and mouse OGH proteins and cDNAs have been deposited in the GenBank database [accession nos. AAO33390 (human protein), AAO33391 (mouse protein), AF467770 (human cDNA), and XM_138061 (mouse cDNA)].
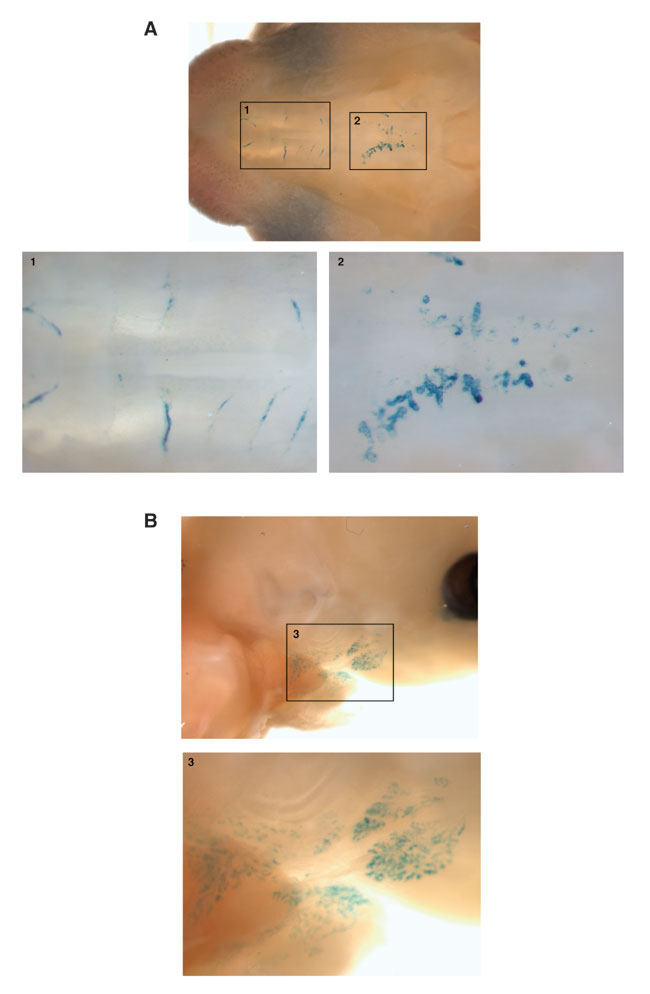
Deletion and Expression Pattern of Endogenous OGH Gene. OGH-/- mice were generated by means of velocigene technology (12) by using bacterial artificial chromosome-based targeting vectors to precisely replace the OGH coding exons with a lacZ-neo cassette (Fig. 1B), and targeting was confirmed by Southern blotting (Fig. 1E). We observed a normal birth ratio of WT, heterozygous (OGH+/-) and homozygous (OGH-/-) mice, and male and female OGH-/- mice appeared grossly normal. To assess expression of the endogenous OGH gene, we performed whole-body lacZ analysis on adult OGH+/- mice. We found no abundant, but a few low-level sites of expression. lacZ was detected over background in a specific region of the brain around the caudal fasciculus retroflexus, in retina and in the seminiferous tubules of the testes (data not shown). The significance of the testicular staining is questionable, because we often observe promiscuous β-gal staining in testis. In contrast to adult animals, newborns showed strong lacZ staining in a subset of salivary glands as well as apparent ducts in the palate (see Fig. 5, which is published as supporting information on the PNAS web site). Although it would seem surprising that a presumptive endocrine factor is strongly expressed in an exocrine gland, there have been numerous reports of other growth factors being expressed in salivary glands (15–19), and it has been shown that endocrine factors expressed after introduction of DNA into these glands are readily secreted into the bloodstream (20, 21). Indeed, a body of opinion contends that salivary glands have a mixed exocrine and endocrine function (22–24). Similarly, we also saw expression in the lacrimal glands of newborn mice. We were unable to determine the functional significance of the observed expression pattern because the OGH-/- mice were of normal body weight, composition, and metabolic parameters (data not shown).
Global OGH Overexpression. Genes inserted into the Rosa26 locus are expressed in virtually every tissue of the body in adult mice and embryos (25–28). Thus, we found a high level of mRNA expression from the Rosa-OGH transgene in F1 heterozygous mice by real-time quantitative RT-PCR analysis of three arbitrarily chosen tissues, heart, kidney, and liver (Fig. 1D). In contrast, the levels of endogenous OGH mRNA were found to be very low (<1 mRNA molecule for every five cells) in these tissues (Fig. 1D) and ≈30 other mouse and examined human tissues (data not shown), consistent with the rare, limited expression of the lacZ reporter gene in OGH-/- mice.
Whereas we observed a normal birth ratio of WT and heterozygous (OGH-TG) mice, all OGH-TG mice had a shortened snout (Fig. 2). This phenotype is first apparent at ≈10 days of age and becomes more prominent as the animals mature. We measured the lengths and widths of facial bones of adult OGH-TG and WT animals and determined that a significant shortening of the nasal (Fig. 2C, graph 1) and frontal (Fig. 2C, graph 2) bones occurs in the transgenic animals, whereas no difference was seen in the length (Fig. 2C, graph 3) or width (Fig. 2C, graph 4) of the parietal bone. Interestingly, the bones of the face differ in their ontogenic origins. The frontal and nasal bones are ectodermal structures arising from the neural crest, whereas the parietal bone, like the other bones of the body, is of mesodermal origin (29). No other skeletal defect was detected in OGH-TG mice.
Fig. 2.

Histomorphometric analysis of skull bones. (A) Bright field (Upper) and x-ray (Lower) photographs of WT and transgenic (OGH-Tg) male animals. (B) Schematic representation of the skull bones. (C) Average lengths and widths of the skull bones from adult WT (four male and four female) and transgenic (OGH-Tg; three male and four female) animals. *, P < 0.05, compared with WT littermate controls.
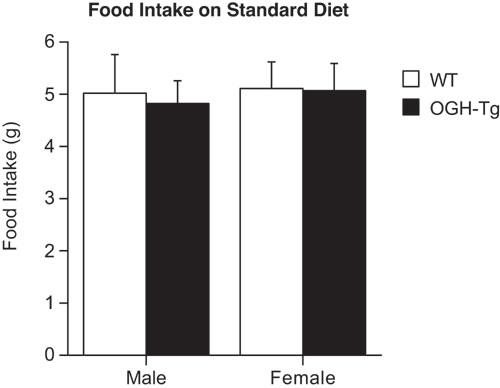
Analysis of Body Weight and Composition. Mice homozygous for the OGH deletion were identical to their WT littermates with respect to overall appearance, body weight, body composition (as measured by pDEXA), metabolic parameters, and response to HFD (data not shown). Unlike the knockouts, the OGH-TG mice exhibited a decreased body weight that first appeared at ≈1 month of age and persisted into adulthood (Fig. 3A). Because there was no overall difference in body length in these animals (data not shown), we investigated whether the body weight difference was the result of altered caloric intake or metabolism. When we measured food consumption, we found that instead of eating less than their heavier WT littermates, the OGH-TG mice actually tended to eat slightly more (see Fig. 6, which is published as supporting information on the PNAS web site). When placed on a HFD, male OGH-TG mice gained significantly less weight (14% increase after 12 weeks) compared with their WT littermates (50% increase after 12 weeks; Fig. 3B). Although this difference was smaller in the female OGH-TG animals, female C57Bl6 mice normally exhibit only a small (<10%) increase in body weight on a HFD. The difference in body weight gain reflected a marked difference in adiposity (Fig. 3D). The percentage of total body weight comprised of adipose in WT male mice increased from 10% on a standard diet to >45% on the HFD, whereas the OGH-TG mice showed a much smaller increase in adiposity (≈10–20% body fat). In female mice, there was no statistical difference in body composition, even though after 12 weeks on the HFD, the WT female mice had twice the relative amount of fat as their OGH-TG littermates (12% vs. 6%).
Fig. 3.

Male transgenic mice weigh less, are resistant to the weight gain on a HFD, and have increased metabolic rate. (A and B) Mice were weighed once a week from birth (A) and, starting at 9 weeks of age, for 12 weeks on a HFD (B). Changes in body weight on a HFD shown as percentage of difference from day 1 of the diet. (C) Photographs of WT and transgenic (OGH-Tg) animals after 6 weeks on a HFD. (D) Measurements of body fat taken immediately before and again after 6 and 12 weeks on a HFD. (E) Metabolic rate before and after 6 weeks on a HFD. Data are expressed as mean ± SEM. *, P < 0.05, compared with WT littermate controls (one-way ANOVA).
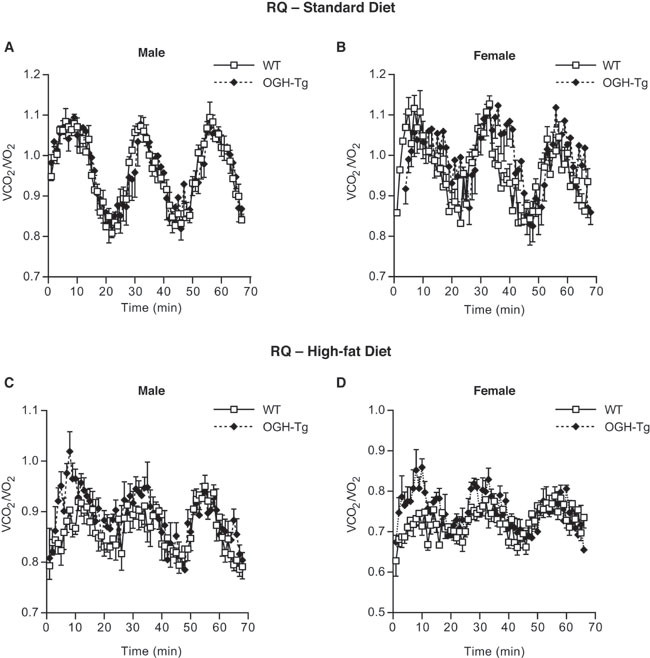
Metabolic Analysis. We assessed metabolic parameters by indirect calorimetry of OGH-TG and age-matched WT mice while on a standard diet and after 6 weeks on a HFD. Both male and female OGH-TG mice consumed more oxygen per unit body weight than did WT controls under both conditions (Fig. 3E). The respiratory quotient was unchanged between genotypes (see Fig. 7, which is published as supporting information on the PNAS web site), indicating that there was no change in metabolic substrate (fat vs. glucose).
We determined levels of glucose, lipids, and several relevant hormones in the blood of 5-month-old OGH-TG mice on a standard diet (Table 1). Male and female OGH-TG mice weighed significantly less than their respective WT littermates. Blood glucose, triglycerides, and insulin levels were reduced in male OGH-TG mice, consistent with reduced body weight. This result suggests an improvement in insulin sensitivity and glucose metabolism and not a diabetogenic effect, as found in patients with hyperthyroidism. Total serum cholesterol was also reduced significantly in both male and female OGH-TG animals. Similar changes in serum chemistry were seen in OGH-TG mice compared with their WT littermates when both were placed on a HFD (see Table 3, which is published as supporting information on the PNAS web site).
Table 1. OGH-TG mice have reduced cholesterol, insulin, and TSH levels and increased levels of thyroid hormone.
| Male
|
Female
|
|||
|---|---|---|---|---|
| Measurement | WT | OGH-TG | WT | OGH-TG |
| Body weight, g | 31.6 ± 3.1 | 26.8 ± 3.6* | 26.4 ± 3.2 | 20.5 ± 2.5* |
| Glucose, mg/dl | 277 ± 10 | 214 ± 25* | 214 ± 10 | 213 ± 17 |
| Triglycerides, mg/dl | 121 ± 15 | 76 ± 7* | 80 ± 11 | 76 ± 6 |
| Cholesterol, mg/dl | 118 ± 9 | 71 ± 7* | 90 ± 5 | 69 ± 6* |
| Insulin, ng/ml | 2.5 ± 0.18 | 1.9 ± 0.15* | ND | ND |
| TSH, ng/ml | (174 ± 13) | (132 ± 5*) | 95 ± 3 | 93 ± 9 |
| T3, ng/dl | (84 ± 5) | (130 ± 10*) | 69 ± 7 | 119 ± 14* |
| T4 μg/dl | (2.8 ± 0.2) | (6.9 ± 1.1*) | 3.5 ± 0.3 | 5.7 ± 1.0* |
Groups of male and female mice were fasted overnight and killed on the next morning for serum collection. Mice were 5 months old, except for values in parentheses, for which the mice were 2 months old. ND, no data. *, P<0.05, compared with WT littermate controls (one-way ANOVA).
Importantly, basal T3 and T4 levels were significantly increased ≈1.5- to 2.5-fold in both male and female OGH-TG mice compared with their WT littermates, whereas TSH levels were reduced or unchanged (Table 1), suggesting mild negative feedback due to the increases in T3 and T4. These increased T3 and T4 are likely to be due to a direct action on the thyroid, as others have reported (10), and we have independently confirmed (data not shown) that OGH/GPB5 is able to activate the TSHR, when coexpressed with a distant homologue of the common glycoprotein hormone α-subunit, termed GPA2 (9) or Zsig51. Thus, we infer that the global overexpression of OGH/GPB5 in OGH-TG animals includes a site, or sites, which also expresses GPA2. Many, if not all, of the phenotypic differences seen in the OGH-TG mice may be attributed to pleiotropic effects resulting from low-level activation of the thyroid axis because of the OGH.
Cardiovascular Telemetry. Most other approaches that activate the thyroid axis for the purpose of promoting weight loss have been associated with undesirable increases in heart rate and blood pressure. Heart rate and blood pressure of OGH-TG animals and WT littermates were determined by means of radiotelemetry in conscious, unrestrained animals. We condensed 3 days of continuous measurement into light- and dark-cycle averages. Although heart rate, diastolic blood pressure, systolic blood pressure, and pulse pressure tended to be slightly elevated in OGH-TG mice, these parameters were within normal limits and the differences failed to reach statistical significance (Table 2).
Table 2. Cardiovascular telemetry in OGH-TG mice and WT littermates.
| Heart rate, bpm
|
Pulse pressure, mmHg
|
Systolic blood pressure, mmHg
|
Diastolic blood pressure, mmHg
|
|||||
|---|---|---|---|---|---|---|---|---|
| Mice | Dark | Light | Dark | Light | Dark | Light | Dark | Light |
| WT | 589 ± 21 | 553 ± 16 | 34.3 ± 1.6 | 32.8 ± 1.3 | 128 ± 7 | 115 ± 6 | 93.7 ± 5.4 | 82.1 ± 4.5 |
| OGH-TG | 600 ± 13 | 550 ± 14 | 37.1 ± 1.6 | 34.8 ± 1.9 | 133 ± 2 | 122 ± 2 | 96.2 ± 2.4 | 86.7 ± 2.8 |
Blood-pressure and heart-rate measurements were recorded in unrestrained, conscious 12-week-old male mice via transmitters implanted in the aortic arch (n = 5 WT, n = 7 OGH-TG). Collected data from each animal were condensed to single values for light- and dark-cycle averages of heart rate and diastolic and systolic blood pressure. bpm, beats per minute; 1 mmHg = 133 Pa.
Discussion
OGH-TG mice are resistant to diet-induced obesity. Although OGH-TG mice gain very little weight on a HFD compared with WT mice, it is not because they eat significantly less food. In fact, on a regular chow diet, they eat more than WT mice, although they weigh less. Instead, their failure to gain weight correlates with an increased metabolic rate as evidenced by their increased consumption of oxygen and production of carbon dioxide. The increased metabolic rate of OGH-TG mice is apparently due to the ability of OGH (GPB5) to heterodimerize with GPA2 and comprise thyrostimulin, which can then activate the TSHR (10), resulting in ≈1.5- to 2.5-fold elevations in circulating T3 and T4 in the OGH-TG mice as compared with WT mice. The role of thyroid hormone in metabolic rate and thermogenesis is well established (30). OGH-TG mice also have significantly lower levels of circulating cholesterol, and thyroid hormone has been shown to lower cholesterol by direct actions in the liver (31). However, unlike mice treated with exogenous thyroid hormone (32, 33), OGH-TG mice do not exhibit significantly elevated heart rates. One possible explanation of this difference may reside in the different levels of thyroid hormone in the two scenarios. For example, in one study (33) in which thyroid hormone treatment of WT mice resulted in a 43% increase in heart rate, the level of circulating T4 was increased >10-fold. Likewise, in another study (32) a 5-fold increase in circulating T3 levels produced a 14–20% increase in heart rate after 3 or 4 days. In contrast, T4 levels in OGH-TG mice are maintained at a level of only ≈2-fold higher than in WT mice, and T3 levels are only ≈50% higher (Table 1). Another possibility is that heart rate is normalized by developmental compensation in OGH-TG mice. Arguing against this possibility is the fact that some other genetic models of hyperthyroidism (34) display tachycardia and that genetic models of hypothyroidism (2) show bradycardia. Last, the activity of OGH/thyrostimulin on extrathyroid TSHRs (35) or even on another, unidentified receptor could contribute to the resistance of OGH-TG mice to diet-induced obesity. It is important to note that direct administration of thyroid hormone would serve to suppress the activation of extrathyroid TSHRs by feedback inhibition of TSH synthesis (Fig. 4), whereas these receptors would be activated in OGH-TG mice.
Fig. 4.

Pathways by which OGH may affect metabolism and weight loss. Stimulatory pathways from OGH-Tg are shown in green, feedback inhibition is shown in red, and normal physiological function is shown in black. Deletion of OGH does not modify thyroid function and hormone levels, but overexpression of OGH results in thyroid stimulation and increased T4/T3 levels. This increased thyroid tone may act on target tissues like muscle, liver, or adipose tissue to induce resistance to diet-induced obesity marked by increased basal metabolic rate, or OGH may act directly on target tissues.
Why the growth of some neural-crest derived skull bones are specifically retarded in OGH-TG mice remains a mystery. Thyroid hormone has known effects on bone formation, as evidenced by the retarded bone growth in mice lacking genes for both thyroid hormone receptors (36). Also, TSH is reported to have a direct effect on bone turnover through TSHRs on osteoblasts and osteoclasts (37). However, other that the shortening of a few facial bones, the skeletons of OGH-TG mice are normal. Although premature craniosynostosis is one of the bone maladies that is associated with congenital hyperthyroidism in humans (38–40), it is not clear at this point how that corresponds to the facial abnormality that we observed. One confounding issue is that, in contrast to our findings, thyroid hormone has been reported to induce an increase in facial length in young rats (41).
Although neither TSH nor the TSHR are required for the initial formation of the thyroid gland during development (42), the lack of signaling through the TSHR leads to thyroid hypoplasia in mature animals (1, 43–45), and overstimulation of cAMP production in the thyroid either artificially (46–48) or by stimulation of the TSHR (49, 50) leads to thyroid hyperplasia. However, we found the thyroid glands of OGH-TG mice to be grossly normal in weight and appearance. Because thyroid growth is mediated directly by stimulation of the TSHR, it is possible that OGH/thyrostimulin activates the receptor to a lesser extent (partial agonism) than either receptor mutations in congenital hyperthyroidism, TSH, or Grave's disease antibodies. Direct pharmacological comparisons using purified proteins will be required to reveal whether OGH/thyrostimulin is a partial agonist. However, because thyroid hyperplasia is known to develop slowly in Grave's disease (51), it remains possible that thyroid hyperplasia will become apparent in OGH-TG mice only in advanced age.
Although global overexpression of OGH has dramatic metabolic effects, the lack of an apparent phenotype of OGH-/- mice means that its normal function remains unknown. Perhaps the more abundant OGH expression in newborn mice, as detected by lacZ analysis, implies that its natural function is constrained to this developmental stage and that challenges of newborns may reveal a phenotype for the knockouts and the normal function of OGH. Regardless of the normal physiologic role of OGH, our findings raise the important possibility that constitutive low-level activation of the thyroid axis (by OGH or other means) may provide a beneficial therapeutic approach for combating diet-induced obesity.
Supplementary Material
Acknowledgments
We thank everyone at Regeneron for support and assistance, especially Karen Garcia, Marshena Moncrieffe, Jennifer Kintner, and Jennifer Griffiths; the velocigene core for blastocyst injections and genotyping of mice; Hilary Cox, Niels Adams, and Nick Gale for help with lacZ staining; Tom DeChiara for the coordinated breeding of KO mice; and Vicki Lan and Scott Staton for artwork. We also thank Dr. Larry Suva for x-ray analysis of OGH-TG skulls and Drs. Len Schleifer and P. Roy Vagelos for continued support and encouragement.
Author contributions: L.E.M., K.E.W., L.C.G., D.M.V., A.N.E., S.J.W., G.D.Y., M.W.S., and A.J.M. designed research; L.E.M., K.E.W., L.C.G., K.D.A., J.D.M., W.T.P., M.V.S., and D.B. performed research; L.E.M., K.E.W., L.C.G., K.D.A., D.M.V., A.N.E., S.J.W., G.D.Y., M.W.S., and A.J.M. analyzed data; and L.E.M., K.E.W., L.C.G., M.W.S., and A.J.M. wrote the paper.
Abbreviations: TSH, thyroid-stimulating hormone; TSHR, TSH receptor; HFD, high-fat diet.
References
- 1.Kendall, S. K., Samuelson, L. C., Saunders, T. L., Wood, R. I. & Camper, S. A. (1995) Genes Dev. 9, 2007-2019. [DOI] [PubMed] [Google Scholar]
- 2.Wikstrom, L., Johansson, C., Salto, C., Barlow, C., Campos Barros, A., Baas, F., Forrest, D., Thoren, P. & Vennstrom, B. (1998) EMBO J. 17, 455-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johansson, C., Vennstrom, B. & Thoren, P. (1998) Am. J. Physiol. 275, R640-R646. [DOI] [PubMed] [Google Scholar]
- 4.Ribeiro, M. O., Carvalho, S. D., Schultz, J. J., Chiellini, G., Scanlan, T. S., Bianco, A. C. & Brent, G. A. (2001) J. Clin. Invest. 108, 97-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor, A. H., Stephan, Z. F., Steele, R. E. & Wong, N. C. (1997) Mol. Pharmacol. 52, 542-547. [DOI] [PubMed] [Google Scholar]
- 6.Trost, S. U., Swanson, E., Gloss, B., Wang-Iverson, D. B., Zhang, H., Volodarsky, T., Grover, G. J., Baxter, J. D., Chiellini, G., Scanlan, T. S. & Dillmann, W. H. (2000) Endocrinology 141, 3057-3064. [DOI] [PubMed] [Google Scholar]
- 7.Grover, G. J., Mellstrom, K., Ye, L., Malm, J., Li, Y. L., Bladh, L. G., Sleph, P. G., Smith, M. A., George, R., Vennstrom, B., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 10067-10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye, L., Li, Y. L., Mellstrom, K., Mellin, C., Bladh, L. G., Koehler, K., Garg, N., Garcia Collazo, A. M., Litten, C., Husman, B., et al. (2003) J. Med. Chem. 46, 1580-1588. [DOI] [PubMed] [Google Scholar]
- 9.Hsu, S. Y., Nakabayashi, K. & Bhalla, A. (2002) Mol. Endocrinol. 16, 1538-1551. [DOI] [PubMed] [Google Scholar]
- 10.Nakabayashi, K., Matsumi, H., Bhalla, A., Bae, J., Mosselman, S., Hsu, S. Y. & Hsueh, A. J. (2002) J. Clin. Invest. 109, 1445-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedrich, G. & Soriano, P. (1991) Genes Dev. 5, 1513-1523. [DOI] [PubMed] [Google Scholar]
- 12.Valenzuela, D. M., Murphy, A. J., Frendewey, D., Gale, N. W., Economides, A. N., Auerbach, W., Poueymirou, W. T., Adams, N. C., Rojas, J., Yasenchak, J., et al. (2003) Nat. Biotechnol. 21, 652-659. [DOI] [PubMed] [Google Scholar]
- 13.Suri, C., Jones, P. F., Patan, S., Bartunkova, S., Maisonpierre, P. C., Davis, S., Sato, T. N. & Yancopoulos, G. D. (1996) Cell 87, 1171-1180. [DOI] [PubMed] [Google Scholar]
- 14.Wortley, K. E., Anderson, K. D., Garcia, K., Murray, J. D., Malinova, L., Liu, R., Moncrieffe, M., Thabet, K., Cox, H. J., Yancopoulos, G. D., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 8227-8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence, A. M., Tan, S., Hojvat, S. & Kirsteins, L. (1977) Science 195, 70-72. [DOI] [PubMed] [Google Scholar]
- 16.Rougeot, C., Rosinski-Chupin, I., Mathison, R. & Rougeon, F. (2000) Peptides (Tarrytown, NY) 21, 443-455. [DOI] [PubMed] [Google Scholar]
- 17.De Matteis, R., Puxeddu, R., Riva, A. & Cinti, S. (2002) J. Anat. 201, 363-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czykier, E., Zabel, M. & Seidel, J. (2003) Folia Histochem. Cytobiol. 41, 29-32. [PubMed] [Google Scholar]
- 19.Nexo, E., Olsen, P. S. & Poulsen, K. (1984) Regul. Pept. 8, 327-334. [DOI] [PubMed] [Google Scholar]
- 20.Goldfine, I. D., German, M. S., Tseng, H. C., Wang, J., Bolaffi, J. L., Chen, J. W., Olson, D. C. & Rothman, S. S. (1997) Nat. Biotechnol. 15, 1378-1382. [DOI] [PubMed] [Google Scholar]
- 21.Kagami, H., O'Connell, B. C. & Baum, B. J. (1996) Hum. Gene Ther. 7, 2177-2184. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa, H. (1956) Allerg. Asthma 2, 42-43. [PubMed] [Google Scholar]
- 23.Arancibia, S. & Assenmacher, I. (1985) J. Biol. Buccale 13, 185-203. [PubMed] [Google Scholar]
- 24.Isenman, L., Liebow, C. & Rothman, S. (1999) Am. J. Physiol. 276, E223-E232. [DOI] [PubMed] [Google Scholar]
- 25.Zambrowicz, B. P., Imamoto, A., Fiering, S., Herzenberg, L. A., Kerr, W. G. & Soriano, P. (1997) Proc. Natl. Acad. Sci. USA 94, 3789-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soriano, P. (1999) Nat. Genet. 21, 70-71. [DOI] [PubMed] [Google Scholar]
- 27.Srinivas, S., Watanabe, T., Lin, C. S., William, C. M., Tanabe, Y., Jessell, T. M. & Costantini, F. (2001) BMC Dev. Biol. 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao, X., Fujiwara, Y. & Orkin, S. H. (1999) Proc. Natl. Acad. Sci. USA 96, 5037-5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang, X., Iseki, S., Maxson, R. E., Sucov, H. M. & Morriss-Kay, G. M. (2002) Dev. Biol. 241, 106-116. [DOI] [PubMed] [Google Scholar]
- 30.Silva, J. E. (1995) Thyroid 5, 481-492. [DOI] [PubMed] [Google Scholar]
- 31.Gullberg, H., Rudling, M., Forrest, D., Angelin, B. & Vennstrom, B. (2000) Mol. Endocrinol. 14, 1739-1749. [DOI] [PubMed] [Google Scholar]
- 32.Johansson, C. & Thoren, P. (1997) Acta Physiol. Scand. 160, 133-138. [DOI] [PubMed] [Google Scholar]
- 33.Weiss, R. E., Korcarz, C., Chassande, O., Cua, K., Sadow, P. M., Koo, E., Samarut, J. & Lang, R. (2002) Am. J. Physiol. 283, E428-E435. [DOI] [PubMed] [Google Scholar]
- 34.Salto, C., Kindblom, J. M., Johansson, C., Wang, Z., Gullberg, H., Nordstrom, K., Mansen, A., Ohlsson, C., Thoren, P., Forrest, D. & Vennstrom, B. (2001) Mol. Endocrinol. 15, 2115-2128. [DOI] [PubMed] [Google Scholar]
- 35.Davies, T., Marians, R. & Latif, R. (2002) J. Clin. Invest. 110, 161-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gothe, S., Wang, Z., Ng, L., Kindblom, J. M., Barros, A. C., Ohlsson, C., Vennstrom, B. & Forrest, D. (1999) Genes Dev. 13, 1329-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abe, E., Marians, R. C., Yu, W., Wu, X. B., Ando, T., Li, Y., Iqbal, J., Eldeiry, L., Rajendren, G., Blair, H. C., et al. (2003) Cell 115, 151-162. [DOI] [PubMed] [Google Scholar]
- 38.Menking, M., Wiebel, J., Schmid, W. U., Schmidt, W. T., Ebel, K. D. & Ritter, R. (1972) Monatsschr. Kinderheilkd. 120, 106-110. [PubMed] [Google Scholar]
- 39.Zimmerman, D. (1999) Thyroid 9, 727-733. [DOI] [PubMed] [Google Scholar]
- 40.Johnsonbaugh, R. E., Bryan, R. N., Hierlwimmer, R. & Georges, L. P. (1978) J. Pediatr. 93, 188-191. [DOI] [PubMed] [Google Scholar]
- 41.Maeda, N., Amano, H., Machino, M., Kaneko, T. & Kumegawa, M. (1986) Anat. Anz. 161, 99-104. [PubMed] [Google Scholar]
- 42.Postiglione, M. P., Parlato, R., Rodriguez-Mallon, A., Rosica, A., Mithbaokar, P., Maresca, M., Marians, R. C., Davies, T. F., Zannini, M. S., De Felice, M. & Di Lauro, R. (2002) Proc. Natl. Acad. Sci. USA 99, 15462-15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stein, S. A., Shanklin, D. R., Krulich, L., Roth, M. G., Chubb, C. M. & Adams, P. M. (1989) Neuroendocrinology 49, 509-519. [DOI] [PubMed] [Google Scholar]
- 44.Stein, S. A., Oates, E. L., Hall, C. R., Grumbles, R. M., Fernandez, L. M., Taylor, N. A., Puett, D. & Jin, S. (1994) Mol. Endocrinol. 8, 129-138. [DOI] [PubMed] [Google Scholar]
- 45.Biebermann, H., Schoneberg, T., Krude, H., Schultz, G., Gudermann, T. & Gruters, A. (1997) J. Clin. Endocrinol. Metab. 82, 3471-3480. [DOI] [PubMed] [Google Scholar]
- 46.Ledent, C., Dumont, J. E., Vassart, G. & Parmentier, M. (1992) EMBO J. 11, 537-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeiger, M. A., Saji, M., Gusev, Y., Westra, W. H., Takiyama, Y., Dooley, W. C., Kohn, L. D. & Levine, M. A. (1997) Endocrinology 138, 3133-3140. [DOI] [PubMed] [Google Scholar]
- 48.Michiels, F. M., Caillou, B., Talbot, M., Dessarps-Freichey, F., Maunoury, M. T., Schlumberger, M., Mercken, L., Monier, R. & Feunteun, J. (1994) Proc. Natl. Acad. Sci. USA 91, 10488-10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaithamana, S., Fan, J., Osuga, Y., Liang, S. G. & Prabhakar, B. S. (1999) J. Immunol. 163, 5157-51564. [PubMed] [Google Scholar]
- 50.Kim-Saijo, M., Akamizu, T., Ikuta, K., Iida, Y., Ohmori, K., Matsubara, K., Matsuda, Y., Suzuki, M., Matsuda, F. & Nakao, K. (2003) Eur. J. Immunol. 33, 2531-2538. [DOI] [PubMed] [Google Scholar]
- 51.Derwahl, M., Huber, G. & Studer, H. (1989) Acta Endocrinol. 121, 389-394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}