

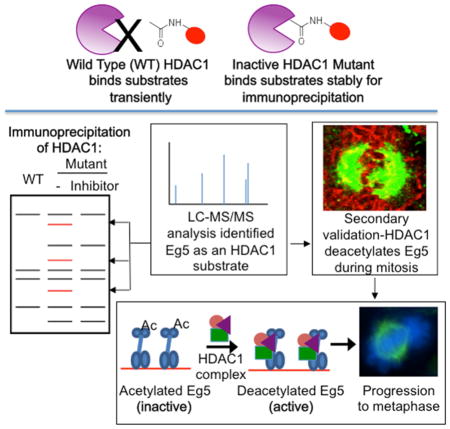
Summary
Histone deacetylase 1 (HDAC1) is an epigenetic enzyme that regulates key cellular processes, such as cell proliferation, apoptosis, and cell survival, by deacetylating histone substrates. Aberrant expression of HDAC1 is implicated in multiple diseases, including cancer. As a consequence, HDAC inhibitors have emerged as effective anti-cancer drugs. HDAC inhibitor-induced G0/G1 cell cycle arrest has been attributed to epigenetic transcriptional changes mediated by histone acetylation. But, the mechanism of G2/M arrest remains poorly understood. Here, we identified mitosis-related protein Eg5 (KIF11) as an HDAC1 substrate using a trapping mutant strategy. HDAC1 colocalized with Eg5 during mitosis and influenced the ATPase activity of Eg5. Importantly, an HDAC1 and 2-selective inhibitor caused mitotic arrest and monopolar spindle formation, consistent with a model where Eg5 deacetylation by HDAC1 is critical for mitotic progression. These findings revealed a previously unknown mechanism of action of HDAC inhibitors involving Eg5 acetylation, and provide a compelling mechanistic hypothesis for HDAC inhibitor-mediated G2/M arrest.
Keywords: Histone Deacetylase, HDAC1, Eg5/KIF11, substrate trapping
eTOC Blurb
HDAC inhibitor drug arrests the cell cycle at the G2/M phase, although the mechanism of arrest has remained elusive. Nalawansha et al. employed a substrate trapping strategy to identify mitosis-related protein Eg5 (KIF11) as an HDAC1 substrate. HDAC1 colocalized with Eg5 during mitosis, influenced the ATPase activity of Eg5, and was critical for mitotic progression. These findings reveal a mechanistic model where HDAC inhibitor drugs arrest cells in mitosis through HDAC1-mediated Eg5 acetylation.
INTRODUCTION
Gene expression is regulated by nucleosomal histone protein modifications, such as acetylation, methylation, and phosphorylation (Khorasanizadeh, 2004). Acetylation is catalyzed by histone acetyltransferases and leads to a less compact chromatin structure, which is associated with transcriptional activation (Kramer et al., 2001). In contrast, histone deacetylase (HDAC) proteins catalyze deacetylation, which induces chromatin condensation and transcriptional repression. Acetylation and HDAC protein activity play important roles in a variety of cellular processes, including proliferation, differentiation, and apoptosis.
The unregulated activities of HDAC proteins are associated with a variety of diseases, such as asthma, arthritis, schizophrenia, and cancer (Kramer et al., 2001). With a causal role in disease, HDAC proteins have emerged as important therapeutic targets for drug development. Currently, four HDAC inhibitors are approved as cancer therapeutics. Vorinostat (SAHA or Suberoyl Anilide Hydroxamic Acid, Zolinza®) and romidepsin (Depsipeptide, FK-228, Istodax®) are approved for the treatment of cutaneous T-cell lymphoma, whereas belinostat (PXD101, Beleodaq®) and panabinostat (LBH-589, Farydak®) are approved to treat peripheral T-cell lymphoma and multiple myeloma, respectively (Taunton et al., 1996, Yang et al., 1996, Yang et al., 1997, Hu et al., 2000). HDAC inhibitors influence proliferation by perturbing cell cycle progression, which ultimately leads to apoptosis (Marks et al., 2000).
HDAC inhibitors arrest cells at G0/G1 and G2/M phases (Richon et al., 2000). HDAC inhibitor-induced G0/G1 cell cycle arrest has been well studied and widely attributed to the expression of the p21 (waf1/cip1) and p27 (kip1) proteins after histone hyperacetylation and transcriptional upregulation (Newbold et al., 2014). In contrast, the mechanism accounting for HDAC inhibitor-induced G2/M arrest is less understood. Similar to G0/G1 arrest, a few reports documented that HDAC inhibitor-induced G2/M arrest is accompanied by transcriptional changes, such as increased expression of p21 and decreased expression of cyclins and retinoblastoma (Anh et al., 2012, Wetzel et al., 2005, Peart et al., 2003). In contrast, several studies reported that HDAC inhibitor-induced G2/M arrest does not correlate with transcriptional changes (Ishii et al., 2008, Warrener et al., 2010), suggesting a mechanism independent of histone acetylation. The limited data suggest that HDAC inhibitor-mediated mitotic arrest involves both histone and non-histone-mediated activities. We hypothesize here that HDAC inhibitors induce mitotic arrest through a mechanism involving non-histone substrates of HDAC proteins.
Histones are unquestionably the most studied substrate of HDAC proteins (Hassig et al., 1998). By studying histone acetylation, the role of HDAC1 in transcriptional regulation has been well characterized. As discussed earlier, the G0/G1 arrest observed with HDAC inhibitors is widely attributed to altered gene expression due to histone acetylation (Peart et al., 2003). However, recent proteomics data revealed that a large number of acetylated proteins exist in cells, in addition to histones (Choudhary et al., 2009, Zhao et al., 2010). Moreover, while many of the eleven HDAC isoform family members are found predominantly in the nucleus near nucleosomal-bound histones, including HDAC1 and HDAC2, several HDAC isoforms are found predominantly in the cytoplasm, such as HDAC6, where histones cannot be their predominant substrates. The available data implicate an expanded role of HDAC proteins in cell biology through non-histone substrates (Zhao et al., 2010, Scholz et al., 2015), which is consistent with the hypothesis that the mitotic arrest observed with HDAC inhibitors involves non-histone targets.
To characterize the complete role of HDAC proteins in cells, here we sought to identify non-histone substrates. We focused on HDAC1 due to its overexpression in multiple cancers (Weichert et al., 2008a, Miyake et al., 2008, Weichert et al., 2008b, Rikimaru et al., 2007, Sasaki et al., 2004, Weichert, 2009) and its association with cell proliferation in knockdown studies (Weichert et al., 2008b, Glaser et al., 2003). Importantly, simultaneous conditional knockout of HDAC1 and 2 led to mitotic defects, indicating that HDAC1 and 2 are neccessary for accurate cell division (Jamaladdin et al., 2014). With this prior evidence, we sought to identify non-histone substrates of HDAC1 that govern the mitotic defects observed in knockdown and inhibitor studies. Given the cell cycle arrest of HDAC inhibitors in cancer cells (Peart et al., 2003), these studies have the potential to reveal an unanticipated mode of action of HDAC inhibitors drugs in cancer treatment.
To identify non-histone targets of HDAC1, we recently developed a substrate trapping and identification strategy using inactive mutants (Nalawansha and Pflum, 2017). Using trapping mutants, here we identified Eg5 (Kinesin like protein 11/KIF11) as a substrate of HDAC1. Eg5 colocalized with HDAC1 during the prophase of the mitosis, suggesting that HDAC1-mediated deacetylation is critical for mitotic progression. Consistent with a role of HDAC1 in mitosis, mutating the K890 site of acetylation in Eg5 resulted in altered ATPase activity and mitotic progression. Importantly, an HDAC1 and 2 selective inhibitor promoted formation of monopolar spindles, similar to an Eg5 inhibitor and prior conditional knockout studies (Jamaladdin et al., 2014). Given that HDAC-targeted anti-cancer drugs lead to cell cycle arrest and apoptosis, the connection between HDAC1 and Eg5 established here suggests that HDAC inhibitors influence the acetylation state and activities of substrates beyond histones. By uncovering novel mechanisms of action of HDAC inhibitors, these studies will be critical to guide the development of more effective and selective anti-cancer drugs.
RESULTS
HDAC1 substrate trapping strategy
Substrate trapping was initially developed to identify substrates of phosphatases using catalytically inactive mutants (Flint et al., 1997). Inactive mutants bind substrates more stably than the wild type protein, allowing the purification of typically transiently bound substrates (Figures 1A and B). Two widely used trapping mutants of protein tyrosine phosphatase 1B (PTP1B) were created by mutating a nucleophilic residue in the active site or a residue that acts as a general acid during catalysis (Zhang et al., 1999). Mutation of these catalytic amino acids produced inactive enzymes that were used to purify novel substrates (Flint et al., 1997, Zhang et al., 1999, Buist et al., 2000, Wu et al., 2006, Motiwala et al., 2010, Merritt et al., 2006). Given the success with phosphatases, we extend the trapping strategy to HDAC1 with the aim of identifying physiological substrates (Figure 1B).
Figure 1.

The Substrate Trapping Strategy. (A) Catalytically active wild type (WT) HDAC enzyme binds substrates transiently. (B) Inactive HDAC mutant enzyme binds substrates more stably, which allows trapped substrates to be isolated by immunoprecipitation. (C) Amino acid residues that were mutated in this study are shown as ball and stick structures in the HDAC1 crystal structure (shown as purple mesh, PBD: 4BKX). The metal ion required for catalysis is shown as a gray sphere. D) FLAG-tagged wild type or mutant HDAC1 were transfected into HEK293 cells, treated with or without SAHA (10 μM) for 24h to induce robust protein acetylation, and then immunoprecipitated with a FLAG antibody, separated by SDS-PAGE, and immunoblotted with FLAG or acetyl-H3K9 antibodies. As controls, lysates prior to immunoprecipitation were separated by SDS-PAGE and immunoblotted with acetyl-H3K9 and GAPDH to assure equal protein content. Additional trials are shown in Figure S1.
The crystal structure of HDAC1 revealed that the active site has a deep, narrow 11 Å channel and a 14 Å internal cavity (Figure 1C) (Millard et al., 2013). Previously, residues in the 11 Å and 14 Å channels of HDAC1 were mutated to alanine to study structure and function (Weerasinghe et al., 2008, Wambua et al., 2014). The majority of these mutants were catalytically inactive, making them potential substrate traps. Among these mutants, we chose three (H141A, F150A and C151A) where the mutated residues are located in the various active site regions (Figure 1C) and conserved among the HDAC family (Weerasinghe et al., 2008, Wambua et al., 2014). H141 acts as a general base during catalysis (Hassig et al., 1998). F150 lies within the 11 Å channel active site near the substrate binding region (Wang et al., 2005, Estiu et al., 2008). C151 is present in the 14 Å internal cavity and is involved in acetate byproduct escape during deacetylation (Wambua et al., 2014). Importantly, all three mutants are stable and associated with the same multi-protein complexes as wild type HDAC1 (Weerasinghe et al., 2008, Wambua et al., 2014). These three mutants are inactive by different mechanisms and may offer differing abilities to trap substrates.
To test substrate trapping by the three inactive HDAC1 mutant proteins, we performed a histone binding experiment. Histones are considered a predominant substrate of HDAC1 (Taunton et al., 1996), making them an appropriate test protein. For this histone binding experiment, HEK293 cells were transfected with FLAG-tagged wild type or mutant HDAC1 (H141A, F150A and C151A) constructs, grown for 48 hr, and then treated with or without SAHA for an additional 24 hr to induce acetylation of cellular proteins. HDAC proteins were immunoprecipitated via the FLAG tag, separated by SDS-PAGE, and immunoblotted with FLAG and acetyl-histone H3 lysine 9 (Ac-H3K9) antibodies. The C151A and F150A mutants immunoprecipitated high levels of Ac-H3K9 compared to wild type HDAC1 (Figure 1D, compare lanes 9 and 10 to lane 7, middle gel). In contrast, the H141A mutant bound the same background level of Ac-H3K9 as wild type (Figure 1D, compare lanes 7 and 8, bottom gel). As a control, SAHA-treated lysates expressing wild type and mutant HDAC1 contained equal levels of Ac-H3K9 (Figure 1D, compare lanes 6 to 7–10, bottom gel), assuring that the histone H3 immunoprecipitated by the HDAC1 mutants was due to stable binding and not variable expression or acetylation. To confirm that acetylation is critical for binding, co-immunoprecipitation of HDAC1 and Ac-H3K9 was more prominent when cells were treated with SAHA to elevate acetylation in the lysates (Figure 1D, compare lanes 9 and 10 with lanes 4 and 5, middle gel), although low levels of Ac-H3K9 binding were observed with C151A (Figure 1D, lane 5). As a further control, SAHA treatment led to similar increased protein acetylation in wild type and mutant-expressing cells (Figure S2), showing that overall acetylation levels in unaffected by overexpression of HDAC1 mutants. In total, this histone H3 binding study established that inactive HDAC mutant proteins bind to acetylated substrates more stably than wild type and can be used for substrate trapping studies.
Substrate trapping with inactive HDAC1 mutants to identify candidate substrates
With the substrate trapping ability of the HDAC1 mutants confirmed, substrate trapping was performed to identify new substrates of HDAC1. In the substrate trapping study, wild type or mutant HDAC1-FLAG proteins were overexpressed and immunoprecipitated in the absence or presence of competitive active site inhibitor, SAHA. An active site inhibitor, such as SAHA, provided a helpful control by distinguishing substrates bound to the active site from associated proteins bound outside of the active site. Bound proteins were separated by SDS-PAGE and visualized with Sypro ruby total protein stain. Proteins present in the mutant but not wild type or SAHA treated immunoprecipitates were putative new substrates (Figure 2A), which were identified using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
Figure 2.

Substrate trapping by HDAC1 mutants. A) Workflow for the substrate trapping study with initial gel analysis of wild type and mutant HDAC1 immunoprecipitates to identify candidate protein bands. HDAC inhibitor (Inh) was included in a control immunoprecipitate to distinguish associated proteins from substrates by competing for active site binding. Candidate proteins present in only the mutant immunoprecipitate were excised from the gel and identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Substrates were then confirmed using a series of secondary experiments. B/C) Wild type (WT) and mutant HDAC1 (indicated below each lane) were expressed as FLAG-tagged proteins in T-Ag Jurkat cells, immunoprecipitated with anti-FLAG agarose, separated by 12% (A) or 16 % (B) SDS-PAGE, and visualized with SyproRuby total protein stain. Arrows indicate immunoprecipitated HDAC1 or possible substrates (p130, p95, p45, or histones) observed in the absence but not presence of competitive active site inhibitor SAHA (0.8 mM).
Substrate trapping was performed with wild type and three inactive HDAC1 mutants (H141A, F150A and C151A) using T-Ag Jurkat cells. All mutants were able to immunoprecipitate histones (Figure 2C, lanes 2–4) and other proteins (Figures 2B and C, lanes 2–4, p45, p95 and p130), consistent with their substrate trapping abilities. The immunoprecipitation of histones and several protein bands (p45, p95 and p130) were decreased upon SAHA treatment (Figures 2B and 2C, compare lanes 2–4 to lanes 6–8), suggesting that these proteins interact directly with the active site of HDAC1 and could be substrates. The data further confirm that inactive HDAC1 mutants display substrate trapping abilities appropriate for substrate identification.
To identify the proteins interacting with the HDAC1 trapping mutants in a SAHA-dependent manner, gel bands were excised, trypsin digested, and subjected to LC-MS/MS analysis. With high confidence, p45 and p130 were identified as γ-actin and Eg5 (also known as Kinesin like protein 11, KIF11), respectively (Table S1 and Figures S3–S6). The protein corresponding to p95 was not identified with high confidence by MS analysis. Prior work reported that HDAC1 interacts with filamentous actin (F-actin), which comprises γ-actin, during mitosis (He et al., 2013). However, a connection between Eg5 and HDAC1 had not been documented previously. Peptides corresponding to Eg5 were observed in all three mutant pull downs, but not wild type (Table S1). Eg5 plays an important role in protein transport and formation of bipolar spindle during the prophase of mitosis (Blangy et al., 1995). Mitotic defects leading to apoptosis are associated with the anti-cancer activities of HDAC inhibitor drugs (Robbins et al., 2005, Warrener et al., 2010, Taddei et al., 2001, Ishii et al., 2008, Stevens et al., 2008), although the mechanism accounting for mitotic arrest is poorly understood. To gain insight into HDAC inhibitor drug action, Eg5 was further studied as an HDAC1 substrate.
Co-immunoprecipitation of HDAC1 and Eg5
To confirm that p130 is Eg5 in the trapping experiment, immunoblotting was performed with an Eg5 antibody after trapping with the C151A mutant. The C151A mutant was selected for these validation studies due to its consistent trapping ability (Figure 1D and 2B). Wild type and C151A mutant HDAC1 immunoprecipitated Eg5 in a SAHA-dependent manner (Figure 3A, top gel, compare lanes 2 and 3 to lane 4), consistent with the MS characterization of p130 as Eg5. In addition, Eg5 acetylation was elevated in immunoprecipitates with the inactive C151A mutant compared to wild type (Figure 3A, middle gel, compare lane 3 to 2), suggesting that Eg5 is a cellular substrate of HDAC1. The elevated levels of acetylation with the mutant also indicate stable binding between acetylated Eg5 and C151A HDAC1, consistent with earlier substrate trapping studies (Figures 1D, 2B, and 2C). Finally, to further establish the trapping ability of C151A, we also tested the Y303F HDAC1 mutant. The homologous mutant, Y306F, in HDAC8 has been characterized thoroughly in terms of structure and activity (Vannini et al., 2007) and was found to be inactive but to maintain substrate binding, as expected of a trapping mutant. Therefore, to test the trapping ability of the C151A mutant, both the C151A and Y303F HDAC1 mutants were used in Eg5 trapping studies. Eg5 was coimmunoprecipitated at comparable level by both the Y303F and C151A HDAC1 mutants (Figure S7), confirming that the C151A mutant is suitable as a trapping mutant for future studies.
Figure 3.

Eg5 interacts with HDAC1. A) Wild type (WT) or C151A HDAC1 were expressed as FLAG-tagged proteins in T-Ag Jurkat cells, immunoprecipitated (IP) with anti-FLAG agarose in the absence (lanes 2 and 3) or presence (lane 4) of SAHA (3 mM), separated by SDS-PAGE, and Western blotted with Eg5 (top), acetyl-lysine (AcLys, middle), or FLAG (bottom) antibodies. B) Endogenous HDAC1 and Eg5 were immunoprecipitated (IP) separately from T-Ag Jurkat cells, separated by SDS-PAGE, and Western blotted with Eg5 (top) or HDAC1 (bottom) antibodies. C) HDAC1-FLAG and Eg5-myc were coexpressed in HEK293 cells, immunoprecipitated (IP) separately with either FLAG or myc antibodies, separated by SDS-PAGE, and Western blotted with myc (top) or FLAG (bottom) antibodies. Agarose beads without bound antibodies were used as an immunoprecipitation controls (lane 1). For all gels, arrows indicate the position of Eg5 and HDAC1 in the gels.
To further confirm the association of HDAC1 with Eg5 in vivo, a co-immunoprecipitation experiment was performed. Endogenous Eg5 and HDAC1 were immunoprecipitated separately from T-Ag Jurkat cells and probed for the presence of the interaction by Western blot. Eg5 was immunoprecipitated with HDAC1 (Figure 3B, lane 2), and vice versa (Figure 3B, lane 3). The data confirm that HDAC1 and Eg5 interact in cellulo. However, the quantities of co-immunoprecipitated proteins were low, which suggests that the interaction between HDAC1 and Eg5 is low affinity or occurring under transient cellular conditions.
To determine whether the Eg5-HDAC1 interaction is cell type specific, HDAC1-FLAG and Eg5-myc were cotransfected into HEK293 cells and immunoprecipitated using FLAG and myc antibodies. The Eg5–HDAC1 interaction was also observed in HEK293 cells (Figure 3C, lanes 2 and 3), confirming that the interaction extends beyond Jurkat cells. Similar to the T-Ag Jurkat study, the low quantities of co-immunoprecipitated proteins suggest that the HDAC1-Eg5 interaction is transient or occurring under specific cellular conditions.
Eg5 is a substrate of HDAC1
To validate that Eg5 is a substrate of HDAC1 in cellulo, we performed in vitro deacetylation assays. First, experiments were performed using recombinant Eg5 and HDAC1. Recombinant Eg5 was chemically acetylated, and then incubated with or without recombinant HDAC1. Eg5 acetylation was significantly reduced in the presence of HDAC1 (Figure 4A, top gel, compare lanes 2 and 1). In the presence of HDAC inhibitor-SAHA, acetylation of Eg5 was restored (Figure 4A, top gel, compare lanes 2 and 3), confirming that deacetylation is HDAC dependent. Quantification also confirmed that HDAC1 deacetylated Eg5 (Figures 4B and S8).
Figure 4.

Eg5 is deacetylated by HDAC1. A) Chemically acetylated full length recombinant Eg5 was incubated in the absence or presence of recombinant HDAC1 and SAHA for 2 hr at 37°C and analyzed by SDS-PAGE and western blotting with acetyl lysine (top) and a combination of Eg5 and HDAC1 (bottom) antibodies. B) Quantification of the Ac-Lys western blot from part A. Mean and standard error of three independent trials are shown (Figures S8A–D). Quantification of the Eg5 blot as a load control is shown in Figures S8E–F. ***p<0.0001. C) Eg5 was immunoprecipitated from T-Ag Jurkat cells (lane 1) and incubated with immunoprecipitated HDAC1 (lane 2) in the presence or absence of 3 mM SAHA (lane 3) for 1 hr at 30°C, followed by SDS-PAGE separation and immunoblotting with acetyl lysine (top), Eg5 (middle), and HDAC1 (bottom) antibodies. D) Quantification of the Ac-Lys western blot from part C. Mean and standard error of three independent trials are shown (Figures S9A–D). Quantification of the Eg5 blot as a load control is shown in Figures S9E–F. ***p<0.0001
To further validate that HDAC1 can deacetylate Eg5, we performed in vitro deacetylation assays using cellular Eg5 and HDAC1. Eg5 was immunoprecipitated from T-Ag Jurkat cells and incubated with immunoprecipitated HDAC1 in the absence or presence of SAHA. Similar to results with recombinant proteins, Eg5 acetylation was significantly reduced in the presence of HDAC1 (Figure 4C, top gel, compare lanes 2 and 1), indicating that HDAC1 was able to deacetylate Eg5. As a control, SAHA inhibited HDAC1-mediated deacetylation of Eg5 (Figure 4C, top gel, compare lanes 2 and 3). Quantification confirmed reproducible HDAC1-dependent Eg5 deacetylation (Figures 4B and S9). Combined with the fact that Eg5 acetylation was elevated in immunoprecipitates with the inactive C151A mutant compared to wild type (Figure 3A), these studies substantiate that Eg5 is a substrate of HDAC1.
HDAC1 deacetylates K890 to influence Eg5 activity
To date three Eg5 acetylation sites (K146, K372 and K376) have been identified. K146 acetylation was observed in a global acetylation proteomics study (Choudhary et al., 2009), whereas K372 and K376 are listed as possible acetylation sites in PhosphoSite Plus®. K146 is present in the motor domain, whereas K372 and K376 reside in the stalk region of Eg5 (Figure 5A). Given that K146 was the only experimentally observed acetylated residue, we studied K146 acetylation by creating the K146A mutant using the full-length myc-tagged Eg5 construct (Blangy et al., 1995). First, we studied the acetylation state of K146A to assess if K146 is a dominant acetylated lysine in Eg5. Wild type Eg5 and K146A were transfected into HEK293 cells, followed by immunoprecipitation and immunoblotting with an acetyl lysine antibody to detect acetylation. The level of acetylation of the K146A mutant was comparable to that of wild type Eg5 (Figure S10A), suggesting that other acetylation sites are present in Eg5. In addition, immunoprecipitation of wild type or K146A mutant Eg5 by HDAC1 was similar (Figure S10B), indicating that K146 is not the dominant acetylation site for HDAC1. Taken together, the data indicate that a site other than K146 is the predominant site for HDAC1-mediated deacetylation on Eg5.
Figure 5.

Eg5 is deacetylated by HDAC1 at K890. A) The domain structure of human Eg5/KIF11, including the three acetylated lysines observed in prior work (K146, K372, and K376), as well as acetylated K890 identified here (red). B) Myc-tagged wild type or K890R mutant Eg5 were transfected into HEK293 cells, treated with HDAC1/2 selective inhibitor (SHI-1:2) for 24h, and immunoprecipitated with a myc antibody. SDS-PAGE separation and immunoblotting was performed with acetyl lysine (top) and myc (bottom) antibodies. C) Quantification of the Ac-Lys western blot from part B. Mean and standard error of four independent trials are shown (Figures S12A–E). Quantification of the myc blot as a load control is shown in Figures S11F–G. D) FLAG-tagged wild type (WT) or C151A mutant HDAC1 were cotransfected with myc-tagged wild type or K890R mutant Eg5 into HEK293 cells. Wild type and C151A mutant HDAC1 were immunoprecipitated using anti-FLAG agarose beads, separated by SDS-PAGE, and immunoblotted with myc (top) and FLAG (bottom) antibodies. E) Myc-tagged wild type or K890R mutant Eg5 were transfected into HEK293 cells, treated with HDAC1/2 selective inhibitor (SHI-1:2) for 24 hr to induce robust acetylation, and immunoprecipitated with a myc antibody. Half of the immunoprecipitate was used for ATPase assay (histogram) and other half was separated by SDS-PAGE and immunoblotted with myc antibody (gel image). Mean fold change and standard error are shown from at least three independent trials (Table S3). NT – non transfected. ***p<0.0001.
To identify the putative acetylation sites in Eg5 regulated by HDAC1, we used a mass spectrometry-based approach. Myc-tagged Eg5 was transfected into HEK293 cells in the absence or presence of wild type HDAC1. After 48 hr, cells were treated with or without HDAC inhibitors for 24 hr. The broad spectrum inhibitor SAHA was used, in addition to the HDAC1/2 selective inhibitor SHI-1:2. After harvesting, immunoprecipitation, and SDS-PAGE separation, protein bands corresponding to Eg5 were excised from the gel, trypsin digested, and subjected to LC-MS/MS analysis. The HDAC 1/2 selective inhibitor SHI-1:2 enhanced acetylation of Eg5 at K890 compared to untreated sample (Table S2 and Figures S11A–D). The site was also unacetylated when Eg5 was cotransfected with HDAC1, suggesting that K890 is the target site for HDAC1-mediated deacetylation (Figure S11G–H). K890 is present in the tail region of Eg5, unlike the other previously identified acetylation sites (Figure 5A).
To probe the acetylation status of Eg5 at K890, we created a K890R mutant using the full length myc-tagged Eg5 construct. The K890R mutant represents an unacetylated mimic of Eg5, which will allow the probing of the role of acetylation in Eg5 binding and function. Following transfection of wild type and K890R mutant Eg5 into HEK293 cells, Eg5 was immunoprecipitated and immunoblotted with an acetyl lysine antibody to detect acetylation. Interestingly, the level of acetylation of K890R was significantly reduced compared that of wild type Eg5 (Figure 5C and S12A–E). The data suggest that K890 is a major acetylation site on Eg5.
To further determine whether K890 is involved in the interaction between Eg5 and HDAC1, wild type or K890R mutant myc-tagged Eg5 were cotransfected with either wild type or C151A mutant FLAG-tagged HDAC1 into HEK293 cells, followed by immunoprecipitation of HDAC1 using a FLAG antibody. The amount of K890R mutant co-immunoprecipitated with wild type or C151A mutant HDAC1 was significantly reduced compared to that of wild type Eg5 (Figure 5D, compare lanes 4 and 5 to 2 and 3, top gel), indicating that K890 is essential for the binding of Eg5 to HDAC1. The data suggest that Eg5 is acetylated predominantly at K890, and K890 is a target site for HDAC1.
Eg5 promotes bipolar spindle formation in the prophase of mitosis through ATP hydrolysis (Blangy et al., 1995). To study the effect of acetylation on Eg5 function, we monitored the ATPase activity of the K890R mutant. Myc-tagged wild type or K890R Eg5 were transfected into HEK293 cells, treated with HDAC1/2 selective inhibitor (SHI-1:2) for 24 hr to induce acetylation, and immunoprecipitated with a myc antibody. Following immunoprecipitation, the Eg5 protein was eluted and subjected to ATPase assays. The ATPase activity of the K890R mutant was elevated by 3.7 ± 0.1-fold compared to wild type Eg5 (set at 1, Figure 5E). The data indicate that the acetylated form of Eg5 (represented by wild type) displays reduced activity compared to the unacetylated form (represented as the K890R mutant). Importantly, the data suggest that HDAC1-mediated deacetylation can influence Eg5 motor activity to regulate mitotic progression.
Eg5 colocalized with HDAC1 during mitosis
For HDAC1 to deacetylate Eg5 in vivo, both proteins must colocalize together in cellulo. HDAC1 is predominantly present in the nucleus (Suzuki et al., 2009), whereas Eg5 is in the cytoplasm (Sawin and Mitchison, 1995). Initially, we analyzed the HDAC1-FLAG fusion protein by microscopy to confirm that the FLAG tag does not affect HDAC1 localization to the nucleus. HEK293 cells expressing FLAG-tagged wild type HDAC1 were grown onto glass cover slips, fixed, and blocked with BSA, followed by visualization of the FLAG tag. As expected (Suzuki et al., 2009, Sawin and Mitchison, 1995), HDAC1-FLAG was predominantly nuclear, whereas Eg5 was cytoplasmic in interphase cells (Figure S13). The data indicate that the HDAC1-FLAG fusion protein maintains the same nuclear localization as endogenous HDAC1.
Eg5 plays a critical role in bipolar spindle formation during mitosis (Blangy et al., 1995). Given that nuclear envelope breakdown occurs during mitosis, we hypothesized that HDAC1 interacts with Eg5 after the initiation of mitosis. To determine the subcellular localization of endogenous HDAC1 and Eg5 under mitotic conditions, HEK293 cells were seeded onto glass cover slips, fixed, and blocked with BSA, followed by visualization of HDAC1 and Eg5 in cells undergoing mitosis. In interphase cells, HDAC1 was predominantly nuclear, whereas Eg5 was in the cytoplasm (Figure 6A). In prophase, the first stage of mitosis, Eg5 was localized to the separating centromeres (Figure 6B), consistent with its role in spindle formation. While HDAC1 was found throughout the cell in prophase, the merged image showed that a population of HDAC1 colocalized with Eg5 (Figure 6B, yellow merged spots). In contrast, Eg5 and HDAC1 did not colocalize during metaphase, anaphase and telophase; HDAC1 was associated with the DNA, which was located between the Eg5-bound spindle poles (Figures 6C–E). These results are consistent with earlier work showing that HDAC1 is excluded from condensed chromatin and is found instead in interchromosomal domains of prophase cells (Kruhlak et al., 2001), similar to Eg5-rich centromeres. These results indicate that HDAC1 partially colocalized with Eg5 during prophase, which suggests that HDAC1-mediated deacetylation of Eg5 occurs at the earliest stages of mitosis.
Figure 6.

Eg5 colocalized with HDAC1 during prophase. HEK293 cells were fixed and stained with HDAC1 (red) and Eg5 (green) antibodies. Cells were counterstained with DAPI (blue). Confocal microscopy was used to visualize HDAC1 and Eg5 in each cell. Cells in interphase (A), prophase (B), metaphase (C), anaphase (D), and telophase (E) are shown. HDAC1 (red) and Eg5 (green) images were used to generate merged images (yellow).
We also assessed Eg5 colocalization with another class I (HDAC3) and a class IIa isoform (HDAC7) during mitosis. HDAC3 was selected because prior work documented localization of HDAC3 to mitotic spindle during prophase (Ishii et al., 2008). However, no association between HDAC7 and mitotic spindles has been reported, making HDAC7 a useful negative control. As expected, HDAC3 partially localized with Eg5 during the prophase of the mitosis (Figure S14). In contrast, HDAC7 failed to colocalize with Eg5 during any mitotic phase (Figure S15). These additional studies with HDAC3 and HDAC7 indicate that only select HDAC isoforms colocalize with Eg5 during prophase to play a role in mitotic progression.
HDAC inhibitors promote mitotic arrest and monopolar spindle formation
HDAC inhibitors arrest the cell cycle at G0/G1 and G2/M phases. HDAC inhibitor-induced cell cycle arrest at G0/G1 has been primarily attributed to histone hyperacetylation, transcriptional upregulation, and overexpression of the p21 (waf1/cip1) and p27 (kip1) proteins (Baradari et al., 2006, Ryu et al., 2006). However, prior reports suggest that HDAC inhibitor-induced G2/M arrest involves both histone and non-histone-mediated mechanisms (Anh et al., 2012, Wetzel et al., 2005, Peart et al., 2003, Ishii et al., 2008, Warrener et al., 2010). Given the role of Eg5 in mitotic progression, we hypothesized that deacetylation of Eg5 by HDAC1 might partially explain the G2/M arrest observed with HDAC inhibitors.
To elucidate a possible role for HDAC1 in mitosis, the HDAC1 and 2 selective inhibitor, SHI-1:2 was used in flow cytometry studies to monitor cell cycle progression. HEK293 cells were treated with SHI-1:2 for 48 hr, before analysis of DNA content and a mitotic marker protein (phospho-H3S10) by flow cytometry. As a positive control, cells were also treated with a specific and potent inhibitor of Eg5 ATPase activity, S-trityl-L-cysteine (STLC), to observe Eg5-mediated mitotic arrest (Skoufias et al., 2006). As expected, both STLC and SHI-1:2 induced mitotic arrest (6.5 ± 0.4 % and 3.9 ± 0.3%, respectively) in contrast to untreated cells (0.45 ± 0.02%, Figure 7A, Figure S16, and Table S4). The elevated mitotic arrest observed with SHI-1:2 is consistent with a role for HDAC1 in mitotic progression.
Figure 7.

Acetylation of Eg5 contributes to SHI-1:2-induced mitotic arrest. A) HEK293 cells were treated with DMSO, STLC (10 μM) or SHI-1:2 (10 μM) for 48 hr followed by fixing and staining with Alexafluoro®488 conjugated phospho-histone H3 (Ser10) antibody and DAPI. Samples were analyzed by flow cytometry (Figure S16) and percent mitotic cells were plotted. Mean and standard error from three independent trials are shown in Table S4. ***p<0.0001. B) Myc-tagged wild type or K890R mutant Eg5 were transfected into HEK293 cells, treated with SHI-1:2 for 48 hr and subjected to staining with Alexa fluoro®488 conjugated phospho-histone H3 (Ser10) antibody and DAPI prior to flow cytometric analysis. A representative trial is shown in Figure S17. Quantification of the mitotic cells is shown as a histogram. Mean and the standard error from three independent trials are shown in Table S5. *p<0.05. ns – not significant. C) HEK293 cells were treated with DMSO, STLC (10 μM) or SHI-1:2 (10 μM) for 48 hr followed by fixing and staining with α-tubulin (green) and DAPI (blue). Fluorescence microscopy was used to visualize tubulin and DAPI in each cell. Scale bar – 10 μm. D) Quantitative analysis of monopolar spindles present in STLC and SHI-1:2 treated cells from part C. Percentage of monopolar spindles was determined after counting at least 100 mitotic cells. Mean and standard error of four independent trials are shown in Table S6. ***p<0.002.
We next studied the role of HDAC1-mediated Eg5 deacetylation in SHI-1:2-induced mitotic arrest. The K890R mutant mimics active, unacetylated Eg5 and is unresponsive to HDAC inhibitors. Consequently, if deacetylation of K890 by HDAC1 is critical for SHI-1:2-induced mitotic arrest, then cells expressing the K890R mutant should demonstrate reduced sensitivity to SHI-1:2 and fewer cells arrested in mitosis. HEK293 cells were transfected with wild type and K890R mutant Eg5, treated with SHI-1:2 for 48 hr, and then analyzed by flow cytometry. Cells expressing the K890R Eg5 mutant displayed reduced mitotic arrest (3.3 ± 0.2%) compared to cells expressing wild type Eg5 (4.6 ± 0.4%, Figure 7B, Figure S17, and Table S5). The small, but significant, effect of the K890R mutant on SHI-1:2-induced mitotic arrest is likely due to the presence of endogenous Eg5 in cells. The reduced mitotic arrest observed with the K890 mutant implicates Eg5 acetylation in HDAC inhibitor activity.
Eg5 inhibition leads to formation of monopolar spindles (Skoufias et al., 2006). If HDAC1 influences mitotic progression through deacetylation of Eg5, then HDAC inhibitors should block Eg5 deacetylation, resulting in reduced activity, monopolar spindle formation, and mitotic arrest. To further explore a role of HDAC1 in mitotic progression via Eg5 activity, spindle formation was monitored as a function of SHI-1:2 treatment. HEK293 cells were seeded onto glass cover slips, treated with SHI-1:2 for 48 hr, fixed, and blocked with BSA, followed by visualization of tubulin to monitor spindle formation. As a positive control for monopolar spindle formation, STLC was also tested. As predicted, monopolar spindles were observed with both STLC and SHI-1:2-treated cells (Figure 7C), in contrast to untreated cells. To quantify the extent of monopolar spindle formation, cells were counted from four independent trials to determine the percentage of cells displaying monopolar spindles. As controls, STLC induced monopolar spindles in 100% of the cells, while untreated cells displayed no population of cells with monopolar spindles. SHI-1:2 induced formation of monopolar spindles in 53 ± 2% of cells (Figure 7D and Table S6). The data provide further evidence that HDAC1 plays a role in mitotic progression via Eg5 deacetylation. Taken together with previous findings documenting that conditional HDAC1 and HDAC2 knockout resulted in mitotic arrest and monopolar spindle formation (Jamaladdin et al., 2014), the data reveal a functional link between HDAC1 and Eg5 activities that governs mitotic progression.
DISCUSSION
Aberrant expression of HDAC1 is implicated in multiple diseases, including cancer (Kramer et al., 2001). Currently, four HDAC inhibitors are approved as anti-cancer therapies, and multiple HDAC inhibitors are in the clinic. HDAC inhibitors induce apoptosis, differentiation, and cell cycle arrest (Marks et al., 2000). The model that has emerged to account for HDAC inhibitor-mediated cancer cell death centers on altered histone acetylation and gene expression (Peart et al., 2003). However, several studies have found that mitotic arrest in the presence of HDAC inhibitors does not correlate with changes in histone acetylation or protein expression (Ishii et al., 2008, Warrener et al., 2010), which suggests that non-histone targets may be influenced by HDAC inhibitors.
We recently developed trapping strategy to identify novel substrates of HDAC1 (Nalawansha and Pflum, 2017). Using the trapping strategy, we provide the first evidence that Eg5 is an HDAC1 substrate. HDAC1 deacetylated Eg5 in vitro using both recombinant and cell-derived proteins (Figure 4), which confirmed that Eg5 is a substrate. Mass spectrometric analysis identified a new acetylation site on Eg5, K890. Studies with the K890R mutant indicated that K890 is a major Eg5 acetylation site regulated by HDAC1 and is critical for binding to HDAC1 (Figure 5). Importantly, the K890R mutant, which mimics the unacetylated state of Eg5, maintained higher ATPase activity than the wild type acetylated form (Figure 5E), indicating that K890 acetylation regulates Eg5 function. In addition, Eg5 colocalized with HDAC1 only during the prophase stage of mitosis (Figure 6). The mitosis-specific interaction is consistent with the low level of coimmunoprecipitiation observed in asynchronous cells (Figures 3B and 3C) and the need for nuclear envelope breakdown to allow HDAC1 and Eg5 to exist in the same cellular location. Further, HDAC1 and 2 selective inhibitor SHI-1:2 caused mitotic arrest and promoted monopolar spindle formation (Figures 7A and 7D), which was dependent on K890 acetylation (Figure 7B). Taken together with prior condiitonal knockout studies of HDAC1 and 2 showing monopolar spindle formation (Jamaladdin et al., 2014), we hypothesize that HDAC1-mediated deacetylation of Eg5 during prophase is an essential step in mitotic progression. The model that emerges suggests that Eg5 is acetylated and inactive prior to mitosis (Figure 8A). Upon entering the prophase of mitosis, the nuclear envelope breaks down to allow HDAC1 to colocalize with Eg5, catalyze deacetylation, and enhance ATPase activity. Then, active and deacetylated Eg5 completes centromere separation and formation of the bipolar spindle to promote the cells into the later stages of mitosis (Figure 8A).
Figure 8.

Model of the role of HDAC1 in Eg5-mediated mitotic progression. A) Eg5 (blue) is acetylated (Ac) and inactive prior to entry into mitosis. Upon entering the prophase of mitosis, HDAC1 (magenta) interacts with Eg5 after nuclear envelope breakdown, which results in Eg5 deacetylation, enhanced activity. Eg5 deacetylation promotes centromere separation and bipolar spindle formation (cell image), leading to metaphase progression and complete cell division. Associated proteins (orange and green) might be involved in the interaction. B) HDAC inhibitors prevent deacetylation of Eg5 by HDAC proteins. Acetylated Eg5 is inactive, which results in mitotic arrest due to monopolar spindle formation (cell image).
HDAC1 is recruited to gene-specific histone targets to regulate transcription through multi-protein complexes, such as Sin3, NuRD and CoREST (Kelly and Cowley, 2013). Based on this prior work, we speculate further that associated proteins are required for HDAC1-mediated Eg5 interaction and deacetylation during prophase (Figure 8A). A recent study showed that HDAC1 is displaced from chromatin during mitosis to interact with filamentous actin (F-actin)(He et al., 2013), which localizes to the mitotic spindles (Woolner et al., 2008). Consistent with this prior data, the trapping mutants studied here also bound γ-actin, a component of F-actin. Given the role of Eg5 in bipolar spindle formation, we hypothesize that HDAC1 interacts with F-actin to facilitate Eg5 deacetylation during mitosis. Eg5 also interacts with NCOR1 (nuclear receptor corepressor 1) (Yoon et al., 2003), which is known to associate with HDAC1 via the mSin3a complex (Cartron et al., 2013). We speculate that Eg5 deacetylation may also be mediated by the mSin3a/NCOR complex. Given the key role of associated proteins in regulating HDAC activity in gene expression, the proposed model also includes associated proteins as players in mediating Eg5 deacetylation (Figure 8A).
HDAC inhibitors block mitotic progression beyond prometaphase (Robbins et al., 2005, Warrener et al., 2010, Taddei et al., 2001, Ishii et al., 2008, Stevens et al., 2008). HDAC inhibitor-induced cell cycle arrest has been primarily attributed to the expression of the p21 (waf1/cip1) and p27 (kip1) proteins after histone hyperacetylation and transcriptional upregulation (Newbold et al., 2014). However, arrest due to mitotic defects as observed rapidly after inhibitor treatment and in the absence of transcription (Blagosklonny et al., 2002, Warrener et al., 2010), suggesting a histone acetylation-independent mechanism. The proposed model (Figure 8A) suggests a novel mechanism for HDAC inhibitor-mediated mitotic arrest. According to the model, HDAC inhibitors are expected to block Eg5 deacetylation, elevate levels of aceylated and inactive Eg5, and prevent bipolar spindle formation, which results in mitotic arrest (Figure 8B). In prior work, maximal mitotic arrest was observed when HDAC inhibitors were present prior to the start of mitosis (Robbins et al., 2005), which is consistent with the proposed model where Eg5 deacetylation occurs in the earliest stage of mitosis, before complete centromere separation. Prior work also reported that HDAC inhibitor-treated cells failed to progress to metaphase partially due to inefficient localization of various centromeric proteins (Robbins et al., 2005, Stevens et al., 2008). However, HDAC inhibitor treatment did not influence the quantities of centromeric proteins, although Eg5 was not tested (Robbins et al., 2005, Taddei et al., 2001, Stevens et al., 2008). The influence of HDAC inhibitors on centromeric protein localization is consistent with the proposed model where HDAC1-mediated Eg5 deacetylation is critical for mitotic progression (Figure 8). Significantly, embryonic stem cells harboring a double conditional knockout of HDAC1 and HDAC2 displayed monopolar spindles and chromosomal segregation defects (Jamaladdin et al., 2014), suggesting that HDAC1 and 2 play an essential role in mitotic progression. Building on these prior results, we show for the first time that the HDAC1 and 2 selective inhibitor SHI-1:2 caused mitotic arrest and promoted monopolar spindle formation, confirming a role for HDAC1 and HDAC2 in Eg5 function. The model provides a compelling mechanistic hypothesis explaining HDAC inhibitor-induced mitotic arrest through elevated Eg5 acetylation, reduction of ATPase activity, and monopolar spindle formation (Figure 8B).
Previous studies implicated HDAC3 in mitotic defects (Ishii et al., 2008, Warrener et al., 2010). HDAC3 knockdown reproduced the mitotic defects observed upon HDAC inhibitor treatment, with spindle instability and prometaphase arrest (Warrener et al., 2010). Like HDAC1, a population of HDAC3 localized to the mitotic spindle during prophase (Ishii et al., 2008). HDAC3 was present in a stable corepressor complex containing NCOR1 throughout mitosis, suggesting it is in an activated form (Ishii et al., 2008). HDAC3 deacetylates NudC (nuclear distribution protein C) (Chuang et al., 2013), which is critical for kinetochore-microtubulin attachment via dynein/dynactin. Here we show that HDAC3 partially colocalized with Eg5 during prophase of the mitosis (Figure S12), which provides further evidence for a role for HDAC3 in mitotic progression. Taken together, HDAC1 and HDAC3 may function together during mitosis to regulate proper centromere separation and spindle formation.
Prior work documented a role for post-translational modifications in regulating Eg5 activity in mitosis. Phosphorylation at T937 promoted Eg5 localization to the spindle (Sawin and Mitchison, 1995). Eg5 is also phosphorylated at T927 to enhance binding to centrosomes during bipolar spindle formation (Blangy et al., 1995). Based on this earlier work on phosphorylation, acetylation might also have an impact on Eg5 activity and/or binding during mitosis. With only three acetylated sites on Eg5 (K146, K372 and K376) previously identified ((Choudhary et al., 2009) and PhosphoSite Plus, Figure 5A), the effect of acetylation on Eg5 function during mitosis has not yet been fully characterized. Here, we identified K890 as a predominant acetylated site on Eg5 and the target for HDAC1-mediated deacetylation. Acetylation of K890 affected the ATPase activity of Eg5 (Figure 5E), as well the affinity of Eg5 for HDAC1 (Figure 5D). Significantly, acetylation of Eg5 at K890 partially contributed to HDAC inhibitor-mediated mitotic arrest (Figure 7B). The work here with HDAC1 suggests that K890 acetylation is a key regulator of mitotic progression by influencing Eg5 activity.
Significance
Histone Deacetylase (HDAC) enzymes are associated with cancer formation, making them exciting new anti-cancer drug targets. While HDAC inhibitor drugs are used in the clinic, their mechanism of action is not yet fully characterized due to the incomplete understanding of HDAC activities in the cell. Here, using inactive trapping mutants, Eg5 was validated as an HDAC1 substrate, which for the first time links HDAC1 activity to the mitotic defects observed with HDAC inhibitors. Given use of HDAC inhibitor drugs in anti-cancer therapy, these studies will have a direct impact on drug design efforts by documenting a new functional role of HDAC1 in mitotic progression through Eg5 acetylation.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
All requests for reagents and resources should be directed to the lead contact, Mary Kay Pflum (pflum@wayne.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human T-Ag Jurkat cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (Life Technologies) and 1% antibiotic/anti-mycotic (Gibco) at 37°C in a 5% CO2 environment. HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% Fetal bovine serum and 1% antibiotic/anti-mycotic.
METHOD DETAILS
Expression plasmids and transient transfections
HDAC1 single point mutants were created using the pBJ5HDAC1-FLAG expression plasmid, as previously described (37). Myc-tagged pRcCMV-Eg5 was a generous gift from Dr. Anne Blangy (Centre de Recherche de Biochimie Macromoléculaire) (42). The Eg5-K146A and K890R mutants were constructed by Quickchange site directed mutagenesis using pRcCMV-Eg5 (K146A Forward primer- 5′CCA CGT ACC CTT CAT CAA ATT TTT GAG GCA CTT ACT GAT AAT GGT ACT G 3′ and K146A Reverse primer- 5′C AGT ACC ATT ATC AGT AAG TGC CTC AAA AAT TTG ATG AAG GGT ACG TGG 3′, K890R Forward primer- 5′ CTT GAT CAG ATG ACT ATT GAT GAA GAT AGA TTG ATA GCA CAA AAT CTA GAA C 3′ and K890R Reverse primer 5′ G TTC TAG ATT TTG TGC TAT CAA TCT ATC TTC ATC AAT AGT CAT CTG ATC AAG 3′). Mutation was confirmed by DNA sequencing. Transfection of DNA into T-Ag Jurkat cells (40 × 106) was performed by electroporation of DNA (20 μg) using a BTX electro cell manipulator. Transfection of DNA (5 μg) into HEK293 cells (5 × 106 cells at 60% confluency) was performed with Jetprime transfection reagent (VWR) according to the manufacturer’s instructions. After a 48 hr growth period, transfected cells were harvested by centrifugation at 1000 rpm, washed with DPBS (Hyclone) 2 times, and either used immediately or stored at −80 °C as a cell pellet.
Substrate trapping and SAHA competition experiments
Following transfection and harvesting, T-Ag Jurkat cells (40 × 106) were lysed in jurkat lysis buffer (JLB; 500 μL; 50 mM Tris-Cl pH 8.0, 150 mM NaCl, 10% glycerol and 0.5% triton-X100) containing 1X protease inhibitor cocktail (GenDEPOT) at 4°C for 30 min with rotation. The soluble fraction of the lysates was collected by centrifugation at 13.2 × 103 rpm for 10 min. HDAC1-FLAG wild type or mutant proteins in the lysates were immunoprecipitated using anti-FLAG agarose beads (20 μL; Sigma) by incubating at 4°C overnight with rocking. For SAHA competition reactions, SAHA (final concentration of 0.8 mM) was added during immunoprecipitation. Bound beads were washed three times at 4°C with JLB containing 500 mM NaCl (1 mL). Bound proteins were separated by SDS-PAGE and visualized with Sypro Ruby total protein stain (Molecular Probes) according to the manufacturer’s instructions.
For histone binding experiments, HEK293 cells (40 × 106) were separately transfected with wild type or mutant HDAC1 plasmids, grown normally for 48 hr, and then treated for another 24 hours with or without SAHA (10 μM) in the growth media. After harvesting and lysis, HDAC proteins were immunoprecipitated using anti-FLAG agarose beads as described. Bound proteins were separated by SDS-PAGE (12%), transferred to PVDF membrane (Immobilon P), and immunoblotted with FLAG or acetyl histone H3 antibodies.
Mass spectrometry analysis
Gel slices containing protein bands were excised from the Sypro Ruby stained gel, washed with destaining buffer (50 μL; 1:1 (v/v) acetonitrile: 50 mM ammonium bicarbonate) for 15 min at room temperature, and dehydrated in acetonitrile (50 μL). The gel pieces were rehydrated with 50 mM ammonium bicarbonate (50 μL) for 5 min and then an equal volume of acetonitrile was added. After incubating at room temperature for 15 min, the gel slices were dehydrated again in acetonitrile (50 μL). After removal of the acetonitrile, the gel slices were dried using a speedvac concentrator (ThermoSavant). Gel slices were swelled with reducing buffer (100 μL; 50 mM TCEP in 25 mM ammonium bicarbonate) for 10 min at 37°C, followed by incubation with alkylation buffer (100 μL; 55 mM iodoacetamide in 25 mM ammonium bicarbonate) for 1 hr at room temperature in the dark while shaking. Then, gel pieces were washed 2 times with destaining buffer (50 μL) for 5 min at room temperature, dehydrated with acetonitrile (50 μL), and dried. To digest the gel-bound proteins, gel slices were incubated with digestion buffer (50 μL; 20 ng/μL trypsin in 40 mM ammonium bicarbonate and 9% acetonitrile) overnight at 37°C. The resultant digest solution was collected and the gel pieces were extracted using extraction buffer (50 μL; 50 % acetonitrile and 0.2 % formic acid (ProteoChem). The combined digestion and extraction solutions were dried and stored at −20°C in preparation for MS analysis at the Wayne State University and Karmanos Cancer Center Proteomics Core faciltiy.
For MS analysis, dried peptide digests were resuspended in a solution of 5% acetonitrile, 0.1% formic acid and 0.005% trifluoroacetic acid. Samples were separated by ultra high pressure reverse phase chromatography using an Acclaim PepMap RSLC column and an Easy nLC 1000 UHPLC system (Thermo). Peptides were analyzed with a Q-Exactive mass spectrometer (Thermo) with a 70,000 resolution MS1 scan over 375–1600 m/z, followed by 17,500 resolution MS2 scans using a 1.6 m/z window and 30% normalized collision energy for HCD. Peak lists were generated with Proteome Discoverer (ver 1.4; Thermo), and peptides scored using Mascot (ver 2.4; Matrix Science). The search parameters included parent and fragment ion tolerances of 15 ppm and 0.02 Da, respectively, fixed modification of +57 on C (carbamidomethylation), variable modifications of +16 on M (oxidation), +42 on K and N-termini (acetylation), +80 on STY (phosphorylation), and a tryptic digest with up to 1 missed cleavage. MS2 spectra were searched against a consensus human protein database from SwissProt, and simultaneously against a scrambled database to calculate the false discovery rate (FDR). Results were imported into Scaffold (ver 4.3; Proteome Software) and a subset database search was performed with X!Tandem using slightly wider search parameters (up to 2 missed cleavages; additional variable modifications of −18 and −17 on N-termini (ammonia loss or conversion to pyro-glutamic acid). Protein identification was considered to be positive if at least 1 unique peptide was scored as ≤1% FDR, and the protein threshold was ≤1% FDR (Table S1).
Deacetylation assays
Recombinant full length Eg5 (Origene, 20 μg) was acetylated by incubating with acetic anhydride (0.2 mM) in ammonium bicarbonate buffer (50 mM, pH 7) for 1 hr at room temperature in a volume of 100 μL. Excess acetic anhydride was removed using a 3 kDa molecular weight cutoff centriprep column (Sigma Aldrich Amicon ultra). For the in vitro deacetylation assay, recombinant HDAC1 (4 μg, BPS Biosciences) was resuspended in HDAC assay buffer (50 μL; 50 mM Tris-Cl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2) and preincubated in the absence (2% DMSO) or presence of SAHA (2 mM in 2% DMSO) for 30 min at 37°C while shaking at 750 rpm. Acetylated Eg5 (1 μg) was then added and the reaction was incubated at 37°C for 2 hr while shaking at 750 rpm. After incubation, the reaction was stopped by adding 5X SDS loading dye (10 μL; 100 mM Tris-Cl pH 6.8, 4% SDS, 20% glycerol, 0.008% bromophenol blue). The protein products were resolved by SDS-PAGE (10%), transferred to PVDF membrane (Immobilon P), and immunoblotted with acetyllysine, HDAC1 and Eg5 antibodies.
Endogenous HDAC1 and Eg5 were immunoprecipitated separately from T-Ag Jurkat cells using their respective antibodies. Briefly, HDAC1 (1 μL) or Eg5 (1 μL) antibodies were incubated separately with lysates (2 mg) at 4°C for 1 h. Then, each lysate was separately added to Protein A/G agarose beads (20 μL, Santa Cruz), incubated overnight at 4°C, and washed three times with JLB (1 mL). Immunoprecipitated HDAC1 was resuspended in HDAC assay buffer (100 μL; 50 mM Tris-Cl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2) and preincubated in the absence (1.2 % DMSO) or presence of SAHA (3 mM in 1.2% DMSO) for 15 min at 30°C while shaking at 750 rpm. Immunoprecipitated Eg5 was resuspended in HDAC assay buffer (50 μL), mixed with the preincubated HDAC1 immunoprecipitate, and then incubated at 30°C for 1 hr while shaking at 750 rpm. After incubation, proteins were eluted from the beads using SDS loading dye (25 μL; 100 mM Tris-Cl pH 6.8, 4% SDS, 20% glycerol, 0.008% bromophenol blue), resolved by 10% SDS-PAGE, transferred to PVDF membrane (Immobilon P), and immunoblotted with acetyl lysine, HDAC1 and Eg5 antibodies.
Co-Immunoprecipitation
T-Ag Jurkat lysates (2 mg) were incubated with either HDAC1 (1 μL) or Eg5 (1 μL) antibodies at 4°C for 1 hr and then further incubated with protein A/G plus agarose beads (20 μL) overnight at 4°C to immunoprecipitate each protein. For co-immunoprecipitation of overexpressed proteins, HDAC1-FLAG and Eg5-myc DNA were cotransfected into HEK293 cells, as described, and the myc tagged Eg5 proteins were immunoprecipitated by incubating lysates (2 mg) with myc antibody (2 μL) for 1 hr at 4°C. Then, the immunocomplex was further incubated with protein A/G plus agarose beads (20 μL) at 4°C overnight. FLAG tagged HDAC1 protein in the lysates (2 mg) were immunoprecipitated using anti-FLAG agarose beads (20 μL; Sigma) by incubating at 4°C overnight with rocking. Bound beads were washed three times with JLB (1 mL) and bound proteins were eluted with SDS loading dye (25 μL), separated by 10% SDS-PAGE, transferred to PVDF membrane (Immobilon P), and immunoblotted with their respective antibodies.
Identification of acetylated lysines in Eg5
To identify the target site of Eg5 regulated by HDAC1, HEK293 cells (5 × 106 cells at 60% confluency) were transfected with myc-tagged Eg5 plasmid alone or cotranfected with FLAG-tagged wild type HDAC1 and allowed to recover under normal growth conditions for 48 hr, as described. After the 48 hr recovery period, cells were treated without (2% DMSO) or with SHI-1:2 (10 μM in 2% DMSO in growth media) or SAHA (10 μM in 2% DMSO in growth media) for an additional 24 hr before harvesting and washing, as described. Cells were lysed and Eg5 was immunoprecipitated with myc antibody, separated on a SDS-PAGE (10%), and visualized by Sypro Ruby total protein stain (Molecular Probes) according to the manufacturer’s instructions. Bands corresponding to Eg5 were excised from the gel and subjected to in gel digestion and mass spectrometry as described above.
ATPase assays
For in vitro ATPase assays, HEK293 cells (5 × 106 cells at 60% confluency) were transfected with myc-tagged wild type or K890R mutant pRcCMV-Eg5 expression plasmids and allowed to recover under normal growth conditions for 48 hr. After the 48 hr recovery period, cells were treated with SHI-1:2 (10 μM in 2% DMSO in growth media) for an additional 24h before harvesting and washing, as described. Cells were lysed, as described, and Eg5 was immunoprecipitated using the myc antibody (3 μg) and protien A/G agarose beads (20 μL of slurry). Bound myc-tagged Eg5 was eluted using a myc peptide (20 μL; 10 μg/mL in 1X TBS buffer) by incubating at 4 °C for 30 min. Eluted wild type or K890R mutant Eg5 was used in the ADP-Glo assay, which measures the amount of ADP produced, according to manufacturer’s instructions. First, the ATPase reaction was performed with immunoprecipitated Eg5 (2 μL or 10% of the immunoprecipitate) and ATP (0.5 mM) in ATPase reaction buffer (25 mM triethanolamine, 13 mM magnesium acetate, 1.8 mM DTT) at 37 °C for 30 min. The final volume of the reaction was 20 μL. Next, the reaction was transferred to 96 well plate, Glo reagent (20 uL) was added, and the mixture incubated at room temperature for 40 min, followed by the addition of kinase detection reagent (40 uL). After 30 min incubation at room temperature, chemiluminescence readings were taken using the GeNiOS plate reader. A reaction containing no immunoprecipitated Eg5 was used for background correction. The background-corrected signal from the wild type Eg5 reaction was set to 1.0 and the IgG control and K890R signals were calculated as fold change relative to wild type. The histogram with mean fold change and standard error is shown in Figure 5E and Table S3.
Indirect Immunofluorescence
For endogenous HDAC1, HDAC3, HDAC7 and Eg5 immunofluorescence experiments, HEK293 cells were grown on glass cover slips in 6 well plates (Corning). At 40% confluency, cells were fixed with ice-cold methanol for 15 min at −20°C, washed three times with PBS (2 mL; 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4), and blocked with 1% bovine serum albumin (GenDEPOT) in PBST (2 mL; PBS containing 0.1% tween-20) for 1 hr at room temperature. After blocking, cells were incubated with a mixture of primary antibodies to HDAC1 (1:200) and Eg5 (1:1000) in PBST overnight at 4°C (1 mL). After washing 3 times with PBS (2 mL), cells were incubated with a mixture of secondary antibodies in PBST (1 mL; Alexa fluoro488 rabbit antibody and Alexa fluoro 647 mouse antibody; 1:500) for 1 hr at room temperature. Then, cells were washed three times with PBS (2 mL) and mounted onto slides using ProLong gold antifade mounting solution. For HDAC1 and Eg5 colocalization studies (Figure 6), slides were visualized using Leica DMi8 inverted confocal microscope, with images prepared using LAS AF analysis software. The Z stacks were recorded using a Leica DMi8 inverted confocal microscope and the LAS AF software automatically generated a merged image from the original images obtained in the separate channels (red and green) at various Z-stacks, thereby avoiding any errors or shifts that may arise from manual editing. For HDAC3/Eg5 and HDAC7/Eg5 localization studies (Figures S13 and S14), slides were visualized using a Nikon Eclipse Ni-U Microscope with motorized epifluorescence. The merged images were prepared using Nikon NIS Elements software.
For HDAC1-FLAG/Eg5 colocalization experiments, the same procedure was followed except transiently transfected HEK293 cells were fixed with 4% paraformaldehyde for 15 min at room temperature, and 0.3% triton-X-100 was used in place of 0.1% tween-20 in the PBST buffer. The slides were visualized using a Nikon NIS Elements software with motorized epifluorescence. The merged images were prepared using Nikon NIS Elements software.
To monitor and quantify monopolar spindle formation, HEK293 cells grown on glass cover slips in 6 well plates (Corning) to 60% confluency were treated with DMSO, STLC (10 μM) or SHI-1:2 (10 μM) for 48 hr. Then, cells were washed with PBS briefly, fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, followed by permeabilizing with 0.3% triton-X-100 for 10 min at room temperature. Cells were washed three times with PBS and then blocked with 1% bovine serum albumin (GenDEPOT) in PBST (2 mL; PBS containing 0.3% triton-X-100) for 1 hr at room temperature. After blocking, cells were incubated with a primary antibody to α-tubulin (1:4000) overnight at 4 °C, followed by secondary antibody in PBST (1 mL; Alexa fluoro488 rabbit antibody) for 1 hr. Then, cells were washed three times with PBS (2 mL) and mounted onto slides using ProLong gold antifade mounting solution. The slides were visualized using an Nikon Eclipse Ni-U Microscope with motorized epifluorescence. Monopolar spindles were counted from at least 100 mitotic cells and presented as a percentage (Figure S6). All cells counted in the STLC-treated samples displayed monopolar spindles (100%, Figure S6).
Cell cycle analysis using Flow Cytometry
HEK293 cells (5 × 106 cells) were treated with DMSO, STLC (10 μM) or SHI-1:2 (10 μM) for 48 hr, harvested, and washed with PBS twice. Cells (2 X106) were fixed with 70 % ethanol overnight at 4 °C. Fixed cells were washed once with cold PBS (500 μL) and permeabilized with 0.25% Triton-X-100 (in PBS) for 15 min on ice, followed by incubation with the antibody to phospho histone H3 (S10) conjugated to Alexa Fluoro 488 (100 μL, 1:50) for 1 hr. After washing with PBS once, cells were stained with DAPI (1 ug/mL in PBS) overnight at 4 °C. Flow cytometry analysis was performed at the Microscopy, Imaging and Cytometry resources core at the Karmanos Cancer Institute, Wayne State University. Samples were run on a BD LSR II analytical flow cytometer (BD Biosciences, San Jose, CA) and analyzed by ModFit LT v4.0 (Verity Software House, Topsham, ME) and FlowJo v10 (FlowJo, LLC, Ashland, OR) software.
To study the effect of wild type and mutant Eg5 on cell cycle arrest, HEK293 cells (5 × 106 cells at 60% confluency) were transfected with myc-tagged wild type or mutant Eg5 plasmid and allowed to recover under normal growth conditions for 48 hr. After the 48 hr recovery period, cells were treated without (2% DMSO) or with SHI-1:2 (10 μM in 2% DMSO in growth media) for an additional 48h. Cells were harvested, washed and fixed overnight at 4 °C with 70% ethanol. Staining of phospho histone H3 and DAPI was performed and analyzed by flow cytometry as described in the above paragraph.
Quantification and Statistical Analysis
All the data were presented as mean ± standard error from at least three independent trials. All data fitting and statistical analysis was performed using GraphPad Prism software (version 5.01).
Supplementary Material
Highlights.
Eg5 (KIF11) was identified as a substrate of HDAC1 using trapping mutants
HDAC1 deacetylated K890 of Eg5
HDAC1 colocalized with Eg5 during mitosis
HDAC inhibitor caused mitotic arrest through HDAC1-mediated Eg5 acetylation
Acknowledgments
We thank Wayne State University for funding, Dr. Xiang Dong Zhang, Dr. Joy Alcedo and Rashmi Chandra (Department of Biology, Wayne State University) for access to the fluorescence microscope facility, Kusal Samarasinghe (Department of Chemistry, Wayne State University) for the support with confocal microscopy, Dr. Anne Blangy (Centre de Recherche de Biochimie Macromoléculaire) for the generous gift of full length myc-tagged pRcCMV Eg5 construct, Dr. Stuart L. Schrieber (Harvard University) for the pBJ5-HDAC1 construct, the Wayne State University and Karmanos Cancer Center Proteomics Core, which is supported by NIH Grants P30 ES020957, P30 CA022453, and S10 OD010700. The Microscopy, Imaging and Cytometry resources core is supported, in part, by NIH center grant P30CA22453 to the Karmanos Cancer Institute, Wayne State University and the Perinatology Research Branch of the National Institute of Child Health and Development, Wayne State University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
M. H. P. conceived the study. D. A. N. performed all experiments and analyzed the data in the paper, except the histone binding experiments, which were performed by I. D. G. M. K. W. performed initial trapping studies. D. A. N. and M. H. P. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ANH TD, AHN MY, KIM SA, YOON JH, AHN SG. The histone deacetylase inhibitor, Trichostatin A, induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells. Oncol Rep. 2012;27:455–60. doi: 10.3892/or.2011.1496. [DOI] [PubMed] [Google Scholar]
- BARADARI V, HUETHER A, HOPFNER M, SCHUPPAN D, SCHERUBL H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr Relat Cancer. 2006;13:1237–50. doi: 10.1677/erc.1.01249. [DOI] [PubMed] [Google Scholar]
- BLAGOSKLONNY MV, ROBEY R, SACKETT DL, DU L, TRAGANOS F, DARZYNKIEWICZ Z, FOJO T, BATES SE. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther. 2002;1:937–41. [PubMed] [Google Scholar]
- BLANGY A, LANE HA, D’HERIN P, HARPER M, KRESS M, NIGG EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–69. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- BUIST A, BLANCHETOT C, TERTOOLEN LG, DEN HERTOG J. Identification of p130cas as an in vivo substrate of receptor protein-tyrosine phosphatase alpha. J Biol Chem. 2000;275:20754–61. doi: 10.1074/jbc.M001626200. [DOI] [PubMed] [Google Scholar]
- CARTRON PF, BLANQUART C, HERVOUET E, GREGOIRE M, VALLETTE FM. HDAC1-mSin3a-NCOR1, Dnmt3b-HDAC1-Egr1 and Dnmt1-PCNA-UHRF1-G9a regulate the NY-ESO1 gene expression. Mol Oncol. 2013;7:452–63. doi: 10.1016/j.molonc.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOUDHARY C, KUMAR C, GNAD F, NIELSEN ML, REHMAN M, WALTHER TC, OLSEN JV, MANN M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- CHUANG C, PAN J, HAWKE DH, LIN SH, YU-LEE LY. NudC deacetylation regulates mitotic progression. PLoS One. 2013;8:e73841. doi: 10.1371/journal.pone.0073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESTIU G, GREENBERG E, HARRISON CB, KWIATKOWSKI NP, MAZITSCHEK R, BRADNER JE, WIEST O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J Med Chem. 2008;51:2898–906. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- FLINT AJ, TIGANIS T, BARFORD D, TONKS NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–5. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLASER KB, LI J, STAVER MJ, WEI RQ, ALBERT DH, DAVIDSEN SK. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun. 2003;310:529–36. doi: 10.1016/j.bbrc.2003.09.043. [DOI] [PubMed] [Google Scholar]
- HASSIG CA, TONG JK, FLEISCHER TC, OWA T, GRABLE PG, AYER DE, SCHREIBER SL. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc Natl Acad Sci U S A. 1998;95:3519–24. doi: 10.1073/pnas.95.7.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HE S, KHAN DH, WINTER S, SEISER C, DAVIE JR. Dynamic distribution of HDAC1 and HDAC2 during mitosis: association with F-actin. J Cell Physiol. 2013;228:1525–35. doi: 10.1002/jcp.24311. [DOI] [PubMed] [Google Scholar]
- HU E, CHEN Z, FREDRICKSON T, ZHU Y, KIRKPATRICK R, ZHANG GF, JOHANSON K, SUNG CM, LIU R, WINKLER J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J Biol Chem. 2000;275:15254–64. doi: 10.1074/jbc.M908988199. [DOI] [PubMed] [Google Scholar]
- ISHII S, KURASAWA Y, WONG J, YU-LEE LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci U S A. 2008;105:4179–84. doi: 10.1073/pnas.0710140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAMALADDIN S, KELLY RD, O’REGAN L, DOVEY OM, HODSON GE, MILLARD CJ, PORTOLANO N, FRY AM, SCHWABE JW, COWLEY SM. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc Natl Acad Sci U S A. 2014;111:9840–5. doi: 10.1073/pnas.1321330111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELLY RD, COWLEY SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–9. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- KHORASANIZADEH S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116:259–72. doi: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- KRAMER OH, GOTTLICHER M, HEINZEL T. Histone deacetylase as a therapeutic target. Trends Endocrinol Metab. 2001;12:294–300. doi: 10.1016/s1043-2760(01)00438-6. [DOI] [PubMed] [Google Scholar]
- KRUHLAK MJ, HENDZEL MJ, FISCHLE W, BERTOS NR, HAMEED S, YANG XJ, VERDIN E, BAZETT-JONES DP. Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J Biol Chem. 2001;276:38307–19. doi: 10.1074/jbc.M100290200. [DOI] [PubMed] [Google Scholar]
- MARKS PA, RICHON VM, RIFKIND RA. Histone Deacetylase Inhibitors: Inducers of Differentiation or Apoptosis of Transformed Cells. Journal of the National Cancer Institute. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- MERRITT R, HAYMAN MJ, AGAZIE YM. Mutation of Thr466 in SHP2 abolishes its phosphatase activity, but provides a new substrate-trapping mutant. Biochim Biophys Acta. 2006;1763:45–56. doi: 10.1016/j.bbamcr.2005.11.013. [DOI] [PubMed] [Google Scholar]
- MILLARD CJ, WATSON PJ, CELARDO I, GORDIYENKO Y, COWLEY SM, ROBINSON CV, FAIRALL L, SCHWABE JW. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67. doi: 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAKE K, YOSHIZUMI T, IMURA S, SUGIMOTO K, BATMUNKH E, KANEMURA H, MORINE Y, SHIMADA M. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas. 2008;36:e1–9. doi: 10.1097/MPA.0b013e31815f2c2a. [DOI] [PubMed] [Google Scholar]
- MOTIWALA T, DATTA J, KUTAY H, ROY S, JACOB ST. Lyn kinase and ZAP70 are substrates of PTPROt in B-cells: Lyn inactivation by PTPROt sensitizes leukemia cells to VEGF-R inhibitor pazopanib. J Cell Biochem. 2010;110:846–56. doi: 10.1002/jcb.22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NALAWANSHA DA, PFLUM MKH. LSD1 substrate binding and gene expression are affected by HDAC1-mediated deacetylation. ACS Chemical Biology. 2017;12:254–264. doi: 10.1021/acschembio.6b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWBOLD A, SALMON JM, MARTIN BP, STANLEY K, JOHNSTONE RW. The role of p21(waf1/cip1) and p27(Kip1) in HDACi-mediated tumor cell death and cell cycle arrest in the Emu-myc model of B-cell lymphoma. Oncogene. 2014;33:5415–23. doi: 10.1038/onc.2013.482. [DOI] [PubMed] [Google Scholar]
- PEART MJ, TAINTON KM, RUEFLI AA, DEAR AE, SEDELIES KA, O’REILLY LA, WATERHOUSE NJ, TRAPANI JA, JOHNSTONE RW. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003;63:4460–71. [PubMed] [Google Scholar]
- RICHON VM, SANDHOFF TW, RIFKIND RA, MARKS PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIKIMARU T, TAKETOMI A, YAMASHITA Y, SHIRABE K, HAMATSU T, SHIMADA M, MAEHARA Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72:69–74. doi: 10.1159/000111106. [DOI] [PubMed] [Google Scholar]
- ROBBINS AR, JABLONSKI SA, YEN TJ, YODA K, ROBEY R, BATES SE, SACKETT DL. Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle. 2005;4:717–26. doi: 10.4161/cc.4.5.1690. [DOI] [PubMed] [Google Scholar]
- RYU JK, LEE WJ, LEE KH, HWANG JH, KIM YT, YOON YB, KIM CY. SK-7041, a new histone deacetylase inhibitor, induces G2-M cell cycle arrest and apoptosis in pancreatic cancer cell lines. Cancer Lett. 2006;237:143–54. doi: 10.1016/j.canlet.2005.05.040. [DOI] [PubMed] [Google Scholar]
- SASAKI H, MORIYAMA S, NAKASHIMA Y, KOBAYASHI Y, KIRIYAMA M, FUKAI I, YAMAKAWA Y, FUJII Y. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer. 2004;46:171–8. doi: 10.1016/j.lungcan.2004.03.021. [DOI] [PubMed] [Google Scholar]
- SAWIN KE, MITCHISON TJ. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci U S A. 1995;92:4289–93. doi: 10.1073/pnas.92.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHOLZ C, WEINERT BT, WAGNER SA, BELI P, MIYAKE Y, QI J, JENSEN LJ, STREICHER W, MCCARTHY AR, WESTWOOD NJ, LAIN S, COX J, MATTHIAS P, MANN M, BRADNER JE, CHOUDHARY C. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol. 2015;33:415–23. doi: 10.1038/nbt.3130. [DOI] [PubMed] [Google Scholar]
- SKOUFIAS DA, DEBONIS S, SAOUDI Y, LEBEAU L, CREVEL I, CROSS R, WADE RH, HACKNEY D, KOZIELSKI F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J Biol Chem. 2006;281:17559–69. doi: 10.1074/jbc.M511735200. [DOI] [PubMed] [Google Scholar]
- STEVENS FE, BEAMISH H, WARRENER R, GABRIELLI B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008;27:1345–54. doi: 10.1038/sj.onc.1210779. [DOI] [PubMed] [Google Scholar]
- SUZUKI J, CHEN YY, SCOTT GK, DEVRIES S, CHIN K, BENZ CC, WALDMAN FM, HWANG ES. Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin Cancer Res. 2009;15:3163–71. doi: 10.1158/1078-0432.CCR-08-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TADDEI A, MAISON C, ROCHE D, ALMOUZNI G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol. 2001;3:114–20. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- TAUNTON J, HASSIG CA, SCHREIBER SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–11. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- VANNINI A, VOLPARI C, GALLINARI P, JONES P, MATTU M, CARFÍ A, DE FRANCESCO R, STEINKÜHLER C, DI MARCO S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex. EMBO Reports. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAMBUA MK, NALAWANSHA DA, NEGMELDIN AT, PFLUM MK. Mutagenesis Studies of the 14 Å Internal Cavity of Histone Deacetylase 1: Insights towards the Acetate Escape Hypothesis and Selective Inhibitor Design. J Med Chem. 2014;57:642–650. doi: 10.1021/jm401837e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG DF, HELQUIST P, WIECH NL, WIEST O. Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem. 2005;48:6936–47. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- WARRENER R, CHIA K, WARREN WD, BROOKS K, GABRIELLI B. Inhibition of histone deacetylase 3 produces mitotic defects independent of alterations in histone H3 lysine 9 acetylation and methylation. Mol Pharmacol. 2010;78:384–93. doi: 10.1124/mol.109.062976. [DOI] [PubMed] [Google Scholar]
- WEERASINGHE SV, ESTIU G, WIEST O, PFLUM MK. Residues in the 11 A channel of histone deacetylase 1 promote catalytic activity: implications for designing isoform-selective histone deacetylase inhibitors. J Med Chem. 2008;51:5542–51. doi: 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEICHERT W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–76. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- WEICHERT W, ROSKE A, GEKELER V, BECKERS T, EBERT MP, PROSS M, DIETEL M, DENKERT C, ROCKEN C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 2008a;9:139–48. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- WEICHERT W, ROSKE A, NIESPOREK S, NOSKE A, BUCKENDAHL AC, DIETEL M, GEKELER V, BOEHM M, BECKERS T, DENKERT C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008b;14:1669–77. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- WETZEL M, PREMKUMAR DR, ARNOLD B, POLLACK IF. Effect of trichostatin A, a histone deacetylase inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J Neurosurg. 2005;103:549–56. doi: 10.3171/ped.2005.103.6.0549. [DOI] [PubMed] [Google Scholar]
- WOOLNER S, O’BRIEN LL, WIESE C, BEMENT WM. Myosin-10 and actin filaments are essential for mitotic spindle function. J Cell Biol. 2008;182:77–88. doi: 10.1083/jcb.200804062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU J, KATREKAR A, HONIGBERG LA, SMITH AM, CONN MT, TANG J, JEFFERY D, MORTARA K, SAMPANG J, WILLIAMS SR, BUGGY J, CLARK JM. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. 2006;281:11002–10. doi: 10.1074/jbc.M600498200. [DOI] [PubMed] [Google Scholar]
- YANG WM, INOUYE C, ZENG Y, BEARSS D, SETO E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc Natl Acad Sci U S A. 1996;93:12845–50. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG WM, YAO YL, SUN JM, DAVIE JR, SETO E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272:28001–7. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- YOON HG, CHAN DW, REYNOLDS AB, QIN J, WONG J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell. 2003;12:723–34. doi: 10.1016/j.molcel.2003.08.008. [DOI] [PubMed] [Google Scholar]
- ZHANG SH, LIU J, KOBAYASHI R, TONKS NK. Identification of the cell cycle regulator VCP (p97/CDC48) as a substrate of the band 4.1-related protein-tyrosine phosphatase PTPH1. J Biol Chem. 1999;274:17806–12. doi: 10.1074/jbc.274.25.17806. [DOI] [PubMed] [Google Scholar]
- ZHAO S, XU W, JIANG W, YU W, LIN Y, ZHANG T, YAO J, ZHOU L, ZENG Y, LI H, LI Y, SHI J, AN W, HANCOCK SM, HE F, QIN L, CHIN J, YANG P, CHEN X, LEI Q, XIONG Y, GUAN KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–4. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.