

Abstract
We herein propose the use of fluoroacetamide and difluoroacetamide moieties as sensitive tags for the detection of sugar–protein interactions by simple 1H and/or 19F NMR spectroscopy methods. In this process, we have chosen the binding of N,N′‐diacetyl chitobiose, a ubiquitous disaccharide fragment in glycoproteins, by wheat‐germ agglutinin (WGA), a model lectin. By using saturation‐transfer difference (STD)‐NMR spectroscopy, we experimentally demonstrate that, under solution conditions, the molecule that contained the CHF2CONH‐ moiety is the stronger aromatic binder, followed by the analogue with the CH2FCONH‐ group and the natural molecule (with the CH3CONH‐ fragment). In contrast, the molecule with the CF3CONH‐ isoster displayed the weakest intermolecular interaction (one order of magnitude weaker). Because sugar–aromatic CH–π interactions are at the origin of these observations, these results further contribute to the characterization and exploration of these forces and offer an opportunity to use them to unravel complex recognition processes.
Keywords: fluorine, molecular modeling, molecular recognition, NMR spectroscopy, noncovalent interactions
Introduction
The study of ligand–receptor interactions from structural and energy perspectives remains a key issue in modern chemistry that requires a precise and specific strategy. In this context, the study of protein–carbohydrate interactions is receiving increased attention due to their implication in many fundamental biological and pathological processes.1 The complete characterization of the molecular determinants at the sugar–protein interface has a pivotal role for the design and development of new chemical probes for the detection2, 3, 4 and controlled modulation of the binding process. For the structurally complex class of N‐glycans, which often present multiple binding epitopes,5 analysis of their interaction parameters is a major challenge. Thus, the use of NMR spectroscopy and dedicated chemical probes can help to overcome the difficulties frequently found in the X‐ray analysis of protein–glycan interactions,6 and may permit us to dissect the fine structural and conformational details of the binding event. To this end, the use of 13C‐labeled glycans7 or paramagnetic lanthanide binding tags has been proposed.8 In addition, recent advances in the synthesis of glycomimetics, which can modify a specific atom or chemical moiety in a regio‐ and stereoselective manner, have opened new avenues for structure‐based studies of molecular recognition events.9 The characterization of the associated binding energy is also of paramount interest for the design.
The introduction of a non‐endogenous atom into the ligand or receptor molecules has proved to be a successful strategy for characterization of the binding process, but also for deduction of the enthalpy contribution of a specific group to the interaction event.10, 11 Although this strategy has been widely applied for the detection of hydrogen‐bond donor/acceptor groups, the use of specific NMR spectroscopy probes to analyze sugar–aromatic interactions has been less explored. A recent analysis of X‐ray crystallographic structures of protein–carbohydrate complexes in the protein data databank (PDB) highlights the key role that these interactions play in the molecular recognition of glycans, and demonstrates the presence of amino acids with electron‐rich aromatic side chains in the receptor binding sites, whereas aliphatic amino acids are underrepresented.12 In addition to the widely known stacking interaction between the more hydrophobic sugar plane and aromatic amino acid side chains,13 there are many examples of the role of methyl/aromatic interactions as additional stabilizing forces in carbohydrate–protein binding events.14 In fact, acetamide sugars are ubiquitous in nature, and GlcNAc, GalNAc, and NeuNAc residues are important recognition elements in many human glycoproteins and glycoconjugates. Recently, we proposed the difluoroacetamide group as a novel 19F‐containing acetamide surrogate for the study of the interactions of lectins with acetylated amino sugars by using NMR spectroscopy.15 Wheat‐germ agglutinin (WGA), a lectin known to bind GlcNAc and Neu5Ac, was used as a model receptor. One of the key interactions involved in the recognition of GlcNAc is the CH–π stacking of the methyl of the acetamide group with the aromatic ring of a tyrosine residue of WGA.14 According to the theoretical calculations, the presence of two fluorine atoms at the acetamide group should enhance the interaction between the corresponding N,N′‐diacetyl chitobiose and N,N′,N′′‐triacetyl chitotriose mimics and WGA, given the polarization of the unique C−H bond at the CHF2CONH‐ function by the electron‐withdrawing fluorine atoms.15, 16, 17, 18 As a further expansion of this idea, herein we analyze the complete series of fluorine‐to‐hydrogen substitutions at the acetamide methyl group in N,N′‐diacetyl chitobiose and describe the NMR spectroscopy analysis of their interactions with WGA. Additionally, we show that the frequently used trifluoroacetamide function (CF3CONH‐) significantly reduces the binding energy due to the absence of any CH–π donor group. On a more general level, we demonstrate that access to various synthetic probes that only differ by a single atom allows the structural and energetic characterization of critical CH–π interactions in sugar–receptor recognition at the atomic level, which thus complements recent advances in the study of direct pyranose/aromatic stacking.16, 17, 18, 19 This approach, together with other already established protocols,4, 5, 6, 7, 8 may permit detailed NMR spectroscopic analyses of the binding events of very complex molecules with their receptors.
Results and Discussion
We have previously demonstrated that difluoroacetamide mimics of N,N′‐diacetyl chitobiose and N,N′,N′′‐triacetyl chitotriose permit a simple characterization of their binding epitopes to WGA with 1H and 19F NMR spectroscopic methods. Although no quantitative analysis of the interaction energy was performed, it was estimated that the corresponding affinities were very similar to those of the natural parent molecules (K D=0.19 and 0.09 mm for N,N′‐diacetyl chitobiose and N,N′,N′′‐triacetyl chitotriose, respectively).15, 20, 21 Herein, we expand our initial findings to analyze quantitatively the interaction between WGA and the complete set of fluoroacetamide analogues of N,N′‐diacetyl chitobiose, with either one, two, or three fluorine atoms at each modified GlcNAc residue, by using NMR spectroscopy. We prove experimentally that the CHF2‐, CH2F‐, and CF3‐ analogues are useful probes to monitor the interaction process by using 19F NMR spectroscopy (for the three mimics) or 1H NMR spectroscopy (for mimics that contain C−H bonds). The synthesis of the mono‐, di‐, and trifluoroacetamide‐containing analogues of N,N′‐diacetyl chitobiose, (1, 2, and 3, Figure 1) is summarized in Scheme 1 and described in detail in the Experimental Section.
Figure 1.
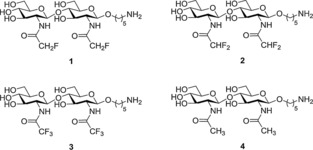
Schematic representation of the different fluorine‐containing glycomimetics (1–3) studied herein and the natural analogue (4).
Scheme 1.
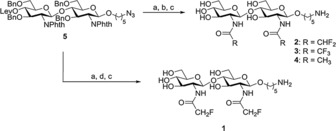
Synthesis of fluorinated derivatives. Reagents and conditions: a) NH2(CH2)2NH2, nBuOH, MW: 3 cycles, 30 min, 120 °C; b) i) (RCO)2O, pyridine; ii) NaOMe, MeOH; c) H2, 10 % Pd/C, MeOH:H2O 9:1, 1 % trifluoroacetic acid (TFA); d) i) CH2FCOOH, NHS, DCC, DMF; ii) NaOMe, MeOH. The synthesis of compounds 2, 4, and 5 have been described previously.12
NMR spectroscopy studies
1H and 19F NMR spectra of novel disaccharides 1 and 3, which contain CH2F‐ and CF3‐ groups, were assigned by using standard NMR spectroscopy techniques as described for 2 (Figures S1–S6 in the Supporting Information).15 For compound 1, the 1H NMR signals of protons at each monofluoroacetamide moiety appear as a doublet of doublets due to a strong heteronuclear coupling (2 J(H,F)=46.2 Hz) and a homonuclear coupling (2 J(H,H)=14.6 Hz), which gave rise to a complex set of 16 NMR signals at around δ=4.9 ppm in a region absent of any other sugar resonance signal. Fittingly, all these 1H nuclei are diastereotopic and differ between the two sugar rings (Figures S1 and S2). Despite this apparent spectral complication, the possibility of distinguishing between the protons at every sugar residue represents an important advantage for structural analysis of the binding mode (see below). The two 19F signals appear as triplets, one centered at δ=−227.12 ppm (reducing end) and the other one at δ=−227.19 ppm (non‐reducing end, Figure S3). For compound 2, as described earlier, the 1H NMR signals of the difluoroacetamide moieties each appear as a triplet with a strong heteronuclear coupling of 2 J(F,H)=53.5 Hz at δ≈6.1 ppm in a spectral region that lacks interference from other signals. In this case, each resonance signal of the 19F nuclei is a doublet of doublets centered at δ≈−127 ppm, with a very large 19F–19F homonuclear coupling of 303 Hz.15 The external components of the multiplet of every 19F signal are barely visible (see Figure S5). Finally, the 19F signals for 3 are singlets at δ=−75.63 and −75.67 ppm for the non‐reducing and reducing ends, respectively (Figure S6). Therefore, the key reporter 1H NMR signals of the fluorine‐containing acetamide moieties of 1 and 2 and the 19F NMR signals of 1–3 appear at significantly different chemical shifts and can be used to monitor possible interaction processes. The 1H NMR signals of the fluoroacetamide moieties of the two residues of 1 and 2 are easily distinguished. Furthermore, those of 2 appear at δ=6.1 ppm, a chemical shift region absent of any other sugar signal in the NMR spectra of N‐glycans and very rarely occupied in the NMR spectra of any putative receptor protein. These fairly diverse chemical shifts may even permit simultaneous evaluation of the relative binding features towards WGA by using a mixture that contains all three molecules.
NMR spectral analysis of the ligand/WGA interaction for 1–3
Our previous NMR spectroscopy study of the interaction between 2 and WGA revealed the key epitope for the molecular recognition process.13 In addition to the existence of a double aromatic–pyranose stacking with both GlcNAc entities, the (fluoro)acetamide moieties are also directly involved in the interaction. Indeed, in this region and in the presence of WGA at a variety of ligand/receptor molar ratios, the 1H and 19F signals assigned to the difluoroacetamide moiety at the non‐reducing end showed a significant line broadening, whereas the corresponding signals at the reducing end were less affected. Thus, use of the CHF2 probe revealed the preferred interaction of the non‐reducing sugar moiety, which is the major interacting ligand epitope.14, 15, 22 Analysis of the spectra of 1 and 3 in the presence of WGA also confirmed the presence of the same binding epitope for these ligands. In all cases, a more pronounced line‐broadening effect for the 1H and 19F NMR signals of the nuclei at the non‐reducing end was observed (Figure 2).
Figure 2.
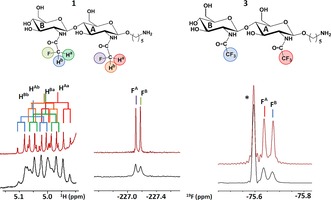
1H and 19F NMR spectra (1H‐decoupled) at 310 K for 1 and 3 (1 mm concentration in deuterated PBS, 50 mm, pH 6) in the absence (top) and presence (bottom) of WGA (60 μm, 17:1 ligand/receptor molar ratio). The existence of different line‐broadening effects for the 1H NMR signals of 1 is evident (left spectra). The presence of distinct line‐broadening properties is also clear in the corresponding 19F NMR spectra, especially for 1. The signal assignment for the 1H and 19F nuclei are color‐coded. An impurity is labeled with *.
As also previously observed for 2,15 the 19F NMR signals of 1 are more sensitive to changes in their local environment in the presence of the receptor than the 1H resonances and the discrimination of the epitope in terms of the GlcNAc residue is straightforward.
Next, we studied the relative affinity of the three fluorinated analogues. Because 3 does not carry any hydrogen atom at the fluorinated acetamide moiety, a first analysis was performed by examining the linewidth of the 19F NMR signals for an equimolar mixture of 1–3.23 The addition of WGA to the sample that contained 1–3 gave rise to observable line‐broadening effects for all 19F NMR signals, although to a different extent depending on the molecule and on the particular fluorine substitution pattern (Figure 3). It is known that these effects are due to the existence of a faster transverse relaxation rate of the ligand nuclei due to the free–bound chemical exchange process in the presence of the protein. These effects can be correlated with the exchange‐rate binding event and indirectly with the relative affinity. For example, at a 10:1 ligand/protein molar ratio, all of the 19F NMR signals were rather broad and the correlation with the existence of different affinities for the three molecules was not evident. The same applies for the largest employed ligand/receptor molar ratio (33:1), for which the line‐broadening effects were more limited and thus conclusions should be made with caution. For the 17:1 molar ratio, the observed line‐broadening effects permitted both modified GlcNAc residues of every N,N′‐diacetyl chitobiose analogue to be differentiated. Consistently, analysis of the 19F NMR spectra (Figure 3) and the linewidths (Table 1) shows a very significant line broadening of the 19F signals at the non‐reducing moiety for compounds 1–3. Therefore, it is evident that the major binding epitope of the disaccharide‐mimetic ligands to WGA remains unaltered upon fluorine substitution at the acetamide moiety.
Figure 3.
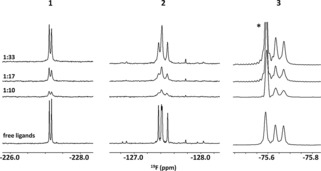
Compound‐specific regions of the 19F NMR spectra of a mixture of 1, 2, and 3 (from left to right, respectively) in the absence (bottom) and presence of WGA at decreasing ligand/WGA ratios (from top to bottom). The existence of reversible binding to WGA in the fast‐intermediate exchange regime on the chemical shift timescale is evident for the three molecules, as deduced from the observed broadenings of the 19F NMR resonance signals. The smallest degree of perturbation takes place for trifluoroacetamide‐containing analogue 3. The spectra were acquired at 298 K with a 30 μm protein concentration and by increasing the ligand concentration from 0.3 to 1.0 mm.
Table 1.
19F NMR signal linewidths at half height for 1–3 as function of the protein/ligand (P/L) concentration.[a]
| P/L molar ratio | NMR signal linewidth [Hz] | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| ‐CH2F (red) | ‐CH2F (non‐red) | ‐CHF2 (red) | ‐CHF2 (non‐red) | ‐CF3 (red) | ‐CF3 (non‐red) | |
| free | 8.0 | 8.0 | 6.9 | 6.2 | 6.4 | 6.4 |
| 1:10 | 32.7 | 34.2 | 22.0 | 31.4 | 9.7 | 12.0 |
| 1:17 | 20.2 | 26.5 | 18.3 | 23.4 | 8.0 | 10.3 |
| 1:33 | 13.5 | 15.3 | 11.7 | 16.9 | 7.7 | 8.4 |
[a] Red=reducing end, Non‐red=non‐reducing end.
This analysis also permitted us to deduce that the linewidth values obtained for 3 were much smaller than those for 1 and 2. The measured differences between these last two molecules were less significant, which precluded the derivation of any conclusion in an unambiguous manner. In fact, at a 1:33 protein/ligand molar ratio, the linewidths of the 19F signals at the non‐reducing end of molecule 3 are similar to those for the protein‐free molecule (i.e., 8.4 and 6.4 Hz, respectively). In contrast, even at a 1:33 protein/ligand ratio, the linewidths of the 19F NMR signals for both 1 and 2 are twofold larger than for the free form (Table 1).
Although this superficial analysis suggests that the binding affinity of 3 towards WGA is lower than that of 1 and 2, additional NMR spectroscopy experiments were performed to clarify the relative stability of the complexes of WGA with 1–3, and to estimate the corresponding dissociation constants (K D). 1H saturation‐transfer difference NMR spectroscopy (STD) experiments were carried out by taking advantage of the distinct chemical shifts of the 1H NMR signals for the protons of 1 and 2 at the fluoroacetamide moieties, which did not overlap with the sugar ring hydrogen atoms.
Therefore, STD experiments were first performed on a mixture of 1–3 with WGA (50:1 ligand/lectin molar ratio). The obtained data additionally supported the notion that the non‐reducing end is always the major binding epitope (Figure 4). In fact, the strongest STD signal for compound 2 corresponded to the C−H proton (100 %) of the difluoroacetamide moiety of the non‐reducing GlcNAc moiety, followed by the corresponding C−H proton at its reducing end (40 %). The corresponding STD signals for 1 were considerably weaker (20 and 15 % for the acetamide protons at the non‐reducing and reducing ends, respectively), which strongly suggested that 2 is the best binding partner for WGA.
Figure 4.

600 MHz 1H NMR STD spectra obtained for a 50:1 mixture of 1–3 with WGA (conditions: 30 μm protein and 1.5 mm ligand at 310 K). The extracted STD values are gathered in the schematic drawings of 1 and 2 (top). The epitope mapping is also described with the corresponding color code. The STD control of the ligand without WGA is shown in the bottom right panel. The on‐resonance frequency for generation of the STD effects was set at δ=7.3 ppm, which corresponds to the aromatic protein resonance region.
The dissociation constants (K D) of the complexes of WGA with 1 and 2 were quantitatively estimated from competition STD experiments with 4, the natural compound (see Figure 5 and Table S1), as described in the Experimental Section. The dissociation constant obtained for monofluoroacetamide 1 was similar (≈150 μm) to the K D of the natural compound 4 (K D≈190 μm), whereas the value obtained for difluoroacetamide analogue 2 was lower (≈50 μm). Therefore, the STD data indicate that, of the studied compounds, chitobiose derivative 2 is the best binder for WGA. We also estimated the K D for compound 3. In this case, the competition STD experiments were carried out by using the inverse strategy, with compound 3 as the competitor in a mixture of WGA and natural ligand 4. The obtained results (Table S2) indicate that derivative 3 is the weaker binder with a K D value of approximately 650 μm.
Figure 5.
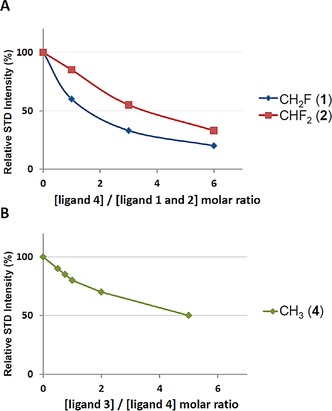
1H NMR STD spectroscopy competition experiments used for K D determination. A) STD intensity of methyl proton signals in ligand 1 (blue) and 2 (red), as a function of the concentration of natural ligand 4. The absolute concentration of compounds 1 and 2 was 0.5 mm and that of the WGA protein was 10 μm. B) STD intensity of methyl proton signals of natural ligand 4 as a function of the concentration of ligand 3. The absolute concentration of compound 4 was 1.5 mm and that of the WGA protein was 30 μm.
Glycan arrays
Multivalent display on dendrimers, nanoparticles, or surfaces is a common strategy to enhance the affinity of sugar–lectin interactions by engaging simultaneously with more than a single receptor or by rapid rebinding.24 However, multivalent presentation of a ligand can alter the binding epitope accessibility. In fact, it is not unlikely that some structural features recognized on the ligand in dilute solution might be hidden in dense presentations on surfaces and/or on nanoparticles. To assess the effect of the fluorine modification on WGA binding in a high‐density presentation of the ligands, we printed aliquots of the four chitobiose derivatives onto NHS (N‐hydroxysuccinimide)‐activated glass slides. The microarrays were then incubated with different concentrations (0.1–3.5 nm) of fluorescently labeled WGA. As seen in Figure 6, the four chitobiose analogues displayed high and consistent affinities at all protein concentrations and provided similar fluorescence values. Under these experimental conditions, we did not observe any effect on the observed macroscopic binding affinity owing to the presence of the fluorine atoms.
Figure 6.
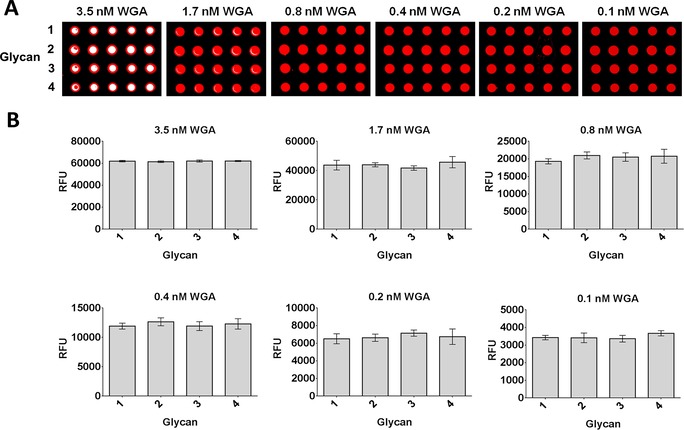
Glycan microarray analysis of compounds 1–4 after binding with Alexa Fluor®‐647‐labeled WGA at increasing protein concentrations. A) Fluorescence images after incubation with different concentrations of WGA‐647. B) Fluorescence quantification after incubation with WGA‐647. Each histogram represents the average RFU values for five replicates with the standard deviation of the mean.
Discussion
The obtained experimental results in solution demonstrate that, for the fluorine‐containing mimics and natural chitobiose (compounds 1–4), the number of fluorine atoms significantly affects the binding affinity (approximately fourfold between 2 and 4) towards WGA. In particular, the binding affinity increased as the hydrogen atom of the (fluoro)acetamide group becomes more polarized by the electron‐withdrawing effect of the fluorine. Previous theoretical calculations15 carried out for simple models (benzene and N‐methyl(fluoro)acetamide) predicted an increased binding enthalpy for the CH–π interaction as the polarization of the CH bond increased. Together, these results allow us to relate the observed increased binding affinities to the magnitude of the CH–π interaction between the acetamide moiety and its aromatic partner at the WGA binding pocket. Moreover, trifluoroacetamide analogue 3, with only C−F bonds at the interacting group epitope, showed the lowest affinity, even lower than that of natural compound 4. In contrast, 1 and 2, with one or two fluorine atoms, presented a higher affinity than 4. These findings can be explained by the degree of polarization of the interacting C−H bond, which was increased in the presence of the highly electronegative fluorine atoms and produced a stronger CH donor group to the aromatic system. This observation is supported by ab initio calculations that simulate the charge distribution for the model N‐methyl(fluoro)acetamides (Table S3, Figure S7).
Despite the difference in C−H bond polarization for 1 and 2, the relative binding energy between the two complexes is only approximately 0.67 kcal mol−1. Fittingly, this value is in satisfactory agreement with our previous ab initio calculations (≈0.3 kcal mol−1) for simple acetamide and fluoroacetamides that interact with a single aromatic ring.15 According to the K D estimations, the relative binding energy between 1 (with one fluorine atom) and natural molecule 4, with no fluorine, is approximately 0.14 kcal mol−1. These figures are of the order of those estimated for the role of C−H polarization in sugar–aromatic stacking.12, 18, 25
Compound 3 lacks any CH–π donor atoms on the acetamide moiety, but presents an electron‐rich group at the interface with the aromatic residue in the complex with WGA. This unfavorable contact considerably reduces the binding affinity.26 In fact, CF3‐containing compound 3 shows a relative binding energy 0.76 kcal mol−1 lower with respect to the natural compound 4.
The use of different CHxFyCO‐ appendages has allowed the analysis and quantification of the energy contribution of polarized CH donor groups27 in CH–π stacking interactions, with implications for the binding affinity. We have found that fluorine substitution at the acetamide moiety enhances the binding to the receptor, in which the ‐CHF2 group provides the best interaction, followed by ‐CH2F. In contrast, the ligand with the ‐CF3 group showed the weakest binding constant. These results offer the opportunity to modulate carbohydrate‐protein interactions in solution by the introduction of fluorine atoms.
The four chitobiose analogues were bound under the glycan microarray conditions, which confirmed that the introduction of fluorine also does not introduce deleterious effects in the molecular recognition process under multivalent presentation conditions. However, the affinity differences found in solution are absent in the microarray experiment. In fact, the binding affinities estimated from the microarray experiment are in the nanomolar range (1.98, 1.86, 2.01, and 1.81 nm for compounds 1, 2, 3, and 4, respectively, see Figure S8) and almost identical. The reason for this behavior is not fully evident. One plausible explanation resides in the particular multivalent effects that take place on the surface and that are absent in the solution state. Indeed, for WGA, a significant increase in the affinity has been observed when the saccharide ligands are presented in a multivalent display form with respect to the monovalent form in solution.28 Moreover, it has been also observed that the binding affinity of saccharides for other lectins (Con A) may increase by more than three orders of magnitude on going from the solution state to the immobilized presentation of a glycan array. In particular, the dissociation constant of mannose (Man1), and the corresponding tetra‐, octa‐, and nona‐mannosides (Man4, Man8, Man9) versus Con A are 250, 55, 0.42, and 0.13 μm in solution, respectively.29 These significant differences are basically abolished in the microarray experiment, in which all the molecules display nanomolar affinities (83, 80, 76, and 73 nm, respectively). A similar process probably takes place for the natural and fluoroderivatives presented herein. The affinity differences observed in solution are lost under the surface presentation conditions, in which additional mechanisms30 are responsible for the increased affinity and which veil the subtle differences observed at the atomic level in solution.
Conclusions
The presence of fluoroacetamide moieties in GlcNAc‐type sugars provides a simple NMR spectroscopy‐based strategy to detect the interaction between these types of glycan and lectin in solution. The interaction of the complete set of chitobiose derivatives with different fluorination patterns at the acetamide moiety (CHxFy‐CO‐) with WGA has been analyzed. STD‐NMR spectroscopy experiments have shown that the CH moiety at the CHxFy‐CO‐ group with either one or two fluorine atoms provides an efficient interaction point for the protein. This intermolecular contact is likely based on sugar–aromatic interactions. Our results demonstrate that the strength of the CH–π interaction can be modulated through the substitution of the hydrogen atoms at the acetamide function by electron‐withdrawing fluorine atoms. Specifically, the substitution of one or two hydrogen atoms by fluorine leads to the polarization of the remaining hydrogen and thus to an increased interaction, as shown by the measured binding constants. Conversely, the complete substitution of all hydrogen atoms on the acetamide function by electron‐rich fluorine leads to an unfavorable contact with the aromatic residue on the receptor and consequently to a decrease in the measured affinity. There is one order of magnitude of difference between the binding affinities determined for compounds 1 and 3 (≈1.4 kcal mol−1). Although not extremely large, these subtle differences contribute to the characterization of the electrostatic term in CH–π interactions and may offer an opportunity to modulate sugar–protein interactions in a site‐specific way. It is also important to note that these modifications do not alter the interaction of the disaccharide with the lectin, as exemplified by using glycan arrays.
Furthermore, given the distinct 1H and 19F NMR spectral features of the different analogues, as exemplified by compounds 1–4, the concurrent use of different CHxFyCO‐ appendages at different positions of one complex molecule (i.e., multiantenna glycans) could provide specific fingerprints for the characterization of binding epitopes at the residue level, which includes the possibility of detecting the existence of multiple binding epitopes that are engaged at the same time. The combination of this fluorine‐based protocol with other chemical approaches (i.e., the use of paramagnetic lanthanides)4, 8 may provide the chemical tools required to advance the complete understanding of sugar–protein recognition events. Advances in chemoenzymatic carbohydrate synthesis permit access to ever more complex target glycans and will finally enable the study of their interactions with natural receptors in great detail by using NMR spectroscopy. This strategy is currently under development in our laboratories.
Experimental Section
NMR spectroscopy analysis
All NMR spectroscopy experiments were recorded at 298 or 310 K by using a 600 MHz Bruker Avance spectrometer equipped with a 19F,1H SEF dual probe optimized for direct 19F detection. Complete 1H signal assignment for molecules 1–4 was obtained by standard TOCSY (60 and 100 ms mixing times), NOESY (300 and 500 ms mixing times), and 1H,13C HSQC experiments. The disaccharide concentration was 1 mm in PBS (50 mm) at pH 6 in D2O and H2O/D2O (90:10). 19F signals were assigned from 2D heteronuclear 1H,19F and homonuclear 19F,19F correlation experiments. STD experiments were performed at 310 K with 30 μm of WGA and 1.5 mm of molecules 1–3. The on‐resonance frequency was set at δ=7.5 ppm and the off‐resonance frequency was at δ=−25 ppm with a 2 s irradiation time, and a PC9 pulse shape without water suppression was used. A T1ρ of 50 ms was used for filtering the protein signals. Negative control STD spectra, in the absence of WGA, were recorded under the same conditions. The STD competition experiments were acquired under the same experimental conditions with increasing concentrations of natural ligand molecule 4 as the competitor for the estimation of the K D values of 1 and 2. The K D value of compound 3 was determined by using an increasing concentration of inhibitor 3 in a solution of WGA/4 at 1:50 molar ratio. The detailed ligand/competitor molecular ratio, relative STD intensity, and the equation used to derive the K D constants are reported in the Supporting Information (Tables S1, S2).
Chemical synthesis: general methods
Chemicals were purchased from Sigma–Aldrich or Acros Organics and used without further purification. All reaction solvents were dried over activated 4 or 3 Å molecular sieves. Microwave irradiation was performed by using a Biotage Initiator monomode oven (Biotage AB, Uppsala, Sweden). All organic solvents were concentrated by using rotary evaporation. Hydrogenation reactions were performed in continuous‐flow hydrogenation reactor H‐Cube® from ThalesNano Nanotechnology (Budapest, Hungary). Glycans were lyophilized by using an ALPHA‐2–4 LSC freeze dryer (Christ, Osterode, Germany). 1H and 13C spectra were acquired by using a Bruker 500 MHz spectrometer and chemical shifts (δ) are given in ppm relative to the residual signal of the solvent used (D2O, δ=4.79 ppm). Splitting patterns are designated as s (singlet), d (doublet), t (triplet), m (multiplet). Coupling constants (J) are reported in Hz. The mass spectrometric data were obtained by using a Micromass®Q‐Tof Premier™ instrument from Waters (Manchester, UK) by direct injection.
1. ‐Aminopentyl‐2‐deoxy‐2‐fluoroacetamido‐β‐d‐glucopyranosyl‐(1→4)‐2‐deoxy‐2‐fluoroacetamido‐β‐d‐glucopyranoside (1)
A solution of 5 (125 mg, 0.107 mmol), 1,2‐ethylenediamine (0.2 mL), and nBuOH (0.8 mL) was heated to 120 °C under microwave irradiation (3 cycles, 30 min each). The solvents were evaporated to dryness and the crude product was used in the next step without further purification. Solutions of fluoroacetic acid (0.5 m), N‐hydroxysuccinimide (0.5 m), and N,N′‐dicyclohexylcarbodiimide (0.5 m) were mixed in a 1:1:1 ratio for 15 min and then centrifuged. The supernatant (8.9 mL) was added to a solution of the crude product in DMF (5 mL) and the resulting mixture was stirred overnight at RT. The crude product was concentrated and dissolved in MeOH (5 mL) and NaOMe (20 μL) was added. The crude product was concentrated and filtered through a plug of silica gel, then dissolved in MeOH/water (9:1) that contained 1 % TFA, and the solution was hydrogenated by passing it twice through a H‐Cube® reactor (0.5 mL min−1, 50 °C, full hydrogen). The reaction mixture was evaporated to dryness, then the crude product was purified by using a Bond Elut carbon cartridge (yield: 22 mg, 30 % over three steps). 1H NMR (500 MHz, D2O): δ=5.07–4.83 (m, 4 H; 2×CH2F), 4.68 (d, J=8.5 Hz, 1 H; H1′), 4.58 (d, J=8.0 Hz, 1 H; H1), 3.96–3.73 (m, 7 H), 3.70–3.58 (m, 4 H), 3.56–3.46 (m, 3 H), 2.98 (t, J=7.7 Hz, 2 H; CH2NH2), 1.71–1.62 (m, 2 H; CH2 linker), 1.63–1.55 (m, 2 H; CH2 linker), 1.43–1.34 ppm (m, 2 H; CH2 linker); 13C NMR (126 MHz, D2O): δ=171.39 (d, J=18.5 Hz), 171.16 (d, J=18.5 Hz), 101.0 (C1′), 100.6 (C1), 80.6, 79.3, 79.1, 76.0, 74.5, 73.2, 72.2, 70.1, 69.7, 60.5, 60.1, 55.2, 54.6, 39.3, 28.0, 26.3, 22.1 ppm; HRMS (Q‐TOF): m/z calcd for C21H37F2N3NaO11: 568.2288 [M+Na]+; found: 568.2240.
2. ‐Aminopentyl‐2‐deoxy‐2‐trifluoroacetamido‐β‐d‐glucopyranosyl‐(1→4)‐2‐deoxy‐2‐trifluoroacetamido‐β‐d‐glucopyranoside (3)
A solution of 5 (125 mg, 0.107 mmol), 1,2‐ethylenediamine (0.15 mL), and nBuOH (0.6 mL) was heated at 120 °C under microwave irradiation (3 cycles, 30 min each). The solvents were evaporated to dryness, then the crude was purified by using Sephadex LH‐20 and eluted with MeOH. The product was dissolved in pyridine, cooled to 0 °C, and trifluoroacetic anhydride was added dropwise. After stirring for 2 h at RT, the mixture was quenched with EtOH and diluted with EtOAc. The organic layer was washed with saturated aqueous CuSO4, water, and saturated aqueous NaHCO3, dried over anhydrous MgSO4, and concentrated. The crude product was dissolved in MeOH (5 mL) and NaOMe (20 μL) was added dropwise. The mixture was concentrated and filtered through a plug of silica gel, then the product was dissolved in MeOH/water (9:1) that contained 1 % TFA and the solution was hydrogenated by passing it twice through a H‐Cube® reactor (0.5 mL min−1, 50 °C, full hydrogen). The reaction mixture was evaporated to dryness, and the crude product was purified by using a Bond Elut carbon cartridge (yield: 22 mg, 33 % over three steps). 1H NMR (500 MHz, D2O): δ=4.71 (d, J=8.4 Hz, 1 H; H1′), 4.62–4.56 (m, 1 H; H1), 3.96–3.89 (m, 2 H), 3.89–3.79 (m, 4 H), 3.76 (dd, J=12.4, 5.4 Hz, 1 H; H6b′), 3.72–3.63 (m, 3 H), 3.60 (dt, J=10.2, 6.4 Hz, 1 H; CH 2O), 3.56–3.47 (m, 3 H), 2.96 (t, J=7.8 Hz, 2 H; CH2NH2), 1.69–1.62 (m, 2 H; CH2 linker), 1.62–1.55 (m, 2 H; CH2 linker), 1.41–1.33 ppm (m, 2 H; CH2 linker); 13C NMR (D2O, 126 MHz): δ=100.3, 78.7, 76.0, 74.5, 72.8, 71.7, 70.2, 69.7, 60.5, 60.0, 56.2, 55.6, 39.3, 38.5, 28.0, 26.4, 22.1 ppm. HRMS (Q‐TOF): m/z calcd for C21H33F6N3NaO11: 640.1911 [M+Na]+; found: 640.1863.
Glycan microarray printing
Ligand solutions (1, 2, 3, and 4) with a final concentration of 50 μm were prepared in sodium phosphate buffer (300 mm, pH 8.5, 0.005 % Tween‐20). These solutions (1.25 nL) were spatially arrayed by using a robotic noncontact spotter sciFLEXARRAYER S11 (Scienion AG, Berlin, Germany) onto NHS‐functionalized glass slides (Nexterion® H, Schott AG, Mainz, Germany). After printing, the slides were placed in a 75 % humidity chamber (saturated NaCl solution) at 25 °C for 18 h. The remaining NHS groups were quenched by placing the slide in a solution of ethanolamine (50 mm) in sodium borate buffer (50 mm, pH 8.0) for 1 h at RT.
Incubation with WGA
The subarrays were compartmentalized by using a 16‐well gasket (Fast Frame® incubation chambers, Whatman®). Solutions of Alexa Fluor® 647‐labeled WGA (100 μL; DOL (degree of labeling):0.1) at different concentrations (0.1, 0.2, 0.4, 0.8, 1.7, 3.5 nm) were incubated in the dark for 1 h in lectin binding buffer (PBS with 5 mm CaCl2, 5 mm MgCl2, and 0.05 % Tween‐20). The slide was washed with PBST (PBS with 0.05 % Tween‐20), PBS, and water, then dried in a slide spinner. The fluorescence was analyzed by using an Agilent G265BA microarray scanner system at 100 PMT (photomultiplier tube). Quantification was achieved by using ProScanArray® Express software (Perkin–Elmer, Shelton, USA) by using an adaptive circle quantification method from 50 (minimum spot diameter) to 300 μm (maximum spot diameter). Average RFU (relative fluorescence units) values with local background subtraction of five replicates and standard deviation of the mean were presented as histograms by using GraphPad Prism 6 software.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge funding by the Spanish Ministry of Economy and Competiveness, MINECO (CTQ2014‐58779‐R, CTQ2015‐64597‐C2‐1P and 2P). This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska‐Curie grant agreements (no. 642870 (ETN‐Immunoshape), no. 642157 (ETN‐Tollerant), and no. 675671 (ETN‐GlycoVAX)). A.A. also thanks MINECO for a Ramón y Cajal contract.
L. Unione, M. Alcalá, B. Echeverria, S. Serna, A. Ardá, A. Franconetti, F. J. Cañada, T. Diercks, N. Reichardt, J. Jiménez-Barbero, Chem. Eur. J. 2017, 23, 3957.
Contributor Information
Dr. Niels Reichardt, Email: nreichardt@cicbiomagune.es
Prof. Dr. Jesús Jiménez‐Barbero, Email: jjbarbero@cicbiogune.es
References
- 1. Solís D., Bovin N. V., Davis A. P., Jiménez-Barbero J., Romero A., Roy R., K. Smetana, Jr. , Gabius H. J., Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 186–235. [DOI] [PubMed] [Google Scholar]
- 2. Yamamoto S., Yamaguchi T., Erdélyi M., Griesinger C., Kato K., Chem. Eur. J. 2011, 17, 9280–9282. [DOI] [PubMed] [Google Scholar]
- 3. Yamamoto S., Zhang Y., Yamaguchi T., Kameda T., Kato K., Chem. Commun. 2012, 48, 4752–4754. [DOI] [PubMed] [Google Scholar]
- 4. Canales A., Mallagaray A., Pérez-Castells J., Boos I., Unverzagt C., André S., Gabius H. J., Cañada F. J., Jiménez-Barbero J., Angew. Chem. Int. Ed. 2013, 52, 13789–13793; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14034–14038. [Google Scholar]
- 5. Ardá A., Blasco P., Varón Silva D., Schubert V., André S., Bruix M., Cañada F. J., Gabius H. J., Unverzagt C., Jiménez-Barbero J., J. Am. Chem. Soc. 2013, 135, 2667–2675. [DOI] [PubMed] [Google Scholar]
- 6. Agirre J., Iglesias-Fernández J., Rovira C., Davies G. J., Wilson K. S., Cowtan K. D., Nat. Struct. Mol. Biol. 2015, 22, 833–834. [DOI] [PubMed] [Google Scholar]
- 7. Kamiya Y., Yanagi K., Kitajima T., Yamaguchi T., Chiba Y., Kato K., Biomolecules 2013, 3, 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Canales A., Mallagaray A., Berbís M. A., Navarro-Vázquez A., Domínquez G., Cañada F. J., André S., Gabius H. J., Pérez-Castells J., Jiménez-Barbero J., J. Am. Chem. Soc. 2014, 136, 8011–8017. [DOI] [PubMed] [Google Scholar]
- 9. Wu L. L., Yang C. L., Lo F. C., Chiang C. H., Chang C. W., Ng K. Y., Chou H. H., Hung H. Y., Chan S. I., Yu S. S., Chem. Eur. J. 2011, 17, 4774–4787. [DOI] [PubMed] [Google Scholar]
- 10. Solís D., Jiménez-Barbero J., Martín-Lomas M., Díaz-Mauriño T., Eur. J. Biochem. 1994, 223, 107–114. [DOI] [PubMed] [Google Scholar]
- 11. Matei E., André S., Glinschert A., Infantino A. S., Oscarson S., Gabius H. J., Gronenborn A. M., Chem. Eur. J. 2013, 19, 5364–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hudson K. L., Bartlett G. J., Diehl R. C., Agirre J., Gallagher T., Kiessling L. L., Woolfson D. N., J. Am. Chem. Soc. 2015, 137, 15152–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vandenbussche S., Diaz D., Fernandez-Alonso M. C., Pan W. D., Vincent S., Cuevas G., Cañada F. J., Jiménez-Barbero J., Bartik K., Chem. Eur. J. 2008, 14, 7570–7578. [DOI] [PubMed] [Google Scholar]
- 14. Espinosa J. F., Asensio J. L., Garcia J. L., Laynez J., Bruix M., Wright C., Siebert H.-C., Gabius H.-J., Cañada F. J., Jiménez-Barbero J., Eur. J. Biochem 2000, 267, 3965–3978. [DOI] [PubMed] [Google Scholar]
- 15. Calle L. P., Echeverria B., Franconetti A., Serna S., Fernández-Alonso M. C., Diercks T., Cañada F. J., Ardá A., Reichardt N.-C., Jiménez-Barbero J., Chem. Eur. J. 2015, 21, 11408–11416. [DOI] [PubMed] [Google Scholar]
- 16. Chávez M. I., Andreu C., Vidal P., Aboitiz N., Freire F., Groves P., Asensio J. L., Asensio G., Muraki M., Cañada F. J., Jiménez-Barbero J., Chem. Eur. J. 2005, 11, 7060–7074. [DOI] [PubMed] [Google Scholar]
- 17. Ramirez-Gualito K., Alonso-Ríos R., Quiroz-Garcia B., Rojas-Aguilar A., Diaz D., Jiménez-Barbero J., Cuevas G., J. Am. Chem. Soc. 2009, 131, 18129–18138. [DOI] [PubMed] [Google Scholar]
- 18. Jiménez-Moreno E., Jiménez-Osés G., Gómez A. M., Santana A. G., Corzana F., Bastida A., Jiménez-Barbero J., Asensio J. L., Chem. Sci. 2015, 6, 6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsu C. H., Park S., Mortenson D. E., Foley B. L., Wang X., Woods R. J., Case D. A., Powers E. T., Wong C.-H., Dyson H. J., Kelly J. W., J. Am. Chem. Soc. 2016, 138, 7636–7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bains G., Lee R. T., Lee Y. C., Freire E., Biochemistry 1992, 31, 12624–12628. [DOI] [PubMed] [Google Scholar]
- 21. Muraki M., Ishimura M., Harata K., Biochim. Biophys. Acta Gen. Subj. 2002, 1569, 10–20. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Beckmann H. S., Möller H. M., Wittmann V., Beilstein J. Org. Chem. 2012, 8, 819–826; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Jiménez-Barbero J., Cañada F. J., Asensio J. L., Aboitiz N., Vidal P., Canales A., Groves P., Gabius H. J., Siebert H. C., Adv. Carbohydr. Chem. Biochem. 2006, 60, 303–354. [DOI] [PubMed] [Google Scholar]
- 23. Fielding L., Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 219–242. [Google Scholar]
- 24. Cecioni S., Imberty A., Vidal S., Chem. Rev. 2015, 115, 525–561. [DOI] [PubMed] [Google Scholar]
- 25. Asensio J. L., Ardá A., Cañada F. J., Jiménez-Barbero J., Acc. Chem. Res. 2013, 46, 946–954. [DOI] [PubMed] [Google Scholar]
- 26. Xu X., Pooi B., Hirao H., Hong S. H., Angew. Chem. Int. Ed. 2014, 53, 1283–1287; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1307–1311. [Google Scholar]
- 27. Jiménez-Moreno E., Gómez A. M., Bastida A., Corzana F., Jiménez-Osés G., Jiménez-Barbero J., Asensio J. L., Angew. Chem. Int. Ed. 2015, 54, 4344–4348; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4418–4422. [Google Scholar]
- 28. Wittmann V., Pieters R. J., Chem. Soc. Rev. 2013, 42, 4492–4503. [DOI] [PubMed] [Google Scholar]
- 29. Liang P.-H., Wang S.-K., Wong C.-H., J. Am. Chem. Soc. 2007, 129, 11177–11184. [DOI] [PubMed] [Google Scholar]
- 30. Lundquist J. J., Toone E. J., Chem. Rev. 2002, 102, 555–578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary