

Abstract
Post-infarction remodeling and expansion of the peri-infarct border zone (BZ) directly correlate with mortality following myocardial infarction (MI); however, the cellular and molecular mechanisms underlying remodeling processes in the BZ remain unclear. Herein, we utilized a label-free quantitative proteomics approach in combination with immunohistochemical analyses to gain a better understanding of processes contributing to post-infarction remodeling of the peri-infarct BZ in a swine model of MI with reperfusion. Our analysis uncovered a significant down-regulation of proteins involved in energy metabolism, indicating impaired myocardial energetics and, possibly mitochondrial dysfunction, in the peri-scar BZ. An increase in endothelial and vascular smooth muscles cells, as well as up-regulation of proteins implicated in VEGF signaling and marked changes in the expression of extracellular matrix and subendothelial basement membrane proteins, are indicative of active angiogenesis in the infarct BZ. A pronounced increase in macrophages in the peri-infarct BZ was also observed and proteomic analysis uncovered evidence of persistent inflammation in this tissue. Additional evidence suggested an increase in cellular proliferation that, concomitant with increased nestin expression, indicates potential turnover of endogenous stem cells in the BZ. A marked up-regulation of proapoptotic proteins, as well as the down-regulation of proteins important for adaptation to mechanical, metabolic, and oxidative stress likely contribute to increased apoptosis in the peri-infarct BZ. The cellular processes and molecular pathways identified herein may have clinical utility for therapeutic intervention aimed at limiting remodeling and expansion of the BZ myocardium, and preventing the development of heart failure post-MI.
Keywords: Label-free proteomics, Myocardial infarction, Ischemia/reperfusion Injury, Immunohistochemistry
Graphical abstract

1. Introduction
Myocardial infarction (MI) is a major cause of morbidity and mortality worldwide, and impaired left ventricular systolic function after MI is a significant risk factor for the development of congestive heart failure (CHF) (1). The loss of functional myocardium after MI results in altered loading conditions and increased ventricular wall stress secondary to LV chamber dilation (2), which serves as a potent stimulus for architectural and functional remodeling that is most severe in the peri-infarct border zone (BZ)—the hypo-contractile myocardium adjacent to the infarct (3-8). Post-infarction remodeling of the hypo-contractile BZ myocardium is of particular importance as this initially isolated region gradually extends to involve a progressive amount of myocardium remote from the infarct (5). Moreover, studies have not only shown that the extent of the peri-infarct BZ is a powerful predictor of post-MI mortality (9), but have also suggested that even partial recovery of contractile function in this region can benefit global cardiac function after MI (10). Therefore, a better understanding of LV remodeling processes in the peri-scar BZ will be essential for improving current therapies and identifying novel strategies to promote and inhibit adaptive and maladaptive processes, respectively, and, ultimately, combat the development of CHF post-MI.
The process of post-infarction ventricular remodeling is arbitrarily divided into an early and a late phase, which correspond to the windows of time prior to and beyond 72 hours following infarction, respectively (11). While the early phase is characterized primarily by expansion of the infarct (11, 12), the late phase of remodeling is highly complex, involving a number of compensatory and pathophysiological alterations within the infarcted and non-infarcted myocardium, including collagen deposition by myofibroblasts (11-13), hypertrophy of remaining viable cardiomyocytes, particularly those within the BZ myocardium (11), profound changes in myocardial energetics (14), vascular remodeling/angiogenesis (15), and cell death (16). Nevertheless, the molecular mechanisms underlying these adaptive and maladaptive processes remain poorly understood. Herein, we utilized label-free quantitative proteomics, which represents a powerful tool for the large-scale identification and quantification of proteins in biological samples (17), to globally quantify changes in protein expression in the peri-infarct BZ myocardium of swine one month after MI. During the first month following MI, patients are at high risk of ventricular rupture and sudden cardiac death, making interventional therapy during this time period an added risk. However, one month following MI, while remodeling of the myocardium is still ongoing, cardiac function has largely stabilized, vital indicators have returned to normal, and the risk of sudden cardiac death decreases dramatically (18). Therefore, since the goal of this study was to gain a better understanding of processes contributing to post-infarction remodeling of the peri-infarct BZ with the aim of identifying potential targets for interventional therapy, we chose to study the myocardium one month after MI as this represents a clinically relevant time point. In addition to proteomic analysis, functional and immunohistochemical analyses were also carried out to link proteomics findings with pathophysiological or compensatory alterations in the peri-scar BZ during post-infarction remodeling.
2. Methods
2.1. Swine Model of Ischemia/Reperfusion (I/R) Injury
Detailed methods have been reported previously (19). Briefly, female Yorkshire swine (Sus scrofa) (∼13 kg, Manthei Hog Farm, MN, USA) were segregated into Sham (n=6) and MI (n=7) experimental groups. I/R injury was surgically induced in MI swine via transient occlusion of the left anterior descending coronary artery for 60 min, followed by reperfusion. The Sham group underwent the same surgical procedure without occlusion of the left anterior descending coronary artery. Following occlusion for 60 min, the hearts were reperfused and the animals were allowed to recover. Cardiac function was assessed four weeks post-MI in Sham and MI swine as described before (8). At 4 weeks post-MI, animals were sacrificed and the BZ myocardium from MI swine, as well as the corresponding tissue in Sham swine (left ventricular myocardium), was snap frozen in liquid nitrogen and stored at -80 °C for later immunohistochemical and proteomic analyses. The BZ was defined as the tissue approximately 3 mm away from the scar, which is easily distinguishable as a thin piece of mature fibrotic tissue. Due to the relatively large size of the swine heart, the BZ tissue was dissected under direction vision.
2.2. Sample Preparation for Proteomic Analysis
As described previously (19, 20), approximately 30-50 mg of cardiac tissue was washed twice with cold PBS buffer containing 1 mg/mL protease inhibitor cocktail (Roche, Basel, Switzerland). The tissue was then immediately homogenized in HEPES buffer (0.25 mM sucrose, 25 mM HEPES at pH 7.4, 50 mM NaF, 0.25 mM Na3VO4, 0.25 mM PMSF, 2.5 mM EDTA, and 1 mg/mL protease inhibitor cocktail) using a Polytron electric homogenizer (Model PRO200, PRO Scientific Inc., Oxford, CT, USA) with short pulses (5-7 seconds). Following homogenization, samples were centrifuged (120,000 g, 30 min, 4 °C). The supernatant, which contains predominately blood proteins, was discarded. The resulting pellet was subsequently re-homogenized in HEPES buffer containing 0.2% of the MS-compatible surfactant, MaSDeS (21). Subsequently, the protein concentration of each sample was determined using the Bradford assay (Bio-Rad, Hercules, CA, USA) in accordance with the manufacturer's protocol. Each protein sample (10 μg) was reduced with 20 mM dithiothreitol at 65 °C and alkylated with 25 mM iodoacetamide at room temperature, with each step requiring 1 h. The samples were all adjusted to a pH of 8, modified trypsin (Trypsin Gold; Promega, Madison, WI, USA) was added to each sample at a ratio of 1:50 (w/w; trypsin/protein), and samples were incubated overnight (20 h) at 37 °C to allow for complete digestion. Following overnight digestion, aqueous solution containing 5% acetonitrile and 1% formic acid was added into each sample to stop the reaction. Samples were then centrifuged at 16,000 g at 4 °C for 1 h before the supernatants were collected for label-free proteomic analysis.
2.3. Label-free Quantitative Proteomics for the Global Profiling of Protein Expression Levels
Label-free quantitative proteomics analysis was carried out using 3 and 4 biological replicates from the Sham and MI animals, respectively, as previously described (19, 20). The in-solution digested samples were separated using a nanoACQUITY (Waters, MA, USA) ultra-high pressure liquid chromatography (LC) system equipped with both a 180 μm × 20 mm trap column (Symmetry C18 trap column, 100Å, 5 μm; Waters, Milford, MA, USA) and a 75 μm × 100 mm ACQUITY UPLC M-Class BEH C18 analytical column (130Å, 1.7 μm; Waters). The pump flow rate was set to 0.35 μL/min, and peptides (1 μg) were contained within the trap column for 10 min before elution of the peptides. Peptide elution was achieved using a linear gradient going from 5-35% B over the first 130 min of the gradient, followed by a rapid increase to 95% B over the subsequent 20 min. Mobile phase A (0.1 % formic acid in water) and B (0.1 % formic acid in acetonitrile) were used. During peptide separation and elution a column temperature of 28 °C was maintained. The nanoACQUITY LC system was coupled directly on-line with a Q Exactive mass spectrometer (Thermo Scientific, Bremen, Germany). An electrospray voltage of 1.9 kV was used. MS1 scans in the Orbitrap were conducted over the range m/z 300–2000 with a mass resolution of 70,000 (at m/z 200). The automatic gain control (AGC) target value in the Orbitrap was set to 1.00E+06 with a maximum injection time of 100 ms. The 15 most intense ions with charge states ≥ 2 were fragmented in the HCD collision cell using a normalized collision energy of 30%. Tandem mass spectra were acquired in the Orbitrap mass analyzer using a mass resolution setting of 17,500 at m/z 200 (AGC target 1.00E+05, 30 ms maximum injection time). The ion selection threshold was 3,300 for MS/MS. Dynamic exclusion of the sequenced peptides for 20 s was used to minimize repeated sequencing of the same peptides. Protein identification and quantification were performed using the SEQUEST-based Proteome Discoverer (Thermo Scientific; version 1.4). Since the swine database provided only limited coverage, searches were performed using a combined human (20,232 entries) and swine (1,411 entries) database obtained from the UniProtKB/Swiss-Prot database (released January, 2013). The search settings used herein allowed for data containing two missed cleavages with mass tolerances of 10 ppm and 0.02 Da for precursor and fragment ions. Trypsin was specified as the enzyme used. The carbamidomethyl of cysteine was specified as a fixed modification, whereas deamidated asparagine and glutamine, as well as oxidized methionine, were set as variable modifications. The data was further searched against a decoy database and filtered using a 1% false discover rate (FDR). Peptides with high confidence, rank 1, and delta Cn < 0.1 were accepted. The function that included distinct proteins in the search was enabled.
For quantification, the area under the curve of each peptide was calculated using Proteome Discoverer and the average of the three most abundant distinct peptides. This value represented the protein intensity. The reproducibility of this method has been previously described (19). All the given protein intensities are presented in Log10 scale. The intensity of each protein in the sample was divided by the median protein intensity for the entire sample for normalization. The normalized protein intensities (in Log10 scale) were used for all heat maps. Protein changes between groups were considered significant if they: (a) had a greater than 1.3fold change in expression between groups and (b) had a p value less than 0.05 as determined using unpaired two-tailed t-test (see Statistical Analysis section below for details). All relevant proteomics data has been uploaded to the MassIVE database and is available at ftp://massive.ucsd.edu/MSV000080621.
2.4. Bioinformatics
To identify potential functional and/or physical interactions among proteins with altered expression in the peri-scar BZ in comparison to Sham left ventricular myocardium, the list of significantly up- and down-regulated proteins was input into the STRING database (http://string-db.org/) (22). Active interaction sources from textmining, experiments, databases, co-expression, neighborhood, gene fusion, and co-occurrence were considered in the analysis. Protein interaction networks consisted of proteins (nodes) and protein-protein interactions (edges). Only interactions with a minimum STRING combined score of 0.700, which represents high-confidence level interactions in STRING, were deemed relevant.
2.5. Immunohistochemistry
For immunohistochemical analyses, tissue from the peri-infarct BZ of MI swine and the left ventricular myocardium of Sham swine were immersed in Tissue Tek optimal cutting temperature compound (Thermo Scientific), snap frozen in liquid nitrogen, and sectioned into approximately 5-7 μM slides. The slides were fixed in 4% paraformaldehyde at room temperature for 20 minutes, permeabilized in 0.1% Triton X-100 at 4°C for 10 minutes, and blocked with secondary antibody serum for 60 minutes. Primary antibodies were added to the 2% secondary antibody serum in PBS at a concentration of 1:100, and the cells were incubated at 4 °C overnight. Subsequently, the labeled sections were washed and incubated with FITC-, and TRITC-conjugated secondary antibodies in Lab Vision UltraV Block (Thermo Scientific) at room temperature for 1 hour, counterstained with DAPI, washed, and visualized under a fluorescence microscope (DP71; Olympus, Tokyo, Japan). Cells staining positive for the indicated proteins were quantified on serial sections using ImageJ (23).
2.6. Angiogenesis
To assess angiogenesis, tissue sections from the peri-infarct BZ were stained with anti-CD31 and anti-SM22α antibodies (Santa Cruz Biotech, TX, USA), and viewed at 20× magnification as described previously (19). CD31+ cells and SM22α+ cells were counted in 2 fields of view from each of 10 sections from the infarct BZ of MI swine and corresponding myocardium in Sham swine.
2.7. Immune Response
To assess the involvement of the immune system in ventricular remodeling four weeks after MI, tissue sections from the peri-infarct BZ of MI swine and the corresponding left ventricular myocardium in Sham swine were stained with anti-CD11b antibody (SouthernBiotech, AL, USA), and the numbers of CD11b+ cells were quantified as previously described (19).
2.8. Cell Proliferation and Apoptosis
To assess cell proliferation and apoptosis, sections from the peri-infarct BZ of MI and analogous tissue in Sham were stained with either TUNEL or Ki67 antibodies (Thermo Scientific), and viewed at 40× magnification. The numbers of TUNEL+ cells or Ki67+, as well as the total number of cells, were counted in 5 fields from each section.
2.9. Statistical Analyses
All data are presented as mean ± SEM. Statistical analysis of the proteomics data was carried out using the multiple t-test function in GraphPad Prism 6 (version 6.07; GraphPad Software, La Jolla, CA, USA) with more power and correction for multiple comparisons using the Holm-Šídák method. For functional and immunohistochemical analyses, group comparisons were carried out using the Wilcoxon rank-sum test. The Software Stata (StataCorp. 2013; College Station, TX, USA) was used to conduct statistical analysis of the functional and immunohistochemical data. For all statistical comparisons, a p value less than 0.05 was considered statistically significant.
3. Results & discussion
3.1. Swine MI model
Cardiac function was assessed in Sham and MI swine during the late phase of left ventricular remodeling four weeks after I/R injury (Fig. 1A). Four weeks after experimentally-induced MI, the left ventricular ejection fraction in MI swine (43.3±2.8%) was significantly decreased in comparison to the ejection fraction in Sham swine (51.2±2.1%) (p<0.05), which is consistent with left ventricular systolic dysfunction secondary to MI (Fig. 1B). In agreement with the findings of previous studies (2), wall stress was significantly elevated in the peri-infarct BZ (∼1-fold) and infarct zone (IZ; ∼3-fold) of MI swine relative to the left ventricular myocardium of Sham swine (Fig. 1C; p<0.05 and p<0.01 for BZ versus Sham and IZ versus Sham, respectively). Although wall stress in the remote non-infarcted myocardium of MI swine was slightly increased relative to that in Sham swine left ventricular myocardium, this increase was not statistically significantly (Fig. 1C). In addition to impaired systolic function, hearts from swine with experimentally-induced MI also showed significant signs of fibrosis within the peri-infarct BZ myocardium (Fig. 1D). Quantification of the area of fibrosis in myocardial sections from Sham swine left ventricular myocardium and the BZ myocardium of MI swine confirmed that fibrosis was significantly increased in the peri-scar BZ (Fig. 1E).
Figure 1. Left ventricular function is impaired one month after MI.
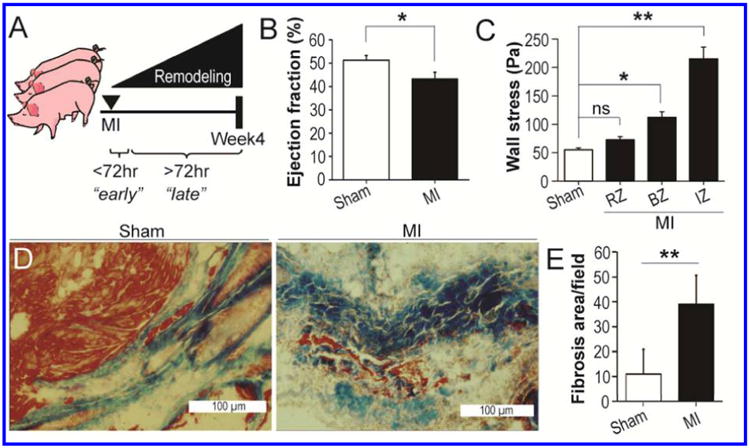
A. Schematic showing experimental timeline with the assessment of cardiac function during ventricular remodeling four weeks after MI. B. Percent ejection fraction in Sham and MI swine determined one month post-MI. C. Left ventricular wall stress in Sham swine and the RZ, BZ, and IZ myocardium in MI swine. D. Masson's trichrome stained myocardial sections showing signs of fibrosis present in the peri-infarct BZ of MI swine that is not present in Sham left ventricular myocardium. Note that the tissue shown in this image was not used for label-free proteomics analysis. E. Quantification of the area of fibrosis in myocardial sections from Sham and MI swine. *p<0.05; **p<0.01; ns, not significant.
3.2. Quantitative proteomic profiling
To globally identify up- and down-regulated proteins in the peri-infarct BZ myocardium of MI swine, towards a better understanding of the compensatory and pathophysiological processes occurring during post-MI remodeling, highly reproducible label-free quantitative proteomic analysis was carried out as previously described (19). Proteins were extracted from the peri-infarct BZ myocardium of MI swine, as well as the corresponding tissue from Sham swine, and subjected to LC-tandem mass spectrometry (MS/MS) analysis with label-free quantification (Fig. 2A). Following quantification, bioinformatics analysis was conducted using the list of proteins with significantly altered expression in the peri-infarct BZ (relative to Sham left ventricular myocardium), and the resulting quantitative and informatics data were combined with data from functional and immunohistochemical analyses to identify the molecular correlates of compensatory and pathophysiological processes in the post-MI remodeled BZ myocardium (Fig. 2A).
Figure 2. Label-free quantitative proteomics profiling of protein expression changes in MI versus Sham swine myocardium.

A. Workflow for label-free quantitative proteomic analysis, which includes (1) preparation of protein extracts from the peri-scar BZ in MI swine and the analogous tissue in Sham, (2) Label-free quantitative proteomic analysis, (3) bioinformatics analysis, (4) functional measures to assess in vivo cardiac function, (5) immunohistochemical analysis of myocardial sections from Sham and MI swine, and (6) correlation of protein changes with immunohistochemical data to identify potential pathways involved in physiological and pathophysiological processes known to occur in the post-MI myocardium. B. Venn diagram showing the number of proteins identified in the Sham (2997; n = 3) and MI (3154; n = 4) groups, as well as the overlap in protein identifications (2485). C. Volcano plot showing the distribution and significance of protein expression changes in the peri-scar BZ versus Sham. D.Heat map showing the relative intensities of significantly up- and down-regulated proteins (MI relative to Sham) identified in both groups. A description of the values used for the heat map is provided in the Methods section.
Using label-free quantitative proteomics, a total of 2997 and 3154 proteins could be confidently identified in the Sham and MI groups, respectively, with 2485 of the proteins being identified in both groups (Fig. 2B). In order to focus on protein changes with a high probability of being important for adaptive and maladaptive processes occurring in the peri-infarct BZ, only proteins exhibiting a 1.3-fold change in expression and having a p value less than 0.05 between groups as determined by group comparison (see 2.8 Statistical Analysis section for details) were selected for further analysis. Of the proteins identified in both groups, 714 had a 1.3-fold or greater change in expression, with 99 and 615 of the proteins increasing and decreasing by 1.3-fold, respectively (Fig. 2C, Table S1). Statistical analysis revealed that the expression of 305 of the proteins exhibiting a 1.3-fold or greater change in expression were altered significantly (p<0.05) in the peri-infarct BZ of MI relative to Sham swine left ventricular myocardium (Fig. 2C). In total, 32 and 273 proteins were significantly up- and down-regulated, respectively, in the peri-infarct BZ of MI swine in comparison to Sham swine left ventricular myocardium (Figs. 2C-D, Table S2).
Based on bioinformatics analysis, the majority of the down-regulated proteins were localized to the cytoplasm and mitochondria (149 and 79 out of 273 significantly down-regulated proteins are localized to the cytoplasm and mitochondria, respectively), while many of the up-regulated proteins were secreted extracellular matrix (ECM)/basement membrane (BM) proteins residing in the extracellular space (10 out of 32 significantly up-regulated proteins) (Fig. S1A, B).
One concern was that the down-regulation of intracellular (cytoplasmic and mitochondrial) proteins and up-regulation of extracellular proteins in samples from MI swine could result from the analysis of predominantly fibrotic tissue from the peri-scar BZ. Indeed, as noted above, specific areas of the peri-scar BZ displayed significant fibrosis (Fig. 1D, E). To determine whether the tissue samples from Sham and MI swine that were analyzed by label-free quantitative proteomics contained significant fibrosis, the normalized protein intensities of all major myofilament proteins were compared in samples from Sham and MI swine. We found that the normalized protein intensities of all major myofilament proteins (with the exception of the myosin heavy chain isoforms) were not significantly different between samples from the Sham and MI groups (Fig. S1C), suggesting that a comparable amount of myocardium was analyzed in both samples. Moreover, the sum of the protein intensities was also not significantly different between samples from both groups (Fig. S1D), which further supports the conclusion that the myocardial samples from the peri-infarct BZ of MI swine that were analyzed herein did not contain significant fibrosis. Therefore, the comparison made in this study was between surviving myocardium from the peri-infarct BZ myocardium of MI swine and the corresponding myocardium in Sham swine. Collectively, these results indicate that protein expression differences between the Sham and MI groups are not simply due to analysis of fibrotic tissue from the infarct BZ of MI swine.
3.3. Myocardial energetics in the peri-scar BZ
Consistent with previous reports of altered myocardial energetics in the infarct BZ myocardium (8), label-free quantitative proteomics analysis uncovered a significant down-regulation of proteins involved in oxidative phosphorylation in the BZ myocardium of MI swine (Fig. 3A; Table S3). Grouping of the down-regulated proteins by STRING showed that, besides core components of electron transport chain complexes I-V, several proteins involved in the assembly of these complexes, as well as ubiquinone biosynthesis, were also significantly down-regulated in the peri-infarct BZ (Fig. 3B; Table S3). Notably, in addition to significantly decreased expression of electron transport chain components, label-free proteomic analysis uncovered a significant down-regulation of proteins involved in a myriad other mitochondrial processes, including branched-chain amino acid catabolism, β-oxidation and fatty acid metabolism, the tricarboxylic acid cycle, and the formation of iron-sulfur clusters (Table S4). These results, in addition to previous work demonstrating mitochondrial DNA damage and decreased activity of electron transport chain complexes in mitochondria purified from the post-MI myocardium (24), indicate that the function of mitochondria in the peri-infarct BZ is markedly impaired.
Figure 3. Decreased expression of proteins in the electron transport chain and high-energy phosphate metabolism may contribute to impaired energy metabolism and contractile dysfunction in the BZ myocardium of MI swine.
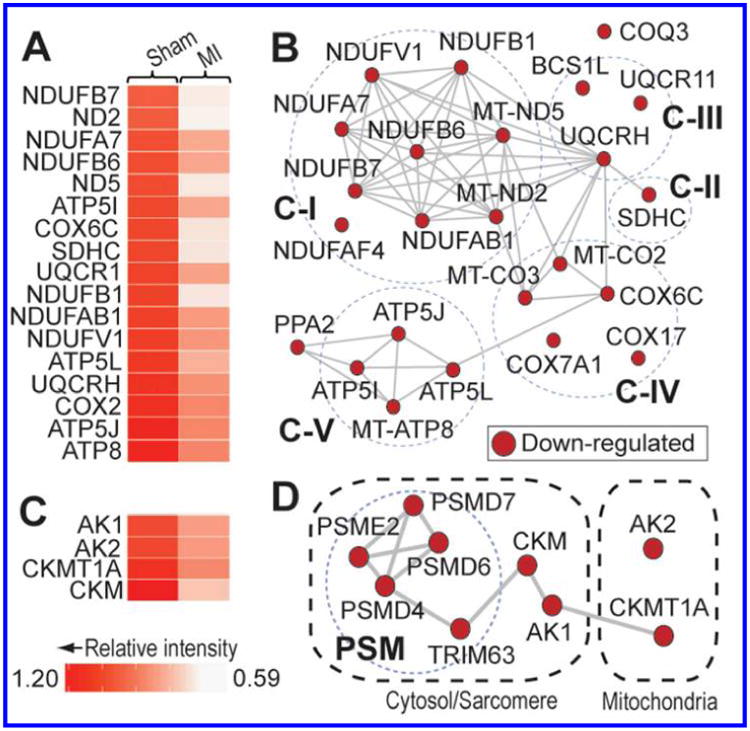
A. Heat map showing the relative intensities of significantly down-regulated proteins (MI relative to Sham) in the electron transport chain. A description of the values used for the heat map is provided in the Methods section. B. Interactome based on the STRING database showing high-confidence interactions among the down-regulated proteins involved in the electron transport chain. Broken circles segregate proteins into the discrete complexes of the mitochondrial respiratory chain. C-I, complex I; C-II, complex II; C-III, complex III; C-IV, complex IV; C-V, complex V. C. Heat map showing the relative intensities of significantly down-regulated proteins (MI relative to Sham) involved in the maintenance of ATP levels at different subcellular localizations. A description of the values used for the heat map is provided in the Methods section. D. High-confidence interactome predicted by STRING showing interactions among the down-regulated proteins involved in the phosphotransfer networks in the mitochondria and cytosol/sarcomeres. Broken circle indicates components of the proteasome (PSM) pathway implicated in CKM turnover.
Several key enzymes involved in glycolysis (Table S5) were also down-regulated in the BZ myocardium of MI swine relative to Sham left ventricular myocardium, implying that a decrease in the supply of intermediates to the tricarboxylic acid cycle may also be, in part, responsible for altered myocardial energetics in the remodeled BZ myocardium. Collectively, these findings implicate global mitochondrial dysfunction and decreased glycolytic flux in decreased ATP production (14), and possibly turnover (8), in the peri-infarct BZ.
Nevertheless, although mitochondrial dysfunction and a reduction in the availability of tricarboxylic acid cycle intermediates can explain decreased ATP turnover kinetics in the peri-infarct BZ (8), quantitative label-free proteomic analysis uncovered a significant down-regulation of ATP-dependent enzymes and enzyme subunits (Table S6), including SERCA and components of the Na+/K+-ATPase, which, together, are responsible for the hydrolysis of a significant proportion of the ATP generated in the heart (second only to the myofilaments).(28) These findings suggest that decreased ATP levels (14), as well as diminished ATP utilization, contribute to the reduction in the kinetics of ATP turnover in the peri-infarct BZ (8).
In addition to ATP production by the electron transport chain, ATP levels are also replenished locally at sites of high ATP turnover through the combined action of the creatine and adenylate kinases (29, 30). Previous determination of the phosphocreatine (PCr)/ATP ratio has revealed that this index of the cardiac energy status is decreased significantly in the peri-scar BZ both at baseline and under high workload conditions (19). Consistent with those findings, label-free quantitative proteomic analysis revealed that both the cytosolic and mitochondrial isoforms of creatine kinase (CKM and CKMT1A, respectively) were significantly down-regulated in the post-MI remodeled BZ (Fig. 3C-D). Furthermore, there was a significant down-regulation of adenylate kinase 1 (AK1) and adenylate kinase 2 (AK2) (Fig. 3C-D), which localize to the cytosol and mitochondria, respectively, and carry out the transfer of a phosphate group from one molecule of ADP to another, forming one molecule of ATP and one molecule of AMP (30). Collectively, down-regulation of the creatine and adenylate kinases suggests impairment of the phosphotransfer network in the peri-infarct BZ (Fig. 3D), which likely contributes to the previously observed decrease in the PCr/ATP ratio in this tissue (19). Moreover, as CKM, AK1, and AK2 all consume ADP to form ATP, this system helps to maintain low intracellular levels of ADP; thereby preventing ADP-mediated impairment of contractile function (31, 32). Consequently, the down-regulation of these enzymes implicates increased intracellular levels of ADP in contractile dysfunction in the peri-infarct BZ.
3.4. Angiogenesis in the peri-scar BZ
Angiogenesis is potently induced by MI and plays an important role in infarct healing (34, 35). To assess angiogenesis four weeks after experimentally-induced MI, myocardial sections from the peri-infarct BZ of MI swine, as well as the corresponding tissue in Sham swine, were stained using antibodies against CD31, an endothelial cell marker, and the number of CD31+ cells in each section was quantified (Fig. 4A). There was a modest 0.8-fold increase in CD31+ cells in myocardial sections from the infarct BZ of MI swine in comparison to Sham (Fig. 4B; p<0.01). Similarly, staining with an anti-SM22α antibody, which marks vascular smooth muscle cells, showed that SM22α+ cells were also increased by approximately 1.7-fold in the peri-scar BZ in comparison to Sham (Fig. 4C,D; p<0.01). Together, these results are suggestive of an increase in capillary density within the infarct BZ four weeks after MI. In accordance with the results of the immunohistochemical analysis, label-free quantitative proteomic analysis revealed a significant up-regulation of collagen α1(VI) chain (COL6A1) and collagen α2(VI) chain (COL6A2), which are important components of the sub-endothelial basement membrane (36), and are enriched in the endothelium in comparison to other tissues (37), in the peri-scar BZ (Fig. 4E). Moreover, both prostacyclin synthase (PTGIS) and caveolin-1 (CAV1), which have previously been implicated in VEGF-mediated angiogenesis (38, 39), were up-regulated in the BZ myocardium of MI swine in comparison to Sham swine left ventricular myocardium (Fig. 4E). Quantitative label-free proteomic analysis further uncovered significant up-regulation of mannose receptor, C type 2 (MRC2), which has previously been found to be up-regulated in angiogenic tumors (Fig. 4E) (37).
Figure 4. Peri-infarct BZ myocardium shows signs of angiogenesis.
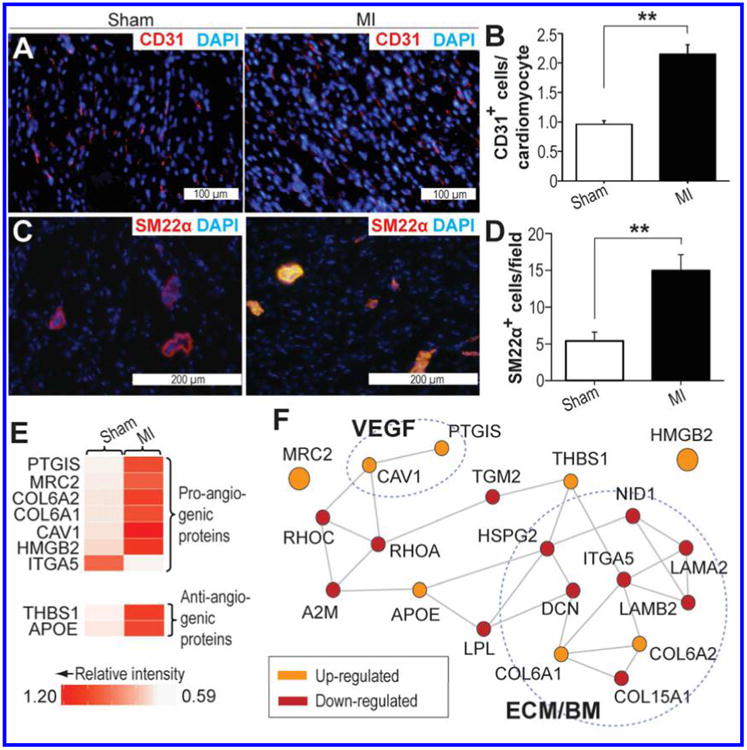
A. Myocardial sections from Sham and MI swine stained with anti-CD31 antibody. Red and blue indicate positive staining for CD31 and nuclei, respectively. B. Quantification of CD31+ cells in the peri-scar BZ of MI swine and the corresponding tissue in Sham. C. Myocardial sections from Sham and MI swine stained with anti-SM22α antibody. Red and blue indicate positive staining for SM22α and nuclei, respectively. D. Quantification of SM22α+ cells in the peri-scar BZ of MI swine and the corresponding tissue in Sham. E. Heat map showing the relative intensities of significantly up-and down-regulated proteins (MI relative to Sham) involved in angiogenesis. A description of the values used for the heat map is provided in the Methods section. F. High-confidence interactome predicted by STRING, which shows potential interactions among the up- and down-regulated proteins involved in angiogenesis. Broken circle indicates components of the extracellular matrix (ECM) and basement membrane (BM), as well as proteins involved in VEGF-mediated angiogenesis (VEGF). *p<0.05; **p<0.01; ns, not significant.
Although the aforementioned findings are highly suggestive of active angiogenesis in the peri-infarct BZ four weeks post-MI, somewhat paradoxically, an up-regulation of anti-angiogenic proteins, including thrombospondin-1(THBS1) and apolipoprotein E (APOE),(40, 41) was identified in the peri-scar BZ (Fig. 4E). Previous work by Frangogiannis et al. has suggested that increased THBS1 in the infarct BZ may help restrict infarct expansion and prevent fibrotic remodeling of non-infarcted myocardium by limiting the expansion of granulation tissue (42). The pro-angiogenic protein integrin α5 (ITGA5) was further found to be down-regulated in the peri-infarct BZ of MI swine (Fig. 4E), which is consistent with the findings of previous studies in mice (43).
STRING analysis revealed that the majority of the aforementioned proteins exist as part of single high-confidence functional interactome with a number of other proteins exhibiting significantly altered expression in the peri-infarct BZ (Fig. 4F). A significant proportion of the proteins in this interactome are components of the ECM/BM indicating potential remodeling of the sub-endothelial BM (Fig. 4F), which is necessary for enhanced vascular permeability and neovascularization (44). Thus, in sum, this information implies a precarious balance between pro- and anti-angiogenic factors, with the outcome (i.e., promotion or inhibition of angiogenesis) likely dependent on the relative levels of these factors in the local environment. Nevertheless, the quantitative proteomics and immunohistochemistry data are strongly indicative of a continued role for active angiogenesis in the peri-infarct BZ one month after I/R injury.
3.5. Inflammation in the peri-infarct BZ
Following MI, there is rapid induction of an inflammatory response that persists to the chronic phase of cardiac injury (45). Macrophages are among the inflammatory cells involved in the response to myocardial ischemic injury, and play an essential and complex role in both the early and chronic phases of the remodeling process following MI (45, 46). Macrophage numbers in the peri-scar BZ of MI swine and left ventricular tissue of Sham swine were assessed by immunohistochemical staining for CD11b, a commonly used marker for macrophages. Immunohistochemical analysis showed a dramatic increase in CD11b+ cells in the infarct BZ of MI swine (Fig. 5A). Quantitative analysis revealed a 19-fold increase in CD11b+ cells in the peri-scar BZ of MI swine in comparison to Sham swine left ventricular myocardium (Fig. 5B; p<0.01), which signifies markedly increased macrophage numbers in this tissue four weeks after MI.
Figure 5. Persistent inflammation in the peri-infarct BZ may contribute to maladaptive ventricular remodeling post-MI.

A. Myocardial sections from Sham and MI swine stained with anti-CD11b antibody. Green and blue indicate positive staining for CD11b and nuclei, respectively. Arrows denote the positions of CD11b+ cells. Insets are zoomed in images and are denoted by (i) and (ii) for Sham and MI images respectively. B. Quantification of CD11b+ cells in the peri-scar BZ of MI swine and the corresponding tissue in Sham. For quantification, cells were counted in 30 microscope fields from 5 locations on each tissue section. C. Heat map showing the relative intensities of significantly up- and down-regulated proteins (MI relative to Sham) associated with inflammation. A description of the values used for the heat map is provided in the Methods section. D. High-confidence interactomes predicted by STRING, which shows potential interactions among the up- and down-regulated proteins involved in inflammation. Broken circles indicate toll-like receptor (TLR) signaling pathway, receptor for advanced glycation endproducts (RAGE) inflammatory pathway, and components of the extracellular matrix (ECM)/basement membrane (BM). *p<0.05; **p<0.01; ns, not significant.
Enhanced vascular permeability is important, not only for angiogenesis, but also for the intravasation of circulating monocytes. Consequently, down-regulation of the laminin subunit α2 (LAMA2), laminin subunit β2 (LAMB2), and basement membrane-specific heparin sulfate proteoglycan core protein (HSPG2), which are components of the sub-endothelial BM, may signify enhanced vascular permeability and contribute to the increased macrophage numbers observed in the peri-scar BZ (Fig. 5C). Moreover, proteomic analysis revealed an up-regulation of PTGIS (Fig. 5C), which has been implicated in VEGF-mediated vascular permeabilization (38).
Early studies provided evidence of a biphasic immune response characterized by monocyte differentiation to pro-inflammatory M1-type macrophages within the first few days after MI, and differentiation (or conversion) to an anti-inflammatory pro-angiogenic M2 phenotype thereafter (47). Although evidence of active angiogenesis in the peri-infarct BZ may indicate that a portion of the macrophages in the peri-infarct BZ are pro-angiogenic M2-type macrophages (Fig. 4), label-free proteomic analysis showed evidence of persistent inflammation in the BZ myocardium four weeks after MI. In particular, the small leucine-rich proteoglycan biglycan (BGN) was significantly up-regulated in the peri-infarct BZ (Fig. 5C). Previous work by Schaefer et al. demonstrated that BGN can activate TLR4 and TLR2 in macrophages, promoting the expression of the pro-inflammatory proteins TNFα and macrophage inflammatory protein-2 (48). Similarly, a significant up-regulation of protein S100-A12 (S100A12) was also identified in the BZ myocardium by label-free quantitative proteomic analysis (Fig. 5C). S100A12 is a ligand for the receptor for advanced glycation end products (RAGE) and can signal through this receptor to induce the production and release of inflammatory factors, including IL-1β and TNFα, from macrophages (49). Notably, activation of RAGE can also promote the display of the cell adhesion molecules such as VCAM-1 and ICAM-1, which are important for the intravasation of monocytes at sites of inflammation (49). Interestingly, expression of S100A12 in mice, which is not normally expressed in this species, induced not only inflammation, but also increased fibrosis, oxidative stress, aortic wall remodeling, and aneurysm formation (50). These findings implicate activation of the RAGE signaling pathway in persistent and detrimental inflammation in the peri-infarct BZ and suggest that this pathway may represent a useful target for the therapeutic modulation of maladaptive inflammation post-MI.
STRING analysis predicted an interactome consisting of the aforementioned proteins, as well as other ECM/BM proteins, including COL6A1, COL6A2, ITGA5, nidogen-1 (NID1), dystrophin (DMD), α-sarcoglycan (SGCA), and γ-sarcoglycan (SGCG) (Fig. 5D). Interestingly, changes in the extracellular matrix/basement membrane were linked to the dystrophin-sarcoglycan complex, which was also found to be significantly down-regulated in the peri-infarct BZ (Fig. 5D). The finding of decreased expression of components of the dystrophin-sarcoglycan complex in the infarct BZ is interesting given that loss of dystrophin is causative in muscular dystrophies that are characterized by chronic muscular inflammation (51).
3.6. Cellular proliferation in the peri-infarct BZ
Immunohistochemical staining for Ki67, a well-established marker of cellular proliferation, revealed an increase in Ki67+ cells in myocardial sections from the peri-infarct tissue in MI swine relative to Sham myocardium (Fig. 6A; p<0.01). Quantification confirmed the increase, with an approximate 5.8-fold elevation in Ki67+ cells in the peri-infarct BZ of MI swine relative to Sham (Fig. 6B), which is suggestive of an increase in cellular proliferation in the infarct BZ. However, despite the discernible increase in cellular proliferation within the peri-infarct BZ, very few proteins with known roles in cellular proliferation were identified by label-free quantitative proteomic analysis. In fact, only a single protein previously implicated in cellular proliferation was determined to be up-regulated in the post-MI remodeled BZ myocardium in comparison to Sham left ventricular myocardium. The intermediate filament protein nestin showed a modest 0.4-fold increase in expression in the peri-infarct BZ of MI swine relative to Sham (Fig. 6C; p<0.05). Nevertheless, this finding is interesting given that nestin is an established marker of neuronal stem cells (52, 53), and evidence indicates that a similar population of resident nestin+ cells possessing a multi-lineage progenitor/stem cell phenotype also exists in the heart (54-56). Although it is tempting to speculate that the observed increase in proliferation may, in part, reflect expansion and differentiation of a nestin+ population of cardiac stem/progenitor cells that participate in regeneration and neovascularization in the injured myocardium, increased numbers of CD11b+ macrophages (Fig. 4) were also observed, and the expansion of this and other cell populations, such as myofibroblasts, likely contribute to the observed increase in cellular proliferation in the peri-scar BZ.
Figure 6. The peri-scar BZ in MI swine shows signs of cellular proliferation.

A. Myocardial sections from Sham and MI swine stained with anti-Ki67 antibody. Red and blue indicate positive staining for Ki67 and nuclei, respectively. Arrows denote Ki67+ cells. Insets are zoomed in images and are denoted by (i) and (ii) for Sham and MI images respectively. B. Quantification of Ki67+ cells in the peri-scar BZ of MI swine and the corresponding tissue in Sham. For quantification, cells were counted in 30 microscope fields from 5 locations on each tissue section. C. Relative intensity of Nestin in Sham and MI myocardium as determined by label-free proteomic analysis. *p<0.05; **p<0.01; ns, not significant.
3.7. Apoptosis in the peri-scar BZ
Although prolonged ischemia and/or I/R injury following percutaneous coronary intervention are well-established causes of cell death within the infarcted myocardium, cell death has also been reported in the normally-perfused peri-infarct BZ and remote non-infarcted myocardium during the post-MI remodeling processes (16, 58). TUNEL staining was used to assess apoptosis in the infarct BZ of MI swine and the corresponding left ventricular myocardium of Sham (Fig. 7A). Quantification of TUNEL+ cells revealed a 5.6-fold increase in TUNEL staining in the peri-infarct BZ of MI swine in comparison to Sham left ventricular myocardium (Fig. 7B; p<0.01), which is consistent with continued cardiomyocyte loss from the infarct BZ four weeks after MI.
Figure 7. Increased expression of pro-apoptotic proteins and reduced expression of proteins involved in the appropriate response to mechanical, metabolic, and oxidative stress may underlie increased apoptosis in the peri-scar BZ.

A. TUNEL-stained myocardial sections from Sham and MI swine. Red, green, and blue indicate positive staining for TUNEL, cardiomyocytes, and nuclei, respectively. CMC, cardiomyocyte. B. Quantification of TUNEL+ cells in the peri-scar BZ of MI swine and the corresponding tissue in Sham. C. Heat map displaying the relative intensities of significantly up- and down-regulated proteins (MI relative to Sham) involved in cell-ECM/BM interactions, protein stabilization/folding, and the cellular response to oxidative stress/clearance of toxic metabolic intermediate. A description of the values used for the heat map is provided in the Methods section. D. Predicted interactomes based on STRING showing predicted high-confidence interactions among the up- and down-regulated proteins. Broken circles indicates DNase I-mediated apoptotic pathway. *p<0.05; **p<0.01; ns, not significant.
As previously shown (59), the pro-apoptotic protein gelsolin (GSN), which induces apoptosis via a DNase I-mediated mechanism, was found to be significantly up-regulated in the BZ myocardium as determined by label-free quantitative proteomic analysis (Fig. 7C). In addition to GSN up-regulation, a number of other protein level changes that could potentially contribute to cell injury and apoptosis in the peri-scar BZ were identified by proteomic analysis. As noted above, components of the dystrophin (DMD)-sarcoglycan complex, including DMD itself, were significantly down-regulated in the peri-infarct BZ (Fig. 7C), which may predispose the sarcolemma to workload-induced damage in response to increased mechanical stress in the infarct BZ (60). A significant down-regulation of the co-chaperone Bcl-2-associated athanogene 3 (BAG3) was also identified by proteomic analysis in the infarct BZ (Fig. 7C). The finding of BAG3 down-regulation in the peri-infarct BZ is particularly interesting as this protein has been shown to interact with Hsc70 and promote tight association of CapZ with F-actin—an association that is critical for the maintenance of myofibrillar integrity under mechanical stress (61). Indeed, BAG3 down-regulation in the peri-infarct BZ may directly contribute to the myofibrillar abnormalities noted in previous studies of the infarct BZ of dogs after MI (62). Collectively, the down-regulation of components of the DMD-sarcoglycan complex and BAG3 in the peri-scar BZ indicates that inability to maintain sarcolemmal and myofibrillar integrity under mechanical stress may contribute to increased cell death in this tissue.
Additionally, label-free proteomic analysis also uncovered down-regulation of a number of enzymes important for neutralizing reactive oxygen species and protecting the cells from oxidative damage, including the mitochondrial isoform of thioredoxin reductase 2 (TXNRD2), peroxiredoxin-6 (PRDX6), the mitochondrial isoform of phospholipid hydroperoxide glutathione peroxidase (GPX4), and superoxide dismutase 1 (SOD1) (Fig. 7C). Interestingly, glutathione peroxidase overexpression in transgenic mice was shown to mitigate post-infarction left ventricular remodeling and decrease the propensity for cardiac failure after MI (63), implicating oxidative injury in maladaptive remodeling and cell death post-MI. Similar findings have been reported in mice overexpressing SOD1 (64). Thus, the observed down-regulation of important enzymes that protect the cell from oxidative damage likely contributes to cell death within the peri-infarct BZ. These findings suggest that antioxidant therapy may be beneficial for reducing oxidative damage and cell death due to an impaired or insufficient oxidative stress response in cardiomyocytes in the infarct BZ.
STRING analysis identified several potentially relevant functional interactions between significantly up-/down-regulated proteins in the post-MI myocardium. High-confidence functional interactions between BAG3 and other down-regulated molecular chaperones/co-chaperones, including DnaJ homolog subfamily B member 4 (DNAJB4), DnaJ homolog subfamily A member 2 (DNAJA2), and heat shock 70 kDa protein 6 (HSPA6) (Fig. 7D), suggests a role for these proteins in mechanical stress-induced cell death in the post-infarct remodeled BZ. Also relevant to mechanical stress-induced cell death were functional interactions between components of the DMD-sarcoglycan complex, nebulin (NEB), myopalladin (MYPN), and ankyrin repeat domain-containing protein 1 (ANKRD1) (Fig. 7D). In addition to roles in the maintenance of the structural integrity of the sarcomere and sarcolemma (60, 65), MYPN and ANKRD1 have previously been implicated in mechanotransduction (66), further supporting a role for the inability to appropriately sense and/or respond to mechanical stress in cell death in the peri-infarct BZ. Of note, up-regulation of ANKRD1 has previously been observed in a variety of pathological cardiac conditions; however, some evidence indicates that this may be a compensatory response by the myocardium (67).
Additional interactions between enzymes important for myocardial protection against oxidative damage were revealed by STRING analysis (Fig. 7D). Interestingly, the down-regulation of nitrilase homolog 2 (NIT2) (Fig. 7D), which is involved in the conversion of the potentially toxic metabolites α-ketoglutaramate and α-ketosuccinamate into non-toxic α-ketoglutarate and oxaloacetate, respectively, in the peri-infarct BZ implies that insufficient clearance of toxic metabolites may also contribute to cell death in the BZ myocardium. The observed up-regulation of glutathione peroxidase 1 (GPX1), which neutralizes H2O2, in the infarct BZ may be a compensatory response to increased oxidative stress in this tissue. Nevertheless, the significant down-regulation of multiple enzymes important for the neutralization of reactive oxygen species suggests that the BZ myocardium may be predisposed to oxidative injury and cell death. Insufficient response to oxidative stress may be particularly relevant given evidence of marked mitochondrial dysfunction (Fig. 3).
5. Conclusion
In this study we utilized a label-free quantitative proteomics approach in combination with immunohistochemical analyses to gain a better understanding of processes contributing to post-infarction remodeling of the peri-infarct BZ one month after MI with the aim of identifying potential targets for interventional therapy. Using label-free quantitative proteomics we identified a myriad of changes suggestive of global mitochondrial dysfunction and impaired shuttling and utilization of high-energy phosphates in the infarct BZ, which may contribute to abnormal myocardial energetics, as well as contractile dysfunction, in the peri-infarct BZ. In accordance with immunohistochemical results showing an increase in endothelial and vascular smooth muscle cells in the BZ myocardium, label-free proteomic analysis revealed an increase in the expression of proteins previously implicated in VEGF-mediated angiogenesis and vascular permeabilization suggesting a role for active angiogenesis in the peri-scar BZ four weeks after MI. Also indicative of enhanced vascular permeabilization were a number of changes in the expression of ECM/BM proteins, which may contribute not only to angiogenesis, but also to enhanced leukocyte infiltration in the peri-infarct BZ. Although some evidence has indicated that macrophages may shift towards an anti-inflammatory phenotype during the later phase of post-infarction ventricular remodeling, signs of persistent inflammation, including the up-regulation of several proteins that have been implicated in pro-inflammatory signaling, were identified in infarct BZ by proteomic analysis. Given the detrimental role of chronic inflammation in the post-MI myocardium, these results suggest that therapeutic modulation of inflammation may represent a treatment option to reduce maladaptive remodeling post-MI. Although increased cellular proliferation in the peri-infarct BZ was demonstrated by immunohistochemical staining for Ki67, proteomic analysis identified relatively few proteins previously implicated in proliferation, with only the intermediate filament protein nestin being up-regulated significantly in the BZ myocardium. Furthermore, an increase in cell death was also observed in the infarct BZ and may be the result of an inability to adapt and respond to mechanical, oxidative, and metabolic stress. Although these findings shed light on potential mechanisms underlying adaptive and maladaptive processes in the infarct BZ myocardium one month after MI, additional studies will be necessary to determine the relative contributions of individual protein/pathway changes to cardiac dysfunction, remodeling, and repair. Moreover, it must be noted that this study represents only a snapshot of these processes without providing information regarding time-dependent changes in these processes following MI, which will be important for a complete understanding of the molecular changes driving cardiac remodeling following MI. Nevertheless, this study lays a foundation for understanding remodeling of the peri-infarct BZ one month after MI, and identifies several potential molecular targets potentially contributing to adaptive and maladaptive remodeling processes for follow-up analyses.
Supplementary Material
Table S1 – List of proteins exhibiting a 1.3-fold or greater change in expression in the peri-infarct BZ of MI swine versus Sham swine left ventricular myocardium; Table S2 – List of proteins exhibiting a 1.3-fold change that was also significant (p<0.05) in the peri-infarct BZ of MI swine versus Sham left ventricular myocardium; Table S3 – List of proteins involved in the electron transport chain that are significantly down-regulated in the peri-infarct BZ in comparison to Sham left ventricular myocardium; Table S4 – List of mitochondrial proteins significantly down-regulated in the peri-scar BZ; Table S5 – Key glycolytic enzymes significantly down-regulated in the peri-infarct BZ in comparison to Sham left ventricular myocardium; Table S6 – List of significantly down-regulated ATP-dependent enzymes in the peri-scar BZ myocardium of MI swine versus Sham swine left ventricular myocardium; Figure S1 – Sub-cellular localization of proteins up- and down-regulated in the peri-scar BZ as well as individual myofilament protein intensities and total protein intensity in Sham and MI samples.
Acknowledgments
The authors would like to thank Thomas Nickel for organizing tissue slides and Rachel Heuer for assistance with the preparation of this manuscript.
Sources of Funding: Financial support was kindly provided by R01 HL114120 (to J.Z.), NIH F31 HL128086 (to Z.R.G.), and R01 HL109810 (to Y.G). YG also would like to acknowledge NIH R01 HL096971, R01 GM 117058, and S10 OD018475.
Abbreviations
- MI
myocardial infarction
- CHF
congestive heart failure
- BZ
border zone
- I/R
ischemia/reperfusion
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- ECM
extracellular matrix
- BM
basement membrane
- PCr
phosphocreatine
- CKM
cytosolic isoform of creatine kinase
- CKMT1A
mitochondrial isoform of creatine kinase
- AK1
cytosolic isoform of adenylate kinase
- AK2
mitochondrial isoform of adenylate kinase
- TRIM63
tripartite motif-containing protein 63
- COL6A1
collagen α1(VI) chain
- COL6A2
collagen α2(VI) chain
- PTGIS
prostacyclin synthase
- CAV1
caveolin-1
- MRC2
mannose receptor, C type 2
- THBS1
thrombospondin-1
- APOE
apolipoprotein E
- ITGA5
integrin α5
- LAMA2
laminin subunit α2
- LAMB2
laminin subunit β2
- HSPG2
basement membrane-specific heparin sulfate proteoglycan core protein
- BGN
biglycan
- S100A12
protein S100-A12
- RAGE
receptor for advanced glycation end products
- NID1
nidogen-1
- DMD
dystrophin
- SGCA
α-sarcoglycan
- SGCG
γ-sarcoglycan
- GSN
gelsolin
- BAG3
Bcl-2-associated athanogene 3
- TXNRD2
thioredoxin reductase 2
- PRDX6
peroxiredoxin-6
- GPX4
mitochondrial isoform of phospholipid hydroperoxide glutathione peroxidase
- SOD1
superoxide dismutase 1
- DNAJB4
DnaJ homolog subfamily B member 4
- DNAJA2
DnaJ homolog subfamily A member 2
- HSPA6
heat shock 70 kDa protein 6
- NEB
nebulin
- MYPN
myopalladin
- ANKRD1
ankyrin repeat domain-containing protein 1
- NIT2
nitrilase homolog 2
- GPX1
glutathione peroxidase 1
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI:
Disclosures: None
References
- 1.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. doi: 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson BM, Gorman JH, Salgo IS, Moainie SL, Plappert T, St John-Sutton M, Edmunds LH, Gorman RC. Border zone geometry increases wall stress after myocardial infarction: contrast echocardiographic assessment. Am J Physiol Heart Circ Physiol. 2003;284:H475–479. doi: 10.1152/ajpheart.00360.2002. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Wilke N, Wang Y, Zhang Y, Wang C, Eijgelshoven MH, Cho YK, Murakami Y, Ugurbil K, Bache RJ, From AH. Functional and bioenergetic consequences of postinfarction left ventricular remodeling in a new porcine model. MRI and 31 P-MRS study. Circulation. 1996;94:1089–1100. doi: 10.1161/01.cir.94.5.1089. [DOI] [PubMed] [Google Scholar]
- 4.Zeng L, Hu Q, Wang X, Mansoor A, Lee J, Feygin J, Zhang G, Suntharalingam P, Boozer S, Mhashilkar A, Panetta CJ, Swingen C, Deans R, From AH, Bache RJ, Verfaillie CM, Zhang J. Bioenergetic and functional consequences of bone marrow-derived multipotent progenitor cell transplantation in hearts with postinfarction left ventricular remodeling. Circulation. 2007;115:1866–1875. doi: 10.1161/CIRCULATIONAHA.106.659730. [DOI] [PubMed] [Google Scholar]
- 5.Jackson BM, Gorman JH, Moainie SL, Guy TS, Narula N, Narula J, John-Sutton MG, Edmunds LH, Gorman RC. Extension of borderzone myocardium in postinfarction dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:1160–1171. doi: 10.1016/s0735-1097(02)02121-6. [DOI] [PubMed] [Google Scholar]
- 6.Shimkunas R, Zhang Z, Wenk JF, Soleimani M, Khazalpour M, Acevedo-Bolton G, Wang G, Saloner D, Mishra R, Wallace AW, Ge L, Baker AJ, Guccione JM, Ratcliffe MB. Left ventricular myocardial contractility is depressed in the borderzone after posterolateral myocardial infarction. Ann Thorac Surg. 2013;95:1619–1625. doi: 10.1016/j.athoracsur.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Homans DC, Asinger R, Elsperger KJ, Erlien D, Sublett E, Mikell F, Bache RJ. Regional function and perfusion at the lateral border of ischemic myocardium. Circulation. 1985;71:1038–1047. doi: 10.1161/01.cir.71.5.1038. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Q, Ye L, Zhang P, Lepley M, Tian J, Li J, Zhang L, Swingen C, Vaughan JT, Kaufman DS, Zhang JF. Functional consequences of human induced pluripotent stem cell therapy: myocardial ATP turnover rate in the in vivo swine heart with post infarction remodeling. Circulation. 2013;127:997–1008. doi: 10.1161/CIRCULATIONAHA.112.000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan AT, Shayne AJ, Brown KA, Gupta SN, Chan CW, Luu TM, Di Carli MF, Reynolds HG, Stevenson W. G, Kwong, R. Y. Characterization of the peri-infarct zone by contrast-enhanced cardiac magnetic resonance imaging is a powerful predictor of post-myocardial infarction mortality Circulatinn. 2006;114:32–39. doi: 10.1161/CIRCULATIONAHA.106.613414. [DOI] [PubMed] [Google Scholar]
- 10.(10) Zhang Z, Sun K, Saloner DA, Wallace AW, Ge L, Baker AJ, Guccione JM, Ratcliffe MB. The benefit of enhanced contractility in the infarct borderzone: a virtualexperiment. Front Physiol. 2012;3:86. doi: 10.3389/fphys.2012.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulatinn. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 12.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction: Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 13.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 14.Hu Q, Wang X, Lee J, Mansoor A, Liu J, Zeng L, Swingen C, Zhang G, Feygin J, Ochiai K, Bransford TL, From AH, Bache RJ, Zhang J. Profound bioenergetic abnormalities in peri-infarct myocardial regions. Am J Physiol Heart Circ Physiol. 2006;291:H648–657. doi: 10.1152/ajpheart.01387.2005. [DOI] [PubMed] [Google Scholar]
- 15.Jugdutt BI, Menon V, Kumar D, Idikio H. Vascular remodeling during healing after myocardial infarction in the dog model: effects of reperfusion, amlodipine and enalapril. J Am Coll Cardiol. 2002;39:1538–1545. doi: 10.1016/s0735-1097(02)01805-3. [DOI] [PubMed] [Google Scholar]
- 16.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Cirmlation. 1997;95:320–323. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 17.Cravatt BF, Simon GM, Yates JR. The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 18.Adabag AS, Therneau TM, Gersh BJ, Weston SA, Roger VL. Sudden death after myocardial infarction. JAMA. 2008;300:2022–2029. doi: 10.1001/jama.2008.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye L, Chang YH, Xiong Q, Zhang P, Zhang L, Somasundaram P, Lepley M, Swingen C, Su L, Wendel JS, Guo J, Jang A, Rosenbush D, Greder L, Dutton JR, Zhang J, Kamp TJ, Kaufman DS, Ge Y. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell. 2014;15:750–761. doi: 10.1016/j.stem.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang YH, Ye L, Cai W, Lee Y, Guner H, Kamp TJ, Zhang J, Ge Y. Quantitative proteomics reveals differential regulation of protein expression in recipient myocardium after trilineage cardiovascular cell transplantation. Proteomics. 2015;15:2560–2567. doi: 10.1002/pmic.201500131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang YH, Gregorich ZR, Chen AJ, Hwang L, Guner H, Yu D, Zhang J, Ge Y. New mass-spectrometry-compatible degradable surfactant for tissue proteomics. J Proteome Res. 2015;14:1587–1599. doi: 10.1021/pr5012679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Cirr Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 25.Suga H. Ventricular energetics. Physiol Rev. 1990;70:247–277. doi: 10.1152/physrev.1990.70.2.247. [DOI] [PubMed] [Google Scholar]
- 26.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(Pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dzeja PP, Vitkevicius KT, Redfield MM, Burnett JC, Terzic A. Adenylate kinase-catalyzed phosphotransfer in the myocardium: increased contribution in heart failure. Circ Res. 1999;84:1137–1143. doi: 10.1161/01.res.84.10.1137. [DOI] [PubMed] [Google Scholar]
- 28.Yamashita H, Sata M, Sugiura S, Momomura S, Serizawa T, Iizuka M. ADP inhibits the sliding velocity of fluorescent actin filaments on cardiac and skeletal myosins. Circ Res. 1994;74:1027–1033. doi: 10.1161/01.res.74.6.1027. [DOI] [PubMed] [Google Scholar]
- 29.Tian R, Nascimben L, Ingwall JS, Lorell BH. Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation. 1997;96:1313–1319. doi: 10.1161/01.cir.96.4.1313. [DOI] [PubMed] [Google Scholar]
- 30.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 31.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 32.Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 33.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, Kinzler KW. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 34.Murohara T, Horowitz JR, Silver M, Tsurumi Y, Chen D, Sullivan A, Isner JM. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- 35.Sonveaux P, Martinive P, DeWever J, Batova Z, Daneau G, Pelat M, Ghisdal P, Grégoire V, Dessy C, Balligand JL, Feron O. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95:154–161. doi: 10.1161/01.RES.0000136344.27825.72. [DOI] [PubMed] [Google Scholar]
- 36.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 37.Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. Critical role of endogenousthrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 39.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 40.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeabilityfactor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 41.Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–129. doi: 10.1038/nri3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lörchner H, Pöling J, Gajawada P, Hou Y, Polyakova V, Kostin S, Adrian-Segarra JM, Boettger T, Wietelmann A, Warnecke H, Richter M, Kubin T, Braun T. Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat Med. 2015;21:353–362. doi: 10.1038/nm.3816. [DOI] [PubMed] [Google Scholar]
- 43.Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circ Res. 2013;112:1624–1633. doi: 10.1161/CIRCRESAHA.113.300890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Götte M, Malle E, Schaefer RM, Gröne HJ. Thematrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 46.Hofmann Bowman M, Wilk J, Heydemann A, Kim G, Rehman J, Lodato JA, Raman J, McNally EM. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ Res. 2010;106:145–154. doi: 10.1161/CIRCRESAHA.109.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–272. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- 48.Cattaneo E, McKay R. Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature. 1990;347:762–765. doi: 10.1038/347762a0. [DOI] [PubMed] [Google Scholar]
- 49.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 50.El-Helou V, Beguin PC, Assimakopoulos J, Clement R, Gosselin H, Brugada R, Aumont A, Biernaskie J, Villeneuve L, Leung TK, Fernandes KJ, Calderone A. The rat heart contains a neural stem cell population, role in sympathetic sprouting and angiogenesis. J Mol Cell Cardiol. 2008;45:694–702. doi: 10.1016/j.yjmcc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 52.Tomita Y, Matsumura K, Wakamatsu Y, Matsuzaki Y, Shibuya I, Kawaguchi H, Ieda M, Kanakubo S, Shimazaki T, Ogawa S, Osumi N, Okano H, Fukuda K. Cardiac neural crest cells contribute to the dormant multipotent stem cell in the mammalian heart. J Cell Biol. 2005;170:1135–1146. doi: 10.1083/jcb.200504061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM, Tikkanen I. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2001;280:H2726–2731. doi: 10.1152/ajpheart.2001.280.6.H2726. [DOI] [PubMed] [Google Scholar]
- 54.Li GH, Shi Y, Chen Y, Sun M, Sader S, Maekawa Y, Arab S, Dawood F, Chen M, De Couto G, Liu Y, Fukuoka M, Yang S, Da Shi M, Kirshenbaum LA, McCulloch CA, Liu P. Gelsolin regulates cardiac remodeling after myocardial infarction through DNase I-mediated apoptosis. Res. 2009;104:896–904. doi: 10.1161/CIRCRESAHA.108.172882. [DOI] [PubMed] [Google Scholar]
- 55.Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G, Des Rosiers C, Petrof BJ. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stressinduced contractile failure and injury. FASEB J. 2001;15:1655–1657. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- 56.Hishiya A, Kitazawa T, Takayama S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ Res. 2010;107:1220–1231. doi: 10.1161/CIRCRESAHA.110.225649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharov VG, Sabbah HN, Ali AS, Shimoyama H, Lesch M, Goldstein S. Abnormalities of cardiocytes in regions bordering fibrous scars of dogs with heart failure. Int J Cardiol. 1997;60:273–279. doi: 10.1016/s0167-5273(97)00117-4. [DOI] [PubMed] [Google Scholar]
- 58.Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H, Takeshita A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2004;109:544–549. doi: 10.1161/01.CIR.0000109701.77059.E9. [DOI] [PubMed] [Google Scholar]
- 59.Wang P, Chen H, Qin H, Sankarapandi S, Becher MW, Wong PC, Zweier JL. Overexpression of human copper, zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc Natl Acad Sci U S A. 1998;95:4556–4560. doi: 10.1073/pnas.95.8.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bang ML, Mudry RE, McElhinny AS, Trombitás K, Geach AJ, Yamasaki R, Sorimachi H, Granzier H, Gregorio CC, Labeit S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153:413–427. doi: 10.1083/jcb.153.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knöll R, Hoshijima M, Chien K. Cardiac mechanotransduction and implications for heart disease. J Mol Med (Berl) 2003;81:750–756. doi: 10.1007/s00109-003-0488-x. [DOI] [PubMed] [Google Scholar]
- 62.Mikhailov AT, Torrado M. The enigmatic role of ankyrin repeat domain 1 gene in heart development and disease. Int J Dev Biol. 2008;52:811–821. doi: 10.1387/ijdb.082655am. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – List of proteins exhibiting a 1.3-fold or greater change in expression in the peri-infarct BZ of MI swine versus Sham swine left ventricular myocardium; Table S2 – List of proteins exhibiting a 1.3-fold change that was also significant (p<0.05) in the peri-infarct BZ of MI swine versus Sham left ventricular myocardium; Table S3 – List of proteins involved in the electron transport chain that are significantly down-regulated in the peri-infarct BZ in comparison to Sham left ventricular myocardium; Table S4 – List of mitochondrial proteins significantly down-regulated in the peri-scar BZ; Table S5 – Key glycolytic enzymes significantly down-regulated in the peri-infarct BZ in comparison to Sham left ventricular myocardium; Table S6 – List of significantly down-regulated ATP-dependent enzymes in the peri-scar BZ myocardium of MI swine versus Sham swine left ventricular myocardium; Figure S1 – Sub-cellular localization of proteins up- and down-regulated in the peri-scar BZ as well as individual myofilament protein intensities and total protein intensity in Sham and MI samples.