

Abstract
Caspases are cysteine proteases with critical roles in apoptosis. The Caenorhabditis elegans caspase CED-3 is activated by autocatalytic cleavage, a process enhanced by CED-4. Here we report that CED-3 zymogen localizes to the perinuclear region in C. elegans germ cells and that CED-3 autocatalytic cleavage is held in check by C. elegans nuclei and activated by CED-4. The nuclear pore protein NPP-14 interacts with the prodomain of CED-3 zymogen, colocalizes with CED-3 in vivo, and inhibits CED-3 autoactivation in vitro. Several missense mutations in the CED-3 prodomain result in stronger association with NPP-14 and reduced CED-3 activation by CED-4 in the presence of nuclei or NPP-14, leading to cell death defects. Those same mutations enhance autocatalytic cleavage of CED-3 in vitro and increase apoptosis in vivo in the absence of npp-14. Our results reveal a critical role for nuclei and nuclear membrane proteins in regulating activation and localization of CED-3.
Apoptotic caspases are a family of conserved cysteine proteases that play central roles in activating and executing apoptosis in diverse organisms1,2. They are initially synthesized as inactive zymogens, containing an N-terminal prodomain or caspase recruitment domain (CARD) and two catalytic subunits known as the large and the small subunits. In cells that receive cell death stimuli, caspase zymogens either undergo autoproteolytic activation through induced dimerization or oligomerization of the zymogens, or are proteolytically activated by upstream caspases or proteases1,2. The activated caspases then act by cleaving a plethora of cellular proteins, leading to self-destruction of the cell, including nuclear membrane breakdown, chromosome fragmentation, apoptotic body formation, and clearance by phagocytes3. Given the critical roles of caspases in apoptosis, the activation and activity of caspases are tightly regulated at multiple levels and by both positive and negative regulators so that they are suppressed in cells that live, but swiftly activated in cells that need to undergo apoptosis1,2.
The C. elegans ced-3 gene is essential for apoptosis and encodes a caspase that is the founding member of the apoptotic caspases4,5. The analysis of CED-3 activation and regulation during apoptosis has provided critical insights into the activation and functions of caspases and apoptosis in general. In C. elegans cells that live, the CED-4 protein, a homolog of the human apoptotic protease activating factor-1 (Apaf-1)6,7, is complexed with the cell death inhibitor CED-9 in a 2:1 stoichiometric ratio and tethered to the surface of mitochondria through CED-98–12. During apoptosis, the cell-death initiator EGL-1 is transcriptionally upregulated, binds to CED-9, and triggers dissociation of the CED-4 dimer from CED-911–13. Released CED-4 dimers subsequently oligomerize and translocate to the perinuclear region to activate apoptosis11,12,14. It is not understood why CED-4 relocates to nuclei during apoptosis.
Related to the dynamic movement of CED-4 during apoptosis, several key questions regarding CED-3 remain unanswered. In particular, the subcellular localization of CED-3 in C. elegans, which will shed light on how CED-3 is activated and how it acts to cause apoptosis, has been a major mystery. In addition, the role of the prodomain of the CED-3 zymogen in C. elegans apoptosis is an enigmatic issue, as the prodomain of CED-3 appears to be completely dispensable for CED-3 activation and activities in vitro15,16, but is critical for the killing activity of CED-3 in vivo4,17.
In this study, we set out to investigate where CED-3 localizes and how the prodomain of CED-3 affects apoptosis. We find that CED-3 also localizes to the perinuclear region in C. elegans germ cells, which provides a causal link for the translocation of CED-4 oligomers to nuclei during apoptosis. Moreover, we identify a nuclear pore protein NPP-14 that is critical not only for the perinuclear localization of CED-3 but also for inhibiting CED-3 zymogen autoactivation in living cells. Finally, we demonstrate that several unique CED-3 prodomain mutations enhance binding of the CED-3 zymogen with NPP-14, leading to inhibition of CED-4-induced CED-3 activation in vitro and apoptosis in vivo.
RESULTS
CED-3 prodomain mutations can enhance CED-3 autoactivation
Several loss-of-function (lf) mutations in ced-3 (n718, n1040 and n2439) result in single amino acid substitutions (G65R, L27F and L30F, respectively) in the prodomain of the CED-3 caspase4,17. These mutations cause strong reduction of apoptosis, as monitored by the number of persistent cell corpses in a ced-1(e1735) mutant background that is defective in cell corpse clearance (Table 1)18. For example, an average of 31 persistent cell corpses were observed in ced-1(e1735) embryos, whereas virtually no cell corpse was detected in ced-1(e1735); ced-3(n718 G65R) embryos (Table 1), indicating that ced-3(n718 G65R) animals are defective in cell death. Similarly, reduced numbers of persistent cell corpses were seen in ced-1(e1735); ced-3(n1040 L27F) and ced-1(e1735); ced-3(n2439 L30F) embryos (13 and 7 cell corpses on average, respectively)(Table 1). We confirmed these observations by quantifying the number of cells that failed to undergo apoptosis in the anterior pharynx of these animals (16 cells are programmed to die in this region of wild-type animals)17. ced-1(e1735); ced-3(n718 G65R), ced-1(e1735); ced-3(n1040 L27F), and ced-1(e1735); ced-3(n2439 L30F) larvae have an average of 12.6, 10.3 and 12.0 extra undead cells, respectively, indicating strong cell death defects (Supplementary Table 1). Because the prodomain of CED-3 appears to be dispensable for CED-3 activity or activation in vitro15,16, these results raise important questions regarding the role of the CED-3 prodomain in regulating CED-3 activation, activity, or apoptosis in vivo.
Table 1.
Loss of npp-14 causes increased cell death in several ced-3 mutants carrying specific prodomain mutations.
| Genotype | No. of cell corpses | Range of cell corpses |
|---|---|---|
| ced-1(e1735) | 31.3±2.2 | 27 – 34 |
| npp-14(sm160) ced-1(e1735) | 29.4±2.1* | 26 – 34 |
|
| ||
| ced-1(e1735); ced-3(n2433 G360S) | 0±0 | 0 – 0 |
| npp-14(sm160) ced-1(e1735); ced-3(n2433 G360S) | 0±0 | 0 – 0 |
|
| ||
| ced-1(e1735); ced-3(n718 G65R) | 0.1±0.4 | 0 – 1 |
| npp-14(sm160) ced-1(e1735); ced-3(n718 G65R) | 2.9±0.8** | 2 – 4 |
|
| ||
| ced-1(e1735); ced-3(n1040 L27F) | 13.2±1.6 | 11 – 16 |
| npp-14(sm160) ced-1(e1735); ced-3(n1040 L27F) | 22.9±1.6** | 21 – 26 |
|
| ||
| ced-1(e1735); ced-3(n2439 L30F) | 7.2±0.9 | 6 – 9 |
| npp-14(sm160) ced-1(e1735); ced-3(n2439 L30F) | 11.3±1.8** | 9 – 15 |
|
| ||
| ced-1(e1735); ced-3(n2449 R51H) | 29.2±2.6 | 26 – 35 |
| npp-14(sm160) ced-1(e1735); ced-3(n2449 R51H) | 28.0±1.5 | 26 – 31 |
Cell corpses were scored in 4-fold stage embryos. Data shown are mean ± s.d. (n = 15 embryos). The significance of difference in cell corpse numbers between two strains with and without npp-14(sm160) was determined by two-sided t-tests.
P < 0.0001.
P < 0.05.
Others have P > 0.05.
We first performed in vitro assays to examine if these prodomain mutations affect CED-3 zymogen autoproteolytic activation or CED-3 activation induced by oligomeric CED-4, the activated form of CED-412. In CED-3 autoactivation assays, 35S-Methionine-labeled CED-3 zymogen synthesized in the rabbit reticulocyte lysate initially existed as an unprocessed precursor and was slowly autocleaved to generate some processed forms, including one that was visible at the 41 kD position by the 90-min time point (Fig. 1a, lanes 1–4, and Supplementary Fig. 1a,c)12,19. In contrast, CED-3(L27F), CED-3(L30F) and CED-3(G65R) zymogens displayed much stronger autoproteolytic activation and were mostly autoprocessed by the 90-min time point (Fig. 1a, lanes 5–12, 17–20, and Supplementary Fig. 1a,c). This is surprising, because in vivo these mutations strongly reduce rather than promote cell death (Table 1 and Supplementary Table 1). Another CED-3 prodomain mutation, n2449 (R51H), caused a barely detectable cell death defect (Table 1 and Supplementary Table 1), and the CED-3(R51H) zymogen was indistinguishable from the wild-type CED-3 zymogen in the CED-3 autoactivation assay (Fig. 1a, lanes 13–16, and Supplementary Fig. 1a,c).
Figure 1.
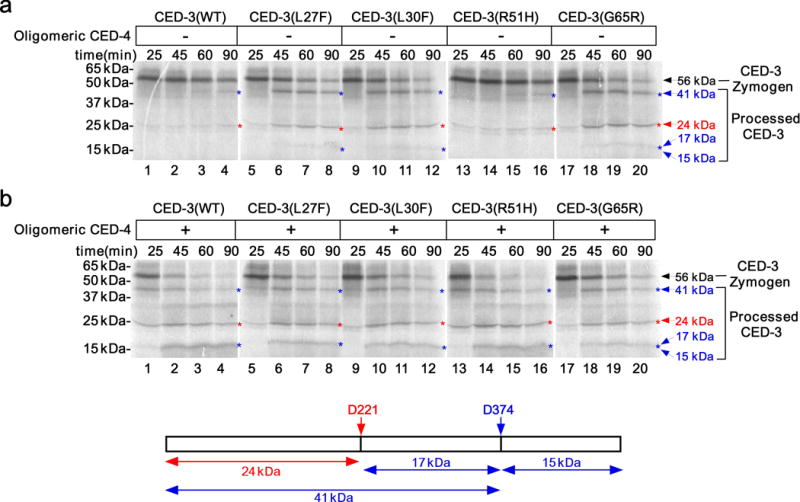
Three unique CED-3 prodomain mutations affect CED-3 autoactivation in vitro. (a,b) Autoradiographs of SDS-PAGE showing time-course of CED-3 activation in vitro. The indicated CED-3 zymogens (wild-type or mutant, synthesized and labeled with 35S-Methionine in vitro, see Online Methods) were incubated for different times before stopping the reaction. In a, samples were incubated with buffer; in b, samples were incubated with oligomeric CED-4 (100 nM final concentration). The cartoon below shows deduced CED-3 processed products derived from cleavage at D221 and D37419. The deduced CED-3 processed bands on gels are indicated with corresponding color arrows. The 15 kDa and 17 kDa bands could not be resolved clearly on the gels. The original gel images are shown in Supplementary Data Set 1.
Prodomain mutations do not impair CED-3 activation by CED-4
Given the strong cell death defects caused by these three CED-3 prodomain mutations, we examined whether they might impair the activation of the CED-3 zymogen induced by CED-4. Oligomeric CED-4 greatly accelerated autoproteolytic activation of both the wild-type CED-3 zymogen and the CED-3(R51H) zymogen (Fig. 1b, lanes 1–4, 13–16 and Supplementary Fig. 1b,c)12,15,20, leading to the activation of some of the CED-3 zymogens at the 25-min time point (Fig. 1b, lanes 1), when there was no sign of any CED-3 activation in the absence of CED-4 (Fig. 1a, lanes 1). In comparison, oligomeric CED-4 only mildly enhanced autoproteolytic activation of CED-3(L27F), CED-3(L30F) and CED-3(G65R) zymogens (Fig. 1b, lanes 5–12, 17–20, and Supplementary Fig. 1b,c), which already self-activated strongly in vitro without CED-4 (Fig. 1a, lanes 5–12, 17–20, and Supplementary Fig. 1a,c). Therefore, in the presence of oligomeric CED-4, all five CED-3 zymogens were activated to a similar level. In contrast, a catalytic-site mutation in CED-3 (G360S) conferred by a strong lf mutation n2433 completely blocked CED-3 zymogen activation with or without CED-4 (Table 1 and Supplementary Fig. 1d). These results indicate that the three CED-3 prodomain mutations (G65R, L27F and L30F) unexpectedly enhance CED-3 autoproteolytic activation and do not affect CED-4-induced CED-3 activation in vitro. These findings are inconsistent with the strong cell death defects caused by these mutations and suggest that they must impair CED-3 activation or activity in vivo through a more complex mechanism that was not recapitulated in the in vitro assays.
CED-3 displays perinuclear localization in germ cells
To understand how CED-3 acts to kill the cell, we examined the subcellular localization pattern of CED-3 in C. elegans using a low-copy integrated transgene expressing a CED-3 translational GFP fusion (Pced-3ced-3::gfp), which was generated by biolistic bombardment21. This transgene fully rescued the cell death defect of the strong ced-3(n2433) mutant (Supplementary Table 2), indicating that this CED-3::GFP fusion is functional and likely localizes to the proper subcellular sites. Interestingly, we observed a perinuclear localization pattern of CED-3::GFP in C. elegans germ cells (Fig. 2a), which was not seen when GFP was expressed in germ cells (Supplementary Fig. 2a,b). This perinuclear localization pattern of CED-3::GFP was lost and became diffuse in apoptotic germ cells (Supplementary Fig. 2c), probably due to breakdown of nuclear membrane during apoptosis3. Although CED-3::GFP expressed from a high-copy number transgene was observed in the cytoplasm of some dying cell and its mother in C. elegans embryos22, we could not detect clearly visible GFP signal in live somatic cells of C. elegans embryos, larvae, or adults carrying this low-copy Pced-3ced-3::gfp transgene. Multiple efforts to generate antibodies to CED-3 failed to produce an antibody that could detect the endogenous CED-3 expression in C. elegans either by immunoblotting or by immunohistochemistry (see Online Methods).
Figure 2.
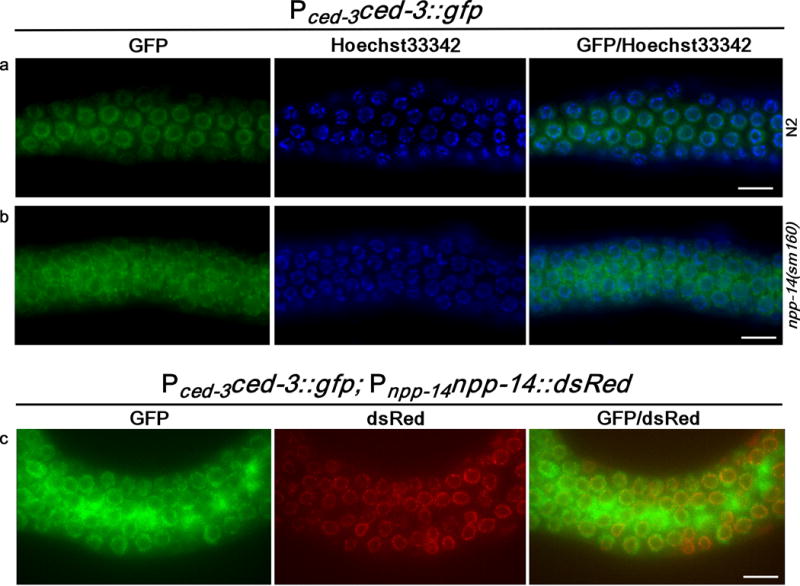
CED-3 localizes to the perinuclear region in C. elegans germ cells. (a,b) Images of GFP, Hoechst33342, and GFP/Hoechst33342 merged from the exposed gonads of hermaphrodite adult animals with the indicated genotypes are shown. (c) Images of GFP, dsRed, and GFP/dsRed merged from the exposed gonad of an N2 hermaphrodite adult carrying the integrated Pced-3ced-3::gfp and Pnpp-14npp-14::dsRed transgenes. Scale bars indicate 10 μm.
C. elegans nuclei inhibit CED-3 autoactivation in vitro
Given the perinuclear localization pattern of CED-3, we examined if C. elegans nuclei might play a role in regulating CED-3 activation or activity. We purified nuclei from wild-type animals (Supplementary Fig. 2d and see Online Methods)23 and included them in CED-3 activation or activity assays. In the absence of oligomeric CED-4, the limited self-activation of the wild-type CED-3 zymogen and the CED-3(R51H) zymogen was inhibited by C. elegans nuclei (Fig. 3a,b, lanes 1–4, 13–16, and Supplementary Fig. 3a,b,d). Similarly, the originally robust autoactivation of CED-3(L27F), CED-3(L30F) and CED-3(G65R) zymogens was markedly suppressed by purified nuclei (Fig. 3a,b, lanes 5–12, 17–20, and Supplementary Fig. 3a,b,d), resulting in loss of the processed catalytic subunits at approximately 15–17 kD positions12,19. On the other hand, purified nuclei did not inhibit cleavage of CED-9, a known CED-3 substrate24, by the active CED-3 protease (Supplementary Fig. 2e), indicating that nuclei do not affect the activity of active CED-3. These results suggest that C. elegans nuclei inhibit autoactivation of both wild-type and mutant CED-3 zymogens in vitro and could serve as a negative regulator to inhibit CED-3 autoproteolytic activation in vivo.
Figure 3.
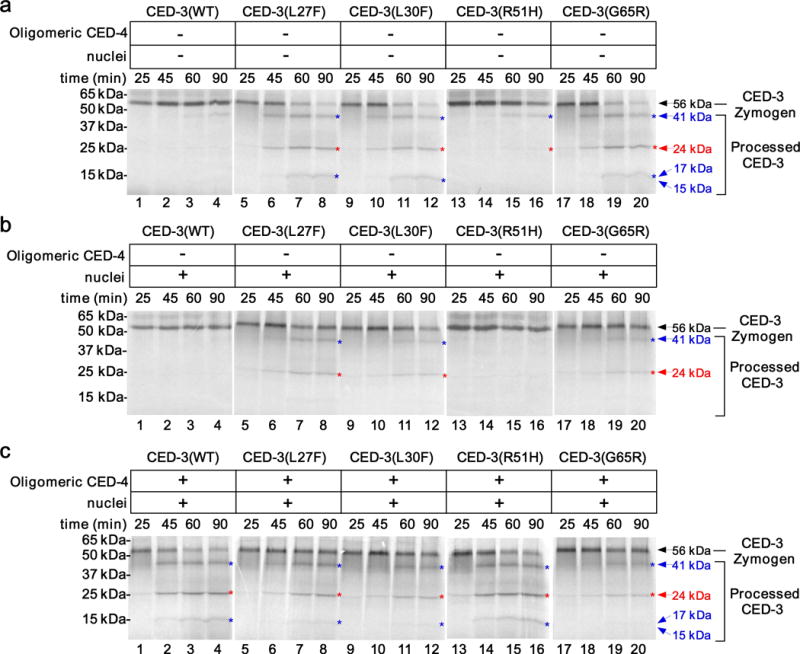
C. elegans nuclei inhibit CED-3 zymogen autoactivation in vitro. (a-c) Autoradiographs of SDS-PAGE showing time-course of CED-3 activation in vitro, performed as described in Fig. 1. CED-3 zymogens were incubated with buffer (a), C. elegans nuclei (b), or oligomeric CED-4 (4 nM final concentration) and C. elegans nuclei (c). The deduced CED-3 processed bands on gels are indicated as described in Fig. 1. The original gel images are shown in Supplementary Data Set 1.
During apoptosis, CED-4 is released from the CED-4 and CED-9 inhibitory complex tethered on the outer mitochondrial membrane and subsequently oligomerizes to promote CED-3 activation and cell killing11,12. When CED-4 oligomers were included in the CED-3 activation assays with the nuclei, both wild-type CED-3 and CED-3(R51H) zymogens were strongly activated (Fig. 3c, lanes 1–4, 13–16, and Supplementary Fig. 3c,d), indicating that CED-4 oligomers overcome the inhibitory effect of nuclei to induce robust activation of the CED-3 and CED-3(R51H) zymogens. In contrast, CED-4 oligomers were unable to reverse the inhibition of nuclei to restore strong activation of the CED-3(L27F), CED-3(L30F) and CED-3(G65R) zymogens, and in particular, generation of catalytic subunits at approximately 15–17 kD positions (Fig. 3c, lanes 5–12, 17–20, and Supplementary Fig. 3c,d). These in vitro results are consistent with the findings that CED-3 prodomain mutations, L27F, L30F and G65R, but not R51H, cause strong cell death defects in C. elegans (Table 1 and Supplementary Table 1).
NPP-14 inhibits cell death in a ced-3 allele-specific manner
We investigated how CED-3 localizes to the perinuclear region and whether inhibition of the CED-3 zymogen activation by nuclei is mediated by a nuclear membrane protein. Because the CED-3 G65R mutation significantly enhanced CED-3 zymogen autoactivation in the absence of C. elegans nuclei (Fig. 3a,b, lanes 17–20), we reasoned that RNA interference (RNAi) knockdown of the nuclear membrane protein that mediates inhibition of CED-3 zymogen autoactivation by nuclei would cause increased cell death in ced-3(n718 G65R) animals. Therefore, we performed a candidate-based RNAi screen on ced-1(e1735); ced-3(n718 G65R) animals and looked for RNAi treatment that increased the number of persistent cell corpses in ced-1(e1735); ced-3(n718 G65R) embryos. Thirty-eight C. elegans genes, encoding nuclear membrane proteins or proteins known to associate with the nuclear envelope23,25–29, were screened (Table 2). RNAi of one gene, npp-14, which encodes a nuclear pore protein, caused appearance of 1–2 cell corpses in 67% of ced-1(e1735); ced-3(n718 G65R) 4-fold stage embryos (n=15), while virtually no cell corpse was seen in ced-1(e1735); ced-3(n718 G65R) embryos treated with RNAi to other genes (Table 2). We confirmed this result using a loss-of-function npp-14 mutation (sm160), which results in substitution of Glutamine 641 of the NPP-14 protein by an Ocher stop codon (see Online Methods). npp-14(sm160) significantly increased the number of persistent cell corpses not only in ced-1(e1735); ced-3(n718 G65R) embryos, but also in ced-1(e1735); ced-3(n1040 L27F) and ced-1(e1735); ced-3(n2439 L30F) embryos (Table 1). For instance, in the ced-1(e1735); ced-3(n1040 L27F) double mutant, a mean of 13 cell corpses was seen in 4-fold stage embryos, but in the npp-14(sm160) ced-1(e1735); ced-3(n1040 L27F) triple mutant, the number of cell corpses almost doubled (a mean of 23 cell corpses; Table 1), indicating markedly increased cell death. In line with these observations, npp-14(sm160) reduced the number of extra cells in the anterior pharynx of ced-1(e1735); ced-3(n718 G65R), ced-1(e1735); ced-3(n1040 L27F), and ced-1(e1735); ced-3(n2439 L30F) animals (Supplementary Table 1). On the other hand, loss of npp-14 did not induce any cell death in ced-1(e1735); ced-3(n2433 G360S) embryos, which have the catalytic-dead CED-3 protein (Table 1 and Supplementary Fig. 1d), and did not increase the number of persistent cell corpses in ced-1(e1735); ced-3(n2449 R51H) embryos, which have a CED-3 zymogen indistinguishable from the wild-type CED-3 zymogen in autoactivation in vitro (Fig. 1a,b). Neither did loss of npp-14 alter the extra cell numbers in ced-1(e1735); ced-3(n2433 G360S) and ced-1(e1735); ced-3(n2449 R51H) animals (Supplementary Table 1).
Table 2.
An RNAi screen to identify the nuclear membrane protein involved in inhibition of CED-3 autoactivation
| Gene | Protein | RNAi phenotype | Cell corpse number | Survival rate |
|---|---|---|---|---|
| npp-1 | Nuclear pore complex protein | − | 0.06 | 37% |
| npp-2 | Nuclear pore complex protein | − | 0 | 4% |
| npp-3 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-4 | Nuclear pore complex protein | − | 0.06 | 6% |
| npp-5 | Nuclear pore complex protein | − | 0.06 | 23% |
| npp-6 | Nuclear pore complex protein | − | 0 | 4% |
| npp-7 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-8 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-9 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-10 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-12 | Nuclear pore complex protein | − | 0.11 | 65% |
| npp-14 | Nuclear pore complex protein | + | 0.73 | 37% |
| npp-15 | Nuclear pore complex protein | − | 0.13 | 13% |
| npp-16 | Nuclear pore complex protein | − | 0.07 | 36% |
| npp-17 | Nuclear pore complex protein | − | 0 | 51% |
| npp-18 | Nuclear pore complex protein | − | 0.07 | 78% |
| npp-19 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-20 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-21 | Nuclear pore complex protein | lethal | ND | 0% |
| npp-23 | Nuclear pore complex protein | − | 0.10 | 78% |
| unc-84 | Inner nuclear membrane protein | − | 0.06 | 71% |
| sun-1 | Inner nuclear membrane protein | − | 0 | 10% |
| unc-83 | Outer nuclear membrane protein | − | 0 | 70% |
| anc-1 | Outer nuclear membrane protein | − | 0.13 | 74% |
| zyg-12 | Outer nuclear membrane protein | − | 0.13 | 2% |
| lem-2 | Lamin-binding protein | − | 0.12 | 73% |
| lem-3 | Lamin-binding protein | − | 0 | 72% |
| emr-1 | Lamin-binding protein | − | 0.06 | 78% |
| ran-1 | Nuclear import/export | lethal | ND | 0% |
| ran-2 | Nuclear import/export | − | 0 | 67% |
| ran-3 | Nuclear import/export | − | 0.07 | 74% |
| ran-4 | Nuclear import/export | − | 0 | 11% |
| ran-5 | Nuclear import/export | − | 0.07 | 61% |
| ima-1 | Importin alpha family | − | 0.18 | 67% |
| ima-2 | Importin alpha family | − | 0 | 13% |
| ima-3 | Importin alpha family | − | 0.07 | 27% |
| imb-2 | Importin beta family | − | 0.07 | 48% |
| imb-6 | Importin beta family | − | 0.13 | 67% |
At least 12 ced-1(e1735); ced-3(n718 G65R) embryos were scored in each RNAi experiment. Animals treated with control RNAi had a mean of 0.07 cell corpses (n=15). “−” indicates RNAi treatment with no detectable effect. “+” indicates RNAi treatment that substantially increased the number of persistent cell corpses. RNAi knockdown of several genes resulted in the “lethal” phenotype with no progeny, which prevented cell corpse counting (not determined or ND).
Similarly, inactivation of npp-14 significantly increased germ cell death in ced-1(e1735); ced-3(n1040 L27F) and ced-1(e1735); ced-3(n2439 L30F) animals (Supplementary Table 3) and resulted in a greater increase of germ cell death when these animals were subjected to ultraviolet (UV) irradiation (Supplementary Table 4). As in somatic cells, loss of npp-14 did not increase germ cell death in ced-1(e1735); ced-3(n2433 G360S) or ced-1(e1735); ced-3(n2449 R51H) animals with or without UV irradiation (Supplementary Tables 3 and 4). Taken together, these results show that npp-14 plays an important, allele-specific cell death inhibitory role in three ced-3 mutants (n718, n1040 and n2439) that have CED-3 zymogens with enhanced autoactivation activity.
NPP-14 is critical for CED-3 perinuclear localization
To investigate if npp-14 affects CED-3 subcellular localization, we introduced the Pced-3ced-3::gfp integrated transgene into the npp-14(sm160) mutant and found that CED-3 largely lost its perinuclear localization (Fig. 2b). This result indicates that NPP-14, a nuclear membrane protein, is critical for localization of CED-3 to the perinuclear region. We generated a low-copy number integrated transgene that expressed npp-14 translational fusion with dsRed from its endogenous promoter (Pnpp-14NPP-14::dsRed) by biolistic bombardment and found that NPP-14::dsRed was colocalized with CED-3::GFP in C. elegans germ cells (Fig. 2c). We then tested if NPP-14 directly interacted with CED-3 using GST fusion protein pull-down assays. GST-NPP-14, but not GST alone, bound full-length CED-3 (residues 1–503; Supplementary Fig. 4a) and the prodomain of CED-3 (residues 1–220)(Fig. 4a, lanes 8 and 9), but failed to interact with a fragment of CED-3 containing both catalytic subunits (residues 206–503; Supplementary Fig. 4a). Interestingly, GST-NPP-14 displayed significantly stronger binding to the CED-3 prodomain harboring the L27F, L30F, or G65R mutation than to the wild-type CED-3 prodomain or the mutant CED-3(R51H) prodomain (Fig. 4a, lanes 10–14, Fig. 4b, and Supplementary Fig. 4b). These results together indicate that NPP-14 plays an important role in tethering the CED-3 zymogen to the nuclear membrane through binding to the CED-3 prodomain. The finding that NPP-14 shows stronger binding to three CED-3 zymogens with prodomain mutations that cause strong cell death defects in vivo is consistent with the observations that these three mutant zymogens were poorly activated by oligomeric CED-4 in the presence of nuclei in vitro (Fig. 3c, lanes 5–12, 17–20, and Supplementary Fig. 3c,d).
Figure 4.
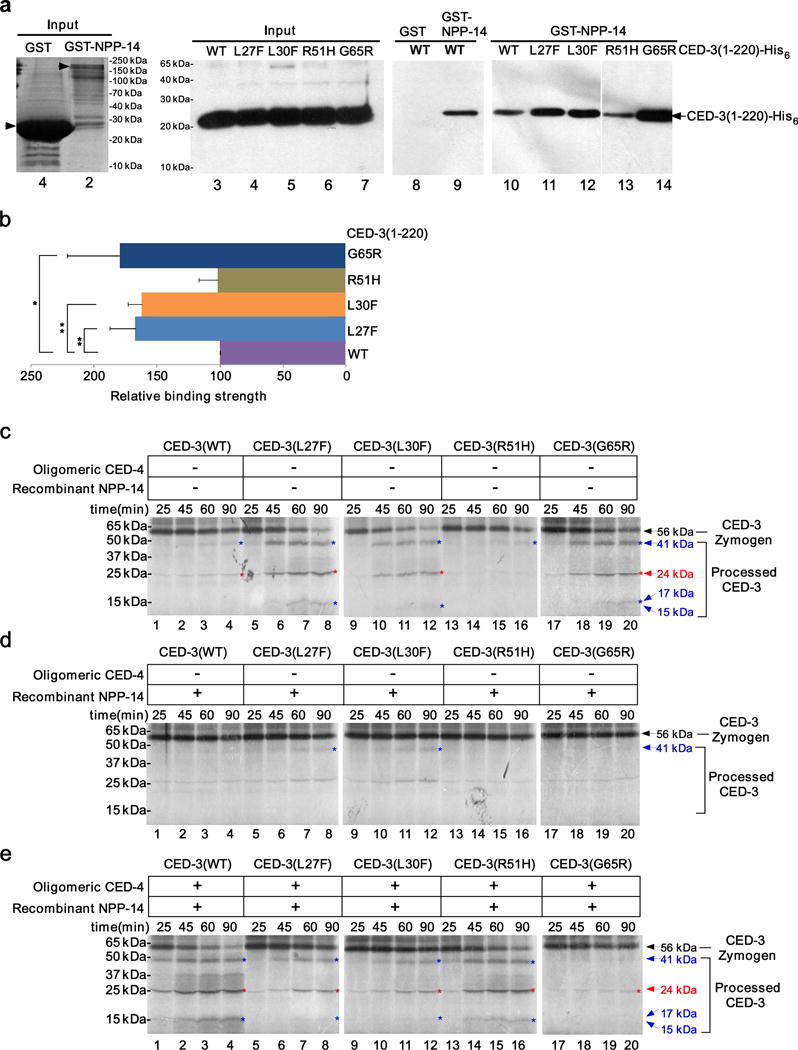
NPP-14 interacts with the prodomain of CED-3 to inhibit CED-3 zymogen activation. (a) Results of pull-down experiments between GST or GST-NPP-14 and 4 nM of purified CED-3(1-220)-His6 (wild-type or mutant, see Online Methods). Lanes 1 and 2 show Coomassie Blue staining. Lanes 3–14 show immunoblotting using an antibody to the His6 tag. (b) The relative binding affinity of NPP-14 to different CED-3 prodomains. The average intensity of the CED-3(1-220)-His6 bands shown in Fig. 4a in 5 independent pull-down experiments was determined by Image J (see Online Methods). In each experiment, the binding between wild-type CED-3(1-220)-His6 and NPP-14 was set as 100. Error bars are s.d. ** P < 0. 01. * P < 0.05 (two-sided t-tests). (c–e) Autoradiographs of SDS-PAGE showing time-course of CED-3 activation in vitro, performed as described in Fig. 1. CED-3 zymogens were incubated with buffer (c), NPP-14 (125 nM final concentration, d), or oligomeric CED-4 (80 nM final concentration) and NPP-14 (125 nM final concentration)(e). The original gel images are shown in Supplementary Data Set 1.
NPP-14 regulates CED-3 autoactivation in vitro
We last tested whether NPP-14 inhibited CED-3 autoactivation in vitro like C. elegans nuclei. When recombinant NPP-14 was included in the CED-3 activation assays, the limited self-activation of CED-3 and CED-3(R51H) zymogens was blocked (Fig. 4c,d, lanes 1–4, 13–16, and Supplementary Fig. 5a,b,d) and the strong self-activation of CED-3(L27F), CED-3(L30F) and CED-3(G65R) zymogens was mostly suppressed (Fig. 4c,d, lanes 5–12, 17–20, and Supplementary Fig. 5a,b,d). Consistent with this in vitro observation, overexpression of NPP-14 in C. elegans inhibited cell death during embryogenesis (Supplementary Fig. 4c). However, when CED-4 oligomers were included in the reactions with NPP-14, the autoproteolytic activation of CED-3 and CED-3(R51H) zymogens was greatly enhanced (Fig. 4e, lanes 1–4, 13–16, and Supplementary Fig. 5c,d). By contrast, the activation of the CED-3(L27F), CED-3(L30F), or CED-3(G65R) zymogen remained largely suppressed (Fig. 4e, lanes 5–12 and lanes 17–20, and Supplementary Fig. 5c,d). Like nuclei, NPP-14 did not inhibit the activity of the active CED-3 protease (Supplementary Fig. 2f). Therefore, NPP-14 mimics nuclei in suppressing CED-4-induced activation of three CED-3 zymogens with prodomain mutations that cause stronger association of CED-3 with NPP-14, but does not affect active CED-3 protease. These results nicely account for the paradoxical observations that CED-3(L27F), CED-3(L30F) and CED-3(G65R) zymogens self-activate strongly in vitro, but have a greatly reduced proapoptotic activity in vivo.
DISCUSSION
Caspases play pivotal roles in the initiation and execution of apoptosis in diverse organisms1–3,30. The activation and activities of caspases need to be tightly controlled to ensure proper animal development and tissue homeostasis1–3,30. As the prototype of the apoptotic caspases and the key cell-killing protein in C. elegans, how the CED-3 caspase is regulated so that it is safely constrained in living cells and specifically activated in apoptotic cells has been a major topic of study. The central unresolved issue is the physical location of the CED-3 zymogen, which governs how it might be activated and how it acts to trigger apoptosis.
In this study, we report that the C. elegans CED-3 zymogen is tethered to the nuclear membrane in live germ cells through its interaction with a nuclear pore component NPP-14. This protein interaction and the nuclear membrane association block autoactivation of the CED-3 zymogen in vitro and appear to protect cells from inadvertent apoptosis in vivo. In cells destined to undergo apoptosis, the activated CED-4 oligomers translocate from mitochondria to the perinuclear region11,12, or accumulate in the perinuclear region14, where they induce robust activation of the CED-3 zymogen by overcoming the inhibitory effect of NPP-14 and nuclei, leading to the activation of apoptosis. These findings unravel the long-standing questions of how autoactivation of the CED-3 zymogen is curbed in cells that live and why CED-4 needs to translocate to the perinuclear region to activate apoptosis. This is also the first report that a caspase zymogen is targeted to the perinuclear region in living cells and that nuclei play a direct and critical role in regulating appropriate activation of a caspase zymogen.
In mammals, caspase zymogens have not been reported to localize to the perinuclear region or associate with the nuclear membrane. However, several caspases, including caspase-2 and caspase-3, have been found in nuclei or in the nuclear fractions31–33, although the importance of such localization to apoptosis and caspase activation is unclear. Moreover, several nuclear pore proteins, such as Nup210, Nup214 and Nup107, have been implicated in suppressing apoptosis in mammalian cells34–36. In particular, Nup210 plays an antiapoptotic role to promote skeletal muscle differentiation by suppressing the caspase cascade induced by endoplasmic reticulum (ER) stress accompanying the differentiation process34. Further investigation is needed to examine if association with nuclear membrane proteins represents a conserved mechanism for regulating apoptosis and caspase activation across organisms.
The prodomains or the CARD domains of caspases in mammals play a critical role in caspase activation by recruiting caspase zymogens to specific death-activation complexes, such as the apoptosome or the death receptor signaling complexes, leading to their autocatalytic activation1,2,30. It is thus intriguing that the prodomain of CED-3 appears to be dispensable for CED-3 activation in vitro15,16, yet multiple mutations in the prodomain, including three examined in this study (n718, n1040, and n2439), cause strong cell death defects in vivo4,17. In this study, we show that these three prodomain mutations result in stronger association of the CED-3 zymogens with NPP-14, which deters not only CED-3 autocatalytic activation but also CED-3 activation induced by CED-4 oligomers (Figs. 3, 4). This finding provides the mechanistic insight into why these mutations cause strong cell death defects in vivo. On the other hand, these three prodomain mutations also cause enhanced CED-3 autoactivation in vitro, and consistent with that finding, increased apoptosis in vivo in the absence of npp-14 (Fig. 1). These findings squarely place NPP-14 and C. elegans nuclei as the key regulator of CED-3 activation and apoptosis in C. elegans and raise the possibility that a similar mechanism may control caspase activation and apoptosis in other organisms.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Strains
Strains of C. elegans were cultured at 20°C using standard procedures37. Most of the alleles used in this study have been described previously17,18, except the npp-14(sm160) allele.
Isolation of the npp-14(sm160) mutation
Ethyl methane sulfonate (EMS) mutagenesis was performed on ced-9(n2812); ced-3(n717) animals carrying an extrachromosomal transgene array (smEx588) containing PhspCED-4::GFP and pRF4, a transgenic marker causing the Rol phenotype. Upon heat-shock treatment, CED-4::GFP localizes to the nuclear membrane due to loss of ced-9. sm160 was isolated as a recessive mutation that prevents CED-4::GFP localization to the nuclear membrane and was found to cause a C to T transition in the npp-14 gene that generates a stop codon at amino acid 641 (Q641Ocher).
Quantification of cell corpses
Somatic cell corpses in the head region of 3-fold transgenic embryos or 4-fold stage embryos were scored using Nomarski optics, as previously described38. The number of germ cell corpses in the pachytene region of one gonad arm of hermaphrodite animals was scored using Nomarski optics. 15 animals were scored at 24 hours or 48 hours post L4 to the adult molt for each strain.
Quantification of extra cells
The number of extra cells in the anterior pharynx of L4 larvae was scored using Nomarski optics, as previously described38. 20 animals were scored for each strain.
Quantification of germ cell death induced by UV irradiation
To assess the effect of UV irradiation on germ cell death, hermaphrodite animals were allowed to develop to the young adult stage (24 hours after L4 to the adult molt) at 20°C and treated with 100 J/m2 UV on non-seeded NGM plates using a Stratalinker (Stratagene, Inc.). Immediately after UV irradiation, animals were transferred to freshly seeded NGM plates and germ cell corpses were scored 6 hours or 24 hours later as inidcated.
Generation of transgenic animals
Phspnpp-14 (25 μg/ml each) and Psur-5gfp (25 μg/ml), which was used as a transgenic marker, were injected into ced-1(e1735) animals to generate transgenic lines. To express GFP in C. elegans germ line under the control of the heat-shock promoters, a mixture of the following DNA constructs were injected into the N2 animals to create complex transgenic arrays that facilitate gene expression in the germ line: the Phspgfp expression constructs linearized with Hind III (1 μg/ml each), the pRF4 construct linearized with EcoR I (1 μg/ml) and the Psur-5mcherry construct (1 μg/ml), which serve as transgenic markers, and wild-type C. elegans genomic DNA digested with Sca I (60 μg/ml). F3, F4 and F5 generations of Roller transgenic animals were examined and imaged for the expression of GFP in the germ line after the heat-shock treatment.
Heat-Shock Treatment
Roller transgenic animals were placed at 33°C for 60 minutes and let recover at 20°C for 3 hours before imaging.
Molecular Biology
To generate the Pced-3ced-3::gfp translational fusion construct for biolistic bombardment, we first replaced the stop codon of the ced-3 coding region in the pJ40 vector with an Nhe I site through site-directed mutagenesis4, which contains 2387 bp of the ced-3 promoter and 2275 bp of 3’ untranslated region. A GFP coding region was then inserted into the modified vector through the newly created Nhe I site. A 10 kb Bam HI/Apa I fragment containing the ced-3::gfp fusion was excised from the vector and inserted into a vector used for biolistic bombardment, which also contains the C. elegans unc-119 gene. To generate the Pnpp-14npp-14::dsRed fusion construct for biolistic bombardment, a 7173 bp PCR fragment containing the npp-14 coding region including 2603 bp of the npp-14 promoter was inserted into a modified pPD95.77 vector, in which the GFP coding region was replaced by the dsRed coding region. A 8.7 kb Pst I/Spe I fragment containing the npp-14::dsRed fusion was excised from the resulting construct and inserted into the same bombardment vector described above.
Protein expression and purification
CED-4 oligomers were expressed and purified as previously described12. A partial cDNA clone (yk1144d07) containing npp-14 and a cDNA fragment containing the part of npp-14 that is missing in the yk1144d07 clone, which was generated by reverse transcription polymerase chain reaction (RT-PCR), were used to assemble the full-length npp-14 cDNA clone. The full-length npp-14 cDNA fragment was subcloned into the pET-15b vector via its Nde I and Xho I sites and the resulting construct was used to express NPP-14 in the bacterial strain BL21(DE3) as an N-terminal His6-tagged protein. After 3 hour induction at 22°C, cells were collected and lysed. His6-NPP-14 was purified from the soluble fraction using a Ni2+-NTA column (GE Healthcare) and further dialyzed by a buffer containing 25 mM Tris (pH8.0), 150 mM NaCl, 1 mM DTT, and 10% (v/v) Glycerol. The ced-3 cDNA fragments encoding amino acids 1–220 (wild-type or with prodomain mutations) with a C-terminal His6 tag were subcloned into the pET-3a vector via its Nde I and BamH I sites to generate the CED-3(1–220)-His6 expression constructs (wild-type or mutant). CED-3(1–220)-His6 proteins (wild-type or mutant) were expressed in the bacterial strain BL21(DE3), and after 4 hour induction at 22°C, cells were collected and lysed. CED-3(1–220)-His6 proteins (wild-type or mutant) were purified from the soluble fraction using a Ni2+-NTA column and were eluted from the column in a buffer containing 100 mM Tris (pH8.0), 250 mM NaCl, 0.5mM Sucrose, 0.04% NP-40, 1 mM DTT, 250mM Imidazole and 10% (v/v) Glycerol. The concentrations of purified proteins were determined by spectroscopic measurement at 280 nm.
CED-3 zymogen activation assays in vitro
The CED-3 expression vector, pTRI-CED-312, was used to generate other pTRI-CED-3 vectors carrying the prodomain mutations, L27F, L30F, R51H, G65R, or G360S, using a QuickChange Site-Directed Mutagenesis kit (Stratagene). All constructs were confirmed by DNA sequencing.
CED-3 zymogens (wild-type or mutant) were synthesized and labeled with 35S-Methionine in the TNT Transcription/Translation coupled system (Promega) at 30°C for 20 min. CED-4 oligomers, C. elegans nuclei, purified NPP-14, or buffer controls were then added to the reactions. At different time points, an aliquot was taken out and SDS sample loading buffer was added to stop the reaction. The samples were resolved by 15% SDS polyacrylamide gels (PAGE), which were fixed, dried and subjected to autoradiography. CED-4 buffer contains 25 mM Tris (pH8.0), 200 mM NaCl, and 10% (v/v) Glycerol. The nuclei buffer contains 20 mM HEPES (pH 7.6), 20 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.5 M sucrose, 10% (v/v) Glycerol, and 1 mM DTT. The NPP-14 buffer contains 25 mM Tris (pH8.0), 150 mM NaCl, 1 mM DTT, and 10% (v/v) Glycerol.
CED-3 activity assays in vitro
CED-9 was synthesized and labeled with 35S-Methionine in the Transcription & Translation coupled system (Promega) for 1 hour at 30°C as described above. 10 ng of purified, active CED-3 were incubated with 35S-Methionine-labeled CED-9 in the presence of C. elegans nuclei, recombinant NPP-14, or the caspase inhibitor (iodoacetic acid) for 1 hour at 30°C. After that, SDS loading buffer was added to stop the reaction. The samples were resolved by 15% SDS-PAGE and subjected to autoradiography. CED-3 buffer contains 100 mM Tris (pH8.0), 250 mM NaCl, 0.5 mM sucrose, 7.5mM beta-mercaptoethanol, 0.04% (v/v) NP-40, and 10% (v/v) Glycerol.
Quantifying the intensity of CED-3 bands from gel images
SDS-PAGE gels were dried and then subjected to autoradiograpghy to obtain corresponding gel images, which were converted by scanner to images that could be opened in Image J software (https://imagej.nih.gov/ij/). The intensity of CED-3 bands of interest was measured and recorded for further quantification.
Isolation of C. elegans nuclei
Isolation of C. elegans nuclei was carried out using a previously described protocol with minor modifications23. Wild-type C. elegans animals carrying an integrated transgene ccIs481039, which expresses a lamin GFP fusion and allows quantification of isolated nuclei, were used to isolate nuclei. Adult ccIs4810 animals were harvested, washed with M9 buffer, resuspended in an equal volume of cold PBS, snap-freeze in liquid nitrogen, and stored at −80°C overnight. The animals were thawed and immediately transferred to a pre-chilled Wheaton stainless-steel tissue grinder and homogenized in an equal volume of cold 2X nuclei preparation buffer [20 mM HEPES (pH 7.6), 20 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.5 M sucrose, 1 mM DTT, and protease inhibitors]. Debris was spun down at 1000 rpm for 1 min to remove unbroken worm bodies and supernatants containing nuclei were collected. Homogenization and collection of nuclei were repeated twice. Supernatants from the first and the second collections were pooled and centrifuged at 3000 rpm for 10 min to obtain the nuclei in the pellets. Nuclei were then washed twice using the nuclei TNT assay buffer [20 mM HEPES (pH 7.6), 20 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.5 M sucrose, 10% (v/v) Glycerol, and 1 mM DTT] and resuspended in 4 volumes of nuclei TNT assay buffer.
Imaging CED-3 and NPP-14 localization in germ cells
The subcellular localization of CED-3::GFP as well as NPP-14::dsRed was examined in dissected gonads from Pced-3ced-3::gfp, npp-14(sm160); Pced-3ced-3::gfp, and Pced-3ced-3::gfp; Pnpp-14npp-14::dsRed adult hermaphrodites. Gonads were gently dissected out from 24-hour old adult hermaphrodites by cutting them at the head region on a slide while immersed in a gonad dissection buffer [60 mM NaCl, 32 mM KCl, 3 mM Na2HPO4, 2 mM MgCl2, 20 mM HEPES, 10 mM Glucose, 33% (w/v) fetal calf serum, and 2 mM CaCl2]40. 5 μM of Hoechst33342 were then added to the buffer containing the dissected gonads. The dissected gonads were mounted on the slide and visualized using a Nomarski microscope equipped with epifluorescence.
RNAi screen
The bacteria feeding protocol was used in all RNAi experiments41. Approximately 30 L4-stage larvae were transferred to RNAi plates. Progeny of RNAi-treated animals were scored for persistent cell corpses at the 4-fold embryonic stage. To determine the survival rate in each RNAi experiment, 30 L3-L4 larvae were transferred from a plate seeded with OP50 bacteria to an RNAi plate. Two days later, 20 adult animals were transferred from the original RNAi plate to a new RNAi plate. The adult animals were allowed to lay eggs for 1 hour before they were removed and the number of eggs laid on the plate was recorded. Three days later, the number of eggs that hatched out to become larvae was scored and used to determine the survival rate. RNAi knockdown of multiple genes resulted in different degrees of embryonic lethality, but allowed scoring of cell corpses in some surviving embryos.
GST fusion protein pull-down assays
GST was expressed from the expression vector, pGEX-4T-1. The full-length npp-14 cDNA was subcloned into the pPGH vector (a derivative of pGEX-4T-2) via its Nde I and Hind III sites to generate the GST-NPP-14 expression construct. cDNA fragments encoding full-length CED-3 or residues 206–503 of CED-3 with an A449V mutation that inactivates the activity of the CED-3 protease19 and a C-terminal FLAG tag were subcloned into the pET-3a vector via its Nde I and BamH I sites. The A449V mutation was generated using a two-step PCR procedure and allows synthesis of CED-3 in bacteria without autoproteolytic cleavage. Bacterial lysate containing GST or GST-NPP-14 was incubated with the glutathione Sepharose beads (GE Healthcare) in the reaction buffer containing 50 mM Tris (pH8.0), 100 mM NaCl, 0.1% (v/v) NP-40, 10% (v/v) Glycerol, and 2 mM DTT at 4°C for 1 hour before washed five times with the binding buffer [50 mM Tris (pH8.0), 300 mM NaCl, 0.2% (v/v) NP-40, 1 mM DTT, and 5 mg/ml BSA]. The glutathione Sepharose beads with immobilized GST or GST–NPP-14 were then incubated with 4 nM of purified wild-type CED-3(1–220)-His6 or CED-3(1–220)-His6 carrying the indicated mutation or bacterial lysate containing 3.5 nM CED-3(A449V)-FLAG or CED-3(206–503, A449V)-FLAG at 4°C for 1 hour in 1 ml binding buffer. Subsequently, the Sepharose beads were washed five times with the washing buffer [50 mM Tris (pH8.0), 300 mM NaCl, 0.2% (v/v) NP-40, and 1 mM DTT] and the bound proteins were resolved by 15% SDS–PAGE, transferred to a PVDF membrane, and detected by immunoblotting.
Immunoblotting
The CED-3(1–220)-His6 proteins (wild-type or mutant) bound to the Sepharose beads in the pull-down assays were resolved by 15% SDS-PAGE and transferred to the PVDF membrane (Millipore). The membrane was first blocked with 5% nonfat dry milk (Bio-Rad) in PBS-T (PBS with 0.1% Tween 20) for 1 hour and then incubated with a mouse anti-His6 antibody (Proteintech, Cat. no. 66005-I-Ig) in 1:6000 dilution for 2 hour at room temperature. The membrane was washed three times with PBS-T for 5 min each. HRP-conjugated goat anti-mouse IgG antibody (Bio-Rad, Cat. no. 170–5047) was then added to the membrane in 1:5000 dilution for 1 hour at room temperature. The membrane was then washed three more times with PBS-T and the proteins were detected with enhanced chemiluminescent substrate (Pierce). For anti-FLAG immunoblotting, a mouse anti-FLAG antibody (CWbiotech, Cat. no. CW0287A) was used in 1:2000 dilution and HRP-conjugated goat anti-mouse IgG (Bio-Rad cat. no. 170–5047) was used in 1:5000 dilution. For immunoblotting analyses to examine purified nuclei, adult ccIs4810 animals were harvested from NGM plates with M9, centrifuged at 500 g to remove bacteria, and sonicated in PBS buffer to generate the worm lysate. 2× SDS loading buffer was added to purified ccIs4810 nuclei and ccIs4810 lysate. The samples were then resolved with 12% SDS-PAGE, transferred to a PVDF membrane, and detected using different antibodies. For the GFP immunoblot (a nuclear membrane marker), a rabbit anti-GFP antibody was used in 1:1500 dilution (Proteintech, Cat. no. 50430-2-AP) and a HRP-conjugated goat anti-rabbit IgG (CWbiotech, Cat. no. CW0103) was used in 1:5,000 dilution. For the α-tubulin immunoblot (a cytoskeletal marker), a mouse anti-α-tubulin antibody (Developmental Studies Hybridoma Bank, Cat. no. AA4.3) was used in 1:5,000 dilution and a HRP-conjugated goat anti-mouse IgG (Bio-Rad, Cat. no. 170-5047) was used in 1:5,000 dilution. For the DLG-1 immunoblot (a plasma membrane marker)42, a mouse anti-DLG-1 antibody (Developmental Studies Hybridoma Bank, Cat. no. DLG1) was used in 1:1000 dilution and a HRP-conjugated goat anti-mouse IgG (Bio-Rad cat. no. 170-5047) was used in 1:5,000 dilution. For the HSP-60 immunoblot (a mitochondrial marker)42, a mouse anti-HSP-60 antibody (Developmental Studies Hybridoma Bank, Cat. no. HSP60) was used in 1:200 dilution and a HRP-conjugated goat anti-mouse IgG (Bio-Rad cat. no. 170-5047) was used in 1:5,000 dilution. For the HDEL immunoblot (an ER marker), a mouse anti-HDEL antibody (Santa Cruz Biotechnology, cat. no. sc-53472, validated by 1DegreeBio) was used in 1:100 dilution and a HRP-conjugated goat anti-mouse IgG (Bio-Rad cat. no. 170-5047) was used in 1:5,000 dilution.
Generation of CED-3 antibodies
Purified CED-3(206-503; A449V)-His6 was used to immunize four rabbits, two rats, and multiple mice to generate either polyclonal or monoclonal antibodies against CED-3. None of the serum, even after affinity purification using CED-3, and none of the monoclonal antibodies from eight different hybridoma cell lines, could detect the CED-3 protein in C. elegans either by immunoblotting or by immunohistochemistry.
Supplementary Material
Acknowledgments
We thank C.L. Sun for generating the Pnpp-14NPP-14::dsRed integrated transgene, X.C. Wang for isolating the sm160 allele, J. Friedman for identifying the sm160 mutation in npp-14, and Y.G. Shi (Tsinghua University) and M. Han (University of Colorado) for providing vectors. This work was supported by National Basic Research Program of China (973 program 2013CB945600), National Institutes of Health (NIH) grants T32GM007135 and F30NS070596 (B.H.), and NIH grants R01 GM59083, R01 GM88241, and R35 GM118188 (D.X.)
Footnotes
AUTHOR CONTRIBUTIONS
X.D.C., Y.W., Y.Z.C., and D.X. conceived the research and designed and analyzed experiments. X.D.C. carried out all CED-3 in vitro assays and some immunoblotting and protein binding assays. Y.W. performed the RNAi screen, microscopy imaging, some protein binding assays, and some of the somatic cell death assays. Y.Z.C. performed germ cell death and some of the somatic cell death assays, microscopy imaging, and some biochemical assays. B.H., A.N., E.S.L, and H.Y.G. assisted in some of experiments. X.D.C., Y.W., Y.Z.C., and D.X. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 2.Salvesen GS, Riedl SJ. Caspase mechanisms. Adv Exp Med Biol. 2008;615:13–23. doi: 10.1007/978-1-4020-6554-5_2. [DOI] [PubMed] [Google Scholar]
- 3.Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu Rev Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 6.Yuan J, Horvitz HR. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development. 1992;116:309–320. doi: 10.1242/dev.116.2.309. [DOI] [PubMed] [Google Scholar]
- 7.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 8.Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO. Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- 9.Chinnaiyan AM, O’Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Wallen HD, Nunez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- 11.Chen F, et al. Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science. 2000;287:1485–1489. doi: 10.1126/science.287.5457.1485. [DOI] [PubMed] [Google Scholar]
- 12.Yan N, et al. Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature. 2005;437:831–837. doi: 10.1038/nature04002. [DOI] [PubMed] [Google Scholar]
- 13.Conradt B, Wu YC, Xue D. Programmed Cell Death During Caenorhabditis elegans Development. Genetics. 2016;203:1533–1562. doi: 10.1534/genetics.115.186247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pourkarimi E, Greiss S, Gartner A. Evidence that CED-9/Bcl2 and CED-4/Apaf-1 localization is not consistent with the current model for C. elegans apoptosis induction. Cell Death Differ. 2012;19:406–415. doi: 10.1038/cdd.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi S, et al. Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell. 2010;141:446–457. doi: 10.1016/j.cell.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Huang W, et al. Mechanistic insights into CED-4-mediated activation of CED-3. Genes Dev. 2013;27:2039–2048. doi: 10.1101/gad.224428.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaham S, Reddien PW, Davies B, Horvitz HR. Mutational analysis of the Caenorhabditis elegans cell-death gene ced-3. Genetics. 1999;153:1655–1671. doi: 10.1093/genetics/153.4.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue D, Shaham S, Horvitz HR. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 1996;10:1073–1083. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- 20.Geng X, et al. Inhibition of CED-3 zymogen activation and apoptosis in Caenorhabditis elegans by caspase homolog CSP-3. Nat Struct Mol Biol. 2008;15:1094–1101. doi: 10.1038/nsmb.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakraborty S, Lambie EJ, Bindu S, Mikeladze-Dvali T, Conradt B. Engulfment pathways promote programmed cell death by enhancing the unequal segregation of apoptotic potential. Nat Commun. 2015;6:10126. doi: 10.1038/ncomms10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue D, Horvitz HR. Caenorhabditis elegans CED-9 protein is a bifunctional cell-death inhibitor. Nature. 1997;390:305–308. doi: 10.1038/36889. [DOI] [PubMed] [Google Scholar]
- 25.Hoelz A, Debler EW, Blobel G. The structure of the nuclear pore complex. Annu Rev Biochem. 2011;80:613–643. doi: 10.1146/annurev-biochem-060109-151030. [DOI] [PubMed] [Google Scholar]
- 26.Adam SA. The nuclear transport machinery in Caenorhabditis elegans: A central role in morphogenesis. Semin Cell Dev Biol. 2009;20:576–581. doi: 10.1016/j.semcdb.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Minn IL, Rolls MM, Hanna-Rose W, Malone CJ. SUN-1 and ZYG-12, mediators of centrosome-nucleus attachment, are a functional SUN/KASH pair in Caenorhabditis elegans. Mol Biol Cell. 2009;20:4586–4595. doi: 10.1091/mbc.E08-10-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorjanacz M, Jaedicke A, Mattaj IW. What can Caenorhabditis elegans tell us about the nuclear envelope? FEBS Lett. 2007;581:2794–2801. doi: 10.1016/j.febslet.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 29.Malone CJ, Fixsen WD, Horvitz HR, Han M. UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development. 1999;126:3171–3181. doi: 10.1242/dev.126.14.3171. [DOI] [PubMed] [Google Scholar]
- 30.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 31.Colussi PA, Harvey NL, Kumar S. Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor. A novel function for a caspase prodomain. J Biol Chem. 1998;273:24535–24542. doi: 10.1074/jbc.273.38.24535. [DOI] [PubMed] [Google Scholar]
- 32.van Loo G, et al. Caspases are not localized in mitochondria during life or death. Cell Death Differ. 2002;9:1207–1211. doi: 10.1038/sj.cdd.4401101. [DOI] [PubMed] [Google Scholar]
- 33.Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- 34.Gomez-Cavazos JS, Hetzer MW. The nucleoporin gp210/Nup210 controls muscle differentiation by regulating nuclear envelope/ER homeostasis. J Cell Biol. 2015;208:671–681. doi: 10.1083/jcb.201410047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharjya S, et al. Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol Cancer. 2015;14:42. doi: 10.1186/s12943-015-0299-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banerjee HN, Gibbs J, Jordan T, Blackshear M. Depletion of a single nucleoporin, Nup107, induces apoptosis in eukaryotic cells. Mol Cell Biochem. 2010;343:21–25. doi: 10.1007/s11010-010-0494-6. [DOI] [PubMed] [Google Scholar]
- 37.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanfield GM, Horvitz HR. The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol Cell. 2000;5:423–433. doi: 10.1016/s1097-2765(00)80437-2. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, et al. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol Biol Cell. 2000;11:3937–3947. doi: 10.1091/mbc.11.11.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, et al. C. elegans mitochondrial factor WAH-1 promotes phosphatidylserine externalization in apoptotic cells through phospholipid scramblase SCRM-1. Nat Cell Biol. 2007;9:541–549. doi: 10.1038/ncb1574. [DOI] [PubMed] [Google Scholar]
- 41.Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- 42.Hadwiger G, Dour S, Arur S, Fox P, Nonet ML. A monoclonal antibody toolkit for C. elegans. PLoS One. 2010;5:e10161. doi: 10.1371/journal.pone.0010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.