

Summary
A detailed understanding of the paths that stem cells traverse to generate mature progeny is vital for elucidating mechanisms governing cell fate decisions and tissue homeostasis. Adult stem cells maintain and regenerate multiple mature cell lineages in the olfactory epithelium. Here we integrate single cell RNA sequencing and robust statistical analyses with in vivo lineage tracing to define a detailed map of the postnatal olfactory epithelium, revealing cell fate potentials and branch points in olfactory stem cell lineage trajectories. Olfactory stem cells produce support cells via direct fate conversion in the absence of cell division, and their multipotency at the population level reflects collective unipotent cell fate decisions by single stem cells. We further demonstrate that Wnt signaling regulates stem cell fate by promoting neuronal fate choices. This integrated approach reveals mechanisms guiding olfactory lineage trajectories and provides a model for deconstructing similar hierarchies in other stem cell niches.
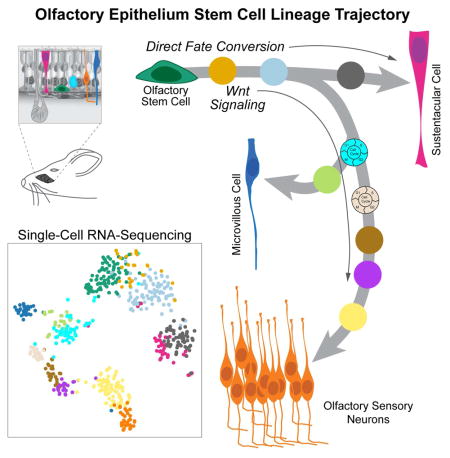
Graphical Abstract
Introduction
A fundamental challenge in stem cell biology is to define both the cell fate potential of a given stem cell and where cell fates are specified along a developmental trajectory. Moreover, detailed lineage trajectory maps are necessary for identifying the regulatory networks that govern the cell fate transitions underlying tissue maintenance and regeneration, and are essential for designing strategies to manipulate cells for therapeutic applications. Lineage tracing – a technique for permanently labeling the descendants of a targeted cell – has long been established as a powerful tool for elucidating the cell fate potential of progenitor cells (Dymecki and Tomasiewicz, 1998; Le Douarin and Teillet, 1974; Price et al., 1987; Weisblat et al., 1978; Zinyk et al., 1998). However, this approach alone cannot readily identify all intermediate stages in a lineage or pinpoint when in a branching lineage multiple cell fates arise.
Whole transcriptome profiling of single cells by RNA sequencing (single-cell RNA-seq) has recently emerged as a powerful method for discriminating the heterogeneity of cell types and cell states in a complex population (Wagner et al., 2016). New statistical approaches have further enabled the ordering of cells along developmental lineages based on gradual changes in gene expression detected at the single cell level (Trapnell et al., 2014). However, current approaches struggle to overcome the challenge of identifying where lineages diverge in more complex branching trajectories of multipotent progenitors, a problem that is only beginning to be addressed (Setty et al., 2016). Importantly, even the most sophisticated analysis of single-cell RNA-seq data can only provide predictions that require independent experimental validation.
The olfactory epithelium maintains a steady state population of mature olfactory sensory neurons via continual neurogenesis in the postnatal animal (Graziadei and Graziadei, 1979b; Mackay-Sim and Kittel, 1991). Olfactory neurogenesis is normally sustained through differentiation of globose basal cells (GBCs), which are the actively proliferating neurogenic progenitor cells in the niche (Caggiano et al., 1994; Graziadei and Graziadei, 1979b; Schwob et al., 1994). Upon targeted destruction of the sensory neurons or more severe injury to the entire tissue, the olfactory epithelium can regenerate (Graziadei and Graziadei, 1979a). Following such injury, the horizontal basal cells (HBCs) – the normally quiescent, reserve stem cells of the niche – become activated to differentiate and reconstitute all major cell types in the epithelium (Iwai et al., 2008; Leung et al., 2007) (Figure 1A).
Figure 1. Experimental Strategy for Olfactory Stem Cell Lineage Analysis with Single-Cell RNA-Seq.

(A) Schematic of the olfactory epithelium describing the constituent cells: horizontal basal cell (HBC, green), globose basal cell (GBC, blue), sustentacular cell (Sus, pink), olfactory sensory neuron (OSN, purple), microvillous cell (MV, dark blue), Bowman’s gland (yellow). (B) Immunohistochemistry for the HBC lineage tracer YFP (green) and SOX2 (magenta) shows basal resting HBCs in the wild type (WT) background (left panel) and asynchronous differentiation following Trp63 conditional knockout (cKO) (center, right). (C) YFP(+) cells were collected by FACS at the indicated times following tamoxifen administration from mice carrying the Krt5-CreER; Rosa26eYFP transgenes and either the Trp63+/+ (WT) or Trp63lox/lox (cKO) alleles. (D) Sox2-eGFP(+)/ICAM1(−)/SCARB1(−)/F3(−) cells were collected by FACS; this enriched for the GBC, INP, and MV fates over Sus cells. (E) Data from both experimental designs were combined, filtered, normalized, clustered, and used in downstream analyses. – Scale bars, 50 microns. See Figure S1.
With its relative simplicity and experimental accessibility, the postnatal olfactory epithelium provides an attractive system for studying the activation and specification events that occur during the differentiation of multiple cell lineages from an adult stem cell. A number of questions relevant to other adult stem cell niches can also be addressed. For example, while lineage tracing suggests that cells arising from HBCs transition through proliferative GBC progenitors to generate olfactory sensory neurons (Leung et al., 2007), it remains unclear whether the epithelium’s other cell types arise via a common GBC intermediate. Similarly, characterizing the transitions in gene expression that occur throughout a developing lineage is a prerequisite to disentangling the gene regulatory networks that underlie cell fate decisions.
In the present study, we combined a statistical approach for making branching lineage assignments from single-cell RNA-seq data with in vivo lineage tracing to map the developmental trajectories of the multiple cell lineages arising from the olfactory epithelium’s HBC stem cell. The first major bifurcation in the HBC lineage trajectory occurs prior to cell division, producing either sustentacular (support) cells or GBCs. The GBC lineage in turn branches to give rise to olfactory sensory neurons, microvillous cells and cells of the Bowman’s gland. Whereas olfactory neurogenesis involves an expansion of the progenitor pool via proliferative GBCs, sustentacular cells instead can arise via direct fate conversion of quiescent HBCs, a process that does not require cell division. Moreover, the multipotency of HBCs as a population reflects independent unipotent cell fate decisions made at the single cell level. Finally, we identified and validated canonical Wnt signaling as a regulator that drives HBCs from quiescence toward neuronal differentiation. Our combined approach serves more generally as a model for illuminating and deconstructing branching lineages that arise from multipotent stem cells.
Results
Experimental Strategy for Analyzing Olfactory Stem Cell Trajectories by Single-Cell RNA-Seq
We applied single-cell RNA-seq to identify the cell state transitions during differentiation of olfactory HBC stem cells into proliferating progenitors and mature cell types. HBCs and their descendants were obtained using two complementary approaches (Figure 1). In the first, we used inducible Cre-lox lineage tracing to label HBCs and their progeny. To obtain resting HBCs (controls), we harvested cell samples 72 or 96 hours following tamoxifen-induced Cre activation in three week-old mice (Figure 1C). To obtain differentiating cells descended from HBCs, we used conditional knockout of Trp63 in HBCs to “release” HBCs from their quiescent state (Figure 1B,C) (Fletcher et al., 2011). In the second approach, using a Sox2eGFP knock-in reporter gene (Arnold et al., 2011), we FACS-purified Sox2-eGFP-positive, ICAM1-negative, SCARB1/F3-negative cells to obtain a population of cells enriched for GBCs, later neuronal intermediates and microvillous cells over sustentacular cells.
Single-cell RNA-seq was carried out on FACS-purified cells using the Fluidigm C1 microfluidics cell capture platform followed by Illumina sequencing (see STAR Methods for these and associated statistical methods). Single cell data from YFP lineage tracing experiments and Sox2-eGFP experiments were combined into one data set, a strategy designed to maximize the representation of cell states along the developmental trajectories. Sequencing data from a total of 687 cells (542 from YFP lineage-traced cells and 145 from Sox2-eGFP-expressing cells) remained after filtering based on various quality control metrics (Figure 1E).
Clustering and Assignment of Differentiating Cells to Branching Cell Lineages
Resampling-Based Ensemble Clustering (RSEC) was applied to the first 50 principal components of the expression matrix to generate stable and tight clusters, yielding a final repertoire of 13 clusters (Figures S1 and S2; see STAR Methods). To visualize cellular heterogeneity, we projected the data onto two dimensions via t-distributed Stochastic Neighbor Embedding (t-SNE; van der Maaten and Hinton, 2008), which confirms that our clustering procedure led to well-defined, distinct groups (Figure 2A). Preliminary cell type assignments were made based on the expression of known and/or validated marker genes for HBCs, GBCs, immediate neuronal precursors (INP1-3), immature and mature olfactory sensory neurons (iOSNs and mOSNs), immature and mature sustentacular cells (iSus and mSus), and microvillous cells (MV1 and MV2) (Figure 2B and S2; see STAR Methods for details). Interestingly, two clusters (ΔHBC1, ΔHBC2) contain cells in which the canonical HBC stem cell markers Trp63, Krt5, and Krt14 appear to be variably down-regulated from cell to cell (Figure S2), suggesting the existence of at least one transition state in which HBCs first begin to differentiate.
Figure 2. Statistical Analysis of Single-Cell RNA-Seq Data Predicts Distinct Cell States and Branch Points in the Olfactory Stem Cell Trajectory.

(A, B) t-SNE plot (perplexity = 10) based on the 500 most variable genes shows the separation of the cells into discrete groups congruent with the clustering. Cluster medoids are displayed as larger circles with initial assignments of cluster identity based on the expression of a small number of marker genes (B). (C) t-SNEs as in (A,B), colored by experimental condition. Differentiation is asynchronous, but cells from the later lineage tracing time-points and the Sox2-eGFP+ cells contribute to more differentiated cell types. (D, E) Three-dimensional representation of single cell gene expression profiles based on principal component analysis (D); cells are colored by cluster. Slingshot predicts an early bifurcation in the lineage trajectories of the neuronal (orange) and sustentacular cell (magenta) lineages whereas the MV lineage (blue) is predicted to branch later off of the neuronal lineage from the GBCs (E). See Figure S2.
Mapping transcriptional changes as cells transition from stem cells to specialized cell types is essential for understanding the mechanisms regulating cell and tissue differentiation. In differentiating lineages, cells are thought to undergo gradual transcriptional changes, where the relationship between states can be represented as a continuous lineage dependent on an underlying spatial or temporal variable. This representation, often referred to as pseudotemporal ordering (Trapnell et al., 2014), can provide a basis for understanding how and when cell fate decisions are made. The task of assigning and ordering cells in a lineage in the olfactory HBC stem cell niche is complicated by the requirement to accommodate multiple, branching cell fate trajectories that give rise to the multiple cell types of the lineage. To address this problem, we applied Slingshot, a statistical framework for inferring branching lineage assignments and developmental distances (STAR Methods). Three distinct trajectories were identified, each starting from the resting HBC stage and leading to the three defined mature cell types (Figure 2D,E). All three trajectories were predicted to pass together through the two transitional HBC stages, at which point the first branching in the lineage occurs. One path leads to immature and then mature sustentacular cells (magenta). The other path connects transitional HBCs to GBCs, from which the remaining two lineages diverge: one to form microvillous cells (blue) and one to form olfactory sensory neurons (orange). The latter trajectory passes through GBCs, three discrete stages of immediate neuronal precursors (INP1-3), and immature olfactory sensory neurons (iOSN), before concluding in mature olfactory sensory neurons (mOSN).
Developmental Ordering of Cells in the Neuronal and Sustentacular Cell Lineages
We next sought to order all cells and analyze transitions in their transcriptional states as they differentiate to become sustentacular cells and olfactory sensory neurons. Slingshot assigned developmental positions of cells along the lineage trajectory (analogous to the concept of pseudotime in Trapnell et al., (2014)) by orthogonal projection of each cell’s principal coordinates onto its respective curve (displayed as one dimensional plots in Figure 3A,C). From this analysis it is evident that the transition from ΔHBC2 to GBCs entails a relatively large jump in gene expression space. Similarly, there is a larger gap between the INP1 and INP2 stages compared to the other transitions following the GBC stage in this lineage. Such jumps in developmental distance may represent major transcriptional changes in which distinct networks of genes are turned on or off.
Figure 3. Patterns of Coordinated Gene Regulation in the Neuronal and Sustentacular Cell Lineages Reveal Different Strategies for Differentiation.

(A, C) Developmental distance of cells within each lineage as inferred by Slingshot. Mean +/− standard deviation of developmental distance is indicated for each cluster. (B, D) Heatmaps display the average scaled expression profile for each gene cluster (numbered at the left of each row) with cells (columns) ordered according to their developmental positions within the neuronal (B) and sustentacular cell (D) lineages. There are numerous step-like transitions in the neuronal lineage but fewer, wave-like changes in the sustentacular cell lineage. The lower row in each heatmap represents a set of 40 cell cycle (CC) genes. See Figures S3 and S4.
To gain insight into the coordinated patterns of gene expression that underlie the cell fate transitions in the neuronal and sustentacular cell lineages, we identified and clustered the most differentially expressed genes within each lineage (Figure S3 and Table S1; see STAR Methods). The average scaled expression profiles for the gene clusters in the neuronal and sustentacular cell lineages are displayed using heatmaps in Figure 3B,D, presented in developmental order according to the predictions made by Slingshot. This comparison highlights the dramatic difference in coordinated gene expression through developmental progression in the two lineages. In the neuronal lineage many modules of genes are transiently turned on and off, patterns that contrast with the more gradual wave-like changes in gene expression exhibited by the sustentacular lineage. Moreover, there is a longer distance traversed in the neuronal lineage both in terms of the number of distinct cell states and the number of genes showing significant changes at each transition (Figure S3 and Table S2).
Consistent with recent studies (Hanchate et al., 2015; Saraiva et al., 2015; Scholz et al., 2016; Tan et al., 2015), our analysis further reveals that low level expression of multiple odorant receptor (OR) genes per cell commences at the INP2/3 stage, culminating with high level expression of a single OR per cell in immature olfactory sensory neurons (Figure S4).
Proliferation Is Restricted to the GBC/Neuronal Lineage
Numerous studies have demonstrated that GBCs represent the major proliferative progenitor cell in the uninjured olfactory epithelium (Caggiano et al., 1994; Graziadei and Graziadei, 1979b; Schwob et al., 1994). We therefore examined the expression of cell cycle-associated genes (Kowalczyk et al., 2015; Whitfield et al., 2002; Figure S3) in order to determine whether and where expansion occurs in the neuronal and sustentacular cell lineages. As judged by expression of cell proliferation markers, GBCs and INP1 cells comprise the two actively proliferating progenitor cell types in the neuronal lineage (Figures 3B and S3). In striking contrast, cells progress through the sustentacular cell lineage without a concerted up-regulation of cell proliferation markers (Figures 3D and S3). Thus, HBCs appear to transdifferentiate into sustentacular cells through a process that does not require cell division.
Lineage Tracing In Vivo Validates Predicted Branch Points in the Olfactory Stem Cell Trajectory
A number of testable predictions can be made from the in silico branching lineage assignments and developmental ordering of differentiating olfactory cells shown in Figures 2 and 3. First, the initial branching of the sustentacular cell and neuronal lineages at the transitional HBC stage and in the absence of active cell proliferation suggests that when stimulated to differentiate, each HBC adopts a unique cell fate – i.e., GBC vs. sustentacular cell. In this scenario, the multipotency of the population of HBCs is driven by independent unipotent differentiation events that occur at the single cell level. To test this prediction, we performed clonal lineage tracing in vivo. HBCs were labeled and stimulated to differentiate by induction of Cre recombinase activity with doses of tamoxifen adjusted to elicit sparse activation of HBCs in Krt5-CreER; Trp63lox/lox; Rosa26Confetti mice. Animals were sacrificed at 7 and 14 days after tamoxifen induction and expression of fluorescent protein reporters encoded by the Rosa26Confetti locus was assessed using an anti-GFP antibody. Representative examples of labeled clones are shown in Figure 4A–D. Consistent with Slingshot’s prediction that individual HBCs initially give rise to single lineages, at 14 days of differentiation 90 of 99 clones contain cells representing either the combined neuronal/microvillous cell lineage (80, comprising 13 GBC-containing, 56 neuron only, 1 neuron + Bowman’s gland, 9 neuron + microvillous cell only, and 1 microvillous cell only) or sustentacular cells (10), but not cells from both lineages (Figure 4E). Clones containing a mixture of sustentacular cells and neurons comprise the remaining 9 clones. Only one labeled Bowman’s gland was detected among the 99 clones scored for this analysis; the scarcity of lineage-traced Bowman’s glands mirrors their apparent absence in our analysis of the single-cell RNA-seq data, suggesting that these cells arise only rarely from HBCs in the normal, uninjured epithelium. Similar results were obtained from clones scored at 7 days of differentiation (Figure S5).
Figure 4. Clonal Lineage Tracing In Vivo Validates Branching Lineage Assignment Predictions.

(A–D) Example images of clones from Krt5-CreER; Trp63lox/lox; Rosa26Confetti transgenic animals analyzed 14 days following tamoxifen induction of the CreER driver: neuronal only (A), neuronal and sustentacular (arrow) (B), sustentacular cell only (C), and neuronal and microvillous cell (arrowhead) (D). (E) Most clones represent unipotent differentiation events, supporting the prediction that the lineages bifurcate early: the majority of clones contain only cells in the GBC/neuronal lineage, and just over half of all sustentacular cell-containing clones are composed of only sustentacular cells. Almost all microvillous cells are found together with neurons. One out of 99 clones contained a labeled Bowman’s gland together with neurons. (F, G, H) Clone size distribution. The number of neurons in neuron-containing clones (F) is larger, validating that the neuronal lineage contains amplification stages whereas the sustentacular cells are usually present as single cells (G). Microvillous cells are usually found as singlets and typically are found in clones together with neurons (H). (I) Percentage of Krt5-CreER; Trp63lox/lox; RosaeYFP lineage-traced cells co-labeled with activated CASPASE3 at 24, 48 and 96 hours and 7 days following tamoxifen injection. The low frequency of apoptosis (0–0.6 %) in the HBC lineage is inconsistent with cell death as a mechanism to generate clones containing single cells. (J, K, L) Ascl1-CreER; Rosa26eYFP lineage tracing at 21 days following tamoxifen induction shows that although Ascl1-positive GBCs usually form neurons (~ 98%) (J), they occasionally form microvillous cells (K). The percentage (mean +/− SD) of each cell type formed from lineage-traced cells (1072 cells from three animals) is summarized in (L). Scale bars, 25 microns. See Figure S5.
A second hypothesis based on our analysis with Slingshot is that sustentacular cells are formed through direct fate conversion from HBCs without cell division, whereas the neuronal lineage expands via proliferation of GBC and INP1 progenitors. Indeed, using our sparse lineage tracing strategy, we observed a marked disparity in the numbers of neurons and sustentacular cells per clone. At 14 days of differentiation, neuron-containing clones contained 13.5 +/− 1.0 neurons (mean +/− SEM) (Figure 4F), whereas sustentacular cells are present at one to two cells per clone (1.3 +/− 0.1 cells; Figure 4G). The average number of neurons per clone increases from 7 days (8.8 +/− 1.0 cells/clone) to 14 days, while the number of sustentacular cells per clone remains essentially unchanged over this interval (1.2 +/− 0.1 cells/clone at 7 days; compare Figure 4F,G to Figure S5). Moreover, most sustentacular cell-containing clones contain only a single sustentacular cell, confirming the assignment of the first major branch point in the HBC-derived lineage to an early, non-proliferative transitional HBC stage. Complementing the predicted lineage trajectories, these observations provide strong evidence that HBCs differentiate directly into sustentacular cells in the absence of cell division. Alternatively, the presence of a single cell per clone may reflect proliferation of a sustentacular cell progenitor followed by apoptotic cell death, leaving on average a single mature daughter cell per differentiating stem cell. However, the very low frequency of activated Caspase3 expression in YFP lineage-traced cells (0–0.6%; Figures 4I and S5) is inconsistent with apoptosis as a significant contributor in the differentiation of mature cell types from HBC stem cell (Fletcher et al., 2011). The presence of two sustentacular cells in a minority fraction of clones may reflect the proliferative capacity of the sustentacular cells themselves (Weiler and Farbman, 1998) and/or the occasional proliferation of resting HBCs (Fletcher et al., 2011) or transitional HBCs. Consistent with these possibilities, rare proliferating cells can be observed in both the HBC and late sustentacular cell stages (Figure 3D).
The third major prediction of our branching lineage assignment is that microvillous cells and olfactory sensory neurons arise from a common sub-lineage that branches at the GBC stage to give rise to these two cell types. In support of this prediction, nine of ten clones containing microvillous cells also contain neurons (Figure 4E). Microvillous cells are rare in comparison to olfactory sensory neurons (Hansen and Finger, 2008; Jia et al., 2013; Ogura et al., 2011; Yamaguchi et al., 2014); this difference is reflected by the small number of microvillous cell-containing clones detected (12% of neuron-containing clones), as well as the low number of microvillous cells present in any given microvillous-containing clone (1.2 +/− 0.1; Figure 4H). These clonal lineage tracing results are complemented by a similar, non-clonal lineage tracing analysis using a knockin Ascl1-CreER driver (Kim et al., 2011), which recapitulates Ascl1’s expression in postnatal GBCs (Figure S4) (Manglapus et al., 2004). Cells lineage-traced 3 weeks after tamoxifen activation of the Ascl1-CreER driver comprise mainly neurons (98% of ~1000 cells scored over 3 animals) (Figure 4J and L), with microvillous cells representing ~1% of the labeled cells (Figure 4K and L). In addition, cells of the Bowman’s gland — identified by their expression of Sox9 and assembly into ducts contiguous with submucosal acini — were also lineage-traced by the Ascl1-CreER driver (Figure S5), suggesting that cells of the Bowman’s gland arise from GBC progenitors. We failed to detect any sustentacular cells labeled by the Ascl1-CreER driver, supporting the prediction that a second branching of the lineage occurs at the GBC stage to give rise to numerous olfactory sensory neurons and occasional microvillous cells, but not sustentacular cells.
Coordinated Transcription Factor Networks Associated with Lineage Progression
To gain a more comprehensive view of the combination of factors that drive HBCs to form the two major OE lineages, we identified the differentially expressed transcription factors in the neuronal and sustentacular cell lineages and displayed their expression profiles by developmental order in their respective lineages (Figure 5A,B and Table S3). The dynamic, step-like changes in gene expression in the neuronal lineage (Figures 3 and S3) are also observed in the expression of transcription factors. Fewer transcription factors show dynamic expression in the sustentacular lineage compared to the neuronal lineage (Figures 5A,B and S6).
Figure 5. Coordinated Transcription Factor Networks Associated with Lineage Progression.

(A, B) Heatmaps of the top DE genes for the neuronal (A) and sustentacular cell (B) lineages. (C, D) Connectivity graphs of the most correlated DE transcription factors for the neuronal (C) and sustentacular cell (D) lineages colored by the cluster in which expression is highest; the size of each node indicates magnitude of expression. (E) Connectivity graph for the neuronal lineage colored by rank across the clusters within the lineage. For each transcription factor, we computed the average expression in each cluster and color-coded the corresponding node to indicate the cluster with the highest average expression. See Figure S6 and Table S3.
We next constructed a co-expression network by correlating differentially expressed transcription factor genes and connecting those with high correlation (Figures 5C,D and S6). The neuronal lineage is composed of three main subnetworks of transcription factors whose expression is enriched either in resting and transitional HBCs, GBCs and INP1 cells, or later-stage neuronal precursors and neurons (Figure 5C, E). In contrast, only one network of transcription factors in the sustentacular cell lineage is identified using the same criteria (Figure 5D), consistent with our observation that the sustentacular cells are closely related to the HBCs at the transcriptional level. Interestingly, the AP1 transcription factors (Karin et al., 1997) – previously demonstrated to regulate keratinocyte differentiation (Eckert et al., 2013) – are enriched in the resting and transitional HBCs (Figure 5 and Table S3), suggesting that similar transcriptional networks may be used to control differentiation in these two divergent epithelial stem cell niches.
Activation of Canonical Wnt Signaling is Necessary and Sufficient to Drive HBC Differentiation into Olfactory Sensory Neurons
Having identified discrete stages in the differentiating olfactory lineages, we next sought to identify specific genes and signaling networks that might govern critical cell state transitions that occur as HBCs differentiate. We applied gene set enrichment analysis (GSEA) with rotation tests (Ritchie et al., 2015) to identify pathways that are enriched in cell clusters based on single-cell RNA-seq data (Figure S6 and Table S4). Among the top hits found in resting HBCs is the canonical Wnt signaling pathway (Figures 6A and S6), which has been shown to regulate stem cell dynamics in other niches (Clevers et al., 2014). Two repressors of canonical Wnt signaling, Dkk3 and Sfrp1 (Kawano and Kypta, 2003), are enriched in resting HBCs and subsequently down-regulated upon HBC differentiation (Figure S6), suggesting that autocrine or paracrine inhibition of Wnt signaling may promote HBC quiescence. To test this hypothesis, we activated Wnt signaling in resting HBCs using the Krt5-CreER driver to conditionally express an activated form of β-catenin (Ctnnb1lox(ex3) allele) (Harada et al., 1999). Compared to controls (Figure 6B), activation from one copy of the β -cateninlox(ex3) allele resulted in the appearance of dysmorphic, globose-like basal cells reminiscent of activated HBCs (Fletcher et al., 2011), as well as occasional differentiated neurons (Figure 6C). Strikingly, a genetic interaction was observed between Trp63 and Wnt signaling in a sensitized trans-heterozygous β -cateninlox(ex3)/+; Trp63lox/+ background, in which HBCs differentiated in a manner similar to conditional ablation of Trp63 alone (compare Figure 6D,E). In this case, however, HBCs predominantly formed neurons and proliferative GBC progenitors, and very rarely produced sustentacular or microvillous cells (Figure 6D,J). Furthermore, neurons derived from these Wnt-activated HBCs failed to fully migrate apically, consistent with the established role of Wnt signaling in stimulating GBCs to differentiate while impeding subsequent neuronal maturation (Chen et al., 2014; Wang et al., 2011).
Figure 6. Wnt Signaling Is Necessary and Sufficient for HBC Activation to Form Neurons.

(A) Wnt signaling pathway gene expression in the resting HBC cluster, color-coded according to log2-fold-change (log2FC) between the resting HBC cluster and the other neuronal lineage clusters (“one vs. all”, see STAR Methods). (B–K) The indicated alleles were on a Krt5-CreER; Rosa26eYFP transgenic background. (B–E, J) Conditionally activating Wnt signaling via removal of exon 3 of β-catenin (C) results in HBCs that change shape and begin to differentiate (compare with cells from heterozygous Trp63lx/+ controls, shown in panel B). Coupled with the removal of one allele of Trp63 (D), activation of Wnt signaling in HBCs induces proliferation and differentiation into neurons, similar to the Trp63lx/lx phenotype (E). (J) Quantitation of the effect of activating β-catenin in the Trp63lx/+ background, n = 4 (Trp63lx/+), n = 3 (Trp63lx/+; β-cateninlxex3/+) at 14–21-days post tamoxifen (DPT); mean is shown in red; p-value = 0.01. (F–I, K) Quiescent HBCs (F) differentiate into neurons (assessed by suprabasal position, cell shape, and lack of SOX2 expression) and support cells after conditional knockout of Trp63 (G). Inhibition of Wnt signaling by conditional knockout of β-catenin in the Trp63lx/lx background decreases the capacity of HBCs to form neurons (H, I, K). (K) There are fewer neurons (assessed by suprabasal position, cell shape, and lack of SOX2 expression) in the double knockout (14 DPT), n = 3 for each genotype, p-value = 0.04. See Figure S6.
To test whether Wnt signaling is necessary for activation of HBC differentiation, we conditionally ablated a floxed β -catenin allele (Ctnnb1lox/lox) in HBCs using the Krt5-CreER driver. Resting HBCs appeared normal in the absence of β-catenin (Figure 6F). Whereas knockout of Trp63 alone resulted in HBC differentiation mostly into neurons (Figure 6G), in the Trp63 and β -catenin double knockout, HBCs produce fewer differentiated cells and had a diminished capacity to produce neurons (Figure 6H,I,K; Figure S6); most of the differentiated cells were sustentacular cells or non-neuronal SOX2+ cells. Taken together, these data indicate that activation of canonical Wnt signaling is both necessary and sufficient to drive the transition of HBCs from resting to an activated neurogenic state in the uninjured epithelium.
Discussion
In the present study, we developed an integrated approach using in silico analysis of single-cell RNA-seq data and in vivo lineage tracing to illuminate the cell fate potentials of individual olfactory stem cells and the locations of branch points in the olfactory lineage trajectory. Our combined analysis identified the trajectories that produce three out of the four major cell types in the olfactory epithelium: the olfactory sensory neurons, microvillous cells and sustentacular cells. Lineage tracing alone further revealed the origins of cells of the Bowman’s gland. One sub-lineage gives rise to the niche’s proliferative GBCs, which in turn generate olfactory sensory neurons, microvillous cells and cells of the Bowman’s gland. The other sub-lineage generates sustentacular cells through a differentiation process characterized by fewer and more gradual transitions that occur in the absence of cell division. The differences in gene expression and cell state transitions in the neuronal and sustentacular cell lineages are likely the result of similarly divergent patterns of coordinated transcription factor expression in the respective lineages. It is possible that additional and more subtle transitions in cell states exist but were not resolved by the present analysis, which was based on sequencing <1000 cells. Nonetheless, our approach enabled an analysis of the olfactory HBC stem cell’s fate potential and the location of branch points in the lineage at a level of detail not possible by either in vivo lineage tracing or single-cell RNA-seq alone, and serves as a model for elucidating complex lineage trajectories in other stem cell niches.
Transitional HBCs Represent a Discrete Cell State in Which Neuronal vs. Sustentacular Cell Fates Are Specified
Our analysis of single-cell RNA-seq data allowed for the identification of previously uncharacterized transitional or activated states that HBCs first enter upon differentiation, during which one of two cell fates is adopted. This first transition – comprising at least two states (ΔHBC1 and ΔHBC2) – is manifested by a subtle shift in gene expression, underscoring the power of single-cell RNA-seq to detect cell state changes that would otherwise elude detection by more conventional approaches based on the expression of a small number of known marker genes. Interestingly, the down-regulation of the transcription factor Trp63 is both necessary (Schnittke et al., 2015) and sufficient (Fletcher et al., 2011) to stimulate HBCs to differentiate. However, transitional HBCs exhibit highly variable expression of Trp63 and other genes normally associated with resting HBCs (e.g., Krt5, and Krt14; Figure S2), suggesting that minor perturbations in Trp63 expression are sufficient to elicit a change in cell state. Together these observations are consistent with a model in which activated HBCs comprise a metastable state that allows for their transition towards a variety of cell fates. We posit that transitional HBCs represent a window along the developmental trajectory during which competing regulatory networks promote progression down one lineage versus another. What are the possible mechanisms underlying such a competition?
We propose a model in which the default state of HBC stem cells is to form sustentacular cells. Accordingly, a signal would be required to repress the default state and drive the HBCs to differentiate into GBCs and subsequently into neurons. Previous studies have established the role of Wnt signaling in GBC proliferation and neurogenesis during injury-induced regeneration in the olfactory epithelium (Chen et al., 2004; Wang et al., 2011). Here we demonstrate that Wnt signaling is both necessary and sufficient earlier in the lineage for HBC activation and specification of GBC neural progenitors under uninjured, steady state conditions. The ability of Wnt signaling to activate quiescent HBCs to differentiate into proliferative neuronal progenitors is consistent with its role in stem cell activation and cell fate specification in other stem cell niches (Choi et al., 2013; Clevers et al., 2014). Interestingly, HBCs are enriched for NFκB signaling components (Table S4), whereas sustentacular cells strongly express IL-33, which is localized to the nucleus of sustentacular cells (Figure S5). In addition to its proposed role as an inflammatory cytokine, IL-33 has been shown to antagonize the transcription factor RelA/p65, an NFκB effector (Ali et al., 2011). Future studies will be required to test the hypotheses that nuclear IL-33 promotes the sustentacular cell fate by antagonizing NFκB signaling.
Multipotency of HBCs at the Population Level Reflects Individual Unipotent Cell Fate Decisions
How do multiple cell types arise from the population of olfactory stem cells? We found that individual HBCs mostly generate clones of cells of a single cell type, with approximately 80% consisting of only cells in the GBC lineage and 10% containing only sustentacular cells (Figure 4). These findings demonstrate a common thread with several other stem cell niches in which individual stem cells adopt singular cell fates that, in aggregate, underlie multipotency at the population level (Snippert et al., 2010).
While the majority of individual HBCs appear to restrict their cell fate choices to a single lineage, about 10% of HBC-derived clones contain a single sustentacular cell amid a group of neurons. What can account for the apparent multipotency of individual HBCs in these cases? Perhaps GBCs occasionally give rise to sustentacular cells in addition to neurons and microvillous cells. However, we failed to detect any sustentacular cells out of ~1000 cells lineage-traced using the GBC-specific Ascl1-CreER driver under steady state conditions, although it is possible that a rare GBC earlier in the lineage may occasionally give rise to sustentacular cells. Alternatively, the occurrence of mixed neuronal/sustentacular cell clones in our analysis could reflect plasticity at the transitional HBC stage, such that a given activated stem cell undergoes a self-renewing cell division, leaving its two daughter cells free to adopt either cell fate independently. This interpretation dovetails with the suggestion that transitional HBCs are metastable based on their variable expression of Trp63, which plays a critical role in maintaining HBCs in the resting, self-renewing state (Fletcher et al., 2011). It is also possible that the multipotent clones reflect symmetric self-renewing HBC mitoses prior to differentiation. Indeed, we observed elevated expression of cell cycle-associated genes in the occasional resting and transitional HBC (Figure 3), in agreement with our previous observation that a small percentage of resting HBCs express the proliferative marker protein Ki67 (Fletcher et al., 2011). Whatever the case, our observations are consistent with the idea that certain cell fate decisions may in fact remain reversible during a limited period early in the trajectory.
Direct Conversion of HBC Stem Cells into Sustentacular Cells
Our analysis also reveals direct conversion of quiescent HBCs to sustentacular support cells without cell division, an unusual mode of stem cell differentiation. In contrast, olfactory neurogenesis involves an expansion through proliferative GBC and INP intermediates. Thus, HBCs generate cells of the two diverging lineages using dramatically different strategies. Our analysis of gene expression at the single cell level further indicates that the developmental distance traversed in the sustentacular cell lineage is shorter than the distance covered in the generation of olfactory sensory neurons (Figures 3 and S3). In addition to expanding the number of cells in this lineage, proliferation of GBCs and INP1 cells may also activate transcriptional networks by inducing larger scale, cell cycle-associated chromatin remodeling (Ma et al., 2015).
Direct reprogramming of cells to replace damaged or diseased cells in a tissue has enormous therapeutic potential. Unlike many other adult stem cells, HBCs are not actively dividing, a characteristic that allowed us to disentangle fate determination of HBC daughter cells from cell division. Moreover, the initiation of HBC differentiation into mature cell types is triggered not by overexpression of a factor but rather by removing one (Trp63). That removing factors would be an effective strategy for reprogramming is also suggested by the conversion of fibroblasts to cardiomyocyte-like cells by miRNA expression (Jayawardena et al., 2012) and pancreatic alpha cells to beta-like cells by inhibiting Arx (Courtney et al., 2013). Our findings provide an alternative way to view cell fate transformation in vivo and strategies for inducing transdifferentiation of cells for therapeutic applications.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John Ngai (jngai@berkeley.edu).
Experimental Model Details
Transgenic Mice
Mice containing the following transgenes or modified alleles were used in this study: Krt5-CreER(T2) driver (Indra et al., 1999), Ascl1-CreER knock-in allele (Kim et al., 2011), Trp63lox/lox conditional knockout allele (Mills et al., 2002), Sox2eGFP knock-in reporter allele (Arnold et al., 2011), the conditional constitutively activated Ctnnb1lxEx3/+ allele (Harada et al., 1999), Ctnnb1lox/lox conditional knockout allele (Brault et al., 2001), Rosa26eYFP reporter (Srinivas et al., 2001), and Rosa26Confetti reporter (Snippert et al., 2010). All experiments were begun on animals at 3–4 weeks of age (P21–P28). We used both male and female mice in our studies; all mice were on a mixed C57BL/6 and 129 background and were assumed to be of normal immune status. Information about sex, age and genotype of animals used in RNA sequencing experiments is included as metadata in GEO record # GSE95601. Animals were housed and maintained according to IACUC guidelines.
Method Details
Fluorescence-Activated Cell Sorting (FACS)
We employed a two-pronged approach to label and isolate cells from the postnatal mouse olfactory epithelium. In one approach, the Krt5-CreER and the Rosa26eYFP transgenes were combined with either the wild-type Trp63 allele (Trp63+/+) or the conditional knockout of Trp63 (Trp63lox/lox). This approach allowed us to label HBCs and lineage-trace their descendants. We collected HBCs that were wild type for Trp63 and the HBC lineage following tamoxifen-induced conditional ablation of Trp63 after 24 hours, 48 hours, 96 hours, 7 days, and 14 days. In the other approach, the Sox2eGFP transgenic was used. This transgene exhibits GFP expression in a pattern faithful to endogenous Sox2 expression in the OE (HBCs, GBCs, sustentacular and microvillous cells), but the GFP perdures in the immediate neuronal precursors and immature neurons.
In the HBC lineage tracing experiments, Krt5-CreER; Rosa26eYFP/eYFP and Krt5-CreER; Trp63lox/lox; Rosa26eYFP/eYFP mice were injected once with tamoxifen (0.25 mg tamoxifen/g body weight) at P21–23 days of age and sacrificed at the specified times after injection (Figure 1). For each experimental time point, the olfactory epithelium was surgically removed, and the dorsal, sensory portion was dissected and dissociated. The dissociation protocol was similar to that used previously (Fletcher et al., 2011), except that papain dissociation was for 25 minutes, and tissue from each animal was processed individually in approximately one mL volume. First, approximately 30 units of papain was dissolved in 5 mL of Neurobasal medium with 1.5 mM Cysteine and 1.5 mM ethylenediaminetetraacetic acid (EDTA) and incubated at 37° C for at least 30 minutes to activate the papain. Then, after dissection of the olfactory epithelium into Neurobasal medium, the tissue samples were added to an equal volume of the activated papain solution along with DNAase I (100 units/mL) and allowed to incubate with gentle nutation for 25 minutes at 37° C. Following papain digestion and dissociation, the tissue was washed multiple times in phosphate buffered saline with 10% fetal bovine serum (PBS-FBS). Prior to loading on the cell sorter, cells were kept in PBS-FBS, and cells were sorted on a BD Influx sorter into PBS-FBS. A negative control (a littermate of the same genotype not injected with tamoxifen) was used to ensure proper gating. Propidium iodide was used to identify and select against dead or dying cells; only viable YFP+ cells were collected. The same dissection and dissociation protocol was followed to purify the Sox2eGFP transgene-labeled cells, except that HBCs were depleted using a phycoerythrin (PE)-conjugated ICAM1 antibody. Upon sequencing several replicates, we noticed that we were collecting many sustentacular cells and few cells of the neuronal lineage from Sox2eGFP animals. We therefore identified two cell surface proteins expressed in the sustentacular cells (SCARB1 and F3) and used immunolabeling with antibodies directed against these proteins to deplete the population of sustentacular cells.
In terms of representation, YFP lineage-traced cells contribute to clusters representing all predicted cell types. Sox2-eGFP cells populate all clusters except those predicted to comprise the earliest HBC stages, immature sustentacular cells and mature olfactory sensory neurons (Figures 2 and S2). These latter observations are consistent with the intentional depletion of HBCs by FACS, Sox2’s documented expression in proliferating neuronal progenitors and sustentacular cells (Guo et al., 2010), and the rarity of sustentacular cell differentiation from HBCs under normal conditions. The contribution of both YFP lineage-traced cells and Sox2-eGFP cells to clusters representing mature and differentiating cells in all identified lineages suggests that these clusters reflect bona fide cell types and not technical artifacts (e.g., batch effects) or other anomalies caused by the different genetic strategies used for cell isolation.
Cell Capture and Single-Cell RNA Sequencing
Labeled cells from the olfactory epithelium were subjected to single-cell RNA-seq. Each FACS collection was considered a biological replicate, and at least two biological replicates were collected for each experimental condition. The Fluidigm C1 system was used to capture single cells, lyse them, and produce cDNA (Wu et al., 2013). For each replicate, a litter of animals were given the same treatment, and each transgenic animal was dissected and processed individually prior to loading on the cell sorter. When possible, all cells were obtained from one animal, but if one animal did not provide the minimum number of cells necessary to load the C1 chip (approximately 2000 cells), then cells from additional animals were sorted into the collection tube. Cells from no more than three animals were pooled in any given FACS purification. Each C1 run was considered a single batch. Upon loading of the C1 chip, each capture site was visually inspected at 100× magnification for fluorescence, debris, and doublets or multiple cells. The following were excluded from future analysis: (1) any capture site scored as having two or more cells (doublets or multiplets), (2) any capture site that included additional debris, and (3) any capture site that did not have an apparently intact, fluorescent cell. The standard Fluidigm C1 Single-Cell auto prep system protocol for cell lysis, cDNA synthesis, and amplification was followed. This incorporates the Clontech SMART-Seq Ultra Low Input RNA reagents (Clontech SMARTer Kit designed for the C1 System) to produce and amplify cDNA. After processing on the C1 chip, 7 μL cell harvest buffer (Fluidigm) was added to each sample, resulting in an approximate final volume of 10 μL. For quality control purposes, any cell with less than 1.7 ng cDNA in 10 μL final volume (quantified using a Qubit fluorometer) was excluded. Illumina Nextera tagmentation-based sequencing library synthesis (Nextera XT DNA Sample Preparation Kit) was performed using Nextera v2 index oligos (Nextera XT DNA Sample Prepration Index Kit). Library size was selected using AMPure XP (Beckman Coulter) beads and confirmed using an Agilent Bioanalyzer. Indexed, single-cell libraries were sequenced on Illumina HiSeq 2500 sequencers to produce 50 nt single-end reads, except for two paired-end read runs (121 cells). Cells were sequenced in multiplex, with approximately 192 cells per lane for most runs. Prior to cell filtering, average sequencing depth was 1.36 million reads per cell (range 0.166 to 4.27 million).
Clonal Lineage Tracing
Krt5-CreER; Trp63lox/lox; Rosa26Confetti transgenic mice were used for clonal lineage tracing of the HBC lineage. A dose-response analysis was performed to determine an optimal dose of tamoxifen to achieve sparse labeling: 0.025 to 0.05 mg tamoxifen/g body weight administered by intraperitoneal injection. At this dose, at 14 days post tamoxifen administration there were only eight occurrences among 127 (6.3%) clones that expressed different reporters (e.g. mCFP and cYFP) with an edge-to-edge distance of 40 microns or less. Because the reporters localize to different compartments, we could distinguish close or overlapping clones expressing different reporters. We expect that the same percentage of clones were within 40 microns of a clone expressing the same reporter; therefore, we estimate that no more than 7% of our scored clones are contaminated by cells derived from multiple independent labeling events. The three animals with the largest and most comparable number of clones were examined for each clonal lineage tracing time point (7 and 14 days post tamoxifen injection, Figure S5). Tissue was fixed overnight (~16 hours), embedded in tissue freezing medium and froze, and then sectioned on a cryostat at 40 micron thickness to allow sampling of the majority if not entirety of each clone. An antibody to GFP was used to visualize the membrane CFP (mCFP), cytosolic YFP (cYFP) and nuclear GFP (nGFP) reporters encoded by the Rosa26Confetti locus. Tissue was also labeled with an antibody to SOX2 to visualize the stem and progenitor cells, as well as the sustentacular and microvillous cells. We visualized SOX2 immunolabeling with an Alexa-568 conjugated secondary antibody (Invitrogen). We did not visualize the RFP reporter signal; there was no interference because the antigen retrieval necessary for optimal SOX2 visualization extinguished the RFP signal. We observed very few nGFP clones and could not judge cell morphology with its restricted localization; therefore, nGFP clones were excluded from our analysis. Identification of lineage-traced cell types was facilitated by cell morphology and position based on mCFP and cYFP expression. Olfactory sensory neurons were identified by their medially located somata and bipolar morphology highlighted by a thin apical dendrite often terminating in a singular dendritic knob and the absence of SOX2 expression. Basal progenitors were identified by position and SOX2 expression. Microvillous cells were assigned based on the more apical position of their cell bodies, tapered, brush-like apical tufts and SOX2 expression. Sustentacular cells were identified by their apical localization, branched processes that span the epithelium, columnar apical shape, and SOX2 expression. Cells of the Bowman’s gland were identified by their organization into multicellular ducts and/or sub-mucosal acini, and absence of SOX2 expression. Any clone that contained an unidentifiable cell was excluded from the analysis (28/127 for 14 DPT, 11/80 for 7 DPT), although inclusion of all clones did not alter the overall conclusions. Confocal z-stacks were obtained using a Zeiss LSM 710 or 780 confocal microscope, and images were processed and quantified using ImageJ (NIH). For a breakdown of clones by animal and reporter, see Figure S5.
Histology
Tissue was fixed at the indicated stages and time points with 4% paraformaldehyde for 16–18 hours at 4° C, washed with PBS, and decalcified in 10% ethylenediaminetetraacetic acid (EDTA) in PBS at 4° C for a minimum of 3–4 days, washed with distilled H2O, and equilibrated in 30% sucrose in PBS overnight at 4° C before mounting and freezing in tissue freezing medium (Fisher). Tissue was sectioned at 12-micron thickness on a cryostat. For immunohistochemistry, tissue sections were incubated with PBS containing 0.1% Triton X-100 with primary antibodies diluted in 10% goat or donkey serum overnight at 4° C followed by at least three 20 minute washes in PBS with 0.1% Triton X-100. Detection was performed by incubating samples with Alexa-488, 555, 568, 594, or 647-conjugated secondary antibodies at room temperature for 2 hours, followed by nuclear counterstaining. We used primary antibodies to the following antigens: SOX2, GFP, P63, NEUROD1, NTUB/TuJ1, SOX9, ICAM1, cleaved CASPASE3, SCARB1, and F3. Nuclei were counterstained with either Hoechst 33342 or DAPI, and/or tissue was visualized with brightfield illumination. Fluorescent confocal slices and z-stacks were obtained using Zeiss LSM 710 or 780 microscopes.
For RNA in situ hybridizations, tissue was fixed for a minimum of 24 hours at 4° C. Probes for RNA in situ hybridization were synthesized with either digoxygenin- or fluorescein-labeled UTP. Supplementary Table 6 includes the oligonucleotides used for PCR amplification of templates for antisense RNA probes. The T7 primer sequence was on the 5-prime end of the reverse sequence oligonucleotide, and T7 RNA polymerase was used for in vitro transcription of probes. In brief, a standard RNA in situ hybridization protocol was used: slides were incubated in a prehybridization buffer for 2 hours. Probes were hybridized overnight at 65° C followed by multiple stringent washes in low salt buffer at 65° C. Subsequently, alkaline phosphatase-conjugated anti-digoxygenin or anti-fluorescein antibodies and BCIP/NBT substrates were used to visualize the hybridized probes. For fluorescent detection, the Perkin-Elmer Tyramide Signal Amplification (TSA) kit was used with additional washes following antibody incubation and fluorophore amplification; both FITC and Cy3 conjugated fluorophores were used. Confocal slices and z-stacks of fluorescent labeling were obtained using Zeiss LSM 710 or 780 microscopes. Brightfield images of colorimetric RNA in situ hybridization were obtained on a Nikon Microphot compound scope and Leica DFC500 camera. For RNA and protein expression profiles, a minimum of biological duplicates were analyzed.
Genetic Mutant and Lineage Analysis
To compare the effects of activating and inhibiting Wnt signaling via genetic manipulation of β-catenin/Ctnnb1, we assessed at least 2 mm of olfactory epithelium and a minimum of 100 cells per animal from a minimum of three animals per experimental condition and quantified different cell-types with the ImageJ cell counter plug-in. The number of animals (n) compared is included in the figure legend to Figure 6. Differences in the percentage of suprabasal, SOX2-negative lineage-traced cells between genotypes was compared with the two-tailed Welch’s t-test assuming unequal variance between groups. For quantitation of the number of HBC lineage traced cells (YFP-positive) that were also positive for activated, cleaved CASPASE3 via immunohistochemistry, between 4.5–5.5 mm of OE was counted from each of nine animals (3 at 24 HPT, 2 each at 48H-, 96H-, and 7 DPT). YFP-positive, CASPASE3-positive, and YFP and CASPASE3 double positive cells were counted using the ImageJ cell counter plug-in. The number of animals (n) is displayed in Figure 4 and Figure S5. For quantitation of the Ascl1 lineage using Ascl1CreER; Rosa26eYFP transgenic lineage tracing, a total of approximately 1000 lineage traced cells were scored from three biological replicates at three weeks following tamoxifen administration. We did not employ randomization or blinding in our analyses.
Quantification of the clonal lineage tracing experiment was performed as described in the main text, the figure legend to Figure 4 and S5, and the methods section on clonal lineage tracing above. We excluded clones that had an undetermined cell type, leaving 99 clones across three animal replicates for the 14 DPT time-point and 69 clones across three animal replicates for the 7 DPT time-point as presented in Figure 4 and S5. A breakdown of the number and types of clones scored by animal are included in Figure S5.
RT-qPCR
Olfactory epithelium was dissected and dissociated and cells were purified with fluorescence-activated cell sorting (FACS). RNA was extracted with Trizol according to the manufacturer’s (Invitrogen) instructions with an additional chloroform extraction and additional ethanol washes. cDNA was synthesized with SuperScript RTIII reverse transcriptase (Invitrogen) at 50 °C. qPCR was performed on a BioRad CFX96 thermocycler. Three technical replicates were averaged, and then ΔCt values were calculated. Gene expression was normalized to Gapdh, and the log2 fold difference in gene expression between conditions is presented. Supplementary Table 6 includes the oligos used for primers.
Wnt Pathway Visualization
PathVisio (Version 3.2.4, pathvisio.org; (Kutmon et al., 2015) was used for pathway visualization. The Mm_Wnt_Signaling_Pathway_and_Pluripotency_WP723_89312.gpml pathway from the WikiPathways Collection (Kutmon et al., 2016) was modified to only include genes that were included in our post-filtering data set. Additionally, a few additional genes were added and removed to highlight the canonical aspect of Wnt signaling. Genes were colored by log2-fold change values derived from one versus all differential expression (DE) (Table S1) performed using clusterExperiment, which uses limma. The modified pathway and the DE data are provided in our GitHub repository.
Quantification and Statistical Analysis
Single-Cell RNA-Seq Alignment and Filtering
Reads were aligned to the GRCm38.3 (mm10, patch release 3) mouse genome assembly with Tophat2 (Version 2.1.1; Kim et al., 2013), and low quality reads were removed with Trimmomatic (Version 0.3.2; Bolger et al., 2014). We used RefSeq transcript annotations, which were modified to contain sequences of specific genes used in our analysis (e.g. CreER and eGFP), and we counted the number of reads aligning to every gene (defined as the union of all splice forms) with featureCounts (Version 1.5.0-p3; Liao et al., 2014). Reads that aligned to more than one gene as well as chimeric fragments were excluded. We also removed the 37 genes that failed to be quantified in at least one sample by Cufflinks (Trapnell et al., 2010).
We implemented a quality control (QC) pipeline that computes an extensive set of quality metrics, relying in part on FastQC (Version 0.3.2) and the Picard suite of alignment metrics (Version 2.5.0 with samtools 1.3.1). We used the open-source R package SCONE (https://github.com/YosefLab/scone; Version 0.0.7) to perform data-adaptive quality metric-based cell filtering. This yielded the following filtering criteria: any cell with fewer than 100,000 aligned reads or a percentage of aligned reads below 88.9% was filtered out. We identified cells that were non-sensory contaminants by expression of Reg3g (Figure S1) and doublets by co-expression of known neuronal (Omp) and sustentacular cell markers (Cyp1a2 or Cyp2g1). After filtering low quality cells (76) and removing doublets (9) and non-sensory epithelial contaminants (78), 687 cells (out of 849) remained. Finally, we retained only those genes having at least 40 reads in at least 5 cells (12,781 genes).
Normalization of Single-Cell RNA-Seq Data
We performed and assessed several normalization schemes using SCONE, based on a set of 9 data-driven performance metrics. These metrics aim to capture two main features of each normalization procedure: the ability to remove unwanted technical variation and the ability to preserve wanted biological variation of interest. The first group of performance metrics includes the correlation of expression measures with factors derived from validated markers of various OE cell types (positive control genes) and the average silhouette width (Rousseeuw, 1987) of the obtained clusters (cluster quality). Metrics in the second group include the correlation of expression measures with factors derived from “housekeeping” negative control genes (obtained from our earlier microarray experiments analyzing injury-induced regeneration of the olfactory epithelium) and the correlation between expression measures and quality control (QC) measures. See the scone package vignettes (available at https://github.com/YosefLab/scone) for more details. According to SCONE, the best performing normalization was full-quantile normalization (Bolstad et al., 2003; Bullard et al., 2010), followed by regression-based adjustment for QC measures (see above). Specifically, principal component analysis (PCA) was applied to the matrix of QC measures, and the first PC was used as a quantitative factor of “unwanted” technical variation across cells. Then, the log-transformed quantile-normalized expression measures (adding a pseudocount of 1 prior to the log transformation) for each gene were modeled as a linear function of this technical covariate. The normalized expression measures were defined as the residuals from the linear model fit, rescaled to have the same mean as the log-transformed quantile-normalized read counts.
Clustering of Single-Cell RNA-Seq Data
We used the clustering framework RSEC (Resampling-based Sequential Ensemble Clustering) to obtain stable and tight cell clusters. The method is implemented in the open-source clusterExperiment R package (Version 0.99.3-9001) available at Bioconductor Project (http://bioconductor.org/packages/clusterExperiment). Briefly, RSEC comprises the following steps. Given a base clustering algorithm, which we chose to be k-means, where the parameter k determines the number of clusters (Hartigan and Wong, 1979), RSEC creates ensemble clusters by the following steps: 1) repeatedly subsampling the observations (cells or genes, respectively), 2) clustering each set of subsampled observations with k-means, and then 3) forming a final clustering determined by clustering samples based on the percentage of subsamples for which two observations were assigned to the same cluster. This procedure was repeated for increasing k (the number of clusters), in order to find the cluster that changed the least; this cluster was deemed the most stable and removed, and then the entire clustering procedure was repeated on the remaining data. This sequential strategy follows that of Tseng and Wong (2005) and ensures that outlying clusters do not dominate the clustering; it also alleviates the dependence on the number of clusters k in lieu of other parameters that define required cluster similarity and stability. RSEC generates a large collection of such cluster sets by repeating the above procedure for different choices of the parameters defining similarity and stability; it then identifies a consensus over the different candidate sets based on the co-clustering of observations. This approach tends to result in a large number of small clusters that sometimes do not differ substantially in terms of gene expression. Hence, the last step of RSEC is to merge closely related clusters that do not exhibit differential expression. The vignette of the clusterExperiment package provides additional details on the RSEC algorithm (available at https://www.bioconductor.org/packages/clusterExperiment).
When applied to the first 50 principal components of the expression matrix RSEC found 18 stable clusters, which were then manually inspected for the presence of marker genes of known cell types in order to make preliminary assignments of cell type identities. Clusters containing fewer than 10 cells (each representing <1.5% of the total population studied) were deemed to be less reliable and therefore set aside to avoid confounding downstream analyses, yielding a final repertoire of 13 clusters (Figures S1 and S2).
Preliminary assignment of cluster identities were made as follows. The resting HBC cluster was annotated based on expression of HBC markers Trp63, Krt5, and Krt14 (Fletcher et al., 2011; Holbrook et al., 1995; Suzuki and Takeda, 1991). Interestingly, two clusters (ΔHBC1, ΔHBC2) contain cells in which the canonical HBC stem cell markers Trp63, Krt5, and Krt14 appear to be variably down-regulated from cell to cell (Figure S2), suggesting the existence of at least one transition state in which HBCs first begin to differentiate. Cells in the GBC cluster express Kit, Ascl1, and high levels of cell cycle genes, markers of GBC progenitors (Goldstein et al., 2014; Manglapus et al., 2004). Clusters representing immediate neuronal precursors (INP1,2,3) and immature neurons (iOSN) within the olfactory neuronal lineage were also identified based on the expression of genes such as Neurod1, Lhx2, Gap43, and the G protein subunit Gγ8 (Gng8) (Hirota and Mombaerts, 2004; Kolterud et al., 2004; Packard et al., 2011; Tirindelli and Ryba, 1996; Verhaagen et al., 1989). The mature olfactory sensory neuron cluster (mOSN) was identified by expression of olfactory marker protein (Omp), the main subunit of the cyclic nucleotide-gated channel (Cnga2) and Gγ13 (Gng13), hallmarks of mature olfactory sensory neurons (Huang et al., 1999; Keller and Margolis, 1975; Sautter et al., 1998). Cells in two clusters (immature and mature sustentacular cells, iSus and mSus, respectively) express increasing levels of Cyp2g1, a marker of sustentacular cells (Gu et al., 1998). We also identified a cluster of microvillous cells (MV2) based on their expression of Ascl3 and Cftr (Pfister et al., 2015). The identity of one small cluster of cells could not be readily classified due to the heterogeneous expression of a few identifying marker genes. Based on the expression of Trpm5, a marker of a subset of microvillous cells (Hansen and Finger, 2008), and Sox9, which we validated as being expressed not only in cells of the Bowman’s gland but also in a subset of microvillous cells (Figure S2), we provisionally assigned this cluster’s identity as microvillous cells (MV1), although we cannot exclude the possibility that it contains precursors of Bowman’s gland. Thus, clustering of our single-cell RNA-seq data successfully identified three of the major mature HBC-derived cell types in the olfactory epithelium (olfactory sensory neurons, sustentacular cells, microvillous cells) as well as intermediate progenitors and putative transition state stem cells. The absence of cells of the Bowman’s gland may reflect their low representation in the overall population and/or their lower survival in our cell isolation procedure.
t-Distributed Stochastic Neighbor Embedding
To display the relative distances between cells in a lower-dimensional representation of gene expression space, we employed t-distributed stochastic neighbor embedding (t-SNE; (Van Der Maaten, 2014; van der Maaten and Hinton, 2008). t-SNE is a method of dimensionality reduction that excels at representing distances on multiple scales. We use it here to visualize our data independently from how we generated the cell clusters (for clustering, see RSEC above). We use the Barnes-Hut implementation, as available in the Rtsne R package. We chose as input expression data corresponding to the 500 most variable genes, and we set perplexity to 10 with 1000 iterations; varying the perplexity to 30 or 50 and increasing iterations does not alter our conclusions about the congruence between the t-SNE visualization and our clustering results (data not shown).
Cell Lineages and Developmental Distance
We used a recently developed cell lineage inference algorithm, Slingshot (Version 0.0.0.9005, available as an open-source R package slingshot at https://github.com/kstreet13/slingshot), to identify lineage trajectories and bifurcations and to order cells along trajectories. Slingshot takes as input a matrix of reduced dimension normalized expression measures (e.g., PCA) and cell clustering assignments. It infers lineage trajectories and branch points by connecting the cluster medoids using a minimum spanning tree (MST) and identifying the starting cluster or root node. Lineages are defined by ordered sets of clusters beginning with the root node and terminating in the most distal cluster(s) with only one connection. Next, principal curves (Hastie and Stuetzle, 1989) are fit to the subsets of cells making up each lineage, providing a smooth, nonlinear summary of each trajectory. Individual cells are then orthogonally projected to each curve and thereby ordered in a space reflecting developmental distance. The ordering provided by Slingshot, analogous to pseudotime, is referred to herein as developmental order.
Any cluster with fewer than 10 cells was removed prior to applying Slingshot. The cluster representing resting HBCs was chosen as the root node. In addition to assignment of the starting point, slingshot allows for the user to specify known end points of the developmental lineage to better guide its identification. Using this feature, we assigned mature sustentacular cells as an endpoint. Note that this assignment only constrains that cluster; all other clusters are allowed to be placed anywhere on a lineage, and additional endpoints can be found beyond this one, as was the case in our data. slingshot then generated principal curves and cell developmental distances for each lineage. slingshot was applied to the first five principal components of the normalized expression matrix. Five components were chosen based on examining the separation of cells along individual PCs (Figure S2E), although the MST was stable beyond the first 10 PCs (data not shown).
Differential Expression and Gene Clustering
We used limma for differential expression (DE) analysis between clusters within each lineage, applying a one-versus-all approach (i.e., comparing the average of one cluster to the average of all the other clusters in that lineage, Table S1). For each lineage, genes being among the top 500 DE genes (lowest adjusted p-value) for each one-versus-all comparison were retained for clustering. The expression profiles of these genes were centered and scaled (subtracting the mean and dividing by the standard deviation across all cells in a given lineage) and then clustered using clusterExperiment. Partitioning Around Medoids (PAM) (Kaufman and Rousseeuw, 1987) was used as base algorithm for a range of numbers of clusters k, and a consensus clustering was identified for each lineage. Expression profiles for the 40 cell cycle genes were also centered and scaled as described above, but across cells from both lineages.
Gene Set Enrichment Analysis
After clustering the cells, we used limma (Version 3.28.19; Smyth, 2004) for DE analysis as implemented in the clusterExperiment package. We applied limma’s implementation of the romer rotation test to perform gene set enrichment analysis (GSEA) (Ritchie et al., 2015) with 107 rotations contrasting each cluster. Gene sets were taken from the Molecular Signatures Database (MSigDB) at the Broad Institute and included all Hallmark gene sets (H) in addition to the Canonical pathways, KEGG, and Reactome gene sets from the curated (C2) gene sets.
Volcano Plots
We used clusterExperiment’s wrapper around limma for DE analysis across all lineages applying a pairwise approach (i.e., comparing the average of one cluster to the average of each other cluster, Table S2). We plotted the –log10 adjusted p-value (Benjamini-Hochberg correction) vs. log2 fold-change in expression for each pairwise comparison corresponding to a transition predicted by Slingshot. Volcano plots highlight genes that are down-regulated or up-regulated with a fold-change greater than two (log2 > 1) and an adjusted p-value less than 0.01.
Transcription Factor Co-Expression Networks
Transcription factor network diagrams were based upon transcription factor genes that were among the 500 most differentially expressed genes within each lineage, using the previously mentioned one-versus-all approach (Table S3). A list of transcription factors was obtained from the Animal Transcription Factor Database (http://www.bioguo.org/AnimalTFDB/species.php?spe=Mus_musculus). Only transcription factors that had a correlation of at least 0.3 with at least 5 other differentially expressed transcription factors along the lineage were included in the correlation network. This correlation threshold was chosen to be greater than the maximum pairwise correlation after random permutation, as previously done in Treutlein et al., (2016).
Odorant Receptor Expression Analysis
Since odorant receptors (ORs) are typically expressed at one allele per mature olfactory sensory neuron, we chose to investigate transcripts per million (TPM) output from RSEM (Version 1.2.31; Li and Dewey, 2015) to assess OR expression in the neuronal lineage instead of filtered counts because most olfactory receptors would have been filtered out. For this analysis, we aligned reads to the transcriptome with Bowtie2 (Version 2.2.9; Langmead and Salzberg, 2012) and quantified gene expression with RSEM, while setting the alignment parameters to the ones recommended by RSEM’s authors. Analysis of OR expression using Kallisto (Bray et al., 2016) TPMs gave similar results (data not shown).
Data and Software Availability
Accession Numbers
RNA-seq data have been deposited in Gene Expression Omnibus (accession number GSE95601).
Software
Analysis scripts for this dataset can be found at https://github.com/rufletch/p63-HBC-diff. The R software packages scone, clusterExperiment, and slingshot are available on GitHub and Bioconductor as described above.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Armenian Hamster: phycoerythrin (PE)-conjugated ICAM1 | BD Pharmingen | Catalog No. 565615 |
| Rabbit: phycoerythrin (PE)-conjugated SCARB1 | Novus Biologicals | Catalog No. NB400-104PE |
| Rabbit: F3 | Bioss Antibodies | Catalog No. BS-4690R |
| Chicken: GFP/YFP | Abcam | Catalog No. ab13970 |
| Goat: SOX2 | Santa Cruz Biotechnology | Catalog No. SC17320 |
| Donkey anti-Goat: Alexa-594 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-11058 |
| Donkey anti-Chicken: Alexa-488 conjugated secondary antibody | Jackson ImmunoResearch | Catalog No. 703-545-155 |
| Donkey anti-Rabbit: Alexa-555 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-31572 |
| Donkey anti-Rabbit: Alexa-568 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-21207 |
| Donkey anti-Mouse IgG: Alexa-568 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-10037 |
| Donkey anti-Mouse IgG: Alexa-647 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-31571 |
| Donkey anti-Goat: Alexa-647 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-21447 |
| Goat anti-Chicken: Alexa-488 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-11039 |
| Goat anti-Mouse IgG: Alexa-633 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-21050 |
| Goat anti-Rabbit: Alexa-568 conjugated secondary antibody | Thermo Fisher Scientific | Catalog No. A-11011 |
| Goat anti-Armenian Hamster: Dylight 594 conjugated secondary antibody | Jackson ImmunoResearch | Catalog No. 127-515-099 |
| Mouse: P63 (4A4) | BioCare | Catalog No. CM163B |
| Goat: NEUROD1 | Santa Cruz Biotechnology | Catalog No. SC1084 |
| Mouse: NTUB/TuJ1 | Neuromics | Catalog No. MO-15013 |
| Rabbit: SOX9 | Milipore | Catalog No. AB5535 |
| Armenian Hamster: CD54/ICAM1 | BD Pharmingen | Catalog No. 550287 |
| Alkaline phosphatase-conjugated anti-digoxygenin | Sigma | Catalog No. 11093274910 |
| Alkaline phosphatase-conjugated anti-fluorescein | Sigma | Catalog No. 11426338910 |
| Bacterial and Virus Strains | ||
| None. | ||
| Biological Samples | ||
| None. | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| tamoxifen | Sigma | T5648 |
| Neurobasal medium | Thermo Fisher Scientific | 21103049 |
| papain | Worthington Biochemical Corporation | LS003119 |
| fetal bovine serum | Thermo Fisher Scientific | 26140-087 |
| propidium iodide | Sigma | 81845 |
| tissue freezing medium | Fisher | 15-183-13 |
| goat serum | Jackson ImmunoResearch | 005000121 |
| donkey serum | Jackson ImmunoResearch | 017000121 |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 |
| DAPI in Vectashield | Vector Laboratories | H-1500 |
| 5-ethynyl-2′-deoxyuridine (EdU) | Thermo Fisher Scientific | A10044 |
| Dig RNA labeling mix | Roche (sold by Sigma) | 1277073 |
| RNA Labeling Mix, Fluorescein | Roche (sold by Sigma) | 11685619910 |
| BCIP/NBT color development substrate | Promega | s3771 |
| SuperScript RTIII reverse transcriptase | Invitrogen (now Thermo Fisher Scientific) | 18080093 |
| Trizol | Invitrogen (now Thermo Fisher Scientific) | 10296-010 |
| DNase1 | Roche | 03539121103 |
| Critical Commercial Assays | ||
| C1 IFC for mRNA-seq (5–10 μM) | Fluidigm | 1005759 |
| C1 IFC for mRNA-seq (10–17 μM) | Fluidigm | 1005760 |
| C1 Single-Cell Auto Prep Kit for mRNA Seq | Fluidigm | 100-6209 |
| C1 Single-Cell Auto Prep Reagent Kit for mRNA Seq | Fluidigm | 100-6201 |
| SMARTer Ultra Low RNA Kit for the Fluidigm C1 System | Clontech | 634833 |
| Advantage 2 PCR Kit | Clontech | 639207 |
| Agilent High Sensitivity DNA Kit Reagents | Agilent | 5067-4626 |
| Nextera XT DNA Sample Preparation Kit | Illumina | FC-131-1096 |
| Nextera XT DNA Sample Preparation Index Kits A–D | Illumina | FC-131-2001-4 |
| Agencourt AMPure XP beads | Beckman Coulter | A63880 |
| Qubit dsDNA High Sensitivity Assay Kit | Life Technologies Molecular Probes | Q32854 |
| Tyramide Signal Amplification kit cy3 | Perkin-Elmer | NEL744001KT |
| Tyramide Signal Amplification kit FITC | Perkin-Elmer | NEL741001KT |
| Click-iT EdU Imaging Kit, Alex Fluor 647 | Thermo Fisher Scientific | C10340 |
| Deposited Data | ||
| Raw data, normalized counts matrix | GEO | GEO accession # GSE95601 |
| Scripts for analysis | GitHub | https://github.com/rufletch/p63-HBC-diff |
| Experimental Models: Cell Lines | ||
| None. | ||
| Experimental Models: Organisms/Strains | ||
| Mus musculus: CD-1 | Charles River | Strain code 022 |
| Mus musculus: C57BL/6 | JAX mouse services | Stock no. 000664 |
| Mus musculus: B6.129-Krt5-CreER(T2) | Indra et al., 1999 | N/A |
| Mus musculus: B6.129-Ascl1-CreER(T2) | JAX mouse services; Kim et al., 2011 | Stock no. 012882 |
| Mus musculus: B6.129-Trp63lox/lox | Mills et al., 2002 | N/A |
| Mus musculus: B6.129-Sox2eGFP | JAX mouse services; Arnold et al., 2011 | Stock no. 017592 |
| Mus musculus: B6.129-Ctnnb1lxEx3/+ | Harada et al., 1999 | N/A |
| Mus musculus: B6.129-Ctnnb1lox/lox | JAX mouse services; | Stock no. 004152 |
| Brault et al., 2001 | ||
| Mus musculus: B6.129-Rosa26eYFP | JAX mouse services; Srinivas et al., 2001 | Stock no. 006148 |
| Mus musculus: B6.129-Rosa26Confetti | JAX mouse services; Snippert et al., 2010 | Stock no. 013731 |
| Oligonucleotides | ||
| See Table S6 for sequences. | ||
| Recombinant DNA | ||
| None. | ||
| Software and Algorithms | ||
| ImageJ (Version 1.48v) | Schneider et al., 2012 | https://imagej.nih.gov |
| Bowtie2 (Version 2.2.9) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Cufflinks (Version 2.2.1) | Trapnell et al., 2010 | cole-trapnell- lab.github.io/cufflinks/ |
| Trimmomatic (Version 0.3.2) | Bolger et al., 2014 | http://www.usadellab.org/cms/index.php?page=trimmomatic |
| Tophat2 (Version 2.1.1) | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/ |
| RSEM (Version 1.2.31) | Li and Dewey, 2015 | https://deweylab.github.io/RSEM/ |
| Kallisto (0.43.0) | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| featureCounts (Version 1.5.0-p3) | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| FastQC (Version 0.3.2) | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| Picard Tools (Version 2.5.0) | https://broadinstitute.github.io/picard/ | |
| samtools (Version 1.3.1) | Li et al., 2009 | http://www.htslib.org/ |
| R (Version 3.3.0) | https://www.R-project.org/ | |
| SCONE (Version 0.0.7) | This paper; Risso et al. (in preparation) | https://github.com/YosefLab/scone |
| clusterExperiment (Version 0.99.3-9001) | This paper; Purdom et al. (in preparation) | http://bioconductor.org/packages/clusterExperiment |
| Slingshot (Version 0.0.0.9005) | This paper; Street et al. (in preparation) | https://github.com/kstreet13/slingshot |
| limma (Version 3.28.19) | Smyth, 2004 | https://bioconductor.org/packages/limma |
| PathVisio (Version 3.2.4) | Kutmon et al., 2015; van Iersel et al., 2008 | https://www.pathvisio.org/ |
| Other | ||
| None. | ||
Supplementary Material
Related to Figure 1.
Related to Figure S4 and STAR Methods.
Acknowledgments
We are grateful to Hector Nolla and Alma Valeros in the Cancer Research Laboratory Flow Cytometry Facility for their expertise and assistance with fluorescence-activated cell sorting, Shana McDevitt, Karen Lundy, Anett Schmittfull, and Minyong Chung in the QB3 Genomics Sequencing and Functional Genomics Laboratories for their tireless efforts on RNA sequencing, Burke Bundy for his gracious assistance with cluster computing, and members of the Ngai laboratory for invaluable advice and suggestions over the course of this project. We also thank Makoto Taketo for providing the Ctnnb1lxE3 mouse. This work was supported by grants from the National Institute on Deafness and Other Communication Disorders (RO1DC007235), the National Institute of Mental Health (MH105979), the National Center for Research Resources (S10RR029668) and the Siebel Foundation. R.B.F. was supported by a grant from the National Institute on Aging (K01AG045344). D.D. and K.N.S. were supported by a training grant from the National Human Genome Research Institute (T32HG000047), L.G. by a training grant from the National Institute of General Medical Sciences (T32GM098218), and Q.F. by a MARC National Research Service Award from the National Institute of General Medical Sciences (T34GM092702). R.B.F. and D.D. were also supported by training grant TG2-01164 from the California Institute of Regenerative Medicine CIRM Scholars program.
Footnotes
Author Contributions
R.B.F., L.G., Y.G.C., D.R., E.P., S.D. and J.N. designed the single-cell RNA-seq experiments; R.B.F., D.D., K.N.S., A.W. and M.B.C. analyzed the single-cell RNA-seq data; R.B.F., L.G., A.B. and Q.F. designed and performed all other experiments; N.Y., E.P., S.D. and D.R. supervised statistical and computational analyses; R.B.F. and J.N. wrote the manuscript with input from the co-authors; J.N. oversaw all aspects of the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU. The Dual Function Cytokine IL-33 Interacts with the Transcription Factor NF- B To Dampen NF- B-Stimulated Gene Transcription. The Journal of Immunology. 2011;187:1609–1616. doi: 10.4049/jimmunol.1003080. [DOI] [PubMed] [Google Scholar]
- Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, Hochedlinger K. Adult Stem and Progenitor Cells Are Important for Tissue Regeneration and Survival of Mice. Stem Cell. 2011;9:317–329. doi: 10.1016/j.stem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano M, Kauer JS, Hunter DD. Globose basal cells are neuronal progenitors in the olfactory epithelium: a lineage analysis using a replication-incompetent retrovirus. Neuron. 1994;13:339–352. doi: 10.1016/0896-6273(94)90351-4. [DOI] [PubMed] [Google Scholar]
- Chen M, Tian S, Yang X, Lane AP, Reed RR, Liu H. Wnt-responsive lgr5+ globose Basal cells function as multipotent olfactory epithelium progenitor cells. J Neurosci. 2014;34:8268–8276. doi: 10.1523/JNEUROSCI.0240-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Fang H, Schwob JE. Multipotency of purified, transplanted globose basal cells in olfactory epithelium. J Comp Neurol. 2004;469:457–474. doi: 10.1002/cne.11031. [DOI] [PubMed] [Google Scholar]
- Choi YS, Zhang Y, Xu M, Yang Y, Ito M, Peng T, Cui Z, Nagy A, Hadjantonakis AK, Lang RA, et al. Distinct functions for Wnt/β-catenin in hair follicle stem cell proliferation and survival and interfollicular epidermal homeostasis. Cell Stem Cell. 2013;13:720–733. doi: 10.1016/j.stem.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Loh KM, Nusse R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346:1248012–1248012. doi: 10.1126/science.1248012. [DOI] [PubMed] [Google Scholar]
- Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas-Gervais S, et al. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013;9:e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymecki SM, Tomasiewicz H. Using Flp-recombinase to characterize expansion of Wnt1-expressing neural progenitors in the mouse. Developmental Biology. 1998;201:57–65. doi: 10.1006/dbio.1998.8971. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Adhikary G, Young CA, Jans R, Crish JF, Xu W, Rorke EA. AP1 transcription factors in epidermal differentiation and skin cancer. J Skin Cancer. 2013;2013:537028–537029. doi: 10.1155/2013/537028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher RB, Prasol MS, Estrada J, Baudhuin A, Vranizan K, Choi YG, Ngai J. p63 regulates olfactory stem cell self-renewal and differentiation. Neuron. 2011;72:748–759. doi: 10.1016/j.neuron.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ, Goss GM, Hatzistergos KE, Rangel EB, Seidler B, Saur D, Hare JM. Adult c-Kit(+) progenitor cells are necessary for maintenance and regeneration of olfactory neurons. J Comp Neurol. 2014;523:15–31. doi: 10.1002/cne.23653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziadei GA, Graziadei PP. Neurogenesis and neuron regeneration in the olfactory system of mammals. II Degeneration and reconstitution of the olfactory sensory neurons after axotomy. J Neurocytol. 1979a;8:197–213. doi: 10.1007/BF01175561. [DOI] [PubMed] [Google Scholar]
- Graziadei PP, Graziadei GA. Neurogenesis and neuron regeneration in the olfactory system of mammals. I Morphological aspects of differentiation and structural organization of the olfactory sensory neurons. J Neurocytol. 1979b;8:1–18. doi: 10.1007/BF01206454. [DOI] [PubMed] [Google Scholar]
- Gu J, Zhang QY, Genter MB, Lipinskas TW, Negishi M, Nebert DW, Ding X. Purification and characterization of heterologously expressed mouse CYP2A5 and CYP2G1: role in metabolic activation of acetaminophen and 2,6-dichlorobenzonitrile in mouse olfactory mucosal microsomes. J Pharmacol Exp Ther. 1998;285:1287–1295. [PubMed] [Google Scholar]
- Guo Z, Packard A, Krolewski RC, Harris MT, Manglapus GL, Schwob JE. Expression of Pax6 and Sox2 in adult olfactory epithelium. J Comp Neurol. 2010;518:4395–4418. doi: 10.1002/cne.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchate NK, Kondoh K, Lu Z, Kuang D, Ye X, Qiu X, Pachter L, Trapnell C, Buck LB. Single-cell transcriptomics reveals receptor transformations during olfactory neurogenesis. Science. 2015;350:1251–1255. doi: 10.1126/science.aad2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, Finger TE. Is TrpM5 a reliable marker for chemosensory cells? Multiple types of microvillous cells in the main olfactory epithelium of mice. BMC Neurosci. 2008;9:115. doi: 10.1186/1471-2202-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. Embo J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan JA, Wong MA. Algorithm AS 136: A K-Means Clustering Algorithm. Applied Statistics. 1979;28:100. [Google Scholar]
- Hastie T, Stuetzle W. Principal Curves. Journal of the American Statistical Association. 1989;84:502–516. [Google Scholar]
- Hirota J, Mombaerts P. The LIM-homeodomain protein Lhx2 is required for complete development of mouse olfactory sensory neurons. Pnas. 2004;101:8751–8755. doi: 10.1073/pnas.0400940101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook EH, Szumowski KE, Schwob JE. An immunochemical, ultrastructural, and developmental characterization of the horizontal basal cells of rat olfactory epithelium. J Comp Neurol. 1995;363:129–146. doi: 10.1002/cne.903630111. [DOI] [PubMed] [Google Scholar]
- Huang L, Shanker YG, Dubauskaite J, Zheng JZ, Yan W, Rosenzweig S, Spielman AI, Max M, Margolskee RF. Ggamma13 colocalizes with gustducin in taste receptor cells and mediates IP3 responses to bitter denatonium. Nat Neurosci. 1999;2:1055–1062. doi: 10.1038/15981. [DOI] [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai N, Zhou Z, Roop DR, Behringer RR. Horizontal Basal Cells Are Multipotent Progenitors in Normal and Injured Adult Olfactory Epithelium. Stem Cells. 2008;26:1298–1306. doi: 10.1634/stemcells.2007-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012;110:1465–1473. doi: 10.1161/CIRCRESAHA.112.269035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Hayoz S, Hutch CR, Iqbal TR, Pooley AE, Hegg CC. An IP3R3- and NPY-Expressing Microvillous Cell Mediates Tissue Homeostasis and Regeneration in the Mouse Olfactory Epithelium. PLoS ONE. 2013;8:e58668. doi: 10.1371/journal.pone.0058668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Liu ZG, Zandi E. AP-1 function and regulation. Current Opinion in Cell Biology. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Kaufman L, Rousseeuw P. Clustering by means of medoids. North-Holland; 1987. [Google Scholar]
- Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. Journal of Cell Science. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- Keller A, Margolis FL. Immunological studies of the rat olfactory marker protein. Journal of Neurochemistry. 1975;24:1101–1106. doi: 10.1111/j.1471-4159.1975.tb03883.x. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Ables JL, Dickel LK, Eisch AJ, Johnson JE. Ascl1 (Mash1) Defines Cells with Long-Term Neurogenic Potential in Subgranular and Subventricular Zones in Adult Mouse Brain. PLoS ONE. 2011;6:e18472–e18476. doi: 10.1371/journal.pone.0018472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolterud A, Alenius M, Carlsson L, Bohm S. The Lim homeobox gene Lhx2 is required for olfactory sensory neuron identity. Development. 2004;131:5319–5326. doi: 10.1242/dev.01416. [DOI] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Research. 2015;25:1860–1872. doi: 10.1101/gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutmon M, Riutta A, Nunes N, Hanspers K, Willighagen EL, Bohler A, Mélius J, Waagmeester A, Sinha SR, Miller R, et al. WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016;44:D488–D494. doi: 10.1093/nar/gkv1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutmon M, van Iersel MP, Bohler A, Kelder T, Nunes N, Pico AR, Evelo CT. PathVisio 3: an extendable pathway analysis toolbox. PLoS Comput Biol. 2015;11:e1004085. doi: 10.1371/journal.pcbi.1004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin NM, Teillet MA. Experimental analysis of the migration and differentiation of neuroblasts of the autonomic nervous system and of neurectodermal mesenchymal derivatives, using a biological cell marking technique. Developmental Biology. 1974;41:162–184. doi: 10.1016/0012-1606(74)90291-7. [DOI] [PubMed] [Google Scholar]
- Leung CT, Coulombe PA, Reed RR. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat Neurosci. 2007;10:720–726. doi: 10.1038/nn1882. [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification fromRNA-Seq data with or without a reference. 2015;12:323–338. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Ma Y, Kanakousaki K, Buttitta L. How the cell cycle impacts chromatin architecture and influences cell fate. Front Genet. 2015;6:19. doi: 10.3389/fgene.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay-Sim A, Kittel P. Cell dynamics in the adult mouse olfactory epithelium: a quantitative autoradiographic study. J Neurosci. 1991;11:979–984. doi: 10.1523/JNEUROSCI.11-04-00979.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglapus GL, Youngentob SL, Schwob JE. Expression patterns of basic helix-loop-helix transcription factors define subsets of olfactory progenitor cells. J Comp Neurol. 2004;479:216–233. doi: 10.1002/cne.20316. [DOI] [PubMed] [Google Scholar]
- Mills AA, Qi Y, Bradley A. Conditional inactivation of p63 by Cre-mediated excision. Genesis. 2002;32:138–141. doi: 10.1002/gene.10067. [DOI] [PubMed] [Google Scholar]
- Ogura T, Szebenyi SA, Krosnowski K, Sathyanesan A, Jackson J, Lin W. Cholinergic microvillous cells in the mouse main olfactory epithelium and effect of acetylcholine on olfactory sensory neurons and supporting cells. Journal of Neurophysiology. 2011;106:1274–1287. doi: 10.1152/jn.00186.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard A, Giel-Moloney M, Leiter A, Schwob JE. The progenitor cell capacity of NeuroD1-expressing globose basal cells in the mouse olfactory epithelium. J Comp Neurol. 2011;519:3580–3596. doi: 10.1002/cne.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister S, Weber T, Härtig W, Schwerdel C, Elsaesser R, Knuesel I, Fritschy JM. Novel role of cystic fibrosis transmembrane conductance regulator in maintaining adult mouse olfactory neuronal homeostasis. J Comp Neurol. 2015;523:406–430. doi: 10.1002/cne.23686. [DOI] [PubMed] [Google Scholar]
- Price J, Turner D, Cepko C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proceedings of the National Academy of Sciences. 1987;84:156–160. doi: 10.1073/pnas.84.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw PJ. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. Journal of Computational and Applied Mathematics. 1987;20:53–65. [Google Scholar]
- Saraiva LR, Ibarra-Soria X, Khan M, Omura M, Scialdone A, Mombaerts P, Marioni JC, Logan DW. Hierarchical deconstruction of mouse olfactory sensory neurons: from whole mucosa to single-cell RNA-seq. Scientific Reports. 2015;5:18178–18195. doi: 10.1038/srep18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautter A, Zong X, Hofmann F, Biel M. An isoform of the rod photoreceptor cyclic nucleotide-gated channel beta subunit expressed in olfactory neurons. Proceedings of the National Academy of Sciences. 1998;95:4696–4701. doi: 10.1073/pnas.95.8.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittke N, Herrick DB, Lin B, Peterson J, Coleman JH, Packard AI, Jang W, Schwob JE. Transcription factor p63 controls the reserve status but not the stemness of horizontal basal cells in the olfactory epithelium. Proceedings of the National Academy of Sciences. 2015;112:E5068–E5077. doi: 10.1073/pnas.1512272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz P, Kalbe B, Jansen F, Altmueller J, Becker C, Mohrhardt J, Schreiner B, Gisselmann G, Hatt H, Osterloh S. Transcriptome Analysis of Murine Olfactory Sensory Neurons during Development Using Single Cell RNA-Seq. Chem Senses. 2016;41:313–323. doi: 10.1093/chemse/bjw003. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Huard JM, Luskin MB, Youngentob SL. Retroviral lineage studies of the rat olfactory epithelium. Chemical Senses. 1994;19:671–682. doi: 10.1093/chemse/19.6.671. [DOI] [PubMed] [Google Scholar]
- Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, Choi K, Bendall S, Friedman N, Pe’er D. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nature Biotechnology. 2016;34:637–645. doi: 10.1038/nbt.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3–Article25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Takeda M. Keratins in the developing olfactory epithelia. Brain Res Dev Brain Res. 1991;59:171–178. doi: 10.1016/0165-3806(91)90097-3. [DOI] [PubMed] [Google Scholar]
- Tan L, Li Q, Xie XS. Olfactory sensory neurons transiently express multiple olfactory receptors during development. Molecular Systems Biology. 2015;11:844–844. doi: 10.15252/msb.20156639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirindelli R, Ryba NJ. The G-protein gamma-subunit G gamma 8 is expressed in the developing axons of olfactory and vomeronasal neurons. Eur J Neurosci. 1996;8:2388–2398. doi: 10.1111/j.1460-9568.1996.tb01202.x. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, et al. Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature. 2016;534:391–395. doi: 10.1038/nature18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, Wong WH. Tight clustering: a resampling-based approach for identifying stable and tight patterns in data. Biometrics. 2005;61:10–16. doi: 10.1111/j.0006-341X.2005.031032.x. [DOI] [PubMed] [Google Scholar]
- Van Der Maaten L. Accelerating t-SNE using tree-based algorithms. Journal of Machine Learning Research 2014 [Google Scholar]
- van der Maaten L, Hinton G. Visualizing Data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- Verhaagen J, Oestreicher AB, Gispen WH, Margolis FL. The expression of the growth associated protein B50/GAP43 in the olfactory system of neonatal and adult rats. Journal of Neuroscience. 1989;9:683–691. doi: 10.1523/JNEUROSCI.09-02-00683.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nature Biotechnology. 2016;34:1145–1160. doi: 10.1038/nbt.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YZ, Yamagami T, Gan Q, Wang Y, Zhao T, Hamad S, Lott P, Schnittke N, Schwob JE, Zhou CJ. Canonical Wnt signaling promotes the proliferation and neurogenesis of peripheral olfactory stem cells during postnatal development and adult regeneration. Journal of Cell Science. 2011;124:1553–1563. doi: 10.1242/jcs.080580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler E, Farbman AI. Supporting cell proliferation in the olfactory epithelium decreases postnatally. Glia. 1998;22:315–328. doi: 10.1002/(sici)1098-1136(199804)22:4<315::aid-glia1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Weisblat DA, Sawyer RT, Stent GS. Cell lineage analysis by intracellular injection of a tracer enzyme. Science. 1978;202:1295–1298. doi: 10.1126/science.725606. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 2013;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Yamashita J, Ohmoto M, Aoudé I, Ogura T, Luo W, Bachmanov AA, Lin W, Matsumoto I, Hirota J. Skn-1a/Pou2f3 is required for the generation of Trpm5-expressing microvillous cells in the mouse main olfactory epithelium. BMC Neurosci. 2014;15:13. doi: 10.1186/1471-2202-15-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinyk DL, Mercer EH, Harris E, Anderson DJ, Joyner AL. Fate mapping of the mouse midbrain-hindbrain constriction using a site-specific recombination system. Current Biology. 1998;8:665–668. doi: 10.1016/s0960-9822(98)70255-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 1.
Related to Figure S4 and STAR Methods.