

Abstract
N-Aryl phenoxazines have been synthesized and introduced as strongly reducing metal-free photoredox catalysts in organocatalyzed atom transfer radical polymerization for the synthesis of well-defined polymers. Experiments confirmed quantum chemical predictions that, like their dihydrophenazine analogs, the photoexcited states of phenoxazine photoredox catalysts are strongly reducing and achieve superior performance when they possess charge transfer character. We compare phenoxazines to previously reported dihydrophenazines and phenothiazines as photoredox catalysts to gain insight into the performance of these catalysts and establish principles for catalyst design. A key finding reveals that maintenance of a planar conformation of the phenoxazine catalyst during the catalytic cycle encourages the synthesis of well-defined macromolecules. Using these principles, we realized a core substituted phenoxazine as a visible light photoredox catalyst that performed superior to UV-absorbing phenoxazines as well as previously reported organic photocatalysts in organocatalyzed atom transfer radical polymerization. Using this catalyst and irradiating with white LEDs resulted in the production of polymers with targeted molecular weights through achieving quantitative initiator efficiencies, which possess dispersities ranging from 1.13 to 1.31.
Graphical abstract
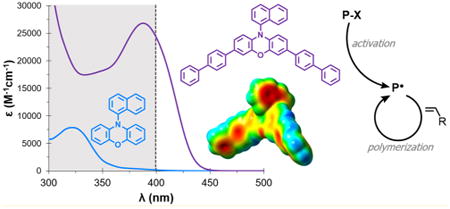
Introduction
Atom transfer radical polymerization (ATRP) is the most used controlled radical polymerization (CRP) for the synthesis of polymers with controlled molecular weight (MW), dispersity (Đ), architecture, and composition.1 Traditionally, metal catalysts have been employed to mediate the equilibrium between an alkyl halide and a carbon centered radical, produced by reduction of the halide, and deter bimolecular termination pathways.2 Concerns about metal contamination of polymers intended for biomedical or electronic applications have motivated efforts to lower metal catalyst loadings and enhance purification methods.3 Although CRPs exist that are mediated by organic catalysts and which thus entirely circumvent the issue of metal contamination,4 organic catalysts capable of mediating an organocatalyzed ATRP (O-ATRP) are limited because of the required significant reducing power required to reduce alkyl bromides commonly used for ATRP (∼ −0.6 to −0.8 V vs SCE).5
Photoredox catalysis presents a strategy to drive chemical transformations under mild conditions through the generation of reactive open-shell catalysts via photoexcitation.6 Recently, work in this field has heavily focused on polypyridal ruthenium and iridium complexes because such metal complexes efficiently absorb visible light, possess sufficiently long excited state lifetimes as a result of metal to ligand charge transfer (MLCT), and have tunable redox properties. However, most photoredox catalysts (PCs) do not possess the reducing power to directly reduce an alkyl bromide through an outer sphere electron transfer mechanism. Commonly, supplemental sacrificial electron donors are required for alkyl bromide reduction through a reductive quenching pathway. The addition of sacrificial electron donors, however, introduces potential side-reactions7 that impede the ability to synthesize polymers with low Đ.8 Select strongly reducing iridium9 or copper10 PCs can directly reduce an alkyl bromide through an oxidative quenching pathway, and elimination of the sacrificial electron donor can facilitate the synthesis of well-defined polymers.11 Light mediated CRPs further introduce spatial and temporal control as an attractive interactive feature for the incorporation of added synthetic complexity.12 However, the concerns of metal contamination and the sustainability of iridium or ruthenium metal PCs motivate the use of organic PCs.14,13
In accord with transition metal PCs, few organic PCs are able to directly reduce an alkyl bromide without the addition of a sacrificial electron donor.14 Strongly reducing organic catalysts, including perylene,15 N-aryl phenothiazines,16 and N,N-diaryl dihydrophenazines17 have been demonstrated as organic PCs capable of mediating O-ATRP (Figure 1). Continued progress in this field is required to further understand the mechanism of this polymerization to realize even more efficient PCs and access a broader application landscape.
Figure 1.
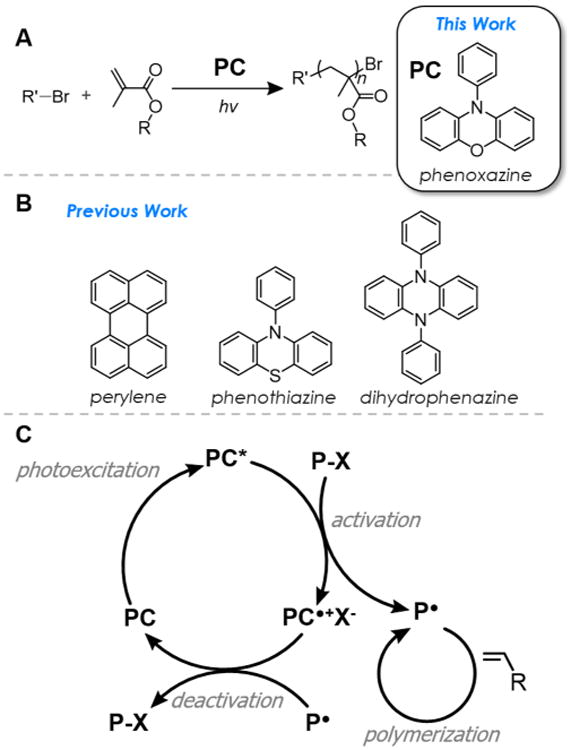
(A) O-ATRP mediated by organic PCs using alkyl bromide initiators and aryl phenoxazines studied in this work. (B) Organic PCs examined in previous work. (C) Proposed, general photoredox catalytic cycle of O-ATRP.
A proposed general photoredox O-ATRP mechanism involves photoexcitation of the PC to an excited state PC (PC*) which is capable of reducing alkyl bromides via an oxidative quenching pathway to generate the active radical for polymerization propagation, while yielding the radical cation PC (PC•+) and Br− ion pair complex, PC•+Br− (Figure 1C). Efficient deactivation is central to the production of well-defined polymers. Deactivation requires the PC•+Br− complex to be sufficiently oxidizing relative to the propagating radical to regenerate the alkyl bromide and ground state PC; subsequent photoexcitation of the PC reinitiates the catalytic cycle. Here, we report N-aryl phenoxazines as a new class of PCs for O-ATRP which produce well-defined polymers with low dispersities. Through following our maturing catalyst design principles, we report a visible light phenoxazine PC that produces polymers with Đ ranging from 1.13 to 1.31 over a range of polymer MWs while achieving quantitative initiator efficiency (I*).
To accelerate our progress in developing O-ATRP, we previously used quantum chemical calculations to guide the discovery and design of strongly reducing diaryl dihydrophenazines PCs for O-ATRP.17 We based our computationally directed strategy on the hypothesis that photoexcitation of the PC delivers, through intersystem crossing (ISC) from the singlet excited state PC (1PC*), a triplet excited state PC (3PC*) which is responsible for the alkyl bromide reduction. This hypothesis hinges on the necessity of the photoexcited species to possess a sufficiently long lifetime for photoredox catalysis.
Our continued work in this field has been piqued by the impressively strong excited state reduction potentials (E0* = E0(2PC•+/PC*)) of N,N-diaryl dihydrophenazines and N-aryl phenothiazines (∼ −2 V vs SCE), which are even more reducing than commonly used metal PCs, such as fac-Ir(ppy)3 (−1.73 V vs SCE).18 These strong E0*s and the success of the diaryl dihydrophenazines in O-ATRP further drew our attention toward N-aryl phenoxazines as a potential class of organic PCs for O-ATRP. We hypothesized that phenoxazines possessed characteristic traits that would distinguish them as organic PCs and make them successful catalysts for O-ATRP. Interestingly, these N, S, and O containing heterocycles are found in biologically relevant molecules19 and organic electronic applications.20
Results and Discussion
DFT calculations predict that N-aryl phenoxazines possess similarly strong E0*s (∼ −2 V vs SCE) in their lowest lying triplet excited state as dihydrophenazines and phenothiazines, which was corroborated experimentally.21 Although dihydrophenazines are stronger excited state reductants, the radical cations of phenoxazines and phenothiazines [E0(2PC•+/PC) = ∼0.5 V vs SCE] are more oxidizing than those of dihydrophenazines [E0(2PC•+/PC) = ∼0.0 V vs SCE]; all three classes of PCs possess an oxidation potential capable of deactivating the propagating radical (e.g., ∼ −0.8 V vs SCE for methyl methacrylate), as required for a successful O-ATRP. Lastly, reports on the photophysical properties of phenoxazines suggested their promise as PCs; the phosphorescence quantum yield of 10-phenylphenoxazine (1) at 77 K was reported to be 94% with a lifetime as long as 2.3 s.22 These properties highlight the efficient ISC to the triplet manifold and slow non-radiative decay attributed to small Franck–Condon vibrational overlap factors between 3PC* and the ground state.
Our analysis of exchanging the sulfur in phenothiazines with the oxygen in phenoxazines identified several distinct phenomena that alter the physical properties of these molecules and which we propose manifest in improvements in PC performance for O-ATRP, qualitatively assessed through analysis of the polymer product. The significant distinction between these two systems is the conformation of their heterocyclic rings. The smaller van der Waals radius of oxygen (1.52 Å) relative to sulfur (1.80 Å) permits the ground state phenoxazine (e.g., PC 1) to access a planar geometry similarly to dihydrophenazines (nitrogen, 1.55, Å). In contrast, phenothiazine adopts a bent boat conformation in its ground state, observable in crystal structures23 and predicted by our computations (Figure 2). However, upon oxidation to the radical cation state 2PC•+, all three PCs adopt a planar conformation.
Figure 2.
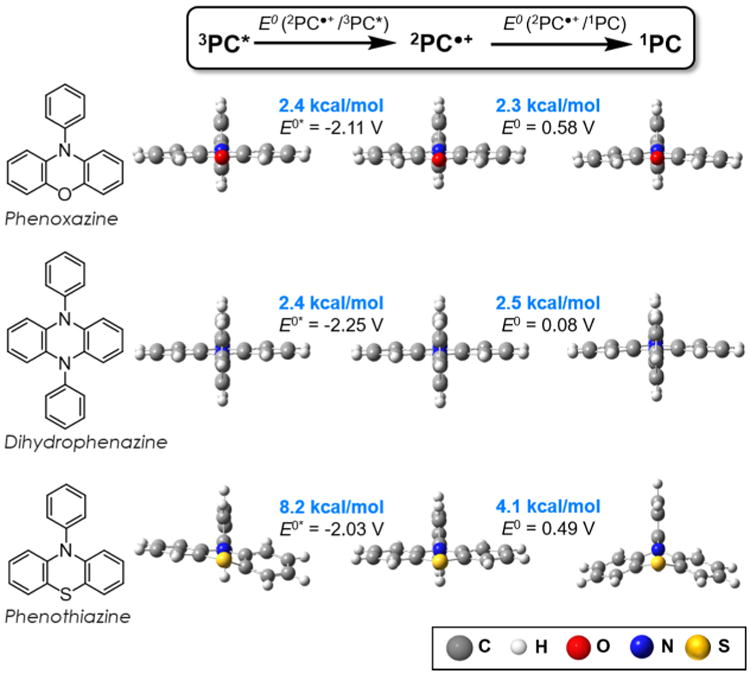
Geometric reorganization energies and reduction potentials (vs SCE) for 10-phenylphenoxazine, diphenyl dihydrophenazine, and 10-phenylphenothiazine (bottom) transitioning from the 3PC* to 2PC•+ to 1PC species involved in the proposed mechanism for photoredox O-ATRP. Reduction potentials were computed here with the improved 6-311+G** basis set compared to 6-31+G** used in the previous report.17 See Supporting Information for further details.
The consequences of phenothiazine adopting bent conformations in the ground and triplet states, but a planar geometry in the radical cation state, introduce larger structural reorganizations during electron transfer (ET) as compared to the consistently planar phenoxazines and dihydrophenazines. We calculated a structural reorganization penalty associated with oxidation of the bent 10-phenylphenothiazine triplet state to the planar radical cation of 8.2 kcal/mol. In contrast, the triplet and radical cation states of 1 are both planar, analogous to diaryl dihydrophenazines, which results in a lower reorganization energy of only 2.4 kcal/mol. As phenoxazine, dihydrophenazine, and phenothiazine derivatives, possess similar E0*s (−2.11, −2.25, and −2.03 V, respectively), we expect a kinetically faster activation (reduction of the alkyl bromide) in O-ATRP by phenoxazines and dihydrophenazines because of their lower reorganization energies for ET.
Polymerization deactivation involves reduction of the planar phenylphenothiazine radical cation to regenerate the bent ground state. We calculate a reorganization energy for this ET of 4.1 kcal/mol. For 1 or diphenyl dihydrophenazine, the same reduction process requires lower reorganization energies of 2.3 or 2.5 kcal/mol, respectively consistent with the conservation of the planarity of the cation radical and ground states. Given the similar ground state oxidation potentials for the phenoxazine and phenothiazine (0.58 and 0.49 V), the radical cation of 1 is likely kinetically faster in deactivation, which imparts better control in O-ATRP (vide infra). How this concept pertains to the less oxidizing dihydrophenazine 2PC•+ requires further investigation, although previous results demonstrated that dihydrophenazines are efficient PCs for O-ATRP.17
Toward the goal of designing phenoxazines as PCs for O-ATRP we applied the concepts conceived from our previous study of diaryl dihydrophenazines, which revealed that PCs with spatially separated singly occupied molecular orbitals (SOMOs) in their 3PC* state yielded PCs with superior performance in O-ATRP in regards to achieving the highest I* and producing polymers with the lowest Đ. As such, we investigated strongly reducing N-aryl phenoxazines with spatially separated SOMOs (with the lower lying SOMO localized on the phenoxazine core and the higher lying SOMO localized on the aryl substituent) and localized SOMOs (with both SOMOs localized on the phenoxazine core), to evaluate their performance as O-ATRP PCs and determine if this concept extends to phenoxazines (Figure 3).
Figure 3.
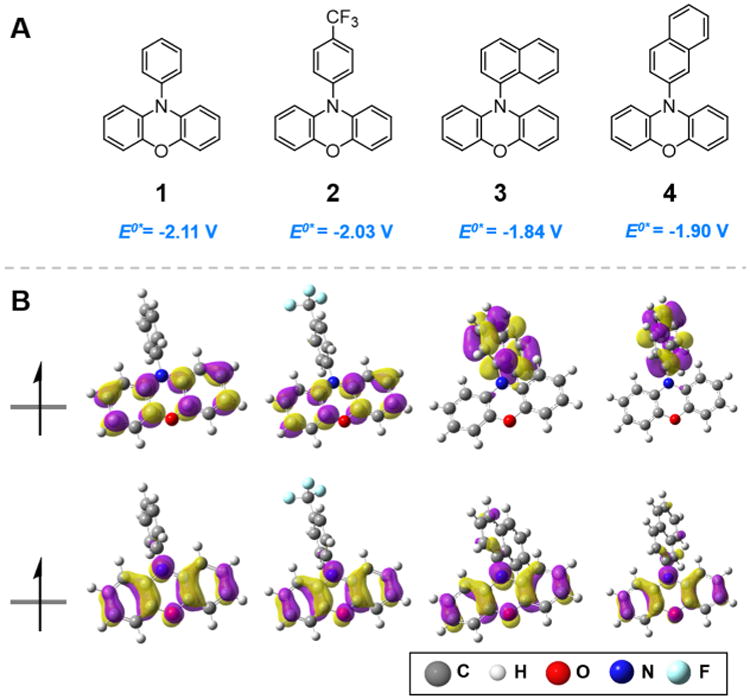
(A) N-Aryl phenoxazines studied in this work along with computed triplet state reduction potentials. (B) Computed triplet state SOMOs of phenoxazine derivatives.
In the cases of diphenyl dihydrophenazine and 1, we calculate that neither exhibits spatially separated SOMOs. In contrast, incorporation of electron withdrawing trifluoromethyl functionalization on the para position of the N-phenyl substituents of the dihydrophenazine yielded spatially separated SOMOs whereas this substitution on phenoxazine (2) results in both SOMOs localized on the phenoxazine core. However, for both dihydrophenazines and phenoxazines, N-aryl functionalization(s) with 1- or 2-naphthalene yielded molecules with spatially separated SOMOs and thus predicted intramolecular charge transfer from the heterocyclic ring to the naphthalene sub-stituent upon photoexcitation and subsequent intersystem crossing to the triplet state.
All four phenoxazine derivatives were synthesized through C–N cross-couplings from commercially available reagents and employed in the polymerization of MMA.21 A screen of common ATRP alkyl bromide initiators revealed that diethyl 2-bromo-2-methylmalonate (DBMM) served as the superior initiator to produce polymers with the lowest Đ while achieving the highest I* (Table S4). To evaluate the PCs, polymerizations using DBMM as the initiator were conducted in dimethylacetamide and irradiated with a 365 nm UV nail curing lamp (54 W) (Table 1). In accord with diaryl dihydrophenazines, N-aryl phenoxazines possessing localized SOMOs (PCs 1 and 2) did not perform as well as the PCs with separated SOMOs (PCs 3 and 4). Specifically, 1 and 2 produced poly(methyl methacrylate) (PMMA) with a relatively high Đ of 1.48 and 1.45, respectively (runs 1 and 2). Polymerization results with PCs 3 and 4 were superior, and produced PMMA with lower dispersities (Đ = 1.22 and 1.11, respectively) while achieving high I*s of 92.6 and 77.3%, respectively (runs 3 and 4).
Table 1. Results of the O-ATRP of MMA Using PCs 1 through 4a.
| run no. | PC | conv (%) | Mw (kDa) | Mn (kDa) | dispersity (Đ) | I* (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | 95.6 | 10.6 | 7.2 | 1.48 | 137 |
| 2 | 2 | 55.3 | 9.5 | 6.5 | 1.45 | 85.5 |
| 3 | 3 | 78.8 | 10.8 | 8.8 | 1.22 | 92.6 |
| 4 | 4 | 80.2 | 11.9 | 10.8 | 1.11 | 77.3 |
[MMA]:[DBMM]:[PC] = [1000]:[10]:[1]; 9.35 μmol PC, 1.00 mL dimethylacetamide, and irradiated with a 54 W 365 nm light source for 8 h.
Further, molecular weight control could be obtained using either PC through modulation of the monomer (runs 5 to 9 for PC 3; runs 13 to 17 for PC 4) or initiator (runs 10 to 12 for PC 3; runs 18 to 20 for PC 4) ratios (Table 2). Overall, PC 3 produced PMMA through higher I* (∼80–100%) while PC 4 produced PMMA with lower Đ (as low as 1.07). This 1-naphthalene versus 2-naphthalene substitution effect influencing high I* or low Đ, respectively was also observed with diaryl dihydrophenazines.
Table 2. Results of the O-ATRP of MMA Using PCs 3 and 4a.
| run no. | PC | [MMA]:[DBMM]:[PC] | conv (%) | Mw (kDa) | Mn (kDa) | dispersity (Đ) | I* (%) |
|---|---|---|---|---|---|---|---|
| 5 | 3 | [500]:[10]:[1] | 80.8 | 5.8 | 4.9 | 1.16 | 86.1 |
| 6 | 3 | [1000]:[10]:[1] | 78.8 | 10.8 | 8.8 | 1.22 | 92.6 |
| 7 | 3 | [1500]:[10]:[1] | 72.2 | 11.4 | 9.5 | 1.19 | 116 |
| 8 | 3 | [2000]:[10]:[1] | 76.5 | 18.4 | 14.6 | 1.26 | 107 |
| 9 | 3 | [2500]:[10]:[1] | 78.4 | 25.9 | 19.8 | 1.31 | 101 |
| 10 | 3 | [1000]:[5]:[1] | 74.6 | 26.4 | 19.1 | 1.38 | 79.6 |
| 11 | 3 | [1000]:[15]:[1] | 74.5 | 8.3 | 6.9 | 1.20 | 75.7 |
| 12 | 3 | [1000]:[20]:[1] | 80.7 | 5.5 | 4.6 | 1.19 | 92.9 |
| 13 | 4 | [500]:[10]:[1] | 85.1 | 5.9 | 5.4 | 1.09 | 84.1 |
| 14 | 4 | [1000]:[10]:[1] | 80.2 | 11.9 | 10.7 | 1.11 | 77.3 |
| 15 | 4 | [1500]:[10]:[1] | 68.9 | 12.2 | 9.8 | 1.25 | 109 |
| 16 | 4 | [2000]:[10]:[1] | 58.2 | 14.7 | 112.5 | 1.17 | 95.2 |
| 17 | 4 | [2500]:[10]:[1] | 65.9 | 21.2 | 17.3 | 1.23 | 96.6 |
| 18 | 4 | [1000]:[5]:[1] | 70.5 | 22.3 | 16.8 | 1.35 | 85.3 |
| 19 | 4 | [1000]:[15]:[1] | 70.9 | 9.3 | 8.3 | 1.12 | 60.6 |
| 20 | 4 | [1000]:[20]:[1] | 76.1 | 6.8 | 6.1 | 1.07 | 64.0 |
See the Supporting Information for experimental details.
Our analysis of the polymerization of MMA by 3 and 4 showed that both PCs imparted control over the polymerization that is becoming expected from O-ATRP. Specifically, a linear growth in polymer molecular weight as well as a low dispersity during the course of polymerization was attained (Figure 4A and B). Additionally, temporal control was demonstrated using a pulsed irradiation sequence (Figure 4C–F). Monomer conversion was only observed during irradiation, which resulted in a linear increase in number-average MW (Mn) while producing PMMA with low Đ.
Figure 4.
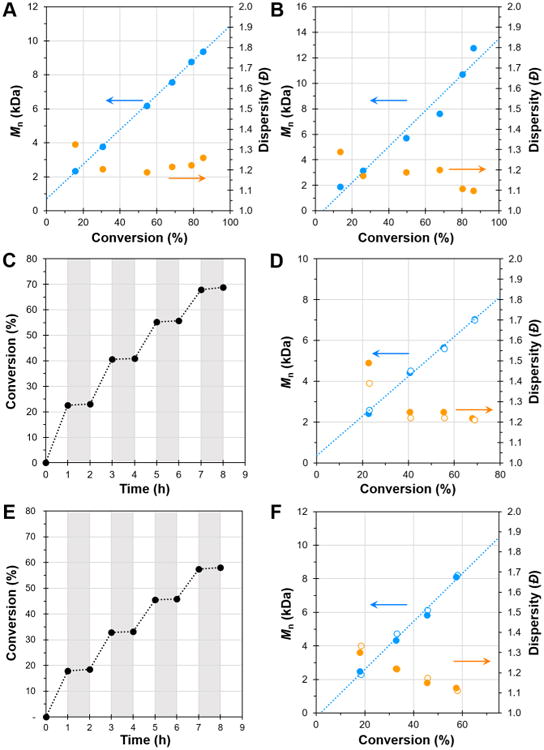
Plots of molecular weight (Mn, blue) and dispersity (Đ, orange) as a function of monomer conversion for the polymerization of MMA catalyzed by 3 (A) and 4 (B). Plots of conversion vs time using 3 (C) or 4 (E) (irradiation in white and dark periods in gray) and plots of molecular weight (Mn, blue) and dispersity (Đ, orange) as a function of MMA conversion using a pulsed-irradiation sequence and PC 3 (D) or 4 (F) (filled markers are data directly after irradiation while open markers are data directly after the dark period) Conditions for all plots: [MMA]:[DBMM]: [PC] = [1000]:[10]:[1]; 9.35 μmol PC, 1.00 mL dimethylacetamide, and irradiated with UV-light.
Both PCs also efficiently polymerized other methacrylates, including benzyl methacrylate (BnMA), isobutyl methacrylate (BMA), and isododecyl methacrylate (IDMA) (Table S2). As such, 3 was used to perform chain extension polymerizations from an isolated PMMA (Mw = 9.9 kDa, Đ = 1.12) macroinitiator because the ATRP mechanism inherently reinstalls the bromine chain end group onto the growing polymer chain (Figure 5). Chain extensions from this PMMA macroinitiator with MMA, DMA, BnMA, and BMA were successful, both confirming high bromine chain end group fidelity and allowing the synthesis of block polymers.
Figure 5.
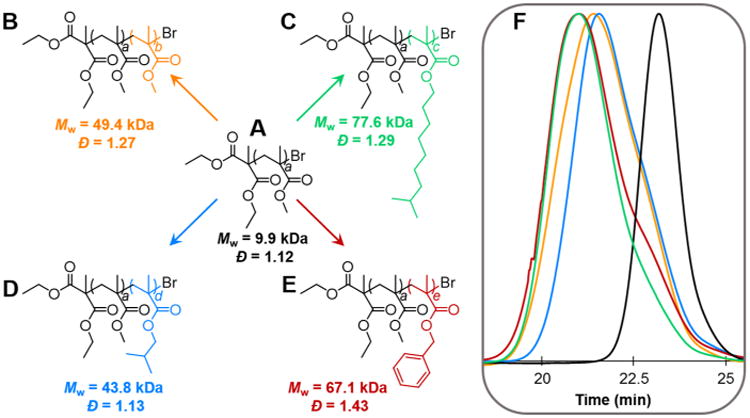
Chain extension polymerizations from a PMMA macroinitiator (A) with MMA (B), IDMA (C), BMA (D), and BnMA (E). Gel permeation chromatography traces of the polymers depicted by the chemical structures with corresponding color schemes (F).
To further establish these naphthalene phenoxazines as efficient PCs, we next directly compared 3 and 1-naphthalene-10-phenothiazine as PCs for O-ATRP under our polymerization conditions (Figure S19). Both catalysts exhibited nearly identical rates of polymerization, achieving 85.1% and 88.4% monomer conversion after 10 h for 3 and the phenothiazine, respectively. Additionally, both PCs achieved high I*s of 93.5% and 95.6%, respectively. However, a significant difference in polymerization performance was observed when comparing the Đ of the resulting PMMA. When using 3, PMMA was produced with Đ = 1.26, while the phenothiazine produced PMMA with comparatively higher Đ = 1.66, consistent with previous reports16a,b using this PC.
As inferred above, the higher Đ of the PMMA produced by the phenothiazine is attributed to the larger reorganization energies of the phenothiazines. Incorporation of O versus S in the core of phenoxazines versus the core of phenothiazines imparts distinct quantitative differences in the electronic and geometric structures of these molecules that affect their performance as PCs for O-ATRP. As such, the planarity of phenoxazines throughout the photoexcitation and ET processes causes them to perform more closely to diaryl dihydrophenazines as PCs for O-ATRP. We hypothesize that the differences between these PCs specifically manifest in each of their abilities to balance the rates of activation and deactivation which results in the differences observed in the Đ of the resulting PMMA produced by each PC.
An additional consideration when comparing phenoxazines, dihydrophenazines, and phenothiazines is that the planar core of phenoxazines and dihydrophenazines promotes intramolecular charge transfer to charge separated SOMOs while the bent phenothiazine core limits electronic coupling between the heterocyclic ring and the N-aryl substituent and consequently the ability to form an intramolecular charge transfer complex.24 The planar phenoxazine core versus the bent phenothiazine core can be visualized in the X-ray crystal structures of the PCs (Figure 6). The electrostatic potential (ESP) mapped electron density of the 3PC* state of these compounds reveal that electron density is transferred to the naphthalene substituent (red region) in phenoxazine upon photoexcitation and ISC from 1PC, even more so with dihydrophenazines, while electron density remains localized on the phenothiazine core (Figure 7).
Figure 6.
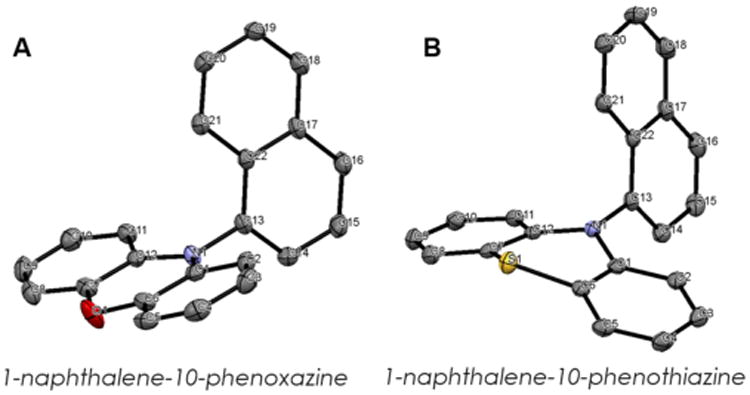
X-ray crystal structures of 1-naphthalene substituted planar phenoxazine (A) and bent phenothiazine (B). Hydrogen atoms
Figure 7.
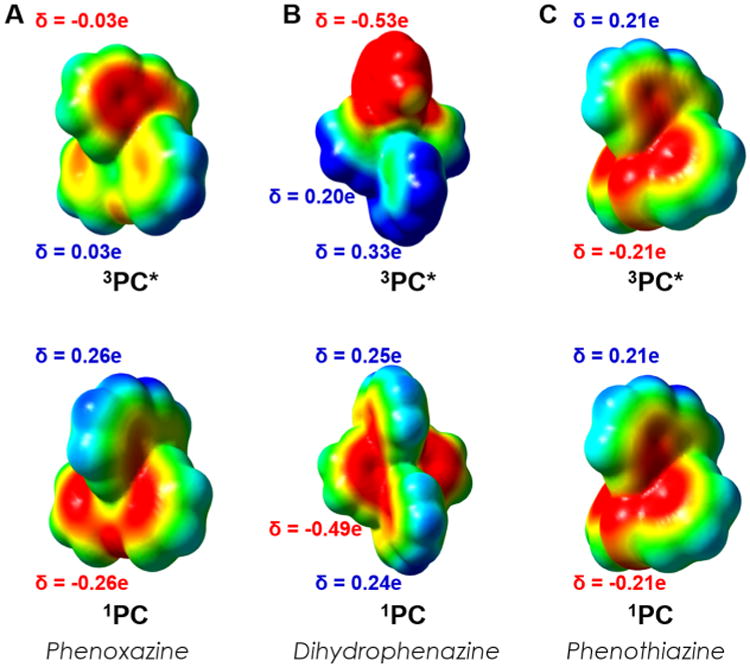
ESP mapped electron density of 1PC and 3PC* of 1-naphthalene substituted phenoxazine (A), dihydrophenazine (B), and phenothiazine (C).
We further envisaged that a visible light absorbing phenoxazine derivative would provide an even more efficient polymerization catalyst, as irradiation of the reaction with high energy UV-light can initiate nondesirable reaction pathways, which may increase the Đ of the produced polymer and lower I*.25 To realize a visible light absorbing PC we explored a core substituted phenoxazine derivative. Computations predicted that PC 5, possessing 4-biphenyl core substitutions, would be an excellent target PC with 3PC* possessing a strong reduction potential and spatially separated SOMOs, while 1PC would exhibit an absorbance profile in the visible spectrum. The visible light absorbing PC 5 was synthesized in high yield from PC 3 through selective bromination at the 3- and 7-positions on the phenoxazine core using N-bromosuccinimide followed by Suzuki cross-coupling.21 A similar synthetic strategy was recently reported to synthesize thiophene core substituted phenothiazines for use as visible light absorbing catalysts for cationic polymerization.26 The absorbance profile of PC 5 was not only red-shifted (Δλmax = 65 nm versus noncore substituted PC 3) into the visible spectrum (λmax = 388 nm), but also exhibited an extremely enhanced molar extinction coefficient (ε = 26635 M−1cm−1 at λmax = 388 nm), making it significantly more efficient at absorbing visible light than the noncore substituted 1-napthalene functionalized phenoxazine, dihydrophenazine, or phenothiazine (Figure 8).
Figure 8.
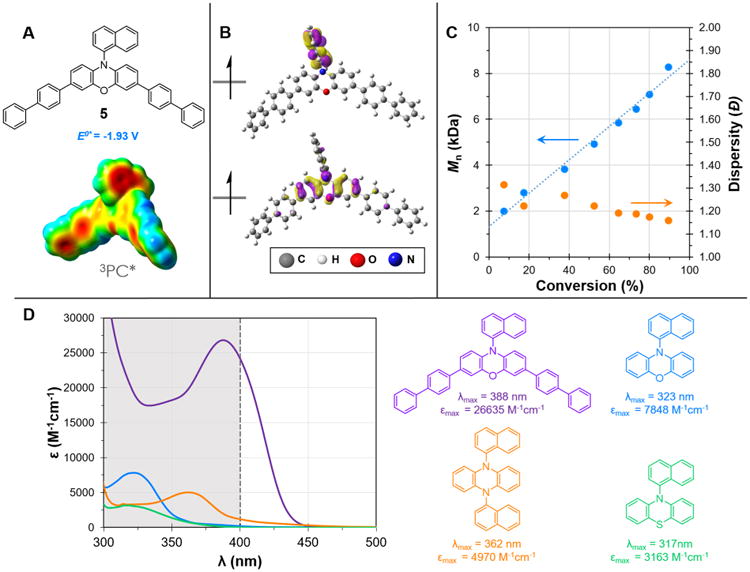
Properties of PC 5. (A) Structure, computed triplet excited state reduction potential, and ESP mapped electron density of 3PC* 5. (B) Computed triplet state SOMOs of PC 5. (C) Plot of Mn and Đ as a function of monomer conversion for the polymerization of MMA by PC 5; [MMA]:[DBMM]:[5] = [1000]:[10]:[1]; 9.35 μmol PC, 1.00 mL dimethylacetamide, and irradiated with white LEDs. (D) UV–vis spectrum of PC 5 and 1-naphthalene functionalized phenoxazine, dihydrophenazine, and phenothiazine, with color coded structures, and extinction coefficients at their respective λmax with the visible absorbance spectrum highlighted in white.
The polymerization performance of PC 5 confirmed our predictions that it would be an excellent PC for O-ATRP, demonstrating superior control over the polymerization than the UV-absorbing phenoxazines or even previously reported dihydrophenazines. The polymerization of MMA using PC 5 irradiated by white LEDs was efficient and showcased characteristics of a controlled polymerization with a linear increase in polymer Mn and a low polymer Đ during the course of polymerization (Figure 8C). Furthermore, the molecular weight of the polymer could be tailored through manipulation of either the monomer or initiator loading, while keeping the polymerization otherwise constant, to produce polymers with Đ of 1.13–1.31 while achieving quantitative I* (Table 3).
Table 3. Results of the O-ATRP of MMA Using PC 5a.
| run no. | [MMA]:[DBMM]:[5] | conv (%) | Mw (kDa) | Mn (kDa) | Dispersity (Đ) | I* (%) |
|---|---|---|---|---|---|---|
| 21 | [500]:[10]:[1] | 67.2 | 4.07 | 3.64 | 1.13 | 99.4 |
| 22 | [1500]:[10]:[1] | 75.2 | 13.7 | 11.8 | 1.16 | 98.0 |
| 23 | [2000]:[10]:[1] | 90.9 | 22.9 | 17.5 | 1.31 | 105 |
| 24 | [2500]:[10]:[1] | 87.5 | 27.5 | 21.3 | 1.29 | 104 |
| 25 | [1000]:[5]:[1] | 89.9 | 23.0 | 18.1 | 1.27 | 101 |
| 26 | [1000]:[15]:[1] | 73.8 | 6.17 | 5.31 | 1.16 | 97.5 |
| 27 | [1000]:[20]:[1] | 72.1 | 4.52 | 3.76 | 1.20 | 103 |
See the Supporting Information for experimental details.
Conclusions
N-Aryl phenoxazines have proven to be efficient PCs for O-ATRP that produce polymers with controlled molecular weights and low dispersity. Through the culmination of computational and experimental results, we report a visible light absorbing phenoxazine photoredox catalyst that produces polymers with controlled molecular weights and low dispersities, achieving quantitative initiator efficiencies that out-compete previously reported organic PCs for O-ATRP. The continued establishment of design principles for PCs capable of mediating O-ATRP will further expand the scope and impact of this polymerization methodology, which we foresee will translate to an additional means for selective small molecule transformations. Our future work will investigate the intricacies of the charge transfer state that is responsible for efficient photoredox catalysis, which we hypothesize provides extended excited state lifetimes and minimizes undesirable back electron transfer.
Supplementary Material
Materials and methods; characterization of photoredox properties; computational details; polymerization data; X-ray crystallography data; coordinates of molecular structures (PDF)
Crystal data for 1-naphthalene phenothiazine (CIF)
Crystal data for 1-naphthalene phenoxazine (CIF)
Acknowledgments
This work was supported by the University of Colorado Boulder, the Advanced Research Projects Agency-Energy (DE-AR0000683), and the American Chemical Society Petroleum Research Fund (56501-DNI7). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. BGM is supported by the University of Colorado Department of Chemistry and Biochemistry Marian Sharrah Fellowship. CBM was supported by NSF grant CHE-1214131. We gratefully acknowledge the use of XSEDE supercomputing resources (NSF ACI-1053575). The authors would like to thank Steven Satur (CU Boulder) for technical assistance.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b08068.
Notes: The authors declare no competing financial interest.
References
- 1.(a) Matyjaszewski K, Tsarevsky NV. J Am Chem Soc. 2014;136:6513–6533. doi: 10.1021/ja408069v. [DOI] [PubMed] [Google Scholar]; (b) Matyjaszewski K, Tsarevsky NV. Nat Chem. 2009;1:276–288. doi: 10.1038/nchem.257. [DOI] [PubMed] [Google Scholar]; (c) Kamigaito M, Ando T, Sawamoto M. Chem Rev. 2001;101:3689–3745. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 2.(a) Boyer C, Corrigan NA, Jung K, Nguyen D, Nguyen TK, Adnan NNM, Oliver S, Shanmugam S, Yeow J. Chem Rev. 2016;116:1803–1949. doi: 10.1021/acs.chemrev.5b00396. [DOI] [PubMed] [Google Scholar]; (b) Ouchi M, Terashima, Sawamoto M. Chem Rev. 2009;109:4963–5050. doi: 10.1021/cr900234b. [DOI] [PubMed] [Google Scholar]; (c) Rosen BM, Percec V. Chem Rev. 2009;109:5069–5119. doi: 10.1021/cr900024j. [DOI] [PubMed] [Google Scholar]
- 3.(a) Tsarevsky NV, Matyjaszewski K. Chem Rev. 2007;107:2270–2299. doi: 10.1021/cr050947p. [DOI] [PubMed] [Google Scholar]; (b) Ding M, Jiang X, Zhang L, Cheng Z, Zhu X. Macromol Rapid Commun. 2015;36:1702–1721. doi: 10.1002/marc.201500085. [DOI] [PubMed] [Google Scholar]; (c) Matyjaszewski K, Jakubowski W, Min K, Tang W, Huang J, Braunecker WA, Tsarevsky NV. Proc Natl Acad Sci U S A. 2006;103:15309–15314. doi: 10.1073/pnas.0602675103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Shanmugam S, Boyer C. J Am Chem Soc. 2015;137:9988–9999. doi: 10.1021/jacs.5b05903. [DOI] [PubMed] [Google Scholar]; (b) Xu J, Shanmugam S, Boyer C. ACS Macro Lett. 2015;4:926–932. doi: 10.1021/acsmacrolett.5b00460. [DOI] [PubMed] [Google Scholar]; (c) Xu J, Shanmugam S, Duong HT, Boyer C. Polym Chem. 2015;6:5615–5624. [Google Scholar]; (d) Chen M, MacLeod MJ, Johnson JA. ACS Macro Lett. 2015;4:566–569. doi: 10.1021/acsmacrolett.5b00241. [DOI] [PubMed] [Google Scholar]; (e) Ohtsuki A, Lei L, Tanishima M, Goto A, Kaji H. J Am Chem Soc. 2015;137:5610–5617. doi: 10.1021/jacs.5b02617. [DOI] [PubMed] [Google Scholar]; (f) Goto A, Zushi H, Hirai N, Wakada T, Tsujii Y, Fukuda T. J Am Chem Soc. 2007;129:13347–13354. doi: 10.1021/ja0755820. [DOI] [PubMed] [Google Scholar]
- 5.(a) Roth GH, Romero NA, Nicewicz DA. Synlett. 2016;27:714–723. [Google Scholar]; (b) Isse AA, Lin CY, Coote ML, Gennaro A. J Phys Chem B. 2011;115:678–684. doi: 10.1021/jp109613t. [DOI] [PubMed] [Google Scholar]
- 6.(a) Corrigan N, Shanmugam S, Xu J, Boyer C. Chem Soc Rev. 2016 doi: 10.1039/C6CS00185H. [DOI] [PubMed] [Google Scholar]; (b) Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Narayanam JMR, Stephenson CR. J Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (e) Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (f) Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, Von Zelewsky A. Coord Chem Rev. 1988;84:85–277. [Google Scholar]
- 7.(a) Wallentin CJ, Nguyen JD, Finkbeiner P, Stephenson CRJ. J Am Chem Soc. 2012;134:8875–8884. doi: 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]; (b) Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010;12:3104–3107. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]
- 8.(a) Liu X, Zhang L, Cheng Z, Zhu X. Polym Chem. 2016;7:689–700. [Google Scholar]; (b) Zhang G, Song IY, Ahn KH, Park T, Choi W. Macromolecules. 2011;44:7594–7599. [Google Scholar]
- 9.Nguyen JD, Tucker JW, Konieczynska MD, Stephenson CRJ. J Am Chem Soc. 2011;133:4160–4163. doi: 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Knorn M, Rawner T, Czerwieniec R, Reiser O. ACS Catal. 2015;5:5186–5193. [Google Scholar]; (b) Paria S, Reiser O. ChemCatChem. 2014;6:2477–2483. [Google Scholar]; (c) Pirtsch M, Paria S, Matsuno T, Isobe H, Reiser O. Chem - Eur J. 2012;18:7336–7340. doi: 10.1002/chem.201200967. [DOI] [PubMed] [Google Scholar]
- 11.(a) Yang Q, Dumur F, Morlet-Savary F, Poly J, Lalevée J. Macromolecules. 2015;48:1972–1980. [Google Scholar]; (b) Treat NJ, Fors BP, Kramer JW, Christianson M, Chiu CY, Read de Alaniz JR, Hawker CJ. ACS Macro Lett. 2014;6:580–584. doi: 10.1021/mz500242a. [DOI] [PubMed] [Google Scholar]; (c) Fors BP, Hawker CJ. Angew Chem, Int Ed. 2012;51:8850–8853. doi: 10.1002/anie.201203639. [DOI] [PubMed] [Google Scholar]
- 12.(a) Chen M, Zhong M, Johnson JA. Chem Rev. 2016 doi: 10.1021/acs.chemrev.5b00671. [DOI] [PubMed] [Google Scholar]; (b) Pan X, Tasdelen MA, Laun J, Junkers T, Yagci Y, Matyjaszewski K. Prog Polym Sci. 2016 doi: 10.1016/j.progpolymsci.2016.06.005. [DOI] [Google Scholar]; (c) Dadashi-Silab S, Doran S, Yagci Y. Chem Rev. 2016 doi: 10.1021/acs.chemrev.5b00586. [DOI] [PubMed] [Google Scholar]; (d) Zivic N, Bouzrati-Zerelli M, Kermagoret A, Dumur F, Fouassier JP, Gigmes D, Lalevée J. ChemCatChem. 2016;8:1617–1631. [Google Scholar]
- 13.(a) Joshi-Pangu A, Lévesque F, Roth HG, Oliver SF, Campeau LC, Nicewicz D, DiRocco DA. J Org Chem. 2016 doi: 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]; (b) Nicewicz DA, Nguyen TM. ACS Catal. 2014;4:355–360. [Google Scholar]; (c) Ravelli D, Fagnoni M, Albini A. Chem Soc Rev. 2013;42:97–113. doi: 10.1039/c2cs35250h. [DOI] [PubMed] [Google Scholar]; (d) Ravelli D, Fagnoni M. ChemCatChem. 2012;4:169–171. [Google Scholar]; (e) Fagnoni M, Dondi D, Ravelli D, Albini A. Chem Rev. 2007;107:2725–2756. doi: 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]
- 14.Romero NA, Nicewicz DA. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 15.Miyake GM, Theriot JC. Macromolecules. 2014;47:8255–8261. [Google Scholar]
- 16.(a) Yan J, Pan X, Schmitt M, Wang Z, Bockstaller MR, Matyjaszewski K. ACS Macro Lett. 2016;5:661–665. doi: 10.1021/acsmacrolett.6b00295. [DOI] [PubMed] [Google Scholar]; (b) Discekici EH, Pester CW, Treat NJ, Lawrence J, Mattson KM, Narupai B, Toumayan EP, Luo Y, McGrath AJ, Clark PG, Read de Alaniz J, Hawker CJ. ACS Macro Lett. 2016;5:258–262. doi: 10.1021/acsmacrolett.6b00004. [DOI] [PubMed] [Google Scholar]; (c) Pan X, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewski K. J Am Chem Soc. 2016;138:2411–2425. doi: 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]; (d) Pan X, Lamson M, Yan J, Matyjaszewski K. ACS Macro Lett. 2015;4:192–196. doi: 10.1021/mz500834g. [DOI] [PubMed] [Google Scholar]; (e) Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Read de Alaniz J, Fors BP, Hawker CJ. J Am Chem Soc. 2014;136:16096–16101. doi: 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]
- 17.Theriot JC, Lim CH, Yang H, Ryan MD, Musgrave CB, Miyake GM. Science. 2016;352:1082–1086. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- 18.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Laursen JB, Nielsen J. Chem Rev. 2004;104:1663–1686. doi: 10.1021/cr020473j. [DOI] [PubMed] [Google Scholar]; (b) Palmer BD, Rewcastle GW, Atwell GJ, Baguley BC, Denny WA. J Med Chem. 1988;31:707–712. doi: 10.1021/jm00399a003. [DOI] [PubMed] [Google Scholar]
- 20.(a) Tanaka H, Shizu K, Miyazaki H, Adachi C. Chem Commun. 2012;48:11392–111394. doi: 10.1039/c2cc36237f. [DOI] [PubMed] [Google Scholar]; (b) Park Y, Kim B, Lee C, Hyun A, Jang S, Lee JH, Gal YS, Kim TH, Kim KS, Park J. J Phys Chem C. 2011;115:4843–4850. [Google Scholar]; (c) Okamoto T, Terada E, Kozaki M, Uchida M, Kikukawa S, Okada K. Org Lett. 2003;5:373–376. doi: 10.1021/ol0274458. [DOI] [PubMed] [Google Scholar]
- 21.See the Supporting Information.
- 22.Huber JR, Mantulin WW. J Am Chem Soc. 1972;94:3755–3760. [Google Scholar]
- 23.(a) Liu N, Wang B, Chen W, Liu C, Wang X, Hu Y. RSC Adv. 2014;4:51133. [Google Scholar]; (b) Klein JCL, Conrad M, III, Morris SA. Acta Crystallogr, Sect C: Cryst Struct Commun. 1985;C41:1202–1204. [Google Scholar]
- 24.(a) Malińska M, Nowacki J, Kapturkiewicz A, Woźniak K. RSC Adv. 2012;2:4318–4328. [Google Scholar]; (b) Borowicz P, Herbich J, Kapturkiewicz A, Anulewicz-Ostrowska R, Nowacki J, Grampp G. Phys Chem Chem Phys. 2000;2:4275–4280. [Google Scholar]; (c) Borowicz P, Herbich J, Kapturkiewicz A, Opallo M, Nowacki J. Chem Phys. 1999;249:49–62. [Google Scholar]
- 25.(a) Frick E, Anastasaki A, Haddleton DM, Barner-Kowollik CJ. Am Chem Soc. 2015;137:6889–6896. doi: 10.1021/jacs.5b03048. [DOI] [PubMed] [Google Scholar]; (b) Ribelli GG, Konkolewicz D, Bernhard S, Matyjaszewski K. J Am Chem Soc. 2014;136:13303–13312. doi: 10.1021/ja506379s. [DOI] [PubMed] [Google Scholar]
- 26.Chao P, Gu R, Ma X, Wang T, Zhao Y. Polym Chem. 2016 doi: 10.1039/C6PY01095D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods; characterization of photoredox properties; computational details; polymerization data; X-ray crystallography data; coordinates of molecular structures (PDF)
Crystal data for 1-naphthalene phenothiazine (CIF)
Crystal data for 1-naphthalene phenoxazine (CIF)