

Abstract
Changes in the lungs due to smoking include inflammation, epithelial damage, and remodeling of the airways. Airway inflammation is likely to play a critical role in the genesis and progression of tobacco smoke-induced airway disease. Soluble epoxide hydrolase (sEH) is involved in the metabolism of endogenous chemical mediators that play an important role in inflammation. Epoxyeicosatrienoic acids (EETs) have demonstrated antiinflammatory properties, and hydrolysis of these epoxides by sEH is known to diminish this activity. To examine whether acute tobacco smoke-induced inflammation could be reduced by a sEH inhibitor, 12-(3-adamantane-1-yl-ureido)-dodecanoic acid n-butyl ester was given by daily s.c. injection to spontaneously hypertensive rats exposed to filtered air or tobacco smoke for a period of 3 days (6 h/day). Acute exposure to tobacco smoke significantly increased by 3.2-fold (P < 0.05) the number of cells recovered by bronchoalveolar lavage. The sEH inhibitor significantly decreased total bronchoalveolar lavage cell number by 37% in tobacco smoke-exposed rats with significant reductions noted in neutrophils, alveolar macrophages, and lymphocytes. A combination of sEH inhibitor and EETs was more significant in its ability to further reduce tobacco smoke-induced inflammation compared with the sEH inhibitor alone. The sEH inhibitor led to a shift in some plasma epoxides and diols that are consistent with the hypothetical action of these compounds. We conclude that an sEH inhibitor, in the presence or absence of EETs, can attenuate, in part, inflammation associated with acute exposure to tobacco smoke.
Keywords: epoxyeicosatrienoic acids, pulmonary, antiinflammatory
Cigarette smoking is associated with a number of pulmonary diseases including bronchitis, airway obstruction, and emphysema. Collectively these pulmonary maladies are referred to as chronic obstructive pulmonary disease (COPD). COPD is prevalent in ≈20 million men and women in the United States and is the fourth leading cause of death (1). The pathology of chronic bronchitis and COPD includes airway mucus gland hyperplasia, mucous hypersecretion, and an influx of inflammatory cells including neutrophils, macrophages, and lymphocytes (2-6). The genesis of this disease is thought to lie in the inflammatory process induced by tobacco smoke, leading to cell injury, cellular hyperplasia, and occasionally neoplasia. Therefore, it is important for us to understand the process by which tobacco smoke induces inflammation in the lungs.
Soluble epoxide hydrolases (sEH) are enzymes that add water to epoxides forming their corresponding 1,2-diols (7, 8). Diols of linoleate epoxides (DiHOMEs) have been implicated in inflammatory disorders, including adult respiratory distress syndrome (9), and may be endogenous regulators of vascular permeability and inflammation (10). Histopathologic evaluation of Swiss-Webster mouse lung after dosing with 12,13-DiHOME show massive alveolar edema and hemorrhage with interstitial edema in vessel walls of the lung. A 50% mortality was observed at a dose of 100 mg/kg within 6.5 h of administration (through the tail vein). Immunohistochemistry of the lung tissue showed epoxide hydrolase concentrated locally in the smooth muscle of small and medium pulmonary vessels (11). Epoxyeicosatrienoic acids (EETs) are metabolites of arachidonic acids that undergo hydrolysis by sEH to form dihydroxyeicosatrienoic acids (DHETs). There is evidence that EETs can prevent vascular inflammation through inhibition of expression of several cell adhesion molecules (CAMs) including E-selectin, vascular cell adhesion molecule 1, and intercellular adhesion molecule 1 (12) and by preventing leukocyte adhesion to the vascular wall (12-14). The role of sEH and EETs in tobacco smoke-induced inflammation and lung disease has not been investigated.
sEH has been identified as a promising pharmacological target, and inhibition of sEH has been proposed as a novel approach for the treatment of diseases, including hypertension (15, 16). We have previously established a model of acute inflammation in the spontaneously hypertensive (SH) rat (17). We tested in this model whether inflammation associated with short-term exposure to tobacco smoke could be altered with the use of a sEH inhibitor, 12-(3-adamantane-1-yl-ureido)-dodecanoic acid n-butyl ester (AUDA-nBE), in the absence or presence of EETs.
Materials and Methods
Reagents, Chemicals, and Analytical Procedures. AUDA-nBE and 1-cyclohexyl-3-tetradecyl urea, 1-phenyl-2-hexanoic acid urea, 20-hydroxyeicosanoic acid, and 1-cyclohexyl-3-dodecanoic acid urea were synthesized in our laboratory. These products were purified by recrystallization and characterized structurally by [1H]NMR and/or [13C]NMR, infrared, and mass spectroscopy. LC-MS/MS analysis was performed by using a Micromass Quattro Ultima triple quadrupole tandem mass spectrometer (Micromass, Manchester, U.K.) described in Supporting Text, which is published as supporting information on the PNAS web site. Conditions for pharmacokinetic analysis are described in Supporting Text. As presented in detail in Supporting Text, EETs were synthesized by using acrachidonic acid methylester and m-chloro-perbenzoic acid, and the regioisomer ratio was determined with LC-MS based on authentic pure standards purchased from Cayman Chemical (Ann Arbor, MI). The composition of the mixture was 10% of 8(9)-EET, 40% of 11(12)-EET, and 50% of 14(15)-EET.
Oxylipid Analysis. EETs, DHETs, DiHOMEs, and linoleate epoxides (EpHOMEs) were analyzed by LC-MS/MS by using negative mode electrospray ionization with a tandem mass spectral detector (Quattro Ultima, Micromass) operated in a multireaction monitoring mode as described in refs. 18 and 19. The acquired data were quantified with the masslynx software package (Micromass).
Formulation of EETs Wax Plug. To create a wax plug, 5 g of beeswax (Aldrich) was melted at 100°C for 20 min by using a hot plate, and EETs (0.5 mg) were added to the molten wax while stirring. The wax-EETs suspension was then poured into a mold made with glass plates and cooled to room temperature. The resultant wax stick containing EETs was cut to suitable size. No degradation of EETs was observed during this process. Release rates of EETs from the wax plug in vitro were investigated as modified from a reported method (20), with details of the method presented in detail in Supporting Text.
Animals. Healthy, 11-week-old male SH (SHR/NCrlBR) rats (derived from WKY rats by phenotypic segregation of the hypertensive trait and inbreeding) were purchased from Charles River Laboratories (Portage, MI) and quarantined for 1 week before exposure to tobacco smoke. Animals were handled in accordance with standards established by the U.S. Animal Welfare Acts set forth in National Institutes of Health guidelines and the University of California, Davis, Animal Care and Use Committee. The rats were housed in plastic cages with TEK-Chip pelleted paper bedding (Harlan Teklad, Madison, WI) and maintained on a 12 h light/12 h dark cycle. All animals had access to water and Laboratory Rodent Diet 5001 purchased from LabDiet (Brentwood, MO) ad libitum before, during, and after exposures.
Treatment of Animals for Pharmacokinetics Study. Animals were selected for pharmacokinetic studies based on a body weight stratified randomization procedure after a 1- to 2-week acclimation period. The body weight of animals was 250-280 g. A 10 mg/kg of body weight dosing of these inhibitors (7 mg/ml corn oil) were s.c. administered to SH rats. The animals were injected one time with AUDA-nBE, and blood concentrations of sEH inhibitor(s) were determined in the experimental animals after 72 h of exposure to filtered air. Plasma (collection described below) concentrations of sEH inhibitor(s) were determined in animals separate from those used in the above study after exposure to tobacco smoke or filtered air for 3 days with the animals treated daily with s.c. injections of AUDA-nBE (three total injections). Blood from these animals was drawn only at necropsy (immediately after exposure on the third day of the study).
Blood Sample Preparation. After administration, serial tail-bled blood samples (<10 μl) were collected at various time points (30 min to 72 h). Blood samples were transferred to 1.5-ml Eppendorf microcentrifuge tubes. Blood samples were weighed with an analytical balance and vortexed with 100 μl of purified water and 25 μl of internal standard in ethyl acetate (500 ng/ml 1-cyclohexyl-3-tetradecyl urea). The samples were extracted with 500 μl of ethyl acetate. The ethyl acetate layer was transferred to a 1.5-ml Eppendorf microcentrifuge tube and dried under nitrogen. The residue was reconstituted in 25 μl of methanol. Aliquots (10 μl) were injected onto the LC-MS/MS system.
Pharmacokinetics Analysis. Pharmacokinetic parameters were obtained by fitting the blood concentration-time data to a noncompartmental model with winnonlin software (Pharsight, Mountain View, CA). Parameters estimated included lambda z (λz), time of maximum concentration (Tmax), maximum concentration (Cmax), elimination half-life (T1/2), area under the concentration-time curve to terminal time (AUCt), area under the concentration-time curve to infinite time (AUC∞) and the mean residence time (MRT). AUCt was calculated by the linear/log trapezoidal rule.
s.c. Implantation of EETs for Tobacco Smoke Exposures. Wax formulations containing EET regioisomers were implanted s.c. 1 day before onset of exposure to filtered air or tobacco smoke. The total dose of EETs in the wax plug implant was 2.5 mg/kg of body weight. Four animals from the control group and four animals from the tobacco smoke-exposed group were implanted with the slow-release formulation of EETs. The approach of a single s.c. implantation for the 3-day study was selected to minimize stress to animals from anesthesia.
s.c. Injection of AUDA-nBE for Tobacco Smoke Exposures. AUDA-nBE (7 mg/ml corn oil) was s.c. administered in SH rats at a dose of 10 mg/kg of body weight. The total volume injected was between 0.36 and 0.46 ml. Animals were injected with AUDA-nBE each day before exposure to tobacco smoke. Doses of AUDA-nBE used in this study were selected based on results from preliminary pharmacokinetic studies in mice and rats. These doses were selected to provide optimal efficacy and minimal toxicity over a 3-day period. Four animals from the control group and four animals from the tobacco smoke-exposed group were injected with AUDA-nBE. Four animals with EET implants from the control group and four animals with EET implants from the tobacco smoke-exposed group were injected with AUDA-nBE. In addition, four control animals and four tobacco smoke-exposed animals were injected with corn oil by using the same protocol as animals injected with AUDA-nBE.
Tobacco Smoke Exposure. Rats were exposed to a mixture of sidestream and mainstream cigarette smoke in a smoking apparatus built in our laboratory (21). The cigarettes were humidified 2R4F research cigarettes (Tobacco Health Research Institute; Lexington, KY). An automatic metered puffer was used to smoke the cigarettes under Federal Trade Commission conditions (35-ml puff, 2-sec duration, 1 puff per min). The smoke was collected in a chimney, diluted with filtered air, and delivered to whole-body exposure chambers. The exposures were characterized for three major constituents of cigarette smoke: nicotine, carbon monoxide, and total suspended particulates (TSP). Animals were exposed for 6 h/day for 3 days. Carbon monoxide was measured every 30 min, total suspended particulates every 2 h, and nicotine daily (about midway through the exposure period). Data for smoke exposure characteristics are presented as mean ± SD.
Bronchoalveolar Lavage (BAL). Established protocols were followed for BAL of animals (22). Eighteen hours after the last exposure to tobacco smoke, animals were anesthetized with an overdose of sodium pentobarbital. This timing was used to ensure a robust inflammatory response. The trachea was cannulated and the lung lavaged with one aliquot of Ca2+/Mg2+-free PBS (pH 7.4). The volume of the aliquot was equal to 35 ml/kg of body weight (≈90% of total lung capacity). The aliquot was instilled into the lungs three times before final collection. The BAL fluid (BALF) was immediately centrifuged at 250 × g for 10 min at 4°C to remove cells. The cell pellet was then resuspended in PBS, and the cells were counted with a hemocytometer. Cell differentials were performed on cytospin preparations (Shandon, Pittsburgh) stained with Hema 3 (Fisher Scientific). Macrophages, neutrophils, lymphocytes, and eosinophils were counted by using light microscopy (1,000 cells per sample).
Plasma. Additional rats were exposed to filtered air or tobacco smoke for 3 days with treatments and exposures as described above, and blood was collected at necropsy for measurement of levels of oxylipids and sEH inhibitor(s) in plasma. Immediately after the last exposure to tobacco smoke, animals were anesthetized with an overdose of sodium pentobarbital, and blood was collected from the caudal vena cava and placed in Vacutainer tubes containing EDTA. This timing was used to ensure that epoxy and diol levels were above limits of detection for the instrumentation. The tubes were placed on ice until collection was completed, then centrifuged at 2,000 × g for 20 min for plasma separation. Plasma was prepared as follows for oxylipid analysis. Eppendorf tubes (2 ml) were spiked with 10 μl of antioxidant solution [0.01 mg of butylated hydroxytoluene (BHT) and EDTA], and 1.5 ml aliquots of plasma were portioned into the tubes and immediately stored at -80°C until oxylipid analysis.
Extraction of Oxylipids from Plasma. Plasma samples were thawed to room temperature. Aliquots (250 μL) were spiked with 10 μl of surrogate solution containing dihydroxynonadecanoic acid and epoxyheptadecanoic acid, then extracted by 60 mg of Oasis HLB (Waters) solid phase extraction cartridges (SPE) according to the following regimen: SPE were washed and preconditioned with 2 ml of HPLC-grade methanol (VWR Scientific) and 2 ml of 2.5 mM H2PO4 with 10% methanol (pH 3.8). Samples were loaded and 250 μl of the phosphoric acid/10% methanol was added. Samples were allowed to extract by gravity, followed by a 2-ml wash with the phosphoric acid/methanol solution. Cartridges were air-dried 15 min by gentle vacuum and then eluted by gravity drip with 2 ml of ethyl acetate (VWR Scientific). Eluate was brought to dryness by evaporation under a gentle blanket of nitrogen and reconstituted in 100 μl of methanol containing 1-phenyl-2-hexanoic acid urea, 20-hydroxyeicosanoic acid, and 1-cyclohexyl-3-dodecanoic acid urea as internal standards for oxylipid analysis by LC-MS/MS.
Data Analysis. All numerical BAL data were calculated as mean ± SD. Comparisons between groups were made by analysis of variance followed by Fisher's protected least significant difference posttest. A P < 0.05 was considered significant. Statistical analysis was performed with statview 5.0.1 (SAS Institute, Cary, NC). Comparisons of mean oxylipid concentrations in plasma were made between groups by using a two-tailed t test, assuming equal variance, with P < 0.05 considered significant.
Results
Release Rate of EETs from Wax Plug in Vitro. The release rate of EETs into water from wax plugs was estimated to be 10 ± 0.4 μg/day.
Tobacco Smoke Exposure Characteristics. Total suspended particulate levels in tobacco smoke during the 3-day study were 76.4 ± 16.0 mg/m3, nicotine levels were 6.8 ± 0.2 mg/m3, and carbon monoxide concentration was 234 ± 2 ppm.
Pharmacokinetics. To estimate blood concentration of AUDA-nBE and 12-(3-adamantane-1-yl-ureido)-dodecanoic acid (AUDA) in SH rats, a pharmacokinetic study was performed with a single dose. Fig. 1 shows blood concentration-time profiles of AUDA-nBE and AUDA in SH rats after s.c. administration of 10 mg/kg AUDA-nBE. AUDA-nBE was rapidly metabolized to AUDA, which is an equally potent inhibitor of sEH. Thus, AUDA-nBE is a pro-drug for AUDA to improve bioavailability, although both compounds show a low nanomolar Ki with the purified recombinant sEH.
Fig. 1.
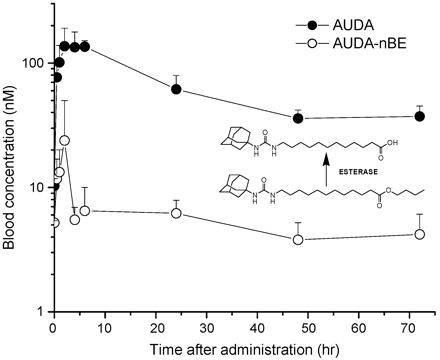
Blood concentration-time profiles of AUDA-nBE and AUDA in SH rats after s.c. administration of 10 mg/kg AUDA-nBE. Data represent the mean ± SD (n = 3 animals). Inset shows structures of the parent (AUDA-nBE) and major active metabolite (AUDA). PK parameters were obtained by fitting the blood concentration-time data to a noncompartmental model. PK parameters are as follows: λz (1/h), 0.0309; Tmax (h), 2.00; Cmax (nM), 136.6; T1/2 (h), 22.4; area under the concentration-time curve to terminal time (AUCt) (nM·h), 4,540; mean residence time (hr), 25.1. The concentration of AUDA after 72 h was 37.3 nM after a single s.c. administration. AUDA concentrations after 3 days of exposure to filtered air or tobacco smoke in rats treated with daily (total of three) s.c. injections of AUDA-nBE were as follows (mean ± SD): filtered air, AUDA-nBE, and EETs, 208 ± 16 nM; filtered air and AUDA-nBE, 152 ± 23 nM; tobacco smoke, AUDA-nBE, and EETs, 298 ± 126 nM; tobacco smoke and AUDA-nBE, 325 ± 117 nM (the last two values had a single high value).
BAL. The volume of BALF recovered was not significantly different between treatment/exposure groups. The total number of cells in the BALF was increased significantly after 3 days of tobacco smoke exposure. s.c. injection of AUDA-nBE before smoke exposure significantly decreased the number of BALF cells compared with treatment with the vehicle (Fig. 2A), and this result was further decreased by treatment of animals with both AUDA-nBE and EETs before tobacco smoke exposure. The number of BALF macrophages was increased significantly after 3 days of tobacco smoke exposure (Fig. 2B) but this result was significantly reduced by AUDA-nBE given before smoke exposure. Treatment with both AUDA-nBE and EETs did not further decrease macrophage number (Fig. 2B). Neutrophil numbers in BALF were also significantly increased after 3 days of tobacco smoke exposure (Fig. 2C) but this result was significantly reduced by AUDA-nBE given before tobacco smoke exposure. Combined treatment with AUDA-nBE and EETs further reduced the number of neutrophils recovered by lavage. Lymphocytes were significantly increased in BALF after exposure to tobacco smoke for 3 days (Fig. 2D), but this result was significantly decreased by AUDA-nBE treatment before smoke exposure to levels similar to those observed in filtered-air control animals. Treatment of animals with AUDA-nBE and EETs before exposure to tobacco smoke did not result in further reduction of BAL lymphocyte numbers compared with treatment with only AUDA-nBE. Numbers of eosinophils were increased in the BALF after 3 days of tobacco smoke exposure but not to a statistically significant level (Fig. 2E). Of note was that the number of total BALF cells and macrophages from rats treated with AUDA-nBE and EETs before exposure to filtered air was significantly lower compared with those recovered from animals treated only with vehicle before exposure to filtered air (Fig. 2 A and B). A similar trend was observed for BAL lymphocytes in animals exposed to filtered air, but statistical significance was not obtained (Fig. 2D). Cell differentials are shown as percentages in Table 1 and exhibit similar trends to the numbers of different cell types recovered in BALF.
Fig. 2.

Bronchoalveolar lavage cell characteristics after exposure of rats to filtered air or tobacco smoke. Numbers of total cells (A), macrophages (B), neutrophils (C), lymphocytes (D), and eosinophils (E) in BAL from rats exposed to filtered air or tobacco smoke for 3 days. Rats were exposed to filtered air (gray bars) after treatment with vehicle, AUDA-nBE, or AUDA-nBE and EETs. Additional rats were exposed to tobacco smoke (black bars) after treatment with vehicle, AUDA-nBE, or AUDA-nBE and EETs. Data are presented as mean ± SD (n = 4). †, P < 0.05 compared with respective filtered air control. ‡, P < 0.05 compared with tobacco smoke and vehicle. §, P < 0.05 compared with tobacco smoke and AUDA-nBE. ¶, P < 0.05 compared with filtered air and vehicle. No eosinophils were observed in BAL from animals treated with vehicle before exposure to filtered air or from animals treated with AUDA-nBE and EETs before exposure to tobacco smoke.
Table 1. Cell differentials in BAL after 3 days of tobacco smoke exposure in rats.
| Vehicle (corn oil)
|
AUDA-nBE
|
AUDA-nBE + EETs
|
||||
|---|---|---|---|---|---|---|
| Filtered air | Tobacco smoke | Filtered air | Tobacco smoke | Filtered air | Tobacco smoke | |
| Macrophages, % | 90.2 ± 2.9 | 48.7 ± 6.9†§ | 92.1 ± 3.2 | 62.9 ± 3.6†‡ | 93.4 ± 3.9 | 72.7 ± 6.8†‡§ |
| Neutrophils, % | 9.0 ± 2.7 | 50.7 ± 6.8†§ | 7.3 ± 3.5 | 36.8 ± 3.2†‡ | 6.2 ± 3.7 | 27.1 ± 6.8†‡§ |
| Lymphocytes, % | 0.80 ± 0.33 | 0.60 ± 0.23§ | 0.55 ± 0.30 | 0.25 ± 0.19‡ | 0.40 ± 0.16¶ | 0.25 ± 0.10‡ |
| Eosinophils, % | 0.00 ± 0.00 | 0.05 ± 0.10 | 0.15 ± 0.19 | 0.10 ± 0.20 | 0.10 ± 0.20 | 0.00 ± 0.00 |
Data are presented as mean ± SD (n = 4). †, P < 0.05 compared with respective filtered air control. ‡, P < 0.05 compared with tobacco smoke plus vehicle. §, P < 0.05 compared with tobacco smoke plus AUDA-nBE. ¶, P < 0.05 compared with filtered air plus vehicle.
Oxylipids in Plasma. 12(13)-EpOME and 9(10)-EpOME (linoleate epoxides), 14(15)-, 11(12)-, 8(9)-, and 5(6)-EET (arachidonate epoxides), 12,13-DiHOME and 9,10-DiHOME (linoleate diols), and 14,15-, 11,12-, 8,9-, and 5,6-DHET (arachidonate diols) concentrations were analyzed in plasma. Internal standard average recoveries ranged between 94% and 100% with the percent of relative SD between 10% and 14%. The diol and epoxy surrogates had average recoveries of 62 and 98% with the percent of relative SD of 22% and 18%, respectively. Control criteria for precision within 25% was met for surrogate recoveries. Arachidonate diol and epoxide concentrations in plasma ranged from 1.03 to 5.17 nM and from 0.00 to 5.05 nM, respectively (Table 2). Linoleate diol and epoxide concentrations ranged from 11.4 to 84.1 nM and from 3.26 to 35.1 nM, respectively. BALF was also extracted for oxylipids according to plasma protocol, but values for many analytes were near or below detection limits. Further method development is necessary for determining oxylipids in BALF.
Table 2. Epoxy and diol oxylipid concentrations in rat plasma after 3 days of tobacco smoke exposure.
| 12(13)-EpOME | 9(10)-EpOME | 14(15)-EET | 11(12)-EET | 8(9)-EET | 5(6)-EET | 12,13-DiHOME | 9,10-DiHOME | 14,15-DHET | 11,12-DHET | 8,9-DHET | 5,6-DHET | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Filtered air | ||||||||||||
| Vehicle | 23.7 ± 3.2 | 10.9 ± 1.4 | 1.91 ± 0.17 | 2.54 ± 0.76 | 0.390 ± 0.46 | 5.05 ± 1.8 | 16.4 ± 0.50 | 17.1 ± 2.8 | 1.31 ± 0.32 | 1.41 ± 0.18 | 3.47 ± 0.15 | 1.19 ± 0.19 |
| AUDA-nBE | 24.2 ± 1.9 | 8.74 ± 0.58† | 2.28 ± 0.46 | 0.670 ± 1.2† | 0.00 ± 0.00 | 4.06 ± 0.67 | 13.5 ± 3.5 | 16.9 ± 2.5 | 1.03 ± 0.15 | 1.67 ± 0.41 | 4.51 ± 1.5 | 1.88 ± 0.37† |
| AUDA-nBE + EETs | 29.2 ± 7.5 | 9.49 ± 2.2 | 2.76 ± 0.35† | 1.78 ± 1.3 | 1.19 ± 1.4 | 4.53 ± 2.1 | 11.4 ± 1.75† | 15.7 ± 1.5 | 1.41 ± 0.32 | 2.87 ± 1.1 | 3.73 ± 0.38 | 1.64 ± 0.17† |
| Tobacco smoke | ||||||||||||
| Vehicle | 8.49 ± 1.2† | 3.26 ± 0.50† | 1.85 ± 0.20 | 1.60 ± 0.31 | 2.70 ± 0.84† | 3.99 ± 0.20 | 84.1 ± 33† | 34.7 ± 11† | 2.56 ± 0.74† | 1.91 ± 0.22† | 4.95 ± 0.57† | 2.01 ± 0.23† |
| AUDA-nBE | 32.0 ± 7.1‡ | 5.13 ± 1.1†‡ | 2.39 ± 0.28†‡ | 1.85 ± 0.27 | 2.69 ± 0.76† | 4.13 ± 0.79 | 19.9 ± 0.89†‡ | 28.7 ± 1.9† | 1.58 ± 0.32 | 2.00 ± 0.22† | 5.17 ± 0.65† | 2.09 ± 0.13† |
| AUDA-nBE + EETs | 35.1 ± 2.2†‡ | 8.61 ± 1.4‡ | 2.65 ± 0.67 | 2.60 ± 0.37‡ | 2.39 ± 1.94 | 4.53 ± 1.2 | 15.0 ± 0.65†‡ | 20.5 ± 1.9‡ | 1.35 ± 0.11‡ | 3.02 ± 0.49†‡ | 4.58 ± 0.85 | 1.23 ± 0.17‡ |
Data are presented in nM as mean ± SD (n = 3-4). †, P < 0.05 compared with filtered air and vehicle. ‡, P < 0.05 compared with tobacco smoke and vehicle (only within tobacco smoke groups).
12(13)- and 9(10)-EpOME plasma concentrations were significantly decreased after 3 days of tobacco smoke exposure in animals treated with vehicle (Table 2). 14(15)-, 11(12)-, and 5(6)-EET were decreased in these animals but not significantly. 8(9)-EET was significantly increased with exposure to tobacco smoke. There was also a significant increase in all arachidonate and linoleate diol concentrations in plasma with exposure to tobacco smoke.
Administration of AUDA-nBE to rats before tobacco smoke exposure significantly increased 12(13)- and 9(10)-EpOME and 14(15)-EET concentrations in plasma compared with levels in rats exposed to tobacco smoke plus vehicle (Table 2). 11(12)- and 5(6)-EET concentrations were increased but not significantly in tobacco smoke-exposed rats treated with AUDA-nBE. 12(13)-EpOME, 9(10)-EpOME, and 11(12)-EET were significantly increased in smoke-exposed animals that received both AUDA-nBE and EETs compared with levels in animals exposed to tobacco smoke and treated only with vehicle. AUDA-nBE alone or in combination with EETs did not significantly change levels of 8(9)-EET or 5(6)-EET in animals exposed to tobacco smoke. Administration of AUDA-nBE to rats before exposure to tobacco smoke significantly decreased 12,13-DiHOME concentrations compared with levels in rats treated with vehicle and exposed to tobacco smoke. In rats that received both AUDA-nBE and EETs before exposure to tobacco smoke, 12,13- and 9,10-DiHOME and 14,15- and 5,6-DHET were significantly decreased compared with levels in tobacco smoke-exposed rats treated with vehicle. 11,12-DHET was significantly increased and 8,9-DHET was not significantly changed in smoke-exposed animals that received both AUDA-nBE and EETs.
Discussion
There is a considerable amount of research to support a key role for inflammation as a driving force to cause the airway epithelium to undergo changes leading to the loss of ciliated cells, hypersecretion of mucin, bronchitis, emphysema, and lung cancer. Smoking causes a local cytokine secretion in the lung, which leads to an infiltration of leukocytes into the airways and alveolar destruction. We have previously demonstrated the ability of a catalytic antioxidant AEOL 10150 to decrease tobacco smoke-induced inflammation in the lungs of rats, suggesting a role of oxygen radicals in the induction of proinflammatory cytokines and chemokines (17), possibly through oxidant-mediated activation of the redox-sensitive transcription factor, NF-κB. However, inflammation induced by tobacco smoke was not resolved to baseline levels by treatment with the antioxidant, suggesting a role of additional mediators of inflammation. Corticosteroids have antiinflammatory properties, making these compounds useful in the treatment of COPD. A review of several important studies does not show evidence of significant improvement in the symptoms of patients with COPD treated with systemic corticosteroids (23), suggesting a need for additional treatment modalities.
sEH is involved in the regulation of EETs and other epoxylipids (24, 25) and may therefore be an important mediator of inflammation in the lung. This enzyme has >90% homology between rodent and human (26) and can be inhibited in vitro by a number of urea, carbamate, and amide derivatives (27, 28). Injection of the sEH inhibitor N,N′-dicyclohexyl urea (DCU) in SH rats resulted in lower blood pressure, an increase in urinary 14(15)-EET, and a decrease in the corresponding urinary diol (DHET). These observations are consistent with in vivo inhibition of sEH by DCU. AUDA and its n-butyl ester are more potent sEH inhibitors than DCU. The blood levels of AUDA after s.c. administration of AUDA-nBE are high enough to suggest that the sEH is significantly inhibited.
In this study, we found pulmonary inflammation to be induced in rats exposed to tobacco smoke for 3 days. Exposure to tobacco smoke significantly increased by 3.2-fold the total number of cells recovered by BAL. s.c. injection of AUDA-nBE significantly decreased the total BAL cell number by 37% in tobacco smoke-exposed rats. Numbers of BAL macrophages, neutrophils, and lymphocytes were also significantly increased with tobacco smoke exposure, whereas AUDA-nBE significantly decreased numbers of these cell types by 18%, 55%, and 74%, respectively, in smoke-exposed animals. The combination of AUDA-nBE and EETs further reduced tobacco smoke-induced inflammation compared with AUDA-nBE alone. Interestingly, treatment of animals with both AUDA-nBE and EETs before exposure to filtered air resulted in significantly lower numbers of total BAL cells and macrophages compared with animals treated only with vehicle before exposure to filtered air. This result may represent a basal effect of these compounds in “control” animals and may contribute to the reduced inflammation during exposure to tobacco smoke.
It is hypothesized that AUDA acts by stabilizing antiinflammatory fatty acid epoxides such as EETs and reducing the formation of proinflammatory fatty acid diols such as the DiHOMEs in the local environment where they are formed. At nanomolar concentrations, the EETs, which are produced by the vascular endothelium, have a variety of antiinflammatory effects including inhibiting expression of several cell adhesion molecules and inhibiting leukocyte adhesion to the vascular wall (12, 14, 29). The data in this study can be interpreted in light of these hypothetical mechanisms of action. Oxylipid concentrations in plasma may reflect local changes in the concentration of these important chemical mediators but are not equivalent to the local concentrations. The results certainly are complicated by changes due to biosynthesis, distribution, and possibly other factors. To fully appreciate the measure of AUDA-nBE and AUDA activity with regard to attenuation of inflammation, some of the results will be discussed in terms of epoxy:diol ratios. As expected, the proinflammatory 12,13-DiHOME, 9,10-DiHOME, and 14,15-DHET increased dramatically by 5.1-, 2.0-, and 2.0-fold, respectively, after exposure to tobacco smoke. After exposure to tobacco smoke, epoxy:diol ratios in plasma were decreased by 14.3-, 6.8-, and 2.0-fold for 12,13-EpHOME:DiHOME, 9,10-EpHOME:DiHOME, and 14,15-EET:DHET, respectively. These effects are largely reversed by both treatments. With administration of AUDA-nBE before exposure to tobacco smoke, 12,13-EpHOME:DiHOME, 9,10-EpHOME:DiHOME, and 14,15-EET:DHET ratios increased by 15.9-, 1.9-, and 2.1-fold, respectively. With s.c. injection of AUDA-nBE before smoke exposure, 12,13-EpHOME:DiHOME, 9,10-EpHOME:DiHOME, and 14,15-EET:DHET ratios were 111%, 28%, and 104% respectively, of those in animals treated with vehicle and exposed to filtered air. This effect is even greater with combined AUDA-nBE and EETs treatment. With coadministration of AUDA-nBE and EETs before exposure to tobacco smoke, 12,13-EpHOME:DiHOME, 9,10-EpHOME:DiHOME, and 14,15-EET:DHET ratios increased by 23.2-, 4.5-, and 2.7-fold, respectively. With administration of both AUDA-nBE and EETs before smoke exposure, 12,13-EpHOME:DiHOME, 9,10-EpHOME:DiHOME, and 14,15-EET:DHET ratios were 162%, 66%, and 135% respectively, of those in animals treated with vehicle and exposed to filtered air. Because 12(13)- and 9(10)-EpOME were not in the wax plug, the mechanism for their overall enhancement by EET administration is not clear. The 11,12-EET:DHET ratio did not follow the trend of the linoleate epoxy:diol ratios because of a significant increase in diol concentrations with coadministration of AUDA-nBE and EETs. 8(9)-EET was increased in animals after exposure to tobacco smoke and did not change significantly with AUDA-nBE treatment alone or in combination with EETs, possibly due to shuttling of 8(9)-EET through the cyclooxygenase pathway when sEH was inhibited. 5(6)-EET is the least preferred substrate for sEH and was not included in the EETs treatment because of lactone formation, so it was not surprising that there were no significant differences between the six treatment groups. However, the 5,6-DHET concentration was significantly increased by exposure to tobacco smoke, and this smoke-induced increase was attenuated in animals treated with the combination of AUDA-nBE and EETs. Because of the high polarity of these diols and the ease of conjugate formation, systemic plasma concentrations probably underestimate the importance of this pathway. Whether the effects of AUDA-nBE on tobacco smoke-induced inflammation are attributable solely to an increase in EETs levels in our animal model is unknown, although the further reduction in inflammation by coadministration of EETs and AUDA-nBE supports this hypothesis.
In summary, s.c. administration of AUDA-nBE, in the presence or absence of EETs, caused marked reduction in tobacco smoke-induced inflammation when given before and during smoke exposure. Our studies suggest that pharmacologic inhibition of sEH with or without the coadministration of EETs might be a mechanism to modulate pulmonary epoxy and diol levels for altering inflammation commonly associated with such common human disorders as COPD and carcinogenesis.
Supplementary Material
Acknowledgments
We thank Kara Schmelzer for advice on oxylipid extraction. This work was supported in part by Grants R01 ES011634 and R37 ES02710, the Superfund Basic Research Program (P42 ES04699), the Environmental Health Sciences Center (P30 ES05707), and the Center for Children's Environmental Health and Disease Prevention (1 P01 ES11269), all from the National Institute of Environmental Health Sciences; and the University of California Systemwide Biotechnology Research and Education Training Grant 2001-07.
Author contributions: K.R.S., K.E.P., T.W., T.L.P., S.J.M., and B.D.H. designed research; K.R.S., K.E.P., T.W., T.L.P., S.J.M., and B.D.H. performed research; K.E.P. and B.D.H. contributed new reagents/analytic tools; K.R.S., K.E.P., T.W., T.L.P., S.J.M., and B.D.H. analyzed data; and K.R.S., K.E.P., T.W., T.L.P., S.J.M., and B.D.H. wrote the paper.
Abbreviations: AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; AUDA-nBE, AUDA n-butyl ester; BAL, bronchoalveolar lavage; BALF, BAL fluid; COPD, chronic obstructive pulmonary disease; DiHOME, diols of linoleate epoxide; DHET, dihydroxyeicosatrienoic acid; EET, epoxyeicosatrienoic acid; EpHOME, linoleate epoxide; sEH, soluble epoxide hydrolase; SH, spontaneously hypertensive.
References
- 1.Snider, G. L. (1996) in Pulmonary and Critical Care Pharmacology and Therapeutics., ed. Leff, A. R. (McGraw-Hill, New York), pp. 821-828.
- 2.Jeffery, P. K. (1998) Thorax 53, 129-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fournier, M., Lebargy, F., Le Roy Ladurie, F., Lenormand, E. & Pariente, R. (1989) Am. Rev. Respir. Dis. 140, 737-742. [DOI] [PubMed] [Google Scholar]
- 4.Saetta, M., Turato, G., Facchini, F. M., Corbino, L., Lucchini, R. E., Casoni, G., Maestrelli, P., Mapp, C. E., Ciaccia, A. & Fabbri, L. M. (1997) Am. J. Respir. Crit. Care Med. 156, 1633-1639. [DOI] [PubMed] [Google Scholar]
- 5.Grashoff, W. F., Sont, J. K., Sterk, P. J., Hiemstra, P. S., de Boer, W. I., Stolk, J., Han, J. & van Krieken, J. M. (1997) Am. J. Pathol. 151, 1785-1790. [PMC free article] [PubMed] [Google Scholar]
- 6.Saetta, M., Di Stefano, A., Turato, G., Facchini, F. M., Corbino, L., Mapp, C. E., Maestrelli, P., Ciaccia, A. & Fabbri, L. M. (1998) Am. J. Respir. Crit. Care Med. 157, 822-826. [DOI] [PubMed] [Google Scholar]
- 7.Hammock, B. D., Storms, D. H. & Grant, D. F. (1997) in Comprehensive Toxicology: Biotransformation (Elsevier, New York), pp. 283-305.
- 8.Oesch, F. (1972) Xenobiotica 3, 305-340. [DOI] [PubMed] [Google Scholar]
- 9.Moghaddam, M. F., Grant, D. F., Cheek, J. M., Greene, J. F., Williamson, K. C. & Hammock, B. D. (1997) Nat. Med. 3, 562-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slim, R., Hammock, B. D., Toborek, M., Robertson, L. W., Newman, J. W., Morisseau, C. H., Watkins, B. A., Saraswathi, V. & Hennig, B. (2001) Toxicol. Appl. Pharmacol. 171, 184-193. [DOI] [PubMed] [Google Scholar]
- 11.Zheng, J., Plopper, C. G., Lakritz, J., Storms, D. H. & Hammock, B. D. (2001) Am. J. Respir. Cell Mol. Biol. 25, 434-438. [DOI] [PubMed] [Google Scholar]
- 12.Campbell, W. B. (2000) Trends Pharmacol. Sci. 21, 125-127. [DOI] [PubMed] [Google Scholar]
- 13.Zeldin, D. C. & Liao, J. K. (2000) Trends Pharmacol. Sci. 21, 127-128. [DOI] [PubMed] [Google Scholar]
- 14.Node, K., Huo, Y., Ruan, X., Yang, B., Spiecker, M., Ley, K., Zeldin, D. C. & Liao, J. K. (1999) Science 285, 1276-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu, Z., Xu, F., Huse, L. M., Morisseau, C., Draper, A. J., Newman, J. W., Parker, C., Graham, L., Engler, M. M., Hammock, B. D., et al. (2000) Circ. Res. 87, 992-998. [DOI] [PubMed] [Google Scholar]
- 16.Sinal, C. J., Miyata, M., Tohkin, M., Nagata, K., Bend, J. R. & Gonzalez, F. J. (2000) J. Biol. Chem. 275, 40504-40510. [DOI] [PubMed] [Google Scholar]
- 17.Smith, K. R., Uyeminami, D. L., Kodavanti, U. P., Crapo, J. D., Chang, L. Y. & Pinkerton, K. E. (2002) Free Radical Biol. Med. 33, 1106-1114. [DOI] [PubMed] [Google Scholar]
- 18.Newman, J. W., Stok, J. E., Vidal, J. D., Corbin, C. J., Huang, Q., Hammock, B. D. & Conley, A. J. (2004) Endocrinology 145, 5097-5105. [DOI] [PubMed] [Google Scholar]
- 19.Newman, J. W., Watanabe, T. & Hammock, B. D. (2002) J. Lipid Res. 43, 1563-1578. [DOI] [PubMed] [Google Scholar]
- 20.Zhang, Y. E. & Schwartz, J. B. (2003) Drug Dev. Ind. Pharm. 29, 131-138. [DOI] [PubMed] [Google Scholar]
- 21.Teague, S. V., Pinkerton, K. E., Goldsmith, M., Gebremichael, A., Chang, S., Jenkins, R. A. & Moneyhun, J. H. (1994) Inhal. Toxicol. 6, 79-93. [Google Scholar]
- 22.Gossart, S., Cambon, C., Orfila, C., Seguelas, M. H., Lepert, J. C., Rami, J., Carre, P. & Pipy, B. (1996) J. Immunol. 156, 1540-1548. [PubMed] [Google Scholar]
- 23.Wood-Baker, R. (2003) Am. J. Respir. Med. 2, 451-458. [DOI] [PubMed] [Google Scholar]
- 24.Zeldin, D. C., Kobayashi, J., Falck, J. R., Winder, B. S., Hammock, B. D., Snapper, J. R. & Capdevila, J. H. (1993) J. Biol. Chem. 268, 6402-6407. [PubMed] [Google Scholar]
- 25.Fang, X., Kaduce, T. L., Weintraub, N. L., Harmon, S., Teesch, L. M., Morisseau, C., Thompson, D. A., Hammock, B. D. & Spector, A. A. (2001) J. Biol. Chem. 276, 14867-14874. [DOI] [PubMed] [Google Scholar]
- 26.Arand, M., Grant, D. F., Beetham, J. K., Friedberg, T., Oesch, F. & Hammock, B. D. (1994) FEBS Lett. 338, 251-256. [DOI] [PubMed] [Google Scholar]
- 27.Morisseau, C., Goodrow, M. H., Dowdy, D., Zheng, J., Greene, J. F., Sanborn, J. R. & Hammock, B. D. (1999) Proc. Natl. Acad. Sci. USA 96, 8849-8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morisseau, C., Goodrow, M. H., Newman, J. W., Wheelock, C. E., Dowdy, D. L. & Hammock, B. D. (2002) Biochem. Pharmacol. 63, 1599-1608. [DOI] [PubMed] [Google Scholar]
- 29.Falck, J. R., Reddy, L. M., Reddy, Y. K., Bondlela, M., Krishna, U. M., Ji, Y., Sun, J. & Liao, J. K. (2003) Bioorg. Med. Chem. Lett. 13, 4011-4014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.