

Abstract
Nature provides many examples of self- and co-assembling protein-based molecular machines, including icosahedral protein cages that serve as scaffolds, enzymes, and compartments for essential biochemical reactions and icosahedral virus capsids, which encapsidate and protect viral genomes and mediate entry into host cells. Inspired by these natural materials, we report the computational design and experimental characterization of co-assembling two-component 120-subunit icosahedral protein nanostructures with molecular weights (1.8–2.8 MDa) and dimensions (24–40 nm diameter) comparable to small viral capsids. Electron microscopy, SAXS, and X-ray crystallography show that ten designs spanning three distinct icosahedral architectures form materials closely matching the design models. In vitro assembly of independently purified components reveals rapid assembly rates comparable to viral capsids and enables controlled packaging of molecular cargo via charge complementarity. The ability to design megadalton-scale materials with atomic-level accuracy and controllable assembly opens the door to a new generation of genetically programmable protein-based molecular machines.
The remarkable forms and functions of natural protein assemblies have inspired many efforts to engineer self- and co-assembling protein complexes (1–24). A common feature of these approaches, as well as the structures that inspired them, is symmetry. By repeating a small number of interactions in geometric arrangements consistent with the formation of regular structures, symmetry reduces the number of unique interactions and subunits required to form higher order assemblies (2, 25). Symmetric complexes can be designed to form through self-assembly of a single type of protein subunit or co-assembly of two or more distinct types of protein subunits. Multi-component materials possess several important advantages, including the potential to control initiation of assembly by mixing independently prepared components. This property could allow, for example, assembly to be performed in the presence of cargo molecules in order to package the cargo inside the designed nanomaterial. Thus far, only relatively small, 24-subunit two-component tetrahedra have been designed with high accuracy (20, 26). Packaging substantial amounts of cargo will require larger assemblies, such as those with icosahedral symmetry; icosahedra possess the highest possible symmetry of any polyhedron in three-dimensional space and, consequently for the purpose of packaging, generate the maximum enclosed volume for symmetric assemblies formed from a given size protein subunit (27, 28).
We set out to design two-component icosahedral protein complexes capable of packaging macromolecular cargo via controlled in vitro assembly. The two-fold, three-fold, and five-fold rotational axes present within icosahedral symmetry provide three possible ways to construct such complexes from pairwise combinations of oligomeric building blocks; we refer to these architectural types as I53, I52 and I32 (fig. S1). The I53 architecture is formed from a combination of twelve pentameric building blocks and twenty trimeric building blocks aligned along the five-fold and three-fold icosahedral symmetry axes, respectively (Fig. 1, A–E; I53 = Icosahedral assembly constructed from 5mers and 3mers). Similarly, the I52 architecture is formed from twelve pentamers and thirty dimers (Fig. 1F), and the I32 architecture is formed from twenty trimers and thirty dimers, each aligned along their corresponding icosahedral symmetry axes (Fig. 1G). To generate novel icosahedral assemblies, 14,400 pairs of pentamers and trimers, 50,400 pairs of pentamers and dimers, and 276,150 pairs of trimers and dimers derived from X-ray crystal structures (tables S1–3) were arranged as described above, with each building block allowed to rotate around and translate along its five-fold, three-fold, or two-fold symmetry axis. These degrees of freedom (DOFs) were systematically sampled to identify configurations suitable for interface design, as assessed by several parameters, including the size and secondary structure content of the newly formed interface, as well as the backbone geometry between pairs of contacting residues. Protein-protein interface design calculations were then carried out on the resulting 66,115 designs of type I53, 35,468 designs of type I52, and 161,007 designs of type I32. The designs were filtered based on a variety of metrics, including interface area, predicted binding energy, and shape complementarity (29). 71 designs of type I53, 44 of type I52, and 68 of type I32, derived from 23 distinct pentameric, 57 distinct trimeric, and 91 distinct dimeric protein scaffolds, were selected for experimental characterization (fig. S2–5, table S4).
Fig. 1. Overview of the design method and target architectures.
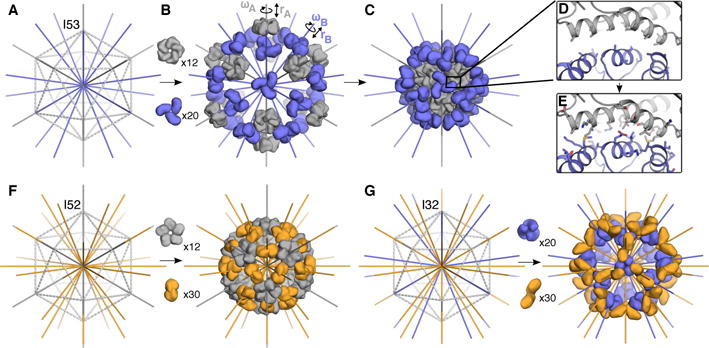
(A–E) A schematic of the design process illustrated with the I53 architecture. (A) An icosahedron is outlined with dashed lines, with the five-fold symmetry axes (grey) going through each vertex and three-fold symmetry axes (blue) going through each face of the icosahedron. (B)12 pentamers (grey) and 20 trimers (blue) are aligned along the 5-fold and 3-fold symmetry axes, respectively. Each oligomer possesses two rigid body DOFs, one translational (r) and one rotational (ω) that are systematically sampled to identify configurations (C) with a large interface between the pentamer and trimer (D) suitable for protein-protein interface design; only the backbone structure and beta carbons of the oligomers are taken into account during this procedure. (E) Amino acid sequences are designed at the new interface to stabilize the modeled configuration. (F) The I52 architecture comprises 12 pentamers (grey) and 30 dimers (orange) aligned along the five-fold and two-fold icosahedral symmetry axes. (G) The I32 architecture comprises 20 trimers (blue) and 30 dimers (orange) aligned along the three-fold and two-fold icosahedral symmetry axes.
Codon-optimized genes encoding each pair of designed sequences were cloned into a vector for inducible co-expression in E. coli, with a hexahistidine tag appended to the N- or C-terminus of one subunit in each pair. The proteins were expressed at small scale, purified by immobilized metal-affinity chromatography (IMAC), and clarified lysates and purification products subjected to gel electrophoresis under denaturing conditions to screen for soluble expression and co-purification of the hexahistidine-tagged and non-tagged subunits (fig. S6A). Designs appearing to co-purify were subsequently analyzed by non-denaturing gel electrophoresis to screen for slowly migrating species as an additional indication of assembly to higher order materials (fig. S6B). Those found to both co-purify and assemble were expressed at larger scale and purified by IMAC followed by size exclusion chromatography (SEC, fig. S7). Ten pairs of designed proteins, four I53 (I53-34, I53-40, I53-47, and I53-50), three I52 (I52-03, I52-32, and I52-33) and three I32 designs (I32-06, I32-19, and I32-28), yielded major peaks by SEC near the elution volumes expected based on the diameters of the design models (Fig. 2, table S4). Two other designs, I53-51 and I32-10, also appeared to form large, discrete assemblies, but their structures could not be verified by subsequent experiments (Supplementary Text, fig. S8 and S9).
Fig. 2. Experimental characterization by size exclusion chromatography and small-angle X-ray scattering.
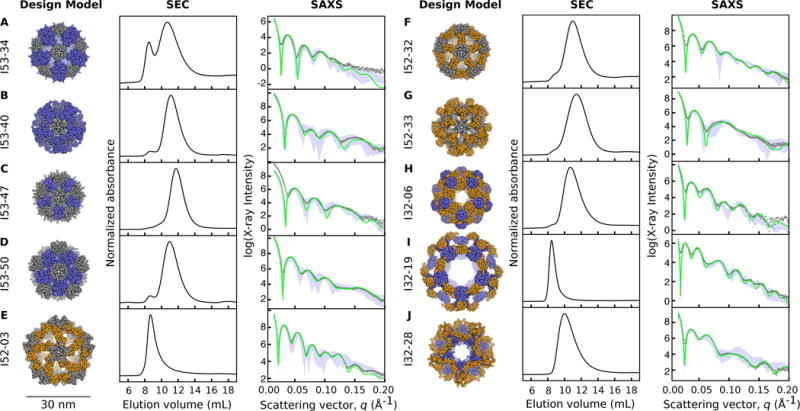
Computational design models (left), SEC chromatograms (middle), and SAXS profiles (right) are shown for (A) I53-34, (B) I53-40, (C) I53-47, (D) I53-50, (E) I52-03, (F) I52-32, (G) I52-33, (H) I32-06, (I) I32-19, and (J) I32-28. Design models (shown to scale relative to the 30 nm scale bar) are viewed down one of the 5-fold symmetry axes with ribbon-style renderings of the protein backbone (pentamers are shown in grey, trimers in blue, and dimers in orange). Co-expressed and purified designs yield dominant SEC peaks near the expected elution volumes for the target 120-subunit complexes and X-ray scattering intensities (grey dots) that match well with profiles calculated from the design models (green). Alternative configurations of the designs, generated by translating and/or rotating the oligomeric building blocks in the design models about their aligned symmetry axes by +/− 10 Å and/or 20 degrees, respectively, generally fit worse with the SAXS data than the original design models (the range of values obtained from fitting the alternative configurations is shown with light blue shading).
Small-angle X-ray scattering (SAXS) performed on the SEC-purified samples indicated all ten designs form assemblies similar to the intended three-dimensional configurations in solution. The experimentally measured SAXS profiles are feature-rich and distinct, with multiple large dips in scattering intensity in the region between 0.015 Å−1 and 0.15 Å−1, each of which is closely recapitulated in profiles calculated from the design models (Fig. 2). In order to further evaluate how accurately and uniquely the design models match the experimental data, each was compared to a set of alternative models generated by systematically perturbing the radial displacements and rotations of the building blocks in each design by +/− 10 Å and/or 20 degrees, respectively. The vast majority of alternative configurations were found to produce worse fits to the experimental data than the original design models (Fig. 2), suggesting that the materials assemble quite precisely in solution.
The information provided by SAXS about the overall ensemble of structures observed in solution for each design was complemented and corroborated by visualization of individual particles by negative stain electron microscopy (EM). Micrographs of I53-34, I53-40, I53-47, I53-50, I52-03, I52-33, I32-06, and I32-28 show fields of particles with the expected size and shape of the design models, and particle averaging yields distinct structures clearly matching the models (Fig. 3). The large trimeric and pentameric voids observed in the I52 and I32 averages, for instance, closely resemble the cavities in projections generated from the corresponding design models when viewed down the three-fold and five-fold symmetry axes, respectively. The turreted morphology of the I53-50 and I52-33 design models and projections, resulting from pentameric and dimeric components that protrude away from the rest of the icosahedral shell, are also readily apparent in the corresponding class averages. Although the results from SEC and SAXS strongly indicate I52-32 and I32-19 form assemblies closely matching the design models in solution, both appear to be unstable under the conditions encountered during grid preparation, yielding broken particles not suitable for further EM analysis (fig. S10).
Fig. 3. Characterization of the designed materials by electron microscopy.
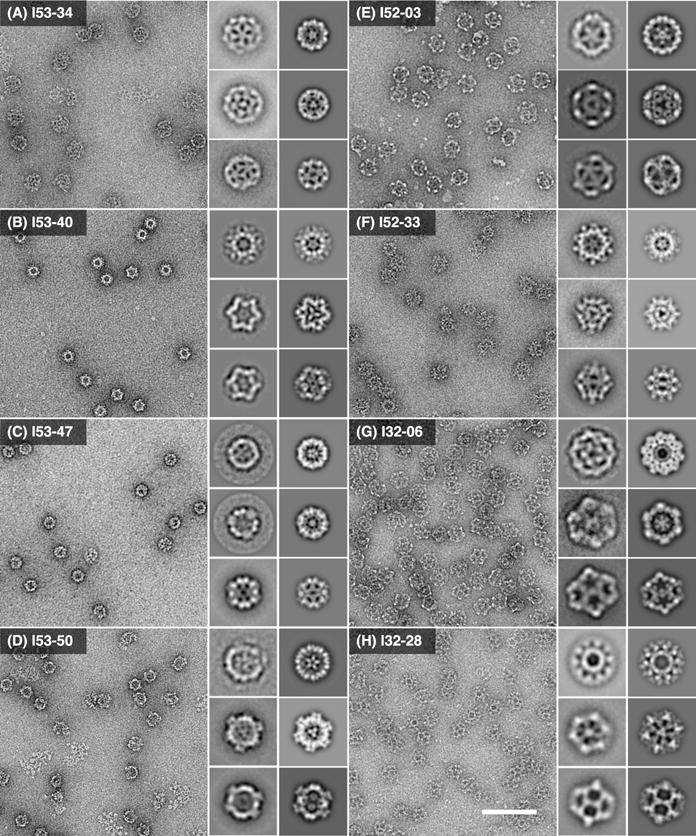
Left: raw negative stain electron micrographs of co-expressed and purified (A) I53-34, (B) I53-40, (C) I53-47, (D) I53-50, (E) I52-03, (F) I52-33, (G) I32-06, and (H) I32-28. All raw micrographs shown to scale relative to 100 nm scale bar in panel (H). Insets: experimentally computed class averages (roughly corresponding to the five-fold, three-fold, and 2-fold icosahedral symmetry axes; left) along with back projections calculated from the design models (right). Each inset box width corresponds to 55 nm.
To further evaluate the accuracy of our designs, X-ray crystal structures were determined for one material from each of the three different architectural types: I53-40, I52-32, and I32-28 (Fig. 4 and table S5). Although the resolution of the structures (3.5 to 5.6 Å) is insufficient to permit detailed analysis of the side chains at the designed interfaces, backbone-level comparisons show the inter-building block interfaces were designed with high accuracy, giving rise to 120-subunit complexes that match the computational design models remarkably well. Comparing pairs of interface subunits from each structure to the design models yields backbone root mean square deviations (r.m.s.d.) between 0.2 and 1.1 Å, while the r.m.s.d. over all 120 subunits in each material ranges from 0.8 to 2.7 Å (Fig. 4, A–C and table S6). With diameters between 26 and 31 nm, over 130,000 heavy atoms, and molecular weights greater than 1.9 megadaltons, these structures are comparable in size to small viral capsids and, to our knowledge, the largest designed biomolecular nanostructures to date to be verified by X-ray crystallography (fig. S11).
Fig. 4. Crystal structures, assembly dynamics, and packaging.
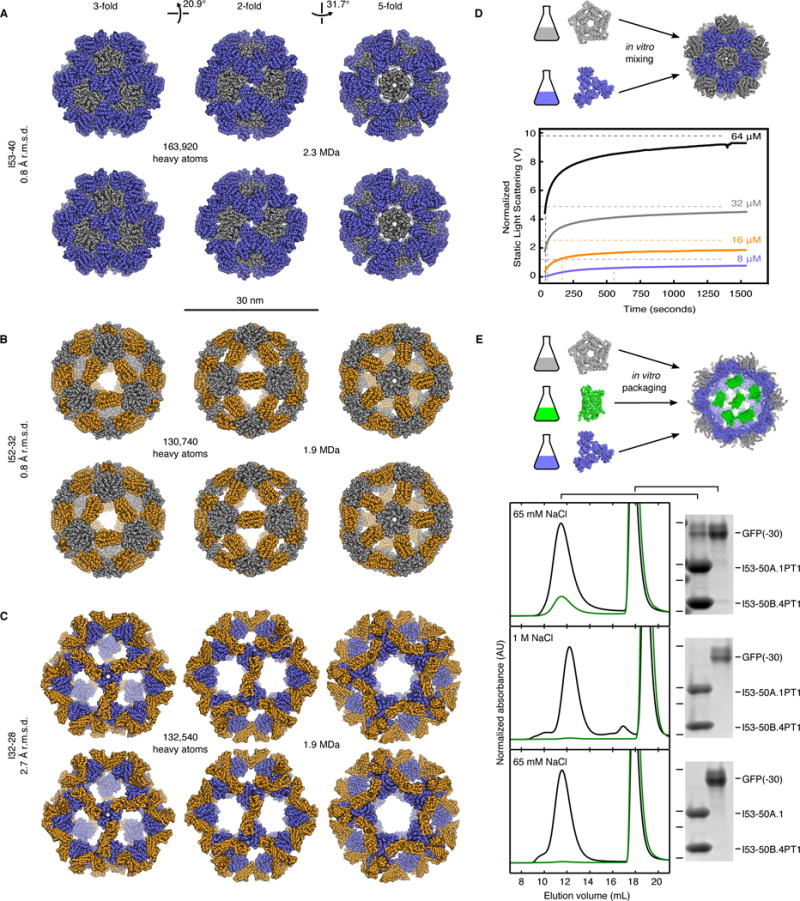
Design models (top) and X-ray crystal structures (bottom) of (A) I53-40, (B) I52-32, and (C) I32-28. Views shown to scale along the 3-fold, 2-fold, and 5-fold icosahedral symmetry axes. Pentamers shown in grey, trimers blue, and dimers orange. R.m.s.d.s are between crystal structures and design models over all backbone atoms in all 120 subunits. (D) In vitro assembly dynamics of I53-50. (Top) Schematic illustration. (Bottom) Normalized static light scattering intensity (detector voltage, solid lines) plotted over time after mixing independently expressed and purified variants of the I53-50 trimer and pentamer in a 1:1 molar ratio at final concentrations of 8, 16, 32, or 64 μM each (blue, orange, grey, and black lines, respectively). Intensities measured from SEC-purified assembly at 8, 16, 32, or 64 μM concentrations indicated with dashed horizontal lines and used as the expected endpoint of each assembly reaction. The midpoint of each reaction is marked with a dashed vertical line. (E) Encapsulation of supercharged GFP in a positively charged I53-50 variant. (Top) Schematic illustration. (Bottom) Superose 6 chromatograms and SDS-PAGE analysis of packaging/assembly reactions performed in buffer containing: (Top Panel) 65 mM NaCl, (Middle Panel) 1 M NaCl, or (Bottom Panel) 65 mM NaCl with a trimer variant without mutations to positively charged residues. In each case, the same buffer used in the packaging/assembly reaction was also used during SEC. Absorbance measurements at 280 nm (black) and 488 nm (green) are shown. Each SEC chromatogram was normalized relative to the 280 nm peak near 12 mL elution volume. Locations of 37, 25, 20, and 15 kDa molecular weight markers on SDS-PAGE gels are indicated by horizontal lines.
The multi-component composition of the materials presents the possibility of controlling their assembly through in vitro mixing of independently produced building blocks (20). Taking advantage of this feature, the assembly kinetics of an I53-50 variant (fig. S12A) with improved individual subunit stability was investigated by light scattering (Supplementary Materials). SEC-purified components were mixed at concentrations of 64, 32, 16, or 8 μM and the change in light scattering monitored over time (Fig. 4D). Assembly is roughly halfway complete within 1 minute at 64 and 32 uM, 3 minutes at 16 uM, and 10 minutes at 8 uM. Similar assembly time scales have been observed for several viral capsids (30, 31). Since our design process focused exclusively on structure without any consideration of kinetics, these results raise the interesting possibility that the rate of assembly of these viral capsids has not been highly optimized during evolution.
The ability to assemble the materials in vitro potentially enables the controlled packaging of macromolecular cargoes. To investigate this possibility, the trimeric and pentameric components of an I53-50 variant with several mutations to positively charged residues on the interior surfaces of the two components (Supplementary Materials) were successively mixed with a supercharged GFP with a net charge of −30 (32), and encapsulation was evaluated using SEC followed by SDS-PAGE of relevant fractions (Fig. 4E and Supplementary Materials). When both the packaging reaction and SEC were performed in a buffer containing low (65 mM) NaCl, GFP(−30) and both I53-50 components co-eluted from the column at the same elution volume previously observed for unmodified I53-50 (Fig. 2D). Mixtures of GFP(−30) with only one of the two components eluted at later volumes, indicating that the observed co-elution requires assembly of I53-50 (fig. S12, B–D). When the packaging reaction was carried out with buffer containing high (1 M) NaCl or using a variant of the trimeric component lacking the positively charged residues on the interior surface, little to no co-elution was observed (Fig. 4E), demonstrating that packaging is driven by the engineered electrostatic interactions between the I53-50 interior and GFP(−30). High salt incubation resulted in disassociation of packaged GFP (fig. S12E), as also observed for an evolved variant of a naturally occurring protein container that packages cargo via electrostatic complementarity (33, 34). Based on measurements of fluorescence intensity and UV/Vis absorbance, we estimate approximately 7 to 11 GFPs are packaged per icosahedral assembly in 65 mM NaCl, occupying roughly 11 to 17% of the interior volume (Supplementary Materials).
How do the architectures of our designs compare to those of virus capsids and other icosahedral protein complexes found in nature? In the nomenclature introduced by Caspar and Klug (27), our designs can be considered T=1 assemblies in which the asymmetric unit is a heterodimer comprising one subunit from each of the two components. The most similar naturally occurring structures of which we are aware are Cowpea Mosaic Virus (CPMV) and related 120-subunit capsids with pseudo T=3 symmetry. Like our I53 designs, CPMV is composed of 60 copies each of two distinct protein subunits, with one type of subunit arranged around the icosahedral 5-folds and a second type of subunit arranged around the 3-folds (fig. S13). However, the two subunits of CPMV are composed of three similar domains occupying spatially equivalent positions to those found in T=3 assemblies formed from 180 copies of a single type of protein subunit (35, 36). Our I53 designs display no such underlying pseudosymmetry and therefore cannot be considered pseudo T=3. Furthermore, we are not aware of any natural protein complexes characterized to date that exhibit I52 or I32 architectures. Our designs thus appear to occupy new regions of the protein assembly universe, which have either not yet been explored by natural evolution or are undiscovered at present in natural systems.
The size and complexity of the materials presented herein, together with the accuracy with which they assemble, push the boundaries of biomolecular engineering into new and exciting territory. The large lumens of our designed materials, combined with their multi-component nature and the ability to control assembly via mixing of purified components, makes them well suited for encapsulation of a broad range of materials including small molecules, nucleic acids, polymers, and other proteins. These features, along with their potential for precisely engineered chemical or genetic modifications, make them attractive starting materials for the design of functional protein nanomaterials for applications in targeted drug delivery, vaccine design, and bioenergy.
Supplementary Material
One Sentence Summary.
A computational approach enables design of 120-subunit icosahedral protein cages capable of packaging macromolecular cargo.
Acknowledgments
We thank Michael Sawaya and Michael Collazo for their assistance with crystallography, conducted at the UCLA-DOE X-ray Crystallization and Crystallography Core Facilities, which are supported by DOE Grant DE-FC02-02ER63421. We thank M. Capel, K. Rajashankar, N.Sukumar, J.Schuermann, I. Kourinov and F. Murphy at NECAT beamlines 24-ID at APS, which are supported by grants from the National Center for Research Resources (5P41RR015301-10) and the National Institute of General Medical Sciences (8 P41 GM103403-10) from the National Institutes of Health. Use of the APS is supported by DOE under Contract DE-AC02-06CH11357. We thank the staff at the SIBYLS beamline at Lawrence Berkeley National Lab, including Kathryn Burnett, Gregory Hura, Michal Hammel, Jane Tanamachi, and John Tainer for the services provided through the mail-in SAXS program, supported by the Department of Energy (DOE) Integrated Diffraction Analysis (IDAT) grant contract number DE-AC02-05CH11231. We also thank Una Nattermann for help with electron microscopy, Yang Hsia for assistance with light scattering experiments, Chris Stafford for mass spectroscopy, Brooke Nickerson for assistance with in vitro assembly experiments, and Gabriel Rocklin for providing scripts used in data analysis. This work was supported by the Howard Hughes Medical Institute (SG, DC, TG, and DB) and the Janelia Research Campus visitor program (SG), the Bill and Melinda Gates Foundation (DB and NPK), Takeda Pharmaceutical Company (NPK), the National Science Foundation (DB and TOY, grant no. CHE-1332907) and the Defense Advanced Research Projects Agency (DB and NPK, grant no. W911NF-14-1-0162). YL was supported by a Whitcome Fellowship through the UCLA Molecular Biology Institute and JBB by an NSF graduate research fellowship (grant no. DGE-0718124). Coordinates and structure factors were deposited in the Protein Data Bank with accession codes 5IM5 (I53-40), 5IM4 (I52-32), and 5IM6 (I32-28). JBB, WS, NPK, DE, and DB have filed a non-provisional patent application, U.S. 14/930,792, related to the work presented herein.
Footnotes
Supplementary Materials:
Materials and Methods
References (37–59)
References and Notes
- 1.Lai YT, King NP, Yeates TO. Principles for designing ordered protein assemblies. Trends Cell Biol. 2012;22:653–661. doi: 10.1016/j.tcb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 2.King NP, Lai YT. Practical approaches to designing novel protein assemblies. Curr Opin Struct Biol. 2013;23:632–638. doi: 10.1016/j.sbi.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Padilla JE, Colovos C, Yeates TO. Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci U S A. 2001;98:2217–2221. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ringler P, Schulz GE. Self-assembly of proteins into designed networks. Science. 2003;302:106–109. doi: 10.1126/science.1088074. [DOI] [PubMed] [Google Scholar]
- 5.Raman S, Machaidze G, Lustig A, Aebi U, Burkhard P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomedicine. 2006;2:95–102. doi: 10.1016/j.nano.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Grueninger D, Treiber N, Ziegler MO, Koetter JW, Schulze MS, Schulz GE. Designed protein-protein association. Science. 2008;319:206–209. doi: 10.1126/science.1150421. [DOI] [PubMed] [Google Scholar]
- 7.Raman S, Machaidze G, Lustig A, Olivieri V, Aebi U, Burkhard P. Design of Peptide Nanoparticles Using Simple Protein Oligomerization Domains. The Open Nanomedicine Journal. 2009;2:15–26. [Google Scholar]
- 8.Usui K, Maki T, Ito F, Suenaga A, Kidoaki S, Itoh M, Taiji M, Matsuda T, Hayashizaki Y, Suzuki H. Nanoscale elongating control of the self-assembled protein filament with the cysteine-introduced building blocks. Protein Sci. 2009;18:960–969. doi: 10.1002/pro.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salgado EN, Radford RJ, Tezcan FA. Metal-directed protein self-assembly. Acc Chem Res. 2010;43:661–672. doi: 10.1021/ar900273t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinclair JC, Davies KM, Venien-Bryan C, Noble ME. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 12.Stranges PB, Machius M, Miley MJ, Tripathy A, Kuhlman B. Computational design of a symmetric homodimer using beta-strand assembly. Proc Natl Acad Sci U S A. 2011;108:20562–20567. doi: 10.1073/pnas.1115124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyle AL, Bromley EH, Bartlett GJ, Sessions RB, Sharp TH, Williams CL, Curmi PM, Forde NR, Linke H, Woolfson DN. Squaring the circle in peptide assembly: from fibers to discrete nanostructures by de novo design. J Am Chem Soc. 2012;134:15457–15467. doi: 10.1021/ja3053943. [DOI] [PubMed] [Google Scholar]
- 14.Brodin JD, Ambroggio XI, Tang C, Parent KN, Baker TS, Tezcan FA. Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat Chem. 2012;4:375–382. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J Am Chem Soc. 2012;134:375–385. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, Gonen T, Yeates TO, Baker D. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai YT, Cascio D, Yeates TO. Structure of a 16-nm cage designed by using protein oligomers. Science. 2012;336:1129. doi: 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- 18.Lanci CJ, MacDermaid CM, Kang SG, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG. Computational design of a protein crystal. Proc Natl Acad Sci U S A. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fletcher JM, Harniman RL, Barnes FR, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, Miles MJ, Sessions RB, Verkade P, Woolfson DN. Self-assembling cages from coiled-coil peptide modules. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO, Baker D. Accurate design of co-assembling multi-component protein nanomaterials. Nature. 2014;510:103–108. doi: 10.1038/nature13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai YT, Reading E, Hura GL, Tsai KL, Laganowsky A, Asturias FJ, Tainer JA, Robinson CV, Yeates TO. Structure of a designed protein cage that self-assembles into a highly porous cube. Nat Chem. 2014;6:1065–1071. doi: 10.1038/nchem.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voet AR, Noguchi H, Addy C, Simoncini D, Terada D, Unzai S, Park SY, Zhang KY, Tame JR. Computational design of a self-assembling symmetrical beta-propeller protein. Proc Natl Acad Sci U S A. 2014;111:15102–15107. doi: 10.1073/pnas.1412768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonen S, DiMaio F, Gonen T, Baker D. Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces. Science. 2015;348:1365–1368. doi: 10.1126/science.aaa9897. [DOI] [PubMed] [Google Scholar]
- 24.Hsia Y, Bale JB, Gonen S, Shi D, Sheffler W, Fong KK, Nattermann U, Xu C, Huang P, Ravichandran R, Yi S, Davis TN, Gonen T, King NP, Baker D. Design of a hyperstable 60-subunit protein icosahedron. Nature. 2016 doi: 10.1038/nature18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 26.Bale JB, Park RU, Liu Y, Gonen S, Gonen T, Cascio D, King NP, Yeates TO, Baker D. Structure of a designed tetrahedral protein assembly variant engineered to have improved soluble expression. Protein Sci. 2015;24:1695–1701. doi: 10.1002/pro.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caspar DL, Klug A. Physical principles in the construction of regular viruses. Cold Spring Harb Symp Quant Biol. 1962;27:1–24. doi: 10.1101/sqb.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- 28.Zandi R, Reguera D, Bruinsma RF, Gelbart WM, Rudnick J. Origin of icosahedral symmetry in viruses. Proc Natl Acad Sci U S A. 2004;101:15556–15560. doi: 10.1073/pnas.0405844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 30.Zlotnick A, Johnson JM, Wingfield PW, Stahl SJ, Endres D. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry. 1999;38:14644–14652. doi: 10.1021/bi991611a. [DOI] [PubMed] [Google Scholar]
- 31.Zlotnick A, Aldrich R, Johnson JM, Ceres P, Young MJ. Mechanism of capsid assembly for an icosahedral plant virus. Virology. 2000;277:450–456. doi: 10.1006/viro.2000.0619. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Worsdorfer B, Woycechowsky KJ, Hilvert D. Directed evolution of a protein container. Science. 2011;331:589–592. doi: 10.1126/science.1199081. [DOI] [PubMed] [Google Scholar]
- 34.Zschoche R, Hilvert D. Diffusion-Limited Cargo Loading of an Engineered Protein Container. J Am Chem Soc. 2015;137:16121–16132. doi: 10.1021/jacs.5b10588. [DOI] [PubMed] [Google Scholar]
- 35.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T, Johnson JE. The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology. 1999;265:20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 36.Lin T, Clark AJ, Chen Z, Shanks M, Dai JB, Li Y, Schmidt T, Oxelfelt P, Lomonossoff GP, Johnson JE. Structural fingerprinting: subgrouping of comoviruses by structural studies of red clover mottle virus to 2.4-A resolution and comparisons with other comoviruses. J Virol. 2000;74:493–504. doi: 10.1128/jvi.74.1.493-504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The PyMOL Molecular Graphics System v. 1.5. (Schrödinger, LLC 2012).
- 38.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 39.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiMaio F, Leaver-Fay A, Bradley P, Baker D, Andre I. Modeling symmetric macromolecular structures in Rosetta3. PLoS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leaver-Fay A, O’Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, Thompson J, Davis IW, Pache RA, Lyskov S, Gray JJ, Kortemme T, Richardson JS, Havranek JJ, Snoeyink J, Baker D, Kuhlman B. Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol. 2013;523:109–143. doi: 10.1016/B978-0-12-394292-0.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Meara MJ, Leaver-Fay A, Tyka MD, Stein A, Houlihan K, DiMaio F, Bradley P, Kortemme T, Baker D, Snoeyink J, Kuhlman B. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J Chem Theory Comput. 2015;11:609–622. doi: 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheffler W, Baker D. RosettaHoles2: a volumetric packing measure for protein structure refinement and validation. Protein Sci. 2010;19:1991–1995. doi: 10.1002/pro.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleishman SJ, Whitehead TA, Strauch EM, Corn JE, Qin S, Zhou HX, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, Moal IH, Li X, Bates PA, Zacharias M, Park H, Ko JS, Lee H, Seok C, Bourquard T, Bernauer J, Poupon A, Aze J, Soner S, Ovali SK, Ozbek P, Tal NB, Haliloglu T, Hwang H, Vreven T, Pierce BG, Weng Z, Perez-Cano L, Pons C, Fernandez-Recio J, Jiang F, Yang F, Gong X, Cao L, Xu X, Liu B, Wang P, Li C, Wang C, Robert CH, Guharoy M, Liu S, Huang Y, Li L, Guo D, Chen Y, Xiao Y, London N, Itzhaki Z, Schueler-Furman O, Inbar Y, Potapov V, Cohen M, Schreiber G, Tsuchiya Y, Kanamori E, Standley DM, Nakamura H, Kinoshita K, Driggers CM, Hall RG, Morgan JL, Hsu VL, Zhan J, Yang Y, Zhou Y, Kastritis PL, Bonvin AM, Zhang W, Camacho CJ, Kilambi KP, Sircar A, Gray JJ, Ohue M, Uchikoga N, Matsuzaki Y, Ishida T, Akiyama Y, Khashan R, Bush S, Fouches D, Tropsha A, Esquivel-Rodriguez J, Kihara D, Stranges PB, Jacak R, Kuhlman B, Huang SY, Zou X, Wodak SJ, Janin J, Baker D. Community-wide assessment of protein-interface modeling suggests improvements to design methodology. J Mol Biol. 2011;414:289–302. doi: 10.1016/j.jmb.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang PS, Ban YE, Richter F, Andre I, Vernon R, Schief WR, Baker D. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One. 2011;6:e24109. doi: 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nannenga BL, Iadanza MG, Vollmar BS, Gonen T. Overview of electron crystallography of membrane proteins: crystallization and screening strategies using negative stain electron microscopy. Curr Protoc Protein Sci. 2013 doi: 10.1002/0471140864.ps1715s72. Chapter 17, Unit17 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 52.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 53.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vagin A, Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. Journal of Applied Crystallography. 1997;30:1022–1025. [Google Scholar]
- 57.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackel C, Bloom JD, Kast P, Arnold FH, Hilvert D. Consensus protein design without phylogenetic bias. J Mol Biol. 2010;399:541–546. doi: 10.1016/j.jmb.2010.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.