

ABSTRACT
Immune checkpoint inhibitors have significantly improved the treatment of several cancers. T-cell infiltration and the number of neoantigens caused by tumor-specific mutations are correlated to favorable responses in cancers with a high mutation load. Accordingly, checkpoint immunotherapy is thought to be less effective in tumors with low mutation frequencies such as neuroblastoma, a neuroendocrine tumor of early childhood with poor outcome of the high-risk disease group. However, spontaneous regressions and paraneoplastic syndromes seen in neuroblastoma patients suggest substantial immunogenicity. Using an integrative transcriptomic approach, we investigated the molecular characteristics of T-cell infiltration in primary neuroblastomas as an indicator of pre-existing immune responses and potential responsiveness to checkpoint inhibition. Here, we report that a T-cell-poor microenvironment in primary metastatic neuroblastomas is associated with genomic amplification of the MYCN (N-Myc) proto-oncogene. These tumors exhibited lower interferon pathway activity and chemokine expression in line with reduced immune cell infiltration. Importantly, we identified a global role for N-Myc in the suppression of interferon and pro-inflammatory pathways in human and murine neuroblastoma cell lines. N-Myc depletion potently enhanced targeted interferon pathway activation by a small molecule agonist of the cGAS-STING innate immune pathway. This promoted chemokine expression including Cxcl10 and T-cell recruitment in microfluidics migration assays. Hence, our data suggest N-Myc inhibition plus targeted IFN activation as adjuvant strategy to enforce cytotoxic T-cell recruitment in MYCN-amplified neuroblastomas.
Keywords: Chemokine, Cxcl10, immunotherapy, infiltration, interferoninfiltration, neuroblastoma, N-Myc, STING
Introduction
Monoclonal antibodies against the negative immune checkpoint molecules PD-1/PD-L1 significantly improve disease outcome in a variety of cancers.1-4 The PD-1 receptor is expressed on the surface of T cells and PD-L1 ligand binding induces an exhausted T-cell state that impedes the elimination of tumor cells by cytotoxic T cells.5,6 Blocking the PD-1/PD-L1 axis restores antitumoral immunity and achieves remissions, but not all patients benefit from this therapy and response rates differ significantly between cancer entities.1,2
Immune checkpoint inhibitors are particularly effective in metastatic melanoma,1 an aggressive skin cancer that derives from pigment producing melanocytes in the epidermis. T-cell infiltration in tumors before treatment has been associated with favorable treatment responses.7-9 This is in line with the concept that checkpoint immunotherapy is most effective against tumors with pre-existing but dampened antitumor immune responses. The interferon (IFN) pathway plays a central role in the orchestration of immune responses and is strongly activated in T-cell-rich melanomas driving the expression of T-cell-recruiting chemokines like CXCL10. Using mouse melanoma models we recently showed that T-cell-poor melanomas respond to PD-1 blockade only after targeted type I IFN activation by the immunostimulatory RNA poly(I:C), because this adjuvant strategy enforced T cell recruitment.10 Similarly, small molecule agonists of the cGAS-STING cytosolic DNA sensing pathway induce innate immune recognition of tumors11 and overcome resistance to PD-1 blockade.12,13
Importantly, recent studies found that the neoantigen load determines tumor immunogenicity and predicts responsiveness to checkpoint inhibitors in melanoma and lung cancer patients.8,9,14-16 Neoantigens arise as a consequence of tumor-specific mutations and their numbers correlate with the exonic mutation frequency. Of note, melanomas and lung cancers have a particular high neoantigen load9,14 due to the mutagenic impact of UV-light irradiation and tobacco smoke in the development of the diseases. These findings suggest that checkpoint inhibitors are less effective in tumors with a low neoantigen load like neuroblastoma, a disease of early childhood, having a more than 20-fold lower exonic mutation frequency.17,18 However, neoantigens are not the only cause of tumor immunogenicity, because cancer testis and cell lineage antigens also elicit strong antitumoral immune responses.
Neuroblastoma is an embryonal malignancy that originates from the developing sympathetic nervous system,19,20 and accounts for 12% of cancer-related deaths in children. Poor disease outcome of high-risk neuroblastoma despite multimodal radiochemotherapy emphasizes the need for additional treatment options like checkpoint inhibitors. As the neoantigen load is low in neuroblastoma, it is likely that additional predictive biomarkers are needed to identify subgroups of patients that could benefit from checkpoint immunotherapy. For this reason, we aimed to identify molecular characteristics of T-cell-infiltrated versus T-cell-poor neuroblastomas, because T-cell infiltration is considered as a marker of pre-existing immune responses that can be enforced by therapeutic checkpoint inhibition.
Using an integrative transcriptomic approach, we report here that a T-cell-poor microenvironment in primary metastatic neuroblastomas is associated with genomic amplification of the MYCN (N-Myc) oncogene. This coincided with a lower interferon pathway activity and reduced chemokine expression in these tumors, and we found that N-Myc suppresses interferon and pro-inflammatory pathway activity in a global manner. Furthermore, N-Myc depletion enhanced targeted interferon pathway activation and Cxcl10 chemokine expression by a small molecule STING agonist, which promoted T-cell recruitment in microfluidics migration assays. Thus, our data delineate an adjuvant strategy to enforce T-cell recruitment and to improve immunotherapy of MYCN-amplified neuroblastomas.
Results
To explore T-cell infiltration in neuroblastoma, we used RNA-seq gene expression data from our previously described patient cohort of 498 primary neuroblastomas with detailed clinical annotation and MYCN-amplification status (Fig. 1A).21 As a measure of T-cell content in neuroblastoma tissues, we calculated the averaged expression level of a T-cell gene signature as described previously.10,22,23 Neuroblastoma samples were subsequently ranked by increasing T-cell signature expression level that separated T-cell-poor from T-cell-rich tumors (Fig. 1B). We also included clinical stage (INSS) and risk (INRG) annotation, genomic MYCN amplification status as well as MYCN mRNA expression level in our analysis. Interestingly, we found that MYCN-amplified neuroblastomas were strongly enriched among the T-cell-poor tumors. Importantly, genomic MYCN amplification and high MYCN mRNA expression were also associated with a T-cell-poor status in the subgroup (n = 181) of stage 4 (INSS) metastatic neuroblastomas (Fig. 1C). This subgroup analysis is important, because the clinical course of neuroblastoma is highly heterogeneous that could confound our results. Next, we repeated the analysis using a gene signature that is highly expressed by different cytotoxic immune cells22 and thus indicative of an antitumor immune response. Again, MYCN-amplified neuroblastomas showed low expression of this cytotoxic immune cell signature within the subgroup of stage 4 metastatic disease (Fig. 1D). We also corroborated the inverse correlation between MYCN amplification and expression of these two immune cell signatures, when we selectively analyzed primary neuroblastomas from the abdomen/pelvis (n = 116) or adrenal gland (n = 197) (Figs. S1 and S2). These are the two most frequent sites of neuroblastoma occurrence comprising 75% of samples in our cohort with available anatomic annotation (n = 420). Hence, this ruled out that contamination or inclusion of lymphoid tissue from metastatic sites such as lymph node or liver confounded our analysis. As MYCN amplification is associated with poor disease outcome, we consistently found that lower expression of the T-cell or cytotoxic immune cell signatures was associated with a reduced overall survival in stage 4 neuroblastoma patients (Fig. 1E). An unbiased median expression cut-off was used for the low versus high classification of the gene expression signatures. Taken together, T-cell or cytotoxic immune cell signatures were stratified by NMYC amplification status and associated with disease outcome.
Figure 1.
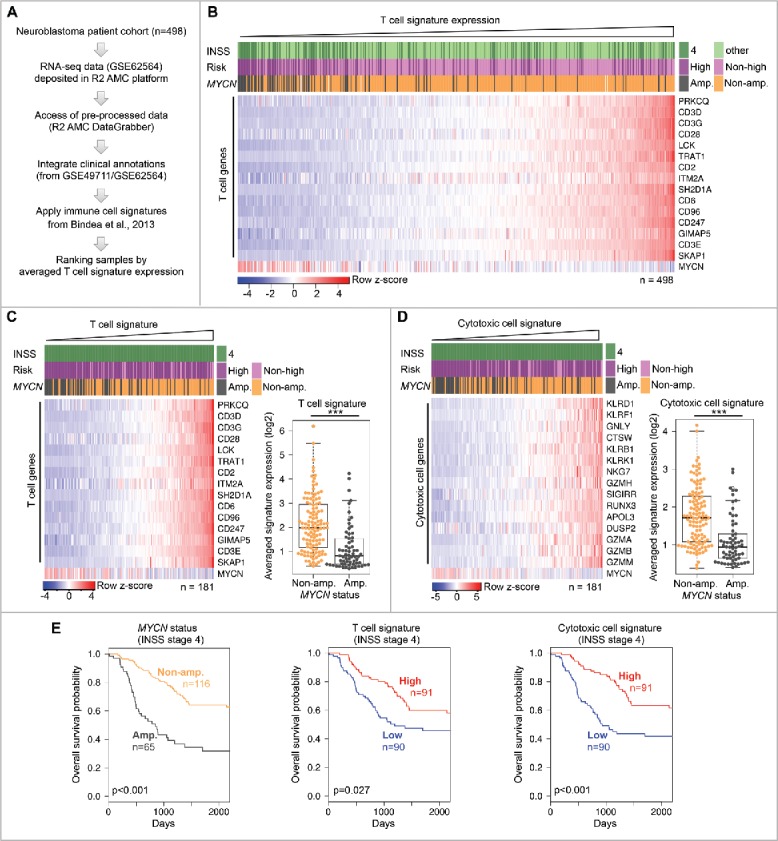
Genomic MYCN amplification is associated with a T-cell-poor microenvironment in metastatic neuroblastoma. (A) Outline of analysis. (B) Expression of T-cell signature genes in entire neuroblastoma cohort. Samples ranked by increasing T-cell signature expression. Log2 gene expression values were z-score transformed for heatmap visualization. Clinical and genomic annotations are indicated. (C) Left panel: The same analysis as in (B), but restricted to INSS stage 4 neuroblastomas. Right panel: T-cell signature expression in MYCN-amplified and non-MYCN-amplified metastatic neuroblastomas. ***p < 0.001; two-sided Wilcoxon rank test. MYCN-amp., n = 65; non-MYCN-amp., n = 116. (D) The same analysis as in (C), but using the cytotoxic immune cell signature. (E) Kaplan–Meier survival plots of INSS stage 4 neuroblastomas stratified by MYCN amplification status (left panel), T-cell signature expression (middle panel) and cytotoxic immune cell signature (right panel). High/low groups were defined by an unbiased median expression value cut-off. p-values determined by two-sided log-rank test.
As these signatures were non-specific for particular subsets of T cells, natural killer T cells (NKT) or NK cells, we aimed to gain more insight by using CIBERSORT, a recently described bioinformatic deconvolution approach to estimate the relative content of immune cell subtypes from whole tissue gene expression data.24,25 First, we ranked the neuroblastoma samples by increasing the expression of a combined T-cell and cytotoxic immune cell signature that included also marker genes for, e.g., NK cells (Fig. S3A). Then, we applied the CIBERSORT method and plotted the estimated fraction of immune cell subtypes using the same ranking of samples (Fig. S3B). Consistently, CIBERSORT reported an increasing cytotoxic CD8+ T cell fraction in line with our analyses (Fig. S3C, upper panel). A concomitant increase in the fraction of regulatory T cells (Tregs) likely reflected feedback inhibition of the immune response (Fig. S3C, lower panel), a notion that was also supported by the elevated expression of negative immune checkpoint genes like CTLA4 or CD274 (Fig. S3A). Interestingly, CIBERSORT revealed opposing trends for the fractions of resting and activated NK cells (Fig. S3D), but this result requires experimental validation and further investigation. Estimated fractions of monocytes and macrophages remained rather constant, besides an increase of pro-inflammatory macrophages (CIBERSORT M1-subtype) (Fig. S3E). Taken together, the independent CIBERSORT approach corroborated our finding that MYCN-amplification is associated with a reduced cytotoxic CD8+ T/immune cell infiltration, also suggesting a poor pre-existing antitumor immune response in this subgroup.
Mutation load emerges as an important determinant of antitumor T-cell responses and responsiveness to checkpoint inhibitors, because it correlates with the number of neoantigens.9,14,26 In lung cancer, a recent study identified a nonsynonymous mutation load ≥ 178 as predictive for good responses to anti-PD-1 therapy14 and similar values have been determined in melanoma.9 Although neuroblastomas have comparably few nonsynonymous mutations, we interrogated the correlation between the T-cell signature expression and the mutation load, which was available for 121 neuroblastomas from our cohort. In these samples, we found that non-MYCN-amplified high-risk neuroblastomas had significantly more nonsynonymous mutations (median 17 mutations, n = 35) than MYCN-amplified high-risk neuroblastomas (median 11 mutations, n = 36) or non-high-risk neuroblastomas (median 4 mutations, n = 50) (Fig. 2A). T-cell signature expression significantly correlated with the mutation load only in high-risk neuroblastomas (Fig. 2B and C). However, this correlation was strictly dependent on the MYCN-status, because no significant correlations were detected within the subgroups of MYCN-amplified and non-MYCN-amplified high-risk neuroblastomas, respectively. Of note, another study found no difference in the mutation load of MYCN-amplified versus non-MYCN-amplified high-risk neuroblastomas.18 Taken together, it cannot be ruled out that the mutation/neoantigen load contributes to immunogenicity and pre-existing immune responses in high-risk neuroblastomas, but our data rather suggest a functional role for N-Myc itself.
Figure 2.
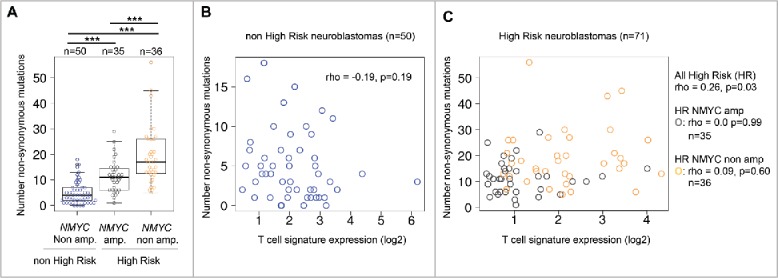
Comparison of T-cell signature expression and mutation load in primary human neuroblastomas. (A) Number of total non-synonymous mutations identified in indicated subgroups of low-risk and high-risk neuroblastomas. ***p < 0.001; pairwise two-sided Wilcoxon rank test with correction for multiple testing (false-discover rate). (B) Correlation analysis of T-cell signature expression level (log2) and mutation load (number of mutations) in non-high-risk neuroblastomas and (C) high-risk neuroblastomas (NMYC-status color-coded). rho: Spearman rank correlation value. p-value determined by two-sided Spearman's rank correlation test with continuity correction.
Alternatively, N-Myc could influence immune cell recruitment to the microenvironment by controlling immune signaling pathways as described by us for c-Myc and Myc-related lineage transcription factors in other tumor entities.27,28 As T-cell-rich melanomas exhibit an activated interferon pathway governing immune cell recruitment,10,29,30 we interrogated the role of N-Myc in the regulation of interferon signaling and chemokine expression. Expression of our melanoma-derived IFN response signature10 positively correlated with the T-cell signature in metastatic neuroblastomas (Fig. 3A). MYCN-amplified neuroblastomas showed a reduced IFN pathway activity (Fig. 3B) and lower CD274 (PD-L1) expression (Fig. 3C). PD-L1 is known to be regulated by the IFN pathway and often predicts treatment responses to anti-PD-1/PD-L1 therapy in several cancers, even though complementary biomarkers will be necessary.31 Furthermore, expression of cytokine and chemokine genes was lower in tissues from MYCN-amplified neuroblastomas (Fig. 3D), which is in line with a T-cell-poor tumor microenvironment. Indeed, a previous study in melanoma correlated the presence of intratumoral T cells with the expression of CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL1032 that were all expressed at higher levels in our non-MYCN-amplified neuroblastomas. Of note, N-Myc has been implicated in the direct transcriptional inhibition of CCL2 chemokine expression.33 However, our data indicated that N-Myc could suppress cytokine and chemokine expression in global manner, similar to what we have described previously for c-Myc in Burkitt's lymphoma.27 In particular, this prompted us to ask whether MYCN amplification determines low IFN pathway activity in cultured neuroblastoma cell lines. For this purpose, we analyzed the expression of the IFN response signature in a panel of human neuroblastoma cell lines with known MYCN amplification status (Fig. 3E). Indeed, baseline IFN pathway activity was significantly lower in MYCN-amplified neuroblastoma cell lines when compared with cell lines lacking this genomic event. Suppression of MYCN expression by two independent siRNAs induced the bona fide IFN response gene OAS1 in the two MYCN-amplified neuroblastoma cell lines SKNBE and NMB, respectively (Fig. 3F and G).
Figure 3.
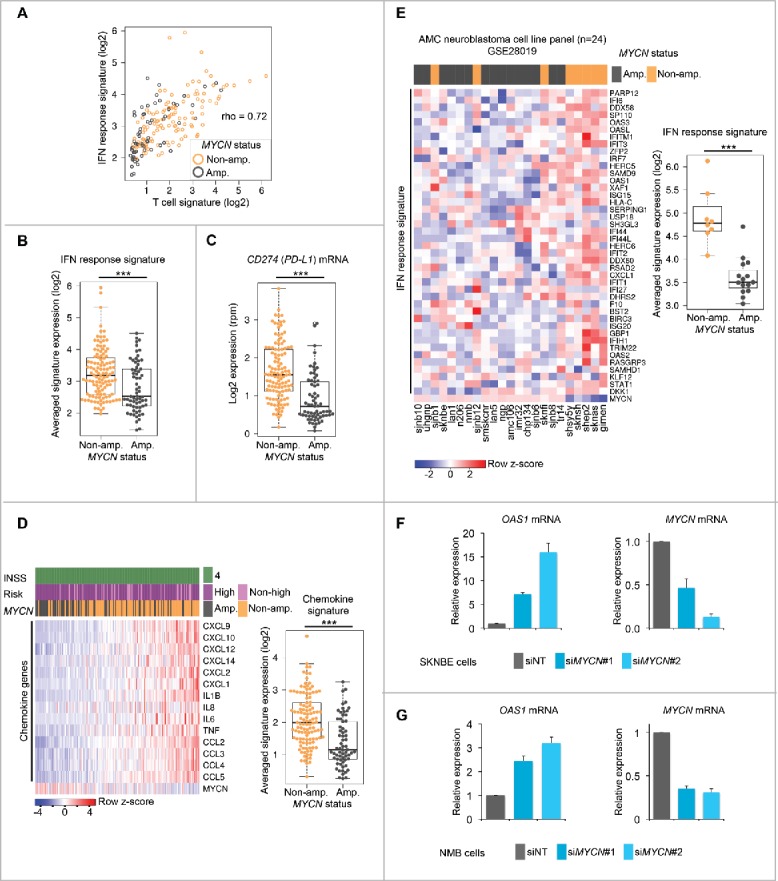
MYCN amplification status is associated with a lower IFN pathway activity in primary metastatic neuroblastomas and cultured neuroblastoma cell lines. (A) Scatter correlation plot of IFN response versus T-cell signature expression in INSS stage 4 neuroblastomas. MYCN amplification status of individual samples is color-coded as indicated. rho; Spearman rank correlation value. (B) IFN response signature expression in MYCN-amplified versus non-MYCN-amplified metastatic neuroblastomas. ***p < 0.001; two-sided Wilcoxon rank test. MYCN-amp., n = 65; non-MYCN-amp., n = 116. (C) The same analysis as in (B), but log2-transformed reads per million (rpm) of CD274 (PD-L1) mRNA expression. (D) Left panel: Expression of selected cytokine and chemokine genes in INSS stage 4 neuroblastoma tissues. Samples ranked by increasing signature expression. Log2 gene expression values were z-score transformed for heatmap visualization. Clinical and genomic annotations as indicated. Right panel: Chemokine signature expression in MYCN-amplified versus non-MYCN-amplified metastatic neuroblastomas. ***p < 0.001; two-sided Wilcoxon rank test. MYCN-amp., n = 65; non-MYCN-amp., n = 116. (E) Left panel: Heatmap clustering of IFN response signature gene expression in human neuroblastoma cell lines. MYCN amplification status is indicated. Right panel: IFN response signature expression in MYCN-amplified versus non-MYCN-amplified human neuroblastoma cell lines. ***p < 0.001; two-sided Wilcoxon rank test. MYCN-amp., n = 16; non-MYCN-amp., n = 8. (F, G) qRT-PCR analysis of relative OAS1 and MYCN mRNA expression (normalized to UBC mRNA expression) in the MYCN-amplified human neuroblastoma cell lines SKNBE (F) and NMB (G) treated with siNT (non-targeting siRNA pool) or two independent siRNAs against MYCN. Error bars indicate standard deviations (s.d.) from three biologic replicates.
Upon recruitment to the tumor tissue T cells become activated by antigen exposure and produce IFN-γ and TNF-α, which evoke a pro-inflammatory microenvironment. Thus, we asked whether N-Myc could impair interferon responses, as this could interrupt feed-forward amplification of inflammatory signals and dampen antitumor immune responses. We treated SKNBE and NMB cells with IFN-γ, IFN-α or TNF-α and detected increased STAT1 phosphorylation and protein levels upon IFN-γ exposure confirming an intact IFN-γ signaling cascade in these cell lines (Fig. 4A). PD-L1 is a well-characterized IFN-γ target and its surface expression was increased by IFN-γ treatment and further augmented by concomitant RNAi knockdown of N-Myc (Fig. 4B). Consistently, RNA-seq expression profiling demonstrated that depletion of N-Myc enhanced the transcriptional response to IFN-γ stimulation (Fig. 4C). Importantly, suppression of N-Myc augmented the induction of chemokine and cytokine genes by IFN-γ including CXCL9 and CXCL10 that are key drivers of T-cell recruitment into the tumor microenvironment (Fig. 4D).34
Figure 4.
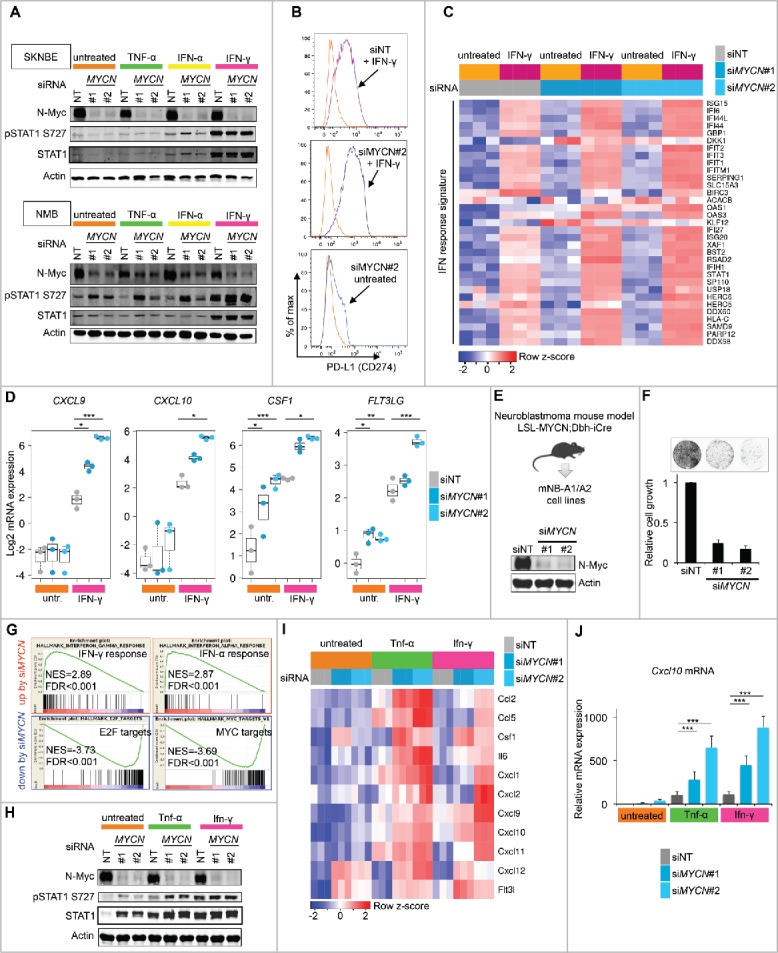
N-Myc restrains INF-γ responses in human and murine neuroblastoma cell lines. (A) Western blot analysis for N-Myc, pSTAT1, total STAT1 and actin protein expression in SKNBE and NMB cells. Transfected siRNAs and interferon/TNF-α stimulation as indicated. (B) FACS analysis of PD-L1 surface expression in SKNBE cells treated as indicated. (C) Expression of IFN response signature genes in SKNBE cells determined by RNA-seq analysis. Transfected siRNAs and INF-γ stimulation as indicated. Log2 expression values were z-score transformed for heatmap visualization. (D) Experiment as described in (C) showing expression of selected chemokine/cytokine genes. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA with Tukey's HSD test for multiple comparisons. (E) Upper panel: Origin of mNB-A1/A2 cell lines. Lower panel: Immunoblot validation of N-Myc depletion in mNB-A1 cells by MYCN siRNAs targeting the human MYCN transgene. (F) Proliferation of mNB-A1 cells treated with non-targeting siRNA pool (siNT) or siRNAs against MYCN. Upper panel: Representative image of crystal violet stained dish. Lower panel: Quantification based on three biologic replicates. Error bars indicate s.d. (G) GSEA showing top hallmark gene sets upregulated or downregulated in mNB-A1 cells by knockdown of MYCN based on RNA-seq gene expression profiling. NES; normalized enrichment score. FDR: False discovery rate q-value. (H) Western blot analysis for N-Myc, pStat1, total Stat1 and actin protein expression in mNB-A1 cells. Transfected siRNAs and Ifnγ/Tnf-α stimulation as indicated. (I) Expression of selected chemokine/cytokine genes in mNB-A1 cells treated as indicated. Visualization of z-score transformed log2 expression values from RNA-seq analysis. (J) qRT-PCR analysis of relative Cxcl10 mRNA expression (normalized to Ubc expression) in mNB-A1 cells treated as described in H/I. Error bars indicate s.d. of biologic triplicates. ***p <0.001, two-sided Wilcoxon rank test.
Based on these results we asked whether N-Myc also impaired IFN-γ responses in neuroblastoma cell lines established from genetically engineered mouse models (GEMMs), as GEMMs are important tools for the pre-clinical evaluation of immunotherapeutic approaches. We described previously the neuroblastoma cell lines mNB-A1 and mNB-A2 isolated from LSL-MYCN;Dbh-iCre transgenic mice.35 In this model, a human MYCN transgene becomes activated in a Cre-conditional manner selectively in dopamine β-hydroxylase (Dbh)-expressing cells and drives neuroblastoma development. Depletion of N-Myc by two independent siRNAs potently impaired proliferation of mNB-A1 cells in culture validating N-Myc oncogene dependency (Fig. 4E and F). We also performed gene expression profiling by RNA-seq analysis to address global signaling pathway alterations in response to acute N-Myc suppression by RNAi. Gene set enrichment analysis (GSEA) using the “hallmark” gene set collection of the Broad molecular signature database (MSigDb) revealed upregulation of IFN pathway activity upon N-Myc depletion in mNB-A1 cells (Fig. 4G and Tables S1 and S2).36,37 In line with the observed growth arrest, MYCN siRNA treated cells also showed downregulation of the proliferation-associated E2F and Myc target gene signatures. Next, we investigated how N-Myc depletion affects the responses of mNB-A1 cells to IFN-γ stimulation. First, IFN-γ exposure increased the levels of phosphorylated and total Stat1 protein confirming intact IFN-γ signaling in mNB-A1 cells (Fig. 4 H). N-Myc depletion by RNAi strongly enhanced IFN-γ and also TNF-α induced transcription of several cytokine and chemokine genes including Cxcl9 and Cxcl10 (Fig. 4I), which was also confirmed by qRT-PCR analysis (Fig. 4J). Altogether, these findings establish N-Myc as a global inhibitor of interferon and pro-inflammatory gene programs in human and murine neuroblastoma cell lines. This provides a mechanistic explanation for the association between MYCN amplifications and a T-cell/immune-cell-poor microenvironment seen in human neuroblastoma tissues.
Targeted IFN pathway activation by stimulating nucleic acid sensing mechanisms of the innate immune system emerges as promising strategy to induce immune responses in T-cell-poor tumors.10,38-40 Indeed, the first pharmacological agents have entered clinical trials. Small-molecule STING agonists potently activate the cGAS-STING cytosolic DNA sensing pathway and trigger IRF3-dependent inflammatory gene transcription,41 including type I IFNs, cytokines and chemokines. DMXAA is a well-characterized small molecule agonist specific for mouse but not human STING.42 Thus, we asked whether inhibition of N-Myc could enhance targeted IFN pathway activation by the STING agonist DMXAA in mouse neuroblastoma cells. Gene expression analysis by RNA-Seq showed that N-Myc depletion strongly augmented the transcriptional response to DMXAA treatment in mNB-A1 cells (Fig. 5A, upper panels). This included IFN inducible genes involved in antigen presentation (Psmb9, Tap1, B2m, H2-K1, and H2-D1) as well as cytokines and chemokines (Flt3l, Cxcl1, and Cxcl10). N-Myc protein level encoded by the human MYCN transgene in mNB-A1 cells was not altered by DMXAA treatment (Fig. 5A, lower panel). Using qRT-PCR, we independently validated our findings by measuring relative mRNA expression of Oas1, Cxcl10 and MYCN in mNB-A2, Nho2a and mNB-A1 cells (Fig. 5B–D). The Nho2a cell line was established from the TH-MYCN transgenic mouse neuroblastoma model and described previously.43 In all three cellular model systems, we consistently observed that MYCN suppression enhanced the transcriptional responses to targeted STING activation by DMXAA. Importantly, N-Myc depletion also strongly augmented Cxcl10 chemokine release into the supernatant induced by DMXAA treatment (Fig. 5E). Several studies have shown that the TH1-type chemokine Cxcl10 plays a pivotal role in the recruitment of T cells to the tumor microenvironment enforcing experimental cancer immunotherapies.34,44 Using a microfluidics-based migration assay, we demonstrated that the conditioned supernatant from N-Myc depleted and DMXAA-treated mNB-A1 cells indeed attracted mouse OT-I T cells (Fig. 5F and G). Within 1 h we tracked the migration of T cells (n = 30 per biologic triplicate) and determined their cell migration speed (velocity) and their forward migration index (yFMI), a measure of the directional migration toward the source of the gradient. Taken together, the data support our concept that inhibition of N-Myc could enhance T-cell recruitment to immune cell-poor MYCN-amplified neuroblastomas upon targeted IFN activation by STING agonists.
Figure 5.
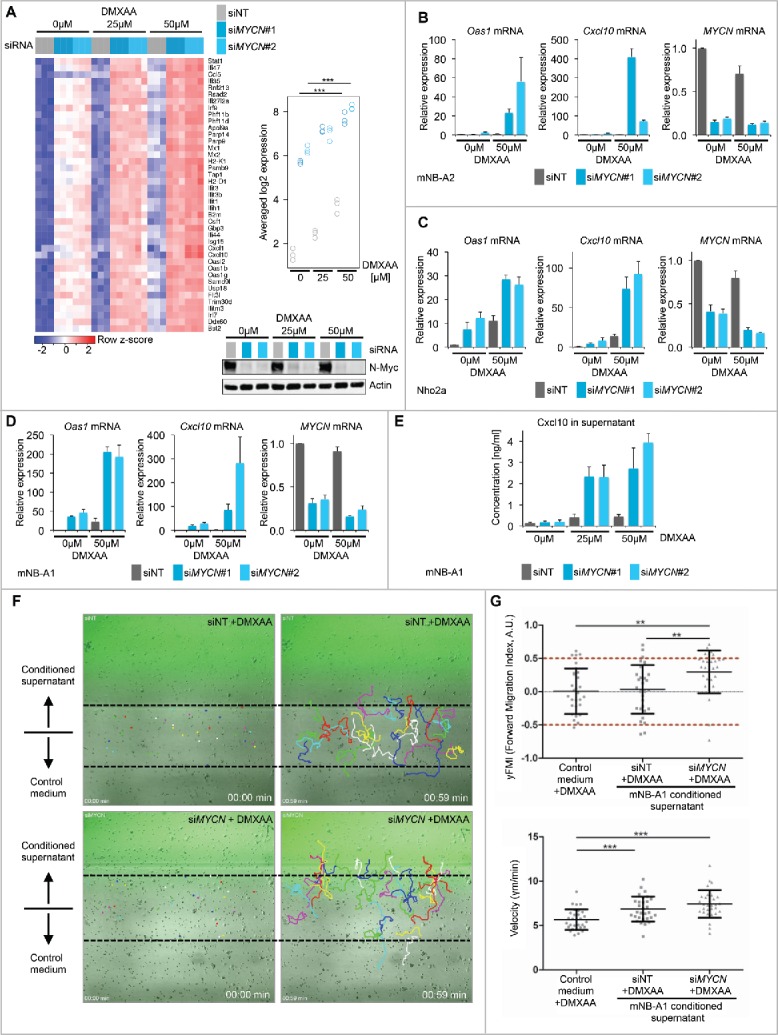
Depletion of N-Myc promotes targeted IFN pathway activation by the STING agonist DMXAA and T-cell recruitment in microfluidics migration assays. (A) Left panel: Expression of IFN-inducible genes in mNB-A1 cells treated with siNT or siMYCN#1/#2 upon exposure to DMXAA or DMSO vehicle control. Log2 expression values were z-score transformed for heatmap visualization. Right upper panel: Quantification of averaged signature expression values shown in the heatmap. ***p < 0.001, ANOVA with Tukey's HSD test for multiple comparisons. Right lower panel: Immunoblot validation of N-Myc depletion in the respective conditions. (B–D) qRT-PCR analysis of relative Oas1, Cxcl10 and MYCN mRNA expression (normalized to Ubc expression) in mNB-A2 (B), Nho2a (C) and mNB-A1 (D) murine neuroblastoma cells treated as described in (A). Error bars indicate s.d. of biologic triplicates. (E) Cxcl10 protein concentration measured by ELISA in supernatant from cultured mNB-A1 cells treated as described in (D/E). Error bars indicate s.d. of biologic triplicates. (F) Representative images (one out of three biologic replicates) showing tracked migration of mouse OT-I T cells in microfluidics gradient chamber. Gradient was established using conditioned supernatant of mNB-A1 cells treated as indicated. (G) Quantitative analyses of T cell (n = 30) migration. Data shown for one representative biologic replicate. Average values (yFMI, velocity) of individual biologic replicates are shown in Fig. S4. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student's t-test.
Discussion
In this study, our transcriptomic analysis demonstrated that genomic MYCN amplification is associated with a T-cell/cytotoxic immune cell poor microenvironment in stage 4 metastatic neuroblastomas. This finding has important implications for clinical efforts that evaluate checkpoint blockade immunotherapy in neuroblastoma patients, because the paucity of intratumoral T cells and lower PD-L1 expression before treatment has been shown to predict poor responses to checkpoint inhibitors in other tumor entities.1,7 Of note, an inverse correlation between NKT cell infiltration and MYCN amplification has been described previously,45 also in line with our cytotoxic cell signature and CIBERSORT analyses. In contrast, a recent immunohistochemical (IHC) study of 84 neuroblastomas (cases with INSS stage 4, n = 20) did not find a significant association with T-cell infiltration and MYCN amplification status in their samples. In concert with our results, T-cell infiltration predicted a better disease outcome, also in the subgroup of MYCN-amplified neuroblastomas.46 Obviously, there is some discrepancy to our study about which we can only speculate, but our analyses were based on a different method (RNA-seq) that is sensitive to capture complex immune cell profiles in whole tumor samples. Our study also involved a significantly larger cohort of patients with metastatic neuroblastoma (cases with INSS stage 4, n = 181). This emphasizes the need to evaluate and compare different methods for quantification of T cell or immune cell content in routine diagnostic setting. Immune signature profiling by the nCounter Nanostring platform,47 cost-effective 3′mRNA-Seq or microarray platforms might be feasible options for this purpose.
Taken together, our data suggest that patients with MYCN-amplified neuroblastomas are less likely to benefit from checkpoint blockade immunotherapy, because their tumors show a T cell/cytotoxic immune cell poor phenotype. Importantly, we provided experimental evidence by showing that the N-Myc driven neuroblastoma cell state impaired IFN pathway activity, chemokine expression and immune cell recruitment in microfluidics migration assays. In vivo, this mechanism could restrain the feed-forward amplification of chemokine responses in the tumor microenvironment and dampen antitumor responses. Our study revealed a potent inhibitory effect of N-Myc on the expression of the TH1-type chemokines Cxcl9 and Cxcl10. Recent experimental studies demonstrated that Cxcl10 expression in tumors promotes T cell-recruitment and enforces T-cell immunotherapy.34,44 Therefore, we hypothesize that N-Myc impairs the establishment of a T-cell-inflamed microenvironment, at least in part, through dampening a feed-forward loop of Cxcl10 expression and recruitment of IFN-γ-producing T cells. However, it is an important finding that N-Myc restrained transcriptional IFN responses on a global level rather than suppressing only individual genes of this pathway. This provides further mechanistic insights in line with previous studies that reported defective antigen processing and presentation in neuroblastoma cells,48 a process that is known to be induced by IFN-γ. Of note, N-Myc and c-Myc have been described as direct suppressors of MHC class I gene transcription.49,50 In summary, N-Myc emerges as a general antagonist of the IFN pathway that regulates antigen processing and presentation as well as chemokine secretion and thus the interaction between neuroblastoma and immune cells. Thereby, N-Myc could also limit the efficacy of immune checkpoint inhibition. However, it is important to consider that IFN signaling can be detrimental to the antitumor immune response due to the activation of multiple negative feedback mechanisms.51 Future studies therefore need to focus on context and scheduling of immunotherapeutic interventions to achieve optimal efficacy.
In contrast to aggressive high-risk neuroblastoma, spontaneous regressions are repeatedly seen among patients with low-risk disease.52,53 This phenomenon is of great interest to researchers and clinicians and various mechanisms have been proposed. Likely, tumor biologic and immunological mechanisms contribute to spontaneous regressions in an interconnected manner.53 For example, insufficient telomerase expression in low-risk neuroblastomas54 may trigger a senescence-like growth arrest that could trigger an immune response,55 as seen in various mouse tumor models.56 Intriguingly, about half of the patients with the neural autoimmune disease opsoclonus myoclonus syndrome (OMS) with antineural autoantibodies have been diagnosed with neuroblastoma.57-59 OMS occurs as a paraneoplastic syndrome in neuroblastoma and is associated with a favorable outcome of the disease. With respect to the rather low neoantigen load in neuroblastoma,17,18,54 this suggests that neuroendocrine lineage antigens substantially contribute to the immunogenicity of neuroblastoma cells. This also implies the caveat of serious neurologic side effects60 that could develop as a consequence of immune checkpoint inhibitor treatment in neuroblastoma patients. Of note, N-Myc is known to impair neuronal differentiation programs,61,62 which could also reduce the expression level of immunogenic neuronal lineage antigens in MYCN-amplified neuroblastomas.
Obviously, a careful pre-clinical evaluation is needed and we believe that MYCN-driven GEMMs are useful tools to explore therapeutic efficacy as well as adverse effects of novel combinatorial treatments. For this reason, we used neuroblastoma cell lines established from two independent MYCN-driven GEMMs and demonstrated that N-Myc antagonized targeted IFN pathway activation by a small molecule STING agonist in both cellular model systems. Thus, our findings provide a scientific basis to evaluate N-Myc inhibition plus targeted IFN activation as adjuvant strategy with immune checkpoint inhibitors in MYCN-driven GEMMs and ultimately also in patients with MYCN-amplified neuroblastomas.
Materials and methods
Cell culture
All cell lines were cultured in a humidified incubator with 5% CO2 at 37 °C. Cells were kept in RPMI 1640 Medium with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin added supplementary. The murine neuroblastoma cell lines mNB-A1 and mNB-A2 additionally received B-27 and N-2 supplement (all from Life Technologies). All used cell lines were negative for mycoplasma contamination and tested on a monthly basis. None of these cell lines appears on the ICLAC list of misidentified cancer cell lines.
Transfection, knockdown and immune stimulation
Cells were seeded at high density in a 12-well plate and reversely transfected with 30 pmol siRNA using 3 µL Lipofectamine RNAiMAX (Life Technologies). Used siRNAs against MYCN were Hs_MYCN_6 (# SI03087518) and Hs_MYCN_7 (SI03113670, both Qiagen). As a negative control, a non-targeting siRNA control pool was used (Dharmacon, # D-001206-13-20). Twenty four hours after knockdown medium was changed and cells were treated with either 25 µM or 50 µM DMXAA (SelleckChem) or equal amounts of DMSO as vehicle control. For cytokine stimulations, murine cells were treated with 100 U/mL murine TNF-α or 250 U/mL murine IFN-γ (Peprotech). Human cell lines were treated with 1,000 U/mL TNF-α, IFN-α or IFN-γ (Peprotech).
Cell growth assay
Cells were seeded at low density and fixed after reaching confluency by covering with 4% formaldehyde. Plates were washed with distilled H2O and stained for 30 min using 0.05% crystal violet. Stained plates were scanned at 800 nm using Odyssey Sa Imaging System (LI-COR Biosciences) and signal intensities were used for quantification.
Immunoblot analysis
For whole cell lysates, cells were lysed in 1X Laemmli buffer (2,000 cells per µL) and incubated for 3 min at 95 °C. Lysed samples were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane (GE Healthcare) by wet blotting for 90 min at 70 V (both systems by BioRad). Membranes were blocked with 5% bovine serum albumin (GE Healthcare) in TBS with 0.5% Tween for 1 h on a shaker and then probed with primary antibodies in 5% BSA in TBS with 0.5% Tween at 4 °C overnight. On the next day membranes were probed with IRDye680LT and IRDye800CW secondary antibodies in 3% BSA in TBS with 0.5% Tween. Proteins were detected by measuring at 700 nm and 800 nm wavelength using Odyssey Sa Imaging system (LI-COR Biosciences). Used antibodies were as follows: Broad Range Markers (#sc-2361), Actin (#sc-47778), N-Myc (#sc-53993), Stat1 (#sc-464, all Santa Cruz Biotechnology), pStat1 (#9177, Cell Signaling Technology).
RNA isolation, cDNA synthesis and quantitative real-time PCR
Treated cells were lysed with RLT buffer (Qiagen) and then frozen at −80°C for at least 15 min. After thawing on ice and adding ethanol lysates were applied to Zymo Spin II columns (Zymo Research). RNA was isolated by washing with RW1 washing buffer (Qiagen) and Zymo RNA washing buffer (Zymo Research) and eluted with RNAse-free H2O. The concentration of the RNA was measured using NanoDrop 2000c (Thermo Scientific). cDNA synthesis was performed using All-in-One cDNA Synthesis SuperMix (Biotools) according to manufacturer's instructions. qRT-PCR reactions were always prepared in technical duplicates in a total volume of 10 µL containing EvaGreen (BioBudget), primers and RNase-free H2O. qRT-PCR was performed using Roche LC480 according to manufacturer's instructions. Samples were quantified by normalization to the housekeeping gene ubiquitin. Sequences of used qRT-PCR primer pairs (all ordered from Microsynth) are listed in Table S3.
3′mRNA RNAseq
Cells were lysed in 200 μL RLT buffer (Qiagen) and total RNA was isolated using Zymo I spin columns (Zymo Research) and eluted in 8 μL of RNAase-free H2O. RNA concentrations were determined using Qubit (LifeTech). 3′mRNA-seq library preparation was performed using the forward QuantSeq 3′mRNA-Seq Library Prep Kit for Illumina (Lexogen GmbH, Austria) according to the manufacturer's protocol. Size distribution and yield of the 3′mRNA-seq library after the PCR step was determined by the D1000 high sensitivity tape station (Agilent) before pooling of the barcoded libraries. The pooled 3′mRNA-Seq libraries were loaded on the Illumina HiSeq2500 platform and analyzed by a 50 cycles rapid run with on-cartridge clustering. Raw data is accessible through ENA project PRJEB20874/ERP023066. Computational 3′mRNA-seq analysis was done with the Bioconductor/R computing environment. FASTQ files were aligned to the hg38 human or mm10 mouse reference genome using the RSubread aligner package.63 The voom method of the limma package was used for normalization and linear modeling.64 mRNA expression values were transformed to log2 values of read counts per million (rpm or cpm). For GSEA non- or very low expressed genes were removed which resulted in ∼6,000 robustly expressed genes.
Gene set enrichment analysis (GSEA)
GSEA36 was performed using the BROAD javaGSEA standalone version (http://www.broadinstitute.org/gsea/downloads.jsp) and the curated hallmark gene set collection (BROAD molecular signature database, MSigDbv5.1). We used the default setting of 1,000 permutations and the gene-set permutation mode. For GSEA we used the eBayes moderated t-test statistics as ranked metric input for the GSEA pre-ranked gene list algorithm. Results from GSEA are provided in Tables S1 and S2.
Analysis of published data sets
Pre-processed and normalized RNA-seq data of primary human neuoblastomas (GSE49711/GSE62564) and Affymetrix (u133p2) microarray data of human neuroblastoma cell lines were accessed through the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). Clinical annotation and MYCN-status were obtained through GSE49711/GSE62564. Data was imported into the R/Bioconductor computing platform and analyzed by standard procedures. Immune cell gene signatures were obtained from Bindea et al.22 and signature expression levels were defined as the mean of log2-transformed gene expression values.
Mutation load analysis
Processed nonsynonymous mutation data and MYCN amplification status for 235 high-risk neuroblastomas was accessed from the recently published study by Pugh et al.18 A full genomic characterization of our neuroblastoma cohort will be described elsewhere (Ackermann et al., The genetic basis of favorable outcome and fatal tumor progression in neuroblastoma, submitted).
Flow cytometry
Seeded out cells were washed with PBS and detached shortly using 0.05% trypsine (Life Technologies). Resuspended cells were then transferred to a high binding 96-well plate (Greiner) and stained with fluorochrome-conjugated monoclonal antibodies (Anti-CD274, #17-5983-41, eBioscience Inc.). Probes were fixed in 4% formaldehyde and kept sealed at 4 °C overnight. Data were acquired using FACSCanto Flow Cytometer (BD Biosciences) and analyzed with FlowJo software (FlowJo LLC, Version 8.7 for Mac).
ELISA
Supernatants of stimulated cells grown to confluency of 80–90% were collected and centrifuged at 300g for 3 min. Samples were stored at −80 °C for a short period and thawed on ice. The murine Cxcl10-Elisa kit (R&D DuoSet #DY466-05) was used according to manufacturer's instructions. The absorbance of the samples was measured using Tecan Infinite M200 Pro. For wavelength correction, measured absorbance at 570 nm was subtracted from the measured 450 nm absorbance. The concentration of chemokines in the supernatants was calculated based on the standard curve received with the provided chemokine standard.
Analysis of T cell chemotaxis
In vitro analysis of T-cell chemotaxis was performed in a microfluidic device for controlled application of dynamic chemokine gradients using µ-slide III 3-in-1 (Ibidi) channel chambers. CD8+ T cells were obtained from OT-1 spleen and lymph node preparations by MACS separation using negative selection according to the manufacturer's instructions (Miltenyi). Subsequently, CD8+ T cells were activated with OVA-pulsed BM-DCs from C57BL/6JRcc mice for 120 h. The slides were coated with 13.0 µg/mL goat anti-human IgG antibody (Dianova) and subsequently were blocked with 1% BSA in PBS at RT for 1 h. 50 µg/mL murine ICAM-1/FC (R&D Systems) was immobilized on the antibody-coated surface at RT for 1 h. 2 × 105 CD8+ T cells in RPMI (+1% FCS, + 50 µM DMXAA, +5 mM Mg2+) were transferred to the channel. Gradient fields were generated by controlled inflow (10 µL/min) of conditioned neuroblastoma cell culture supernatants and control media by the use of separately connected infusion syringe pumps to individual inflow channels. Live cell imaging of adherent CD8+ T cells was performed at 37 °C using a fully automated inverted Olympus Fluoview 1000 confocal microscope equipped with a motorized xyz-stage (Märzhäuser). Chemotaxis of CD8+ T cells was recorded over a period of 60 min by capture of differential interference contrast images every 60 sec with a 0.40 UPLAPO 10× objective (Olympus). A solution of 5 µM FITC-Dextran (10 kDa, Invitrogen) was used to characterize the concentration profile of the cell culture supernatant because of the comparable molecular weight to most relevant chemokines. Migration parameters of motile cells were calculated using the Manual Tracking and Chemotaxis Tool plugins in ImageJ.
Statistical tests
All statistical tests were calculated with R and specified in the figure legends. If necessary, corrections for multiple comparisons were applied.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank K. Keppler from the department of human genetics and the UKB NGS core facility for technical NGS support. We also thank the UKB FACS core facility for support. G.H., T.T., W.K. and M.H. are members of the DFG excellence cluster ImmunoSensation. All authors reviewed the manuscript.
Funding
This work was supported by a grant to J.P.L. and M.H. from the Bonn NeuroImmunology (BonnNI) program funded by the Else-Kröner-Fresenius Stiftung.
References
- 1.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E et al.. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320-30; PMID:25399552; https://doi.org/ 10.1056/NEJMoa141208210.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 2.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E et al.. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015; 373:1627-39; PMID:26412456; https://doi.org/ 10.1056/NEJMoa150764310.1056/NEJMoa1507643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER et al.. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015; 373:1803-13; PMID:26406148; https://doi.org/ 10.1056/NEJMoa151066510.1056/NEJMoa1510665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L et al.. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015; 372:2018-28; PMID:25891174; https://doi.org/ 10.1056/NEJMoa150182410.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 5.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol 2015; 36:265-76; PMID:25797516; https://doi.org/ 10.1016/j.it.2015.02.00810.1016/j.it.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YJ. Subverting the adaptive immune resistance mechanism to improve clinical responses to immune checkpoint blockade therapy. Oncoimmunology 2014; 3:e954868; PMID:25964860; https://doi.org/ 10.4161/21624011.2014.95486810.4161/21624011.2014.954868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; https://doi.org/ 10.1038/nature1395410.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G et al.. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016; 168:35-44; https://doi.org/ 10.1016/j.cell.2016.02.06510.1016/j.cell.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM et al.. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015; 350:207-11; PMID:26359337; https://doi.org/ 10.1126/science.aad009510.1126/science.aad0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U et al.. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 2014; 4:674-87; PMID:24589924; https://doi.org/ 10.1158/2159-8290.CD-13-045810.1158/2159-8290.CD-13-0458 [DOI] [PubMed] [Google Scholar]
- 11.Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA et al.. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014; 41:830-42; PMID:25517615; https://doi.org/ 10.1016/j.immuni.2014.10.01710.1016/j.immuni.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol Res 2016; 4:1061-71; PMID:27821498; https://doi.org/ 10.1158/2326-6066.CIR-16-0104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W et al.. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 2015; 7:283ra52; PMID:25877890; https://doi.org/ 10.1126/scitranslmed.aaa430610.1126/scitranslmed.aaa4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al.. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124-8; PMID:25765070; https://doi.org/ 10.1126/science.aaa134810.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al.. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371:2189-99; PMID:25409260; https://doi.org/ 10.1056/NEJMoa140649810.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J et al.. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515:577-81; PMID:25428507; https://doi.org/ 10.1038/nature1398810.1038/nature13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA et al.. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214-8; PMID:23770567; https://doi.org/ 10.1038/nature1221310.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A et al.. The genetic landscape of high-risk neuroblastoma. Nat Genet 2013; 45:279-84; PMID:23334666; https://doi.org/ 10.1038/ng.252910.1038/ng.2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung N-KV, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 2013; 13:397-411; PMID:23702928; https://doi.org/ 10.1038/nrc352610.1038/nrc3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang M, Stanke J, Lahti JM. The connections between neural crest development and neuroblastoma. Curr Top Dev Biol 2011; 94:77-127; PMID:21295685; https://doi.org/ 10.1016/B978-0-12-380916-2.00004-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Zhang W, Thierry-Mieg D, Wang J, Furlanello C, Devanarayan V, Cheng J et al.. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol 2015; 16:133; PMID:26109056; https://doi.org/ 10.1186/s13059-015-0694-110.1186/s13059-015-0694-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A et al.. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013; 39:782-95; PMID:24138885; https://doi.org/ 10.1016/j.immuni.2013.10.00310.1016/j.immuni.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 23.Hölzel M, Landsberg J, Glodde N, Bald T, Rogava M, Riesenberg S, Becker AJ, Jonsson G, Tüting T. A preclinical model of malignant peripheral nerve sheath tumor-like melanoma is characterized by infiltrating mast cells. Cancer Res 2015; 76:251-63; PMID:26511633; https://doi.org/ 10.1158/0008-5472.CAN-15-109010.1158/0008-5472.CAN-15-1090 [DOI] [PubMed] [Google Scholar]
- 24.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015; 12:453-7; PMID:25822800; https://doi.org/ 10.1038/nmeth.333710.1038/nmeth.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD et al.. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21:938-45; PMID:26193342; https://doi.org/ 10.1038/nm.390910.1038/nm.3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schumacher TN, Hacohen N. Neoantigens encoded in the cancer genome. Curr Opin Immunol 2016; 41:98-103; PMID:27518850; https://doi.org/ 10.1016/j.coi.2016.07.00510.1016/j.coi.2016.07.005 [DOI] [PubMed] [Google Scholar]
- 27.Schlee M, Hölzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, Eick D, Marinkovic D, Wirth T, Rosenwald A et al.. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt's lymphoma. Int J Cancer 2007; 120:1387-95; PMID:17211884; https://doi.org/ 10.1002/ijc.2237210.1002/ijc.22372 [DOI] [PubMed] [Google Scholar]
- 28.Riesenberg S, Groetchen A, Siddaway R, Bald T, Reinhardt J, Smorra D, Kohlmeyer J, Renn M, Phung B, Aymans P et al.. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat Commun 2015; 6:8755; PMID:26530832; https://doi.org/ 10.1038/ncomms975510.1038/ncomms9755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenzel J, Bekisch B, Uerlich M, Haller O, Bieber T, Tüting T. Type I interferon-associated recruitment of cytotoxic lymphocytes: a common mechanism in regressive melanocytic lesions. Am J Clin Pathol 2005; 124:37-48; PMID:15923172; https://doi.org/ 10.1309/4EJ9KL7CGDENVVLE10.1309/4EJ9KL7CGDENVVLE [DOI] [PubMed] [Google Scholar]
- 30.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL et al.. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012; 4:127ra37; PMID:22461641; https://doi.org/ 10.1126/scitranslmed.300368910.1126/scitranslmed.3003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Festino L, Botti G, Lorigan P, Masucci GV, Hipp JD, Horak CE, Melero I, Ascierto PA. Cancer treatment with Anti-PD-1/PD-L1 agents: is PD-L1 expression a biomarker for patient selection? Drugs 2016; 76:925-45; PMID:27229745; https://doi.org/ 10.1007/s40265-016-0588-x10.1007/s40265-016-0588-x [DOI] [PubMed] [Google Scholar]
- 32.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009; 69:3077-85; PMID:19293190; https://doi.org/ 10.1158/0008-5472.CAN-08-228110.1158/0008-5472.CAN-08-2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song L, Ara T, Wu H-W, Woo C-W, Reynolds CP, Seeger RC, DeClerck YA, Thiele CJ, Sposto R, Metelitsa LS. Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. J Clin Invest 2007; 117:2702-12; PMID:17710228; https://doi.org/ 10.1172/JCI3075110.1172/JCI30751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W et al.. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015; 527:249-53; PMID:26503055; https://doi.org/ 10.1038/nature1552010.1038/nature15520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprüssel A, Lindner S, De Preter K, Florin A, Heukamp LC et al.. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015; 34:3357-68; PMID:25174395; https://doi.org/ 10.1038/onc.2014.26910.1038/onc.2014.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102:15545-50; PMID:16199517; https://doi.org/ 10.1073/pnas.050658010210.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015; 1:417-25; PMID:26771021; https://doi.org/ 10.1016/j.cels.2015.12.00410.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo S-R, Lemmens E, Banda T, Leong JJ et al.. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 2015; 11:1018-30; PMID:25959818; https://doi.org/ 10.1016/j.celrep.2015.04.03110.1016/j.celrep.2015.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the Non-T-Cell-Inflamed tumor microenvironment. Semin Oncol 2015; 42:663-71; PMID:26320069; https://doi.org/ 10.1053/j.seminoncol.2015.05.01110.1053/j.seminoncol.2015.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlee M, Hartmann G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 2016; 16:566-80; PMID:27455396; https://doi.org/ 10.1038/nri.2016.7810.1038/nri.2016.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cavlar T, Deimling T, Ablasser A, Hopfner K-P, Hornung V. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J 2013; 32:1440-50; PMID:23604073; https://doi.org/ 10.1038/emboj.2013.8610.1038/emboj.2013.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L et al.. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 2013; 154:748-62; PMID:23910378; https://doi.org/ 10.1016/j.cell.2013.07.02310.1016/j.cell.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 1997; 16:2985-95; PMID:9214616; https://doi.org/ 10.1093/emboj/16.11.298510.1093/emboj/16.11.2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L et al.. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res 2016; 76:275-82; PMID:26567139; https://doi.org/ 10.1158/0008-5472.CAN-15-193810.1158/0008-5472.CAN-15-1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Metelitsa LS, Wu H-W, Wang H, Yang Y, Warsi Z, Asgharzadeh S, Groshen S, Wilson SB, Seeger RC. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med 2004; 199:1213-21; PMID:15123743; https://doi.org/ 10.1084/jem.2003146210.1084/jem.20031462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mina M, Boldrini R, Citti A, Romania P, D'Alicandro V, De Ioris M, Castellano A, Furlanello C, Locatelli F, Fruci D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015; 4:e1019981; PMID:26405592; https://doi.org/ 10.1080/2162402X.2015.101998110.1080/2162402X.2015.1019981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cesano A. nCounter( PanCancer Immune Profiling Panel (NanoString Technologies, Inc., Seattle, WA). J Immunother Cancer 2015; 3:42; PMID:26674611; https://doi.org/ 10.1186/s40425-015-0088-710.1186/s40425-015-0088-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raffaghello L, Prigione I, Bocca P, Morandi F, Camoriano M, Gambini C, Wang X, Ferrone S, Pistoia V. Multiple defects of the antigen-processing machinery components in human neuroblastoma: immunotherapeutic implications. Oncogene 2005; 24:4634-44; PMID:15897905; https://doi.org/ 10.1038/sj.onc.120859410.1038/sj.onc.1208594 [DOI] [PubMed] [Google Scholar]
- 49.Bernards R, Dessain SK, Weinberg RA. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 1986; 47:667-74; PMID:3096575; https://doi.org/ 10.1016/0092-8674(86)90509-X10.1016/0092-8674(86)90509-X [DOI] [PubMed] [Google Scholar]
- 50.Versteeg R, Noordermeer IA, Krüse-Wolters M, Ruiter DJ, Schrier PI. c-myc down-regulates class I HLA expression in human melanomas. EMBO J 1988; 7:1023-9; PMID:3402430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-SaintVictor C, Cucolo L, Lee DSM, Pauken KE, Huang AC et al.. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016; 167:1540-54.e12; PMID:27912061; https://doi.org/ 10.1016/j.cell.2016.11.02210.1016/j.cell.2016.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter H-G, Schwabe D, Schilling FH, Benz-Bohm G, Berthold F. Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 2008; 26:1504-10; PMID:18349403; https://doi.org/ 10.1200/JCO.2007.12.334910.1200/JCO.2007.12.3349 [DOI] [PubMed] [Google Scholar]
- 53.Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol 2014; 11:704-13; PMID:25331179; https://doi.org/ 10.1038/nrclinonc.2014.16810.1038/nrclinonc.2014.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Krämer A, Roncaioli JL, Sand F, Heuckmann JM et al.. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015; 526:700-4; PMID:26466568; https://doi.org/ 10.1038/nature1498010.1038/nature14980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; https://doi.org/ 10.1038/nature0552910.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A et al.. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479:547-51; PMID:22080947; https://doi.org/ 10.1038/nature1059910.1038/nature10599 [DOI] [PubMed] [Google Scholar]
- 57.Antunes NL, Khakoo Y, Matthay KK, Seeger RC, Stram DO, Gerstner E, Abrey LE, Dalmau J. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol 2000; 22:315-20; PMID:10959901; https://doi.org/ 10.1097/00043426-200007000-0000710.1097/00043426-200007000-00007 [DOI] [PubMed] [Google Scholar]
- 58.Cooper R, Khakoo Y, Matthay KK, Lukens JN, Seeger RC, Stram DO, Gerbing RB, Nakagawa A, Shimada H. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: histopathologic features – a report from the Children's Cancer Group. Med Pediatr Oncol 2001; 36:623-9; PMID:11344493; https://doi.org/ 10.1002/mpo.113910.1002/mpo.1139 [DOI] [PubMed] [Google Scholar]
- 59.Rudnick E, Khakoo Y, Antunes NL, Seeger RC, Brodeur GM, Shimada H, Gerbing RB, Stram DO, Matthay KK. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies – a report from the Children's Cancer Group Study. Med Pediatr Oncol 2001; 36:612-22; PMID:11344492; https://doi.org/ 10.1002/mpo.113810.1002/mpo.1138 [DOI] [PubMed] [Google Scholar]
- 60.de Maleissye M-F, Nicolas G, Saiag P. Pembrolizumab-induced demyelinating polyradiculoneuropathy. N Engl J Med 2016; 375:296-7; PMID:27468083; https://doi.org/ 10.1056/NEJMc151558410.1056/NEJMc1515584 [DOI] [PubMed] [Google Scholar]
- 61.Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev 2002; 16:2699-712; PMID:12381668; https://doi.org/ 10.1101/gad.102120210.1101/gad.1021202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shimada H, Stram DO, Chatten J, Joshi VV, Hachitanda Y, Brodeur GM, Lukens JN, Matthay KK, Seeger RC. Identification of subsets of neuroblastomas by combined histopathologic and N-myc analysis. J Natl Cancer Inst 1995; 87:1470-6; PMID:7674334; https://doi.org/ 10.1093/jnci/87.19.147010.1093/jnci/87.19.1470 [DOI] [PubMed] [Google Scholar]
- 63.Liao Y, Smyth GK, Shi W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 2013; 41:e108; PMID:23558742; https://doi.org/ 10.1093/nar/gkt21410.1093/nar/gkt214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15:R29; PMID:24485249; https://doi.org/ 10.1186/gb-2014-15-2-r2910.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.