

Abstract
G-protein-coupled receptors (GPCRs) have been shown to form dimers, but the relevance of this phenomenon in G-protein activation is not known. Among the large GPCR family, metabotropic glutamate (mGlu) receptors are constitutive dimers. Here we examined whether both heptahelical domains (HDs) are turned on upon full receptor activation. To that aim, we measured G-protein coupling efficacy of dimeric mGlu receptors in which one subunit bears specific mutations. We show that a mutation in the third intracellular loop (i3 loop) known to prevent G-protein activation in a single subunit decreases coupling efficacy. However, when a single HD is blocked in its inactive state using an inverse agonist, 2-methyl-6-(phenylethynyl)pyridine (MPEP), no decrease in receptor activity is observed. Interestingly, in a receptor dimer in which the subunit that binds MPEP is mutated in its i3 loop, MPEP enhances agonist-induced activity, reflecting a ‘better' activation of the adjacent HD. These data are consistent with a model in which a single HD is turned on upon activation of such homodimeric receptors and raise important issues in deciphering the functional role of GPCR dimer formation for G-protein activation.
Keywords: allostery, G-protein coupling, metabotropic glutamate receptors, receptor activation
Introduction
G-protein-coupled receptors (GPCRs) are major players in cell–cell communication (Bockaert and Pin, 1999). These receptors are encoded by more than 1% of the mammalian genes and are the target of about 50% of the drugs on the market. Although our knowledge of their activation mechanism, as well as of the various processes involved in their regulation, has expanded extensively within the last 10 years, it is still unclear how these receptors stimulate the GDP–GTP exchange in heterotrimeric G-proteins. For many years, it was assumed that GPCRs are monomers, one receptor molecule being activated by a single ligand and activating one heterotrimeric G-protein. However, recent studies revealed that these receptors can form dimers or higher ordered oligomers, but the functional significance of this phenomenon remains unclear (Kühn, 1984; Salahpour et al, 2000; Bouvier, 2001; Chabre et al, 2003; Fotiadis et al, 2003). Some authors propose that a dimer of GPCRs is required for G-protein activation (Baneres and Parello, 2003; Liang et al, 2003), but monomeric rhodopsins are capable of activating transducin (Kühn, 1984; Jastrzebska et al, 2004). This raises the question of whether both subunits in a dimeric receptor have to be turned on to activate a G-protein.
Several classes of GPCRs have been defined based on their sequence similarity (Kolakowski, 1994; Bockaert and Pin, 1999; Fredriksson et al, 2003). Whereas the rhodopsin-like receptors constitute the most abundant class (class A), the secretin-like and metabotropic glutamate (mGlu)-like receptors constitute smaller classes (B and C, respectively). Class C includes receptors for the two major neurotransmitters, glutamate and γ-aminobutyric acid (GABA), as well as the Ca2+-sensing and some taste and pheromone receptors (Pin et al, 2003). Most of these class C GPCRs are constitutive dimers, with the two subunits being covalently linked by a disulfide bridge (Romano et al, 1996; Tsuji et al, 2000; Pin and Acher, 2002). This has been firmly demonstrated for the mGlu and Ca2+-sensing receptors, and is likely the case for the taste and pheromone receptors, but not for the GABAB receptor (Pin et al, 2003). However, the latter is an obligatory heterodimer composed of the GABAB1 and GABAB2 subunits stabilized by an intracellular coiled-coil interaction (Calver et al, 2001). Such receptors therefore constitute an excellent model to examine the specific role of the two subunits in G-protein activation.
In addition to the heptahelical domain (HD), which is typical for all GPCRs, class C receptors possess a large extracellular domain consisting of a Venus Flytrap domain (VFT). Biochemical and structural studies further demonstrate direct interaction between the two VFTs in these dimeric receptors (Kunishima et al, 2000; Tsuji et al, 2000; Liu et al, 2004). Structural as well as functional analysis indicates that a important change in the relative orientation of the two VFTs resulting from their closure upon agonist binding is a necessary step for receptor activation (Kunishima et al, 2000; Bessis et al, 2002; Tsuchiya et al, 2002; Kniazeff et al, 2004b). As such, the dimeric nature of these receptors appears crucial for the intramolecular transduction, that is, transfer of information from the VFT to the HD.
Despite these differences, HDs of class A and class C GPCRs likely function in a similar manner (Binet et al, 2004; Goudet et al, 2004). Of interest, noncompetitive antagonists that bind in the HD have been identified for class C GPCRs (Pagano et al, 2000; Carroll et al, 2001; Knoflach et al, 2001). As observed for class A antagonists, most of these compounds stabilize the inactive state, as demonstrated by their inverse agonist activity both on full-length receptors (Pagano et al, 2000; Carroll et al, 2001) and receptors deleted of their VFT (Goudet et al, 2004).
In the present study, we examined whether a single HD or two HDs have to be turned on per mGlu1 receptor dimer for full activity. To that aim, we used a system that allows the functional expression of mGlu1 dimeric receptors composed of two well-defined subunits, each bearing specific mutations. Our data indicate that binding of a single inverse agonist per dimer does not affect receptor activity. This is in contrast to the decreased G-protein coupling efficacy observed when a mutation is introduced in the third intracellular loop (i3 loop) of a single subunit. These plus other data are consistent with a model in which a single HD is turned on upon activation of such a dimeric receptor.
Results
Generation of ‘heterodimeric' mGlu1 receptors
In order to analyze the role of each subunit in the activation process of homodimeric mGlu receptors, one need to have access to receptors in which engineered mutations are carried by a single subunit only. To that aim, we used the quality control system of the heterodimeric GABAB receptor. In this receptor dimer, the GB1 subunit does not reach the cell surface alone due to the presence of an endoplasmic reticulum (ER) retention signal (RSRR) in its C-terminal intracellular tail. This signal is masked when associated with the C-terminal tail of GB2 (Couve et al, 1998; Margeta-Mitrovic et al, 2000; Pagano et al, 2001). Accordingly, two chimeric mGlu1 receptors called R1c1 and R1c2 were created by replacing the mGlu1 C-terminal tail by that of GB1 and GB2 subunits, respectively (all constructs used in this study are described in Table I). As expected, R1c1 does not reach the cell surface alone (Figure 1), whereas it does if the ER retention signal RSRR is mutated into ASAR (data not shown). As observed with the GABAB receptor, the C-terminal tail of GB2 in R1c2 did not prevent this receptor from reaching the cell surface alone, but allowed R1c1 to be targeted to the surface (Figure 1).
Table 1.
Nomenclature used for the constructs described in this study
| Symbol | Description of the modification | Effect of the modification |
|---|---|---|
| R1 | mGlu1 receptor-based construct | |
| c1 | C-terminal tail of GB1 | Retained in the ER in the absence of a c2 construct |
| c1ASA | c1 with the ER signal mutated into ASA | Not retained in the ER |
| c2 | C-terminal tail of GB2 | Allows R1c1:R1c2 to reach the surface |
| X | One point mutation (F781P) in the i3 loop | Loss of coupling to Gq |
| M | Three point mutations in HD creating an MPEP site | Inhibited by MPEP |
| B | Two mutations in VFT (Y236A, D318A) | No activation by agonist |
Figure 1.
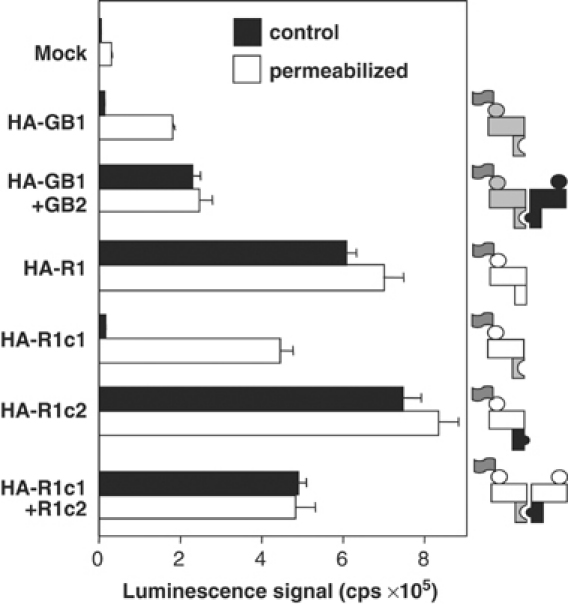
Determination of cell surface expression of GABAB and wild-type or chimeric mGlu1 receptors. ELISA assay was conducted on intact cells (control, black columns) or on cells permeabilized with Triton X-100 (white columns) using an HA antibody. HA-tagged GB1, mGlu1 (R1), R1c1 or R1c2 was transfected alone or together with myc-tagged GB2 or R1c2, as illustrated in the figure. Mock represents the signal obtained with pRK6-transfected cells. Values are means±s.e.m. of triplicate determinations from one representative experiment out of three independent experiments.
Heterodimers R1c1:R1c2 reach the cell surface
In order to demonstrate firmly that R1c1:R1c2 heterodimers exist at the cell surface, time-resolved fluorescence resonance energy transfer (TR-FRET) experiments were performed with an anti-HA (HA: hemagglutinin) antibody labeled with the donor fluorophore EuCryptate, and an anti-myc antibody labeled with the acceptor fluorophore Alexa647. As shown in Figure 2A, a large FRET signal was detected in cells expressing HA-R1c1 and myc-R1c2, as well as in cells expressing both HA-GB1 and myc-GB2. Such a signal was not observed after mixing cells expressing HA-R1c1 and cells expressing myc-R1c2, and only a small signal was obtained in cells coexpressing HA-R1c2 and the myc-tagged V2 vasopressin receptor (Figure 2A) despite a similar expression level of each partner at the cell surface (Figure 2B and C). Moreover, the FRET signal was directly proportional to the amount of HA-tagged subunit expressed at the cell surface (Figure 2E).
Figure 2.
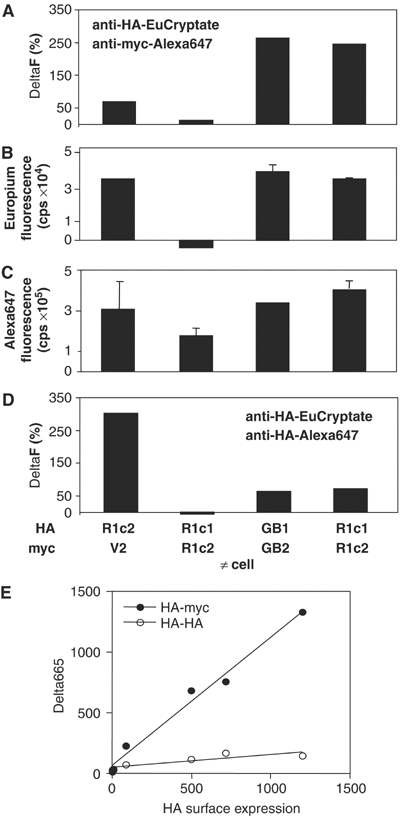
Heterodimerization of chimeric mG1 receptor subunits. (A) The TR-FRET signal was measured using anti-HA-EuCryptate and anti-myc-Alexa647 antibodies using intact cells expressing the indicated subunits. (B) The specific fluorescent signal (signal measured in the indicated cells minus the signal measured in mock-transfected cells) was measured with the anti-HA-EuCryptate antibody, and is used here to estimate the level of surface expression of the HA-tagged subunits. (C) Same as in (B) with the anti-myc-Alexa647 antibody to estimate the surface expression of the myc-tagged subunits. (D) Same as in (A) but using an equimolar amount of anti-HA-EuCryptate and anti-HA-Alexa647 to visualize any possible interaction between the HA-tagged subunits. (E) The TR-FRET signal was measured in cells transfected with 1 μg HA-R1c1 and various amounts of myc-R1c2 (0–1 μg). Anti-HA-EuCryptate and anti-myc-Alexa647 antibodies were used to estimate HA-R1c1:myc-R1c2 heterodimers (open circles), while anti-HA-EuCryptate and anti-HA-Alexa647 antibodies were used to estimate the amount of HA-R1c1:HA-R1c1 homodimers (closed circles). The plot shows the TR-FRET signal as a function of the surface expression of the HA-tagged subunits as determined by the specifically bound anti-HA-EuCryptate antibody. All values are means±s.e.m. of triplicate determinations from a representative experiment out of three. Data shown in (A–D) are from a single experiment.
These data do not exclude the possibility that myc-R1c2 allows targeting of preformed HA-R1c1 homodimers to the cell surface. This is unlikely the case since the FRET signal detected between HA epitopes in cells expressing HA-R1c1 and myc-R1c2 remains low. It is indeed similar to that measured in cells expressing HA-GB1 and myc-GB2 (Figure 2A and E) and thus likely results from an overexpression of the receptors (Maurel et al, 2004). In contrast, a clear signal was detected between HA epitopes in cells expressing HA-R1c2 only, demonstrating that homodimers of HA-mGlu1 can be identified using this method (Figure 2D).
The proportion of both populations of dimers (R1c1:R1c2 heterodimers and R1c2 homodimers) at the cell surface was further examined by quantifying the expression level of the subunits using ELISA on intact cells. Anti-HA antibody was used to detect either a single or both subunits. Our data revealed that the amount of R1c1 at the cell surface is more than one-third of the total amount of subunits (Figure 3). According to these data, we estimated that 72±3% (n=6) of the receptors corresponded to the R1c1:R1c2 combination when an equal amount of plasmid encoding each subunit was used for transfection. This proportion can be increased by augmenting the proportion of plasmid encoding R1c1 (data not shown).
Figure 3.
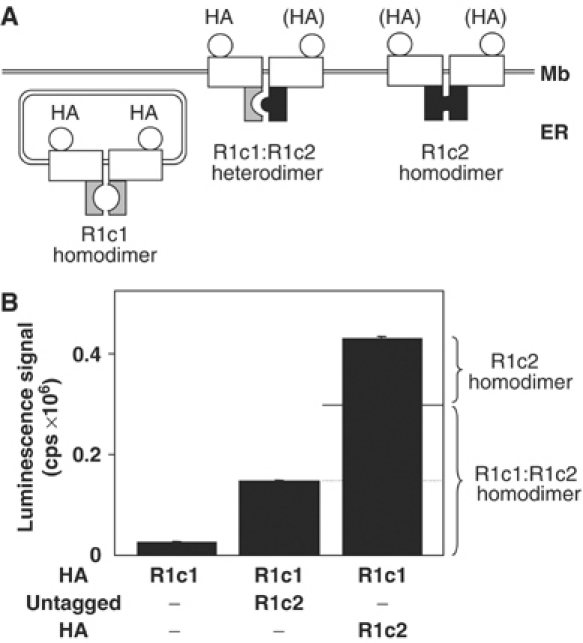
Quantification of the relative expression of R1c1:R1c2 heterodimers and R1c2:R1c2 homodimers by ELISA on intact cells. (A) Schematic representation of the expected surface expression of the chimeric receptors. Mb: plasma membrane; ER: endoplasmic reticulum; HA indicates that the subunit is HA-tagged; (HA) means that this subunit is either HA-tagged or not, as indicated in (B). (B) Determination of the luminescence signal obtained by ELISA using HA antibody measured in cells expressing the indicated subunits, of which one or both are HA-tagged. Values are means±s.e.m. of triplicate determinations from a typical experiment out of three and correspond to the raw values obtained with the luminometer.
Functional expression of R1c1 and R1c2 chimeras
In cells expressing R1c1, no quisqualate response could be measured due to the retention of this subunit in the ER (Figure 4). However, the presence of the C-terminal tail of GB1 in R1c1 does not prevent coupling of this receptor to G-protein, as indicated by the normal functioning of the equivalent chimera in which the ER retention signal RSRR is mutated into ASAR (R1c1ASA; Figure 4). Similarly, replacement of the C-terminal tail of mGlu1 by that of GB2 did not prevent activation of phospholipase C (PLC) (Figure 4). However, a clear decrease in the maximal effect of quisqualate was observed with either R1c1ASA or R1c2 even though care was taken to have a similar level of expression of each at the cell surface, as quantified using ELISA. This suggests that the C-terminal tail of either GB1 or GB2 decreases coupling efficacy of the mGlu1 receptor. Regardless, it is important to note that the C-terminal tails of GB1 and GB2 similarly affect the coupling efficacy of the chimeric receptors.
Figure 4.
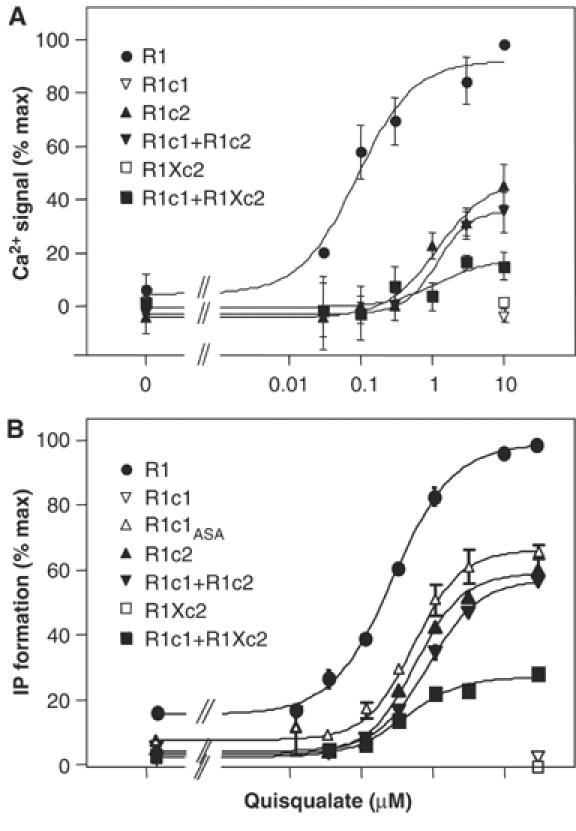
Effect of increasing doses of quisqualate on mutated mGlu1 receptors. Increase in Ca2+ (A) or IP formation (B) in cells expressing the indicated receptor subunits is plotted as a function of quisqualate concentration. Values are normalized to the quisqualate-evoked maximal response obtained with wild-type mGlu1 receptor (100%) and are means±s.e.m. of at least three independent experiments performed in triplicate.
Coexpression of the R1c1 and R1c2 chimeras results in functional heterodimers
When both R1c1 and R1c2 were coexpressed in the same cells, a clear activation of PLC by quisqualate was observed (Figure 4). In order to demonstrate firmly that the R1c1:R1c2 heterodimer was functional, a point mutation was introduced into the i3 loop of R1c2 (R1Xc2) (Table I). This mutation (F781P) is known to suppress the ability of mGlu1 receptor to activate PLC and adenylyl cyclase (Francesconi and Duvoisin, 1998). Mutation of the equivalent residue in the Ca2+-sensing receptor to Ala also suppresses coupling (Chang et al, 2000). As shown in Figure 4, when expressed alone R1Xc2 did not activate PLC as measured either by inositol phosphates (IP) production or Ca2+ release, although it was correctly targeted to the cell surface (data not shown). When R1c1 and R1Xc2 were coexpressed in the same cells, a clear response was observed. Since no response is expected from the R1c1 and R1Xc2 homodimers, this demonstrates a functional coupling of the R1c1:R1Xc2 heterodimer.
In cells expressing R1c1 and R1Xc2, the maximal effect was about one-third of that measured in cells expressing R1c1 and R1c2 for a similar expression level of these constructs at the cell surface. The same is true with other similar combinations of subunits as long as the c2 version is mutated in the i3 loop (Figure 5). Although the absence of functional R1c2 homodimer may explain part of this decrease, only a 30% decrease would be expected since 30% of the receptors are R1c2 homodimers, as described above. The larger decrease observed suggests that the R1c1:R1Xc2 heterodimer is not as efficient as the control combination in activating PLC. Accordingly, this suggests that both HDs must be able to activate G-proteins to obtain a full receptor activity.
Figure 5.
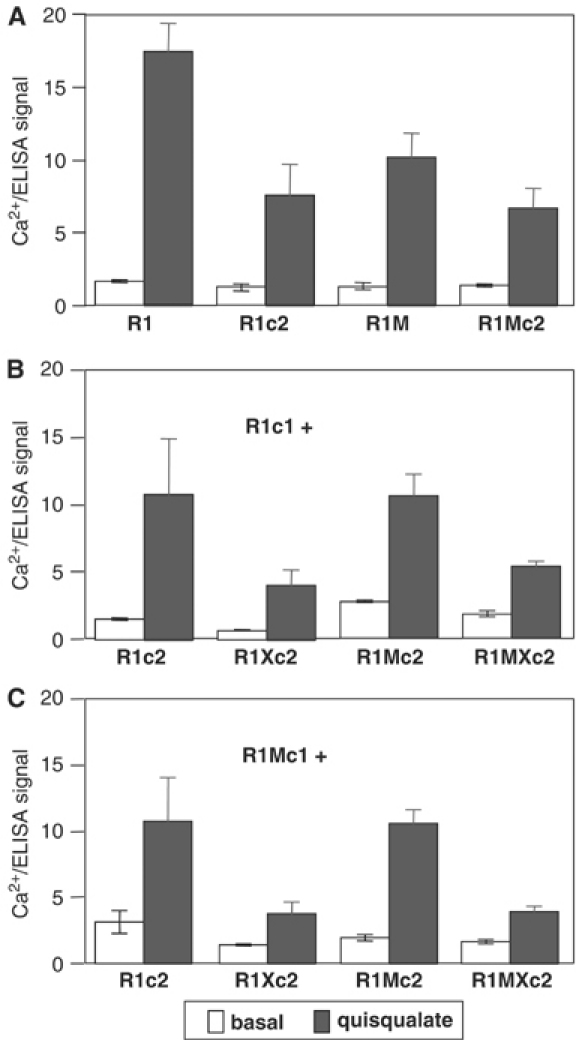
Effect of F781P point mutation within the i3 loop on quisqualate-evoked Ca2+ signal in cells expressing various types of receptor dimer combinations. (A) Response measured in cells expressing wild-type, chimeric or M-mutated receptors alone. (B) Response measured in cells coexpressing R1c1 and the indicated R1c2 constructs. (C) Response measured in cells coexpressing R1Mc1 and the indicated R1c2 constructs. In each case, basal (open columns) and quisqualate-induced (gray columns) responses were determined. For each individual experiment, both the Ca2+ signal and surface expression of the HA-tagged subunit were measured. Values are means±s.e.m. of the Ca2+ signal over the ELISA signals of 3–4 independent experiments performed in triplicate.
A single noncompetitive antagonist does not inhibit activation of dimeric mGlu receptors
In order to examine whether one or both HDs must reach its active state for dimeric receptor activation of G-proteins, we created a mutant mGlu1 receptor sensitive to the mGlu5 selective noncompetitive inverse agonist 2-methyl-6-(phenylethynyl)pyridine (MPEP), as described by others (Pagano et al, 2000). This mutant, named R1M (Table I), displays an agonist-induced activity similar to that of the control receptor (Figure 5). However, in contrast to the wild-type receptor, R1M was fully antagonized by MPEP (IC50 of 3.7±1.3 μM) (Figure 6). Of interest, the mGlu1 selective noncompetitive inverse agonist BAY 36-7620 is still able to antagonize the mutated R1M receptor. The combination R1Mc1:R1Mc2 was also inhibited by MPEP with a similar IC50 (3.4±1.4 μM) (Figures 7 and 8).
Figure 6.
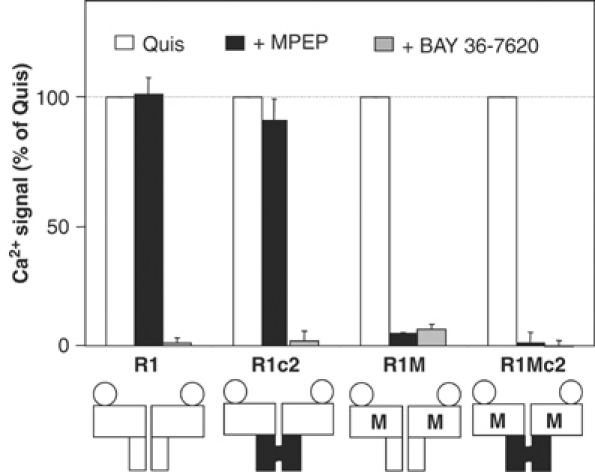
Effect of MPEP and BAY 36-7620 on quisqualate-evoked Ca2+ signal on wild-type, R1c2, R1M and R1Mc2 homodimers. Effect of quisqualate (1 μM) alone (open columns) or together with MPEP (100 μM) (black columns) or BAY 36-7620 (10 μM) (gray columns) on Ca2+ signals was measured in cells expressing the indicated subunits. Values are expressed as percentage of the maximal quisqualate effect and are mean±s.e.m. of three independent experiments performed in triplicate.
Figure 7.
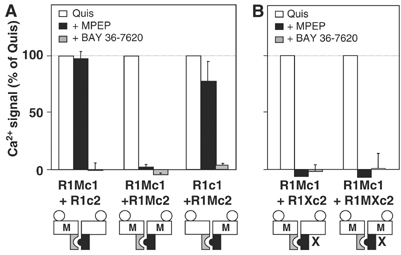
Two MPEP sites per dimer appear necessary for MPEP inhibition of receptor activity. Effect of quisqualate (1 μM) alone (open columns) or together with MPEP (100 μM) (black columns) or BAY 36-7620 (10 μM) (gray columns) on Ca2+ signals was measured in cells expressing the indicated subunits. Values are expressed as a percentage of the maximal quisqualate effect and are means±s.e.m. of three independent experiments performed in triplicate.
Figure 8.
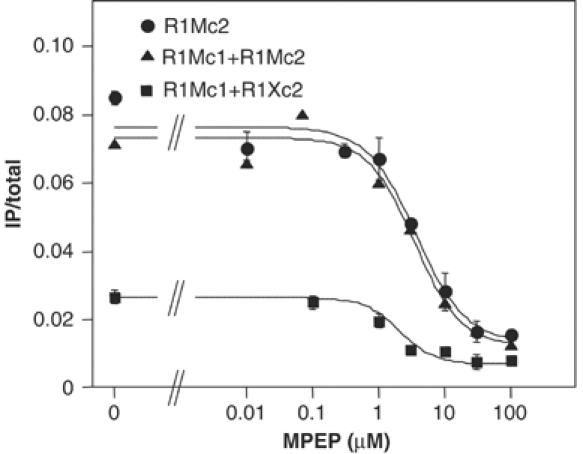
Dose-dependent effect of MPEP on the quisqualate-stimulated IP production. HEK293 expressing R1Mc2 (circles) or coexpressing R1Mc1 with R1Mc2 (triangles) and R1Mc1:R1Xc2 (squares) were monitored for changes in IP formation upon stimulation with quisqualate (1 μM) in the presence of various concentrations of MPEP. Results are expressed as IP production over the total radioactivity remaining in the membrane fraction of the cells. Values are means±s.e.m. of triplicate determinations from a representative experiment out of three independent experiments.
Next, we examined the effect of MPEP on receptor combinations in which a single subunit was sensitive to MPEP (R1Mc1:R1c2 and R1c1:R1Mc2). As shown in Figure 7, no inhibition by MPEP was observed in cells expressing both R1Mc1 and R1c2, although BAY 36-7620, which can bind both subunits, was able to block the response fully. When the MPEP site is included in the R1c2 subunit, MPEP inhibits 20% of the agonist-mediated response. This inhibition likely represents the component of the response mediated by the R1Mc2 homodimers, consistent with the heterodimer not being sensitive to MPEP.
To further confirm that MPEP has no antagonist activity on receptor dimers possessing a single MPEP site, we performed additional experiments with dimer combinations made of R1c1 and an R1c2 subunit that does not form a functional receptor alone (R1Bc2) (Figure 9). The latter possesses two mutations in the agonist binding site (Y236A and D318A). As shown in Figure 9, under such condition, only the R1c1:R1Bc2 heterodimer is functional, allowing the clear analysis of the effect of MPEP in such receptor dimer possessing a single site in either subunit. As shown in Figure 9, whether the MPEP site is introduced in R1Bc2 or R1c1, the effect of quisqualate is not affected by MPEP. Only when both subunits possess the MPEP site can MPEP act as an antagonist. Although these data indicate that the presence of a single MPEP site per dimer is not sufficient to allow this inverse agonist to inhibit receptor activity, it is important to know whether or not MPEP binds in such a site and inhibits activation of this subunit.
Figure 9.
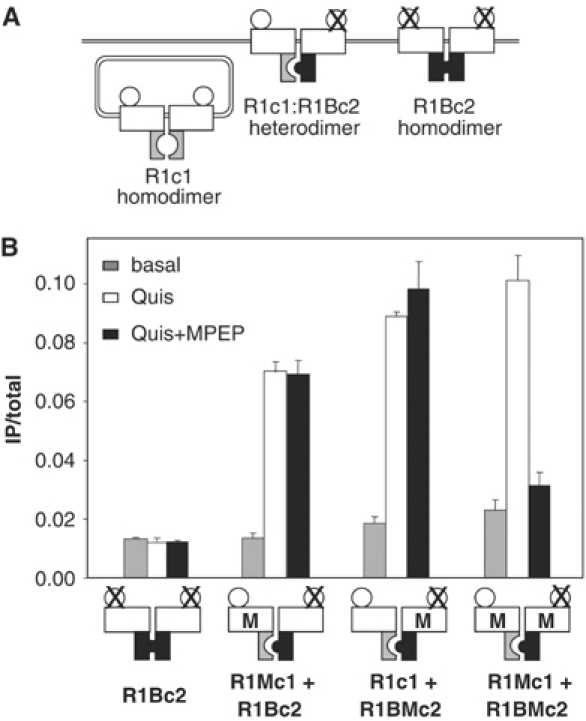
MPEP does not inhibit activity of receptor dimers containing a single MPEP site. (A) Scheme illustrating that in cells expressing R1c1 and R1Bc2, the R1c1 homodimer is retained in the ER and the R1Bc2 homodimer is not functional due to the agonist site mutations, such that only the R1c1:R1Bc2 combination is functional. (B) IP production measured under basal condition (gray columns), in the presence of quisqualate (white columns) or in the presence of both quisqualate and MPEP (black columns) in cells expressing the indicated combinations of subunits. Data are means±s.e.m. of triplicate determinations from a representative experiment out of five.
MPEP fully antagonizes the R1Mc1:R1Xc2 combination
To verify that MPEP can still bind a receptor dimer possessing a single site, binding experiments could be performed. However, the only commercially available radioligand for the MPEP site is [3H]MPEP. Unfortunately, the affinity of MPEP for the mutated R1M subunit (3 μM) is too low to expect any significant binding with this radioligand. As an alternative, we examined the effect of MPEP on a receptor dimer in which one subunit was unable to activate G-proteins, and the other contained an MPEP site (R1Mc1:R1Xc2). Such a receptor combination is supposed to have the same ability as the R1Mc1:R1c2 combination to bind MPEP. Moreover, because the only functional HD is the one that possesses the MPEP site, we will be able to examine whether or not MPEP can stabilize it in an inactive state. As shown in Figure 7, MPEP fully inhibited activation of this receptor combination with an IC50 (1.9±0.2 μM; n=4) and a Hill coefficient (1.24±0.25; n=4) similar to those measured with the combination containing two MPEP sites per dimer (IC50=2.3±0.4 μM; Hill coefficient=1.16±0.21; n=4) (Figures 7 and 8). This demonstrates that MPEP can indeed bind a receptor dimer containing a single MPEP site, and can stabilize the occupied HD in its inactive state. These data also suggest that there is no cooperativity for the MPEP binding in such dimeric receptors. Importantly, these data suggest that for receptor combinations in which one HD is maintained in its inactive state with MPEP, the associated subunit is able to generate the full response of the receptor.
MPEP enhances agonist activity at R1c1:R1MXc2 combination
The effect of intracellular loop mutations indicated that both HDs in a receptor dimer can potentially activate G-proteins. The above results suggest that stabilizing a single HD in an inactive state with an inverse agonist has no effect on the coupling efficacy of a receptor dimer. Taken together, these data suggest that stabilizing one HD in its inactive state favors coupling by the associated subunit.
To test directly this possibility, we examined the effect of MPEP on a receptor dimer in which one HD is wild type and the other possesses the MPEP site and is impaired in its ability to activate G-proteins. If the above proposal is correct, then MPEP binding in one subunit should favor receptor activity mediated exclusively by the second subunit. We therefore coexpressed R1c1 that contains a wild-type HD with R1MXc2 that has both an MPEP site and a mutation in the i3 loop. Note that when these subunits are coexpressed, only the R1c1:R1MXc2 combination is functional, since the other receptor combination reaching the cell surface, R1MXc2 homodimer, is not functional. As expected according to the above proposal, MPEP was found to enhance the effect of quisqualate in a dose-dependent manner (Figure 10). This further suggests that preventing the HD of the R1MXc2 subunit to reach its active state facilitates G-protein activation by the associated subunit.
Figure 10.
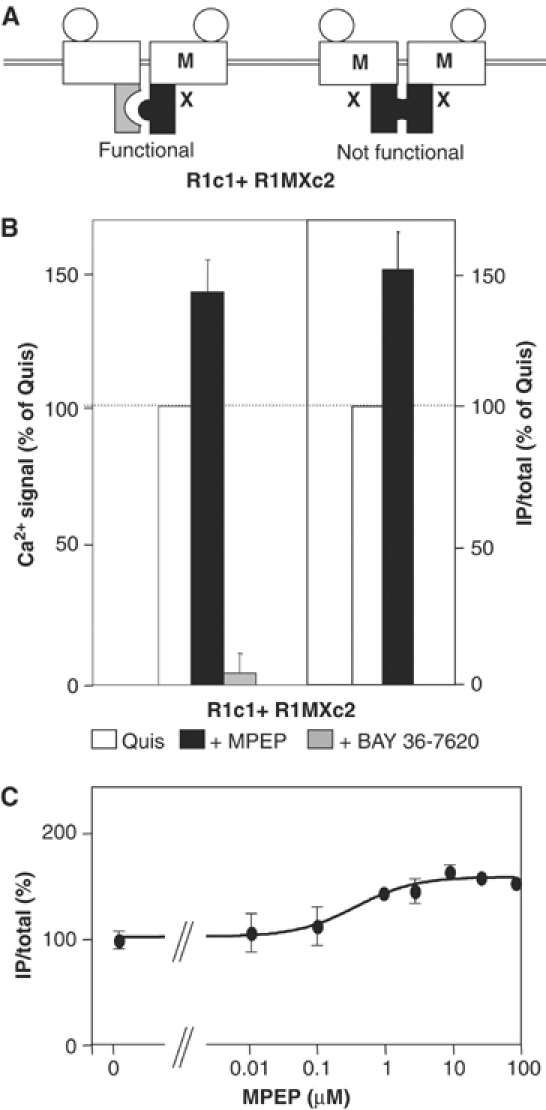
MPEP enhances quisqualate-induced responses in cells expressing R1c1 and R1MXc2. (A) Schematic representation of the expected receptor dimers at the surface of cells expressing both R1c1 and R1MXc2 subunits. Note that only the heteromer can generate a signal upon agonist activation. (B) The Ca2+ signal (left panel) and IP production (right panel) induced by quisqualate alone (open columns) or together with MPEP (black columns) or BAY 36-7620 (gray columns) were measured in cells expressing the indicated subunits. (C) Effect of increasing concentrations of MPEP on IP production induced by quisqualate in cells expressing R1c1 and R1MXc2. Values are expressed as a percentage of the maximal quisqualate effect and are means±s.e.m. of 3–7 independent experiments performed in triplicate.
Discussion
In the present study, we examined whether one or both HDs in a dimeric mGlu1 receptor are turned on during receptor activation. To that aim, we applied the quality control system of the GABAB receptor to control the formation of dimeric mGlu1 receptors composed of wild-type or differentially mutated subunits. Then, we examined the effect of the noncompetitive mGlu5 antagonist MPEP on mGlu1 receptor combinations in which a single subunit is made sensitive to MPEP.
Either HD in homodimeric mGlu receptors can activate G-proteins
We show that mGlu1 receptor dimers in which one HD is impaired in its ability to activate G-proteins are still able to activate PLC. This is nicely illustrated when the i3 loop of R1c2 is mutated (R1Xc2). Indeed, in cells coexpressing this subunit and the R1c1, the R1Xc2 homodimers that are at the cell surface are not functional, such that the measured response can only be generated by the heterodimer in which a single HD is functional. This illustrates that a single HD able to couple to G-protein is sufficient to get a functional dimeric receptor. This is consistent with the finding that the GB2 HD is crucial for G-protein activation by the heterodimeric GABAB receptor. Indeed, a GABAB receptor bearing a single mutation in the i2 or i3 loop of GB2 HD does not activate G-proteins, whereas the equivalent mutation in GB1 HD has a minor effect (Robbins et al, 2001; Duthey et al, 2002).
Although receptor dimers with one subunit mutated in the i3 loop are functional, a large decrease in the maximal response is observed, even when care was taken to control cell surface density of receptors. Indeed, this decrease is larger (about 60%) than that expected from the loss of function of R1Xc2 homodimers, which represent less than 30% of the total number of dimers at the cell surface. This suggests that the R1c1:R1Xc2 heterodimer is less efficient in activating G-proteins than the control R1c1:R1c2 heterodimer. Accordingly, either HD can activate a G-protein in mGlu dimers. This is reminiscent of our observation that in a GABAB receptor combination in which both subunits possess a GB2 HD, either HD can activate G-proteins (Havlickova et al, 2002). This is also consistent with our recent data with the mGlu5 receptor (Kniazeff et al, 2004a). In that case, we used an R5c2 construct (mGlu5 with the C-terminal tail of GB2) made nonfunctional by a mutation that prevents agonist activation of the receptor, and an R5c1 construct with a wild-type agonist binding site. As such, only the R5c1:R5c2 heterodimers are functional. In that case, whether the i3 loop mutation is introduced in either one of the subunits, a two-fold decrease in the maximal response was observed. Although the interpretation of this may be that each HD is capable of activating a G-protein independently of the other, another explanation is also possible. Indeed, in an activated homodimeric GPCR, either one or the other (but not both) HD may be turned on at a time.
Blocking one HD in its inactive state with an inverse agonist does not impair receptor coupling
As reported previously, the simultaneous introduction of three point mutations (one in TM3 and two in TM7) into mGlu1 is sufficient to make it sensitive to the mGlu5 selective inverse agonist MPEP (Pagano et al, 2000). Such mutations did not impair the sensitivity of the receptor to the mGlu1 selective inverse agonist BAY 36-7620. Of interest, if a single subunit within the dimer possesses such a site, no effect of MPEP was observed. However, the receptor was fully antagonized by BAY 36-7620, which can bind in both subunits of the dimer. The absence of effect of MPEP is unlikely due to the inability of MPEP to act in a dimeric receptor possessing a single MPEP site. Indeed, MPEP fully blocks a receptor combination in which one subunit is sensitive to MPEP and the other is impaired in its ability to couple to G-protein by an i3 loop mutation (R1Mc1:R1Xc2 combination). Taken together, these data show that binding of an inverse agonist in one HD within a dimer does not impair G-protein coupling efficacy of the dimer.
Either HD in a dimeric mGlu1 receptor is activated at a time
How can one reconcile the two apparently opposite observations that (i) impairment of G-protein coupling by i3 mutation decreases G-protein activation, whereas (ii) binding of an inverse agonist in one HD does not?
If one accepts that the mutation in i3 loop impairs G-protein activation, but not the ability of the HD to reach an active conformation, then our data can be interpreted by a single HD being turned on per dimer at a time (Figure 11). According to this model, upon activation of the receptor, 50% of the dimers are active due to the active conformation of one HD (white HD on the left scheme in Figure 11A) and 50% due to the active form of the other HD (black HD on the right scheme). If one HD is impaired in its ability to couple to G-proteins (the white one in Figure 11B), then only half of the receptor dimers can couple to G-proteins, and as such the maximal response is decreased two-fold. In contrast, if one HD (the black one in Figure 11B) is blocked in an inactive state due to the presence of MPEP, then activation of the dimer of VFTs has no other possibility than to activate the other HD (the white one), leading to 100% of receptor dimers with the wild-type HD reaching the active state (compare Figure 11A and C). Accordingly, MPEP is not expected to inhibit such a receptor dimer, as observed here.
Figure 11.
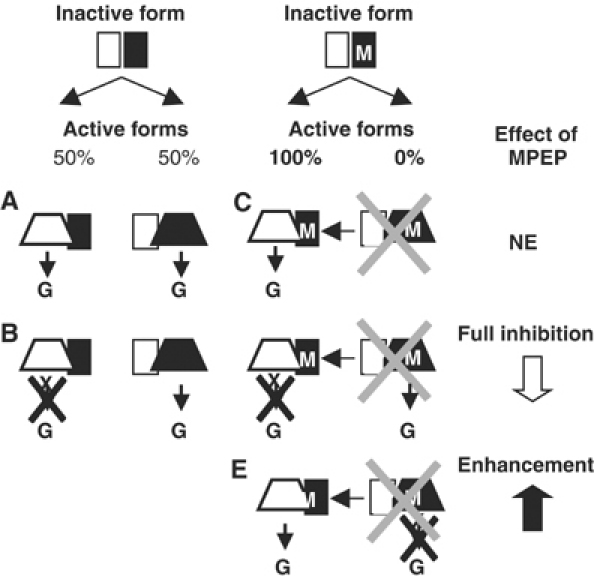
Proposed model: one HD being turned on at a time. The receptor is represented as a dimer of HDs, one in white and the other in black. The inactive conformation of the HD is represented by a rectangle, whereas the active form is represented by a trapezoid. The presence of the i3 loop mutation that prevents G-protein activation is indicated by a star. The presence of an MPEP site is indicated by an M. The expected effect of MPEP, according to our model proposing that only one HD can reach the active state at a time, is indicated on the right. (A) Control condition, with either HD being turned on. (B) One HD is mutated in its i3 loop such that only 50% of the dimers activate the G-protein. (C) When MPEP prevents the black HD from reaching the active state, then only the white HD is turned on in every receptor dimer. (D) MPEP fully blocks receptor dimer activity when bound to the only active subunit. (E) By preventing the black HD from reaching its active state, MPEP increases the probability of the white HD to be turned on, thus leading to an enhancement of the agonist effect. NE: no effect.
In agreement with this proposal, in a receptor dimer comprised of one HD bearing the i3 loop mutation (indicated in white, Figure 11D) and the other possessing an MPEP site (indicated in black, Figure 11D), only the white HD can reach an active conformation in the presence of MPEP. Because this HD is not able to activate G-proteins, MPEP is expected to block receptor activity fully (Figure 11, compare panels B and D). This is what we observed with the R1Mc1:R1Xc2 combination.
This proposal also explains why MPEP enhances agonist effect in the R1c1:R1MXc2 combination. Indeed, in such a receptor dimer, both HDs may reach their active state in the absence of MPEP, but only one is capable of activating the G-protein. By adding MPEP, and thus preventing the R1MXc2 (black HD in Figure 11E) from reaching the active state, we would expect to observe a higher proportion of R1c1 subunits (white HD in Figure 11E) reach the active state, thereby leading to an increase in the maximal response to agonists (Figure 11, compare panels B and E).
Why a receptor dimer for G-protein activation?
Within the last 5 years, accumulating data indicate that class A rhodopsin-like GPCRs can form dimers, including heterodimers (Bouvier, 2001; Angers et al, 2002; Javitch, 2004; Milligan, 2004; Terrillon and Bouvier, 2004). In some cases, functional interactions between the associated receptors have been observed. For example, activation of a single receptor type has been shown to induce or prevent the desensitization of the associated receptor (Jordan et al, 2001; Lavoie et al, 2002). There are also some examples where agonist binding in one subunit decreases the signaling of the other (Jordan et al, 2003), whereas antagonist of one subunit increases agonist affinity or signaling of the associated subunit (Gomes et al, 2004). Although these observations may well be explained by our proposal that a single HD is activated in a receptor dimer, positive synergistic effect resulting from the activation of both receptor subunits has also been observed (Jordan and Devi, 1999). However, such effects can result from a crosstalk between the signaling pathways activated by each individual receptor without a prerequisite for heterodimer formation. In addition, the relative proportion of heterodimers versus monomers and homodimers is generally not known, thus making it difficult to draw a clear conclusion as to the functioning of such heterodimers.
Other observations suggest that our proposal that a single HD reaches the active state in a dimeric mGlu receptor may well be relevant to other GPCRs. Indeed, data reported almost 20 years ago (Kühn, 1984), as well as recently (Jastrzebska et al, 2004), demonstrated that monomeric rhodopsin can activate transducin, suggesting that in a receptor dimer, there is no need for both subunits to be turned on to activate G-proteins. Moreover, it is generally accepted that a single photon can be detected by the retina, such that a single activated rhodopsin can signal. If rhodopsin exists in a dimeric form in disk membranes, as revealed recently (Fotiadis et al, 2003, 2004; Liang et al, 2003), then a single photon will activate a single subunit only, leaving the associated subunit covalently linked with the inverse agonist cis-retinal. Accordingly, a rhodopsin dimer with a single subunit in an active state, and the other stabilized in its inactive state by an inverse agonist is expected to activate transducin. Also of interest, in GPCRs known to function exclusively in a heterodimeric form, such as the GABAB receptor and the sweet and umami taste receptors, only one HD appears to play a pivotal role in G-protein activation (Galvez et al, 2001; Duthey et al, 2002; Havlickova et al, 2002; Xu et al, 2004).
Taken together, these data raise the question of the role of GPCR dimerization in G-protein activation. In the case of the class C GPCRs, structural as well as mutational studies indicate that dimerization in required for intramolecular transduction, that is, transfer of the signal from the VFTs to the HDs. Indeed, it is proposed that a change in the relative orientation of the two VFTs in the dimer (Kunishima et al, 2000; Tsuchiya et al, 2002; Kniazeff et al, 2004a) leads to a different relative position of the HDs (Tateyama et al, 2004) and allows for their activation. As such, a dimeric structure appears intimately linked to function in class C GPCRs. In the case of class A GPCRs, as shown for rhodopsin, although a monomer can activate G-proteins, recent data suggest that the dimeric form couples more efficiently (Jastrzebska et al, 2004). This is supported by recent data obtained with the yeast α-factor receptor illustrating that each receptor subunit in this dimeric GPCR can be activated independently, but function in concert to activate G-proteins (Chinault et al, 2004).
Why is the activated dimer of HDs not symmetric?
The most surprising observation of the present study is that the HD dimer in mGluRs does not function in a symmetrical way. This is particularly surprising when one considers that the dimer of VFTs apparently remains symmetric during the activation process, with full activation being observed only when both VFTs are occupied by an agonist and when both are in a closed state (Kniazeff et al, 2004a). Although other possibilities exist, the simplest way to explain such a rupture of symmetry in a receptor dimer composed of two identical proteins is that there is an external constraint that prevents both HDs from behaving similarly. Recent data suggest that a single heterotrimeric G-protein interacts with a dimer of GPCRs (Baneres and Parello, 2003). Based on the known surface area of the G-protein that contacts the receptor, it has been proposed that both subunits of the receptor dimer contact the G-protein, one HD interacting with the α subunit and the other with βγ (and the N-terminal α helix of the α subunit) (Liang et al, 2003; Filipek et al, 2004). This proposal is supported by biophysical analysis of the dimer of BLT1 receptors associated with one G-protein heterotrimer (Baneres and Parello, 2003), as well as by modeling studies (Liang et al, 2003; Filipek et al, 2004). Accordingly, it is tempting to speculate that the G-protein heterotrimer acts as an external constraint to allow only one of the two HDs to reach the active state. This proposal is in agreement with recent modeling data suggesting that transducin can associate well with a rhodopsin dimer in which only one subunit is in the active state (Liang et al, 2003; Filipek et al, 2004).
Materials and methods
Chemicals
L-quisqualic acid (quisqualate) and MPEP were obtained from Tocris Cookson Ltd (Bristol, UK), and BAY 36-7620 has been synthesized by Bayer. [3H]myo-inositol (23.4 Ci/mol) (1 Ci=37 GBq) was obtained from Amersham Pharmacia (Perkin-Elmer Life Science (NEN), Paris, France). Glutamate-pyruvate transaminase was purchased from Roche (Roche Bioscience, Meylan, France) and used in 1 U/ml concentration.
Mutagenesis and construction of chimeric mGlu1a receptor
Unless noted otherwise, the mutants were generated using the QuickChange site-directed mutagenesis kit from Stratagene (Chemos, Czech Republic). The entire coding region of all point mutants was sequenced from leading strand using the Big Dye Terminator v. 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA).
Chimeric mGlu1 receptors bearing the C-terminal tail of either GB1 (R1c1) or GB2 (R1c2), starting at position Met873 and Gln761, respectively, were constructed by taking advantage of the restriction site SphI (GB1 or GB2 tails inserted after His859) in the mGlu1 sequence.
In most constructs (Table I), an HA or a myc tag was introduced in the N-terminal end after the signal peptide. To that aim, the coding sequence of the mature mGlu1 receptor was introduced after the unique MluI restriction site located after the epitope tag of pRKGB1-HA or pRKGB1-myc (Galvez et al, 2001). The resulting constructs consist of the signal peptide of mGlu5, then either the HA or myc epitope, followed by the mGlu1 coding sequence starting at Ser34. As previously reported for other mGlu (Ango et al, 2000; Havlickova et al, 2003) or GABAB receptors (Pagano et al, 2000; Galvez et al, 2001), the presence of these tags did not modify the functional expression and pharmacological properties of the mGlu1 receptor.
Cell culture and transfection
HEK293 (human embryonic kidney cells) and COS-7 cells were cultured in Dulbecco's modified Eagle's medium (BRL-Life Technologies Inc., Cergy Pontoise, France), without sodium pyruvate, supplemented with 10% fetal calf serum, penicillin and streptomycin (100 U/ml final). Electroporation was performed as described elsewhere (Maurel et al, 2004). For the functional assays, we also added the high-affinity glutamate transporter EAAC1 to prevent the influence of glutamate in the medium.
Quantification of cell surface receptors using ELISA
Cell surface expression level of the N-terminal HA-tagged receptors was determined using ELISA as previously described (Zerangue et al, 1999; Balasubramanian et al, 2004; Goudet et al, 2004). Transfected cells were fixed with 4% paraformaldehyde, then permeabilized or not using 0.05% Triton X-100 (5 min) and incubated for 1 h with rat monoclonal anti-HA antibody coupled to horseradish peroxidase (clone 3F10 (Roche) at 0.5 μg/ml). Antibodies were quantified by chemiluminescence using SuperSignal® ELISA femto maximum sensitivity substrate (Pierce) and a Wallac Victor2 luminescence counter (Molecular Devices).
Time-resolved fluorescence resonance energy transfer analysis
These experiments were conducted as previously described (Maurel et al, 2004). Briefly, COS-7 cells expressing the indicated tagged receptor subunits were labeled with monoclonal anti-HA (12CA5) and/or anti-myc (9E10; American Type Culture Collection no. CRL-1729) carrying either Eu3+-Cryptate PBP or AlexaFluor 647 (provided by Cis Bio International Research). The bound Alexa647 antibodies were quantified after excitation at 640 nm and emission monitored at 682 nm using an Analyst™ reader (Molecular Devices). Eu3+-Cryptate fluorescence and TR-FRET signal were measured 50 μs after excitation at 337 nm at 620 and 665 nm, respectively, using a RubyStar fluorimeter (BMG Labtechnologies, Champigny-sur-Marne, France). The FRET signal was measured either as Delta665 (Delta665=R665pos−R665neg, where R665pos is the fluorescence intensity measured at 665 nm in the presence of both fluorophores and R665neg is that measured in the absence of the acceptor molecule) or DeltaF (%) (DeltaF (%)=((R665/620)pos−(R665/620)neg) × 100/(R665/620)neg, where (R665/620)pos is the ratio of the 665 signal over that at 620 measured in the presence of both antibodies and (R665/620)neg is the same ratio measured in the absence of the acceptor-labeled antibody.
Intracellular Ca2+ mobilization assay
Measurement of intracellular Ca2+ mobilization in transfected cells was performed in 96-well plates using the Ca2+-sensitive fluorescent dye Fluo-4AM (Molecular Probes) as already described (Goudet et al, 2004). The fluorescence signal (excitation 485 nm, emission 525 nm) was measured at 1.5 s intervals for a period of 60 s using the microplate reader FlexStation (Molecular Devices). The effect of added compounds was examined after 20 s of recording.
Determination of inositol phosphates accumulation
The determination of IP accumulation in transfected cells was adapted for a 96-well plate format as previously described (Goudet et al, 2004). Briefly, after an overnight labeling with [3H]myo-inositol, HEK293 cells were stimulated in the presence of 10 mM LiCl and indicated compounds for 30 min. The reaction was stopped with 0.1 M formic acid. IP produced were purified in 96-well plates by ion-exchange chromatography. Radioactivity was measured using a Wallac 1450 MicroBeta microplate liquid scintillation counter (Molecular Devices). Results are expressed as the ratio between IP and the total radioactivity present in the membranes.
Acknowledgments
We thank Drs B Mouillac, P Rondard, T Durroux, C Barberis, C Brock, A Brady (Montpellier, France) and M Chabre (Sophia Antipolis, France) for constructive discussion and critical reading of the manuscript. We also thank Dr C Brock for sharing constructs, F Malhaire for technical help, and Dr A Brady for correction of the English. This work was supported by the Grant Agency of Czech Republic (GACR 301/03/1183 and 309/03/H095), Grant Agency of Academy of Science of Czech Republic (GAAV B5039402), 5th FW EC (QLG3-CT-2001-00929 ‘Epileptosome') and 5th FW EC (ICA1-CT-2000-70028 ‘MEDIPRA') and CEZ (AVOZ 2039906 and AVOZ5008914), to JB, and by grants from the CNRS, the Action Concertée Incitative ‘Molécules et Cibles Thérapeutiques' of the French Ministry of Research and Technology, the Comité Parkinson of the Fondation de France, the fondation Paul Hamel, Addex Pharmaceuticals and the European Community (grant LSHB-CT-200-503337) to JPP. CG was supported by a fellowship from the Fondation pour la Recherche Médicale, JK by a bourse de Docteur Ingénieur from the CNRS and DM s by both Cis Bio International and the French Government (CIFRE fellowship).
References
- Angers S, Salahpour A, Bouvier M (2002) Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol 42: 409–435 [DOI] [PubMed] [Google Scholar]
- Ango F, Pin J-P, Tu JC, Xiao B, Worley PF, Bockaert J, Fagni L (2000) Dendritic and axonal targeting of type 5 metabotropic glutamate receptor (mGluR5) is regulated by homer1 proteins and neuronal excitation. J Neurosci 20: 8710–9716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Teissere JA, Raju DV, Hall RA (2004) Hetero-oligomerization between GABAA and GABAB receptors regulates GABAB receptor trafficking. J Biol Chem 279: 18840–18850 [DOI] [PubMed] [Google Scholar]
- Baneres J-L, Parello J (2003) Structure-based analysis of GPCR function. Evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J Mol Biol 329: 815–829 [DOI] [PubMed] [Google Scholar]
- Bessis A-S, Rondard P, Gaven F, Brabet I, Triballeau N, Prézeau L, Acher F, Pin J-P (2002) Closure of the Venus Flytrap module of mGlu8 receptor and the activation process: insights from mutations converting antagonists into agonists. Proc Natl Acad Sci USA 99: 11097–11102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet V, Brajon C, Le Corre L, Acher F, Pin JP, Prézeau L (2004) The heptahelical domain of GABAB2 is directly activated by CGP7930, a positive allosteric modulator of the GABAB receptor. J Biol Chem 279: 29085–29091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J, Pin J-P (1999) Molecular tinkering of G-protein coupled receptors: an evolutionary success. EMBO J 18: 1723–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier M (2001) Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci 2: 274–286 [DOI] [PubMed] [Google Scholar]
- Calver AR, Robbins MJ, Cosio C, Rice SQ, Babbs AJ, Hirst WD, Boyfield I, Wood MD, Russell RB, Price GW, Couve A, Moss SJ, Pangalos MN (2001) The C-terminal domains of the GABAB receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J Neurosci 21: 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Müller T, Pin JP, Prézeau L (2001) BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol 59: 965–973 [PMC free article] [PubMed] [Google Scholar]
- Chabre M, Cone R, Saibil H (2003) Biophysics: is rhodopsin dimeric in native retinal rods? Nature 426: 30–31 [DOI] [PubMed] [Google Scholar]
- Chang W, Chen TH, Pratt S, Shoback D (2000) Amino acids in the second and third intracellular loops of the parathyroid Ca2+-sensing receptor mediate efficient coupling to phospholipase C. J Biol Chem 275: 19955–19963 [DOI] [PubMed] [Google Scholar]
- Chinault SL, Overton MC, Blumer KJ (2004) Subunits of a yeast oligomeric G protein-coupled receptor are activated independently by agonist but function in concert to activate G protein heterotrimers. J Biol Chem 279: 16091–16100 [DOI] [PubMed] [Google Scholar]
- Couve A, Filippov AK, Connolly CN, Bettler B, Brown DA, Moss SJ (1998) Intracellular retention of recombinant GABAB receptors. J Biol Chem 273: 26361–26367 [DOI] [PubMed] [Google Scholar]
- Duthey B, Caudron S, Perroy J, Bettler B, Fagni L, Pin J-P, Prézeau L (2002) A single subunit (GB2) is required for G-protein activation by the heterodimeric GABAB receptor. J Biol Chem 277: 3236–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipek S, Krzysko K, Fotiadis AD, Liang Y, Saperstein DA, Engel A, Palczewski K (2004) A concept for G protein activation by G protein coupled receptor dimers: the transducin/rhodopsin interface. Photochem Photobiol Sci 3: 628–638 [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K (2003) Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature 421: 127–128 [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K (2004) The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett 564: 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM (1998) Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J Biol Chem 273: 5615–5624 [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63: 1256–1272 [DOI] [PubMed] [Google Scholar]
- Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prézeau L, Pin J-P (2001) Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO J 20: 2152–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA (2004) A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 101: 5135–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C, Gaven F, Kniazeff J, Vol C, Liu J, Cohen-Gonsaud M, Acher F, Prezeau L, Pin JP (2004) Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc Natl Acad Sci USA 101: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havlickova M, Blahos J, Brabet I, Liu J, Hruskova B, Prezeau L, Pin JP (2003) The second intracellular loop of metabotropic glutamate receptors recognizes C termini of G-protein a-subunits. J Biol Chem 278: 35063–35070 [DOI] [PubMed] [Google Scholar]
- Havlickova M, Prezeau L, Duthey B, Bettler B, Pin J-P, Blahos J (2002) The intracellular loops of the GB2 subunit are crucial for G-protein coupling of the heteromeric gamma-aminobutyrate B receptor. Mol Pharmacol 62: 343–350 [DOI] [PubMed] [Google Scholar]
- Jastrzebska B, Maeda T, Zhu L, Fotiadis D, Filipek S, Engel A, Stenkamp RE, Palczewski K (2004) Functional characterization of rhodopsin monomers and dimers in detergents. J Biol Chem 279: 54663–54675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitch JA (2004) The ants go marching two by two: oligomeric structure of G-protein-coupled receptors. Mol Pharmacol 66: 1077–1082 [DOI] [PubMed] [Google Scholar]
- Jordan BA, Devi LA (1999) G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399: 697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Gomes I, Rios C, Filipovska J, Devi LA (2003) Functional interactions between mu opioid and alpha 2A-adrenergic receptors. Mol Pharmacol 64: 1317–1324 [DOI] [PubMed] [Google Scholar]
- Jordan BA, Trapaidze N, Gomes I, Nivarthi R, Devi LA (2001) Oligomerization of opioid receptors with beta 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc Natl Acad Sci USA 98: 343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Bessis A-S, Maurel D, Ansanay H, Prezeau L, Pin J-P (2004a) Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol 11: 706–713 [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Saintot P-P, Goudet C, Liu J, Charnet A, Guillon G, Pin J-P (2004b) Locking the dimeric GABAB G-protein coupled receptor in its active state. J Neurosci 24: 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci USA 98: 13402–13407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF (1994) GCRDb: a G-protein-coupled receptor database. Receptors Channels 2: 1–7 [PubMed] [Google Scholar]
- Kühn H (1984) Interactions between photoexcited rhodopsin and light-activated enzymes in rods. In Progress in Retinal Research, Osborne N, Chader J (eds) pp 123–156. Oxford and New York: Pergamon Press [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407: 971–977 [DOI] [PubMed] [Google Scholar]
- Lavoie C, Mercier JF, Salahpour A, Umapathy D, Breit A, Villeneuve LR, Zhu WZ, Xiao RP, Lakatta EG, Bouvier M, Hebert TE (2002) Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacy. J Biol Chem 277: 35402–35410 [DOI] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem 278: 21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JF, Maurel D, Etzol S, Brabet I, Ansanay H, Pin JP, Rondard P (2004) Molecular determinants of the allosteric control of agonist affinity in GABAB receptor by the GABAB2 subunit. J Biol Chem 279: 15824–15830 [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY (2000) A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27: 97–106 [DOI] [PubMed] [Google Scholar]
- Maurel D, Kniazeff J, Mathis G, Trinquet E, Pin J-P, Ansanay H (2004) Cell surface detection of membrane protein interaction with homogeneous time-resolved fluorescence resonance energy transfer technology. Anal Biochem 329: 253–262 [DOI] [PubMed] [Google Scholar]
- Milligan G (2004) G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol 66: 1–7 [DOI] [PubMed] [Google Scholar]
- Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, Ristig D, Schuler V, Meigel I, Lampert C, Stein T, Prézeau L, Blahos J, Pin J-P, Froestl W, Kuhn R, Heid J, Kaupmann K, Bettler B (2001) C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J Neurosci 21: 1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A, Rüegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prézeau L, Carroll F, Pin J-P, Cambria A, Vranesic I, Flor PJ, Gasparini F, Kuhn R (2000) The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 275: 33750–33758 [DOI] [PubMed] [Google Scholar]
- Pin J-P, Acher F (2002) The metabotropic glutamate receptors: structure, activation mechanism and pharmacology. Curr Drug Targets—CNS & Neur Dis 1: 297–317 [DOI] [PubMed] [Google Scholar]
- Pin J-P, Galvez T, Prezeau L (2003) Evolution, structure and activation mechanism of family 3/C G-protein coupled receptors. Pharmacol Ther 98: 325–354 [DOI] [PubMed] [Google Scholar]
- Robbins MJ, Calver AR, Filippov AK, Hirst WD, Russell RB, Wood MD, Nasir S, Couve A, Brown DA, Moss SJ, Pangalos MN (2001) GABAB2 is essential for G-protein coupling of the GABAB receptor heterodimer. J Neurosci 21: 8043–8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano C, Yang W-L, O'Malley KL (1996) Metabotropic glutamate receptor 5 is a disulfide-linked dimer. J Biol Chem 271: 28612–28616 [DOI] [PubMed] [Google Scholar]
- Salahpour A, Angers S, Bouvier M (2000) Functional significance of oligomerization of G-protein-coupled receptors. Trends Endocrinol Metab 11: 163–168 [DOI] [PubMed] [Google Scholar]
- Tateyama M, Abe H, Nakata H, Saito O, Kubo Y (2004) Ligand-induced rearrangement of the dimeric metabotropic glutamate receptor 1alpha. Nat Struct Mol Biol 11: 637–642 [DOI] [PubMed] [Google Scholar]
- Terrillon S, Bouvier M (2004) Roles of G-protein-coupled receptor dimerization. EMBO Rep 5: 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K (2002) Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc Natl Acad Sci USA 99: 2660–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Shimada Y, Takeshita T, Kajimura N, Nomura S, Sekiyama N, Otomo J, Usukura J, Nakanishi S, Jingami H (2000) Cryptic dimer interface and domain organization of the extracellular region of metabotropic glutamate receptor subtype 1. J Biol Chem 275: 28144–28151 [DOI] [PubMed] [Google Scholar]
- Xu H, Staszewski L, Tang H, Adler E, Zoller M, Li X (2004) Different functional roles of T1R subunits in the heteromeric taste receptors. Proc Natl Acad Sci USA 101: 14258–14263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron 22: 537–548 [DOI] [PubMed] [Google Scholar]