

Abstract
Mitochondrial dysfunction is a hallmark of many diseases. The retrograde signaling initiated by dysfunctional mitochondria can bring about global changes in gene expression that alters cell morphology and function. Typically, this is attributed to disruption of important mitochondrial functions, such as ATP production, integration of metabolism, calcium homeostasis and regulation of apoptosis. Recent studies showed that in addition to these factors, mitochondrial dynamics might play an important role in stress signaling. Normal mitochondria are highly dynamic organelles whose size, shape and network are controlled by cell physiology. Defective mitochondrial dynamics play important roles in human diseases. Mitochondrial DNA defects and defective mitochondrial function have been reported in many cancers. Recent studies show that increased mitochondrial fission is a pro-tumorigenic phenotype. In this paper, we have explored the current understanding of the role of mitochondrial dynamics in pathologies. We present new data on mitochondrial dynamics and dysfunction to illustrate a causal link between mitochondrial DNA defects, excessive fission, mitochondrial retrograde signaling and cancer progression.
Graphical abstract
I. Introduction
An essential function of mitochondria in mammalian cells is to supply most of the cellular ATP through oxidative phosphorylation. In addition, mitochondria integrate various metabolic pathways and through this process synthesize intermediates needed for the synthesis of cellular biomass. The more specialized functions of mitochondria include maintenance of Ca2+ homeostasis and regulation of apoptosis. These organelles also produce reactive oxygen species as byproducts of respiration coupled oxidative metabolism, which in turn act as physiological signaling molecules, but induce toxicity when produced in excess under pathological conditions. It has been proposed that mitochondria evolved from symbiotic aerobic bacteria engulfed by pre-eukaryotic cells more than a billion years ago (1;2). While this symbiosis likely gave a survival advantage to the host cell in an oxygen rich environment, it was necessary for the cell to stringently control function of the endosymbiont, not only to meet its energy needs but also to protect itself from any toxic metabolites produced by the organelle. This was achieved during the course of evolution by transferring most of the bacterial DNA to the host nucleus (2). The mitochondrial DNA (mtDNA) encodes only 13 polypeptide subunits, all of which are part of the electron transport chain. However, normal mitochondrial function requires well over 1000 proteins (3) and large number of metabolic substrates, which is maintained by constant bidirectional communication with the nucleus.
II. Factors affecting mitochondrial functions
Generally, mitochondrial function is designed to meet the cell s energy and metabolic demands. The efficiency of the process however varies widely due to genetic polymorphism of mtDNA and tissue specific variations in nuclear encoded mitochondrial proteins. Several pathologies and adverse environmental conditions disrupt mitochondrial function in multiple ways. MtDNA mutations, deletions or impaired DNA replication are the most common cause of mitochondrial dysfunction (4;5). Mitochondria contain multiple copies of their own DNA ranging from 100 to 10,000 per cell. However, many diseases such as mtDNA depletion syndrome, several cancers including breast, prostate and colon cancers, and age related pathologies are characterized by significantly low mtDNA copy number that affect normal mitochondrial function (6–13). MtDNA is susceptible to damage and mutation due to both its proximity to a high concentration of toxic metabolites and also relatively inefficient repair mechanism (14). The overall effect on mitochondrial function depends on the extent of heteroplasmy of mtDNA (15;16). Mitochondrial dysfunction can also result from exposure to xenobiotics and adverse environmental conditions.
Hypoxia is a common factor in many pathological conditions such as the interior of solid tumors, tissue ischemia and inflammation (17;18). Hypoxia not only depletes oxygen, which is the terminal acceptor of electrons from the electron transport chain, but also causes permanent damage to the proteins, lipids and mitochondrial nucleic acid components by inducing oxidative stress. Xenobiotics or their metabolites can cause mitochondrial dysfunction by multiple mechanisms including disruption of membrane potential, direct interaction with mitochondrial DNA, and proteins, affecting their function (19–21). The mitochondrial dysfunctions range from reduced ATP production by oxidative phosphorylation, inability to modulate production of excessive reactive oxygen and nitrogen species, dysregulation of calcium to opening of permeability transition pore and initiation of apoptosis.
Many chemotherapeutic drugs used for the treatment of primary tumors, including doxorubicin, cisplatin, other DNA damaging anthracycline derivatives, damage mtDNA and impair mitochondrial functions in cancer patients (22). Doxorubycin, has been shown to cause cardiomyopathy by targeting mitochondria. It increases ROS production, reduces ATP synthesis and inhibits CcO by directly binding to the complex (23). Environmental toxin TCDD is shown to disrupt mitochondrial membrane potential and mitochondrial transcription translation mechanisms in a time-dependent manner and induce stress signaling (24). Similarly, benzo[a]pyrene a common cigarette smoke carcinogen has been shown to be metabolized in mitochondria and form mtDNA adducts. Benzo[a]pyrene treatment disrupts mitochondrial respiration and induces oxidative stress (25). Anti-retroviral drugs such as Zidovudine affect mitochondrial function by inhibiting mtDNA polymerase and causing mtDNA depletion (26). Defects in mitochondrial DNA and membrane components affect mitochondrial function depending on the extent of damage. Reduced ATP production from oxidative phosphorylation is seen in mtDNA depletion and heteroplasmic mutations in mtDNA or components of electron transport chain.
Excessive production of reactive free radicals is one of the common effects of mitochondrial dysfunction. Mitochondrial electron transport chain is a source of reactive oxygen and nitrogen species that are produced as byproducts of oxidative phosphorylation. Hypoxia and mutations in subunits have been associated with increased ROS production from complex I. Mitochondria play an important role in Calcium homeostasis. Dysregulation of calcium can be attributed to both overload of mitochondria with calcium or their inability to uptake calcium. Mitochondrial calcium overload has been shown to be one of the hallmarks of reperfusion injury after ischemic episode. Excessive mitochondrial calcium and reactive oxygen species have been shown to be important triggers for mitochondrial permeability transition. Loss of membrane potential causes decreased calcium uptake and increased cytosolic calcium levels [Ca2+]c.
III. Mitochondrial Morphological Alterations in response to stress
Mitochondrial morphology in different cells can vary from long tubular structures seen in fibroblasts and myocytes to smaller spherical and ovoid structures found in macrophages. Mitochondrial stress and dysfunction resulting from hypoxia, exposure to mitochondrial toxins and metabolic diseases affect mitochondrial morphology. Excessive ROS production induced by photo-excitation of cytochrome c oxidase in ASTC-1 and COS7 cells resulted in increased fragmentation of mitochondria (27). Similarly, hypoxia-reoxygenation stress in H9C2 cells resulted in fragmented, donut-shaped mitochondria (28). We observed fragmented, circular mitochondrial structures in C2C12 cells expressing shRNA to cytochrome c oxidase subunits (29). As shown in Figures 1A–C, electron micrographs of C2C12 cells with more than 80% depleted mtDNA content show small, circular mitochondria. In control cells mitochondria are densely packed in the periphery of the nuclear envelope, whereas in cells with reduced mtDNA content (either by ethidium bromide treatment or silencing Tfam mRNA), the mitochondria are fewer in number, markedly smaller in size, appear fragmented and distributed throughout the cellular cytoplasm (Fig 1B). In contrast to control cells, the mitochondrial cristae appear disorganized in mtDNA-depleted cells. Furthermore, we observed that loss of Tfam mRNA has more severe effect on mitochondrial structural integrity indicated by fragmented mitochondria (Fig 1C) possibly due to the function of TFAM as a mitochondrial nucleoid component.
Figure 1. Altered mitochondrial ultrastructure in response to mtDNA depletion.

Electron micrographs of control, EtBr-treated mtDNA-depleted and Tfam shRNA-mtDNA depleted cells. Left panel: Scale Bar 2μm, magnification 10000x; Right panel: Scale Bar 100nm, magnification 75000x.
IV. Mitochondrial defects deregulate mitochondrial dynamics
Mitochondrial dynamics is regulated by cellular bioenergetic demands. Although several studies report effect of mitochondrial dysfunction on mitochondrial morphology (27–30), the role of mitochondrial dynamics in cell signaling has not been well studied. Healthy mitochondria are dynamic and morphologically fluid which facilitate both the efficiency of mitochondrial function and turnover. The mitochondrial morphology is the result of the interplay between rapid fusion and fission events and is brought about by large dynamin family GTPases (31). Mitochondrial fusion is a two-step process that begins with the initial joining of outer membrane, followed by fusion of the inner membrane. Outer membrane fusion is mediated by the mitofusin proteins (Mfn1 and Mfn2), anchored on the mitochondrial outer membrane (31–33). OPA1 mediates the fusion of inner membrane (31;32). The mechanism of fusion is similar for both the outer and inner membrane, in which the fusion proteins on the opposing membranes form interlocking coiled coils, which are then powered to fuse by GTP hydrolysis (31). Under normal conditions, the fusion of outer and inner membranes is coordinated (31). However, mutations targeted to these genes show that the two events can be independent of each other with the inner membrane fusion dependent on mitochondrial membrane potential (34;35). Mitochondrial fission is mediated by a highly conserved dynamin related GTPase, Drp1 (36). Drp1 is a cytoplasmic protein that is maintained in the cytosol by PKA mediated phosphorylation at Ser 637 (37). Drp1 is recruited to the outer membrane of mitochondria by membrane anchored proteins Mff and Fis1, where it forms a constricting spiral around the outer and inner membrane to fragment the mitochondrion (38–40). Mitochondrial fusion results in extended mitochondrial networks, which provides advantage to cells under high energy demands and disruption of mitochondrial fusion has been shown to result in mitochondrial dysfunction.
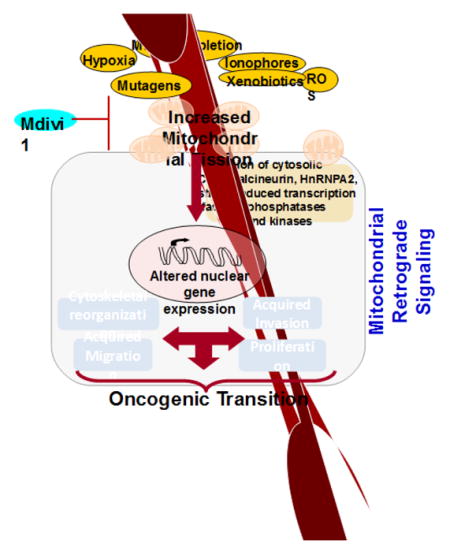
In immortalized as well as primary cells, in response to induction of mitochondrial dysfunction, we observed lower expression of the mitochondrial fusion marker protein OPA1 (Fig 2A, B). This correlated with increased expression of the mitochondrial fission marker protein DRP1 (Fig 2C). Additionally inhibiting mitochondrial fission by Mdivi1 (Drp1 inhibitor) treatment reverses cellular morphology in mtDNA-depleted cells (Fig 2D). Reorganization of cytoskeleton is associated with cell migratory potential and aggressive tumor cells are highly migratory in nature. Using a scratch-wound healing assay we tested the migration pattern of mtDNA depleted C2C12 cells as a function of their metastatic potential. We observed that mtDNA depleted C2C12 cells acquired migratory capacity which was markedly reduced in response to treatment with Mdivi1 (Fig 3 and Movie files 1–3). The Windrose plots of cell movement, where all cell tracks are placed at the same starting point, clearly demonstrate a markedly different migratory pattern of mtDNA-depleted cells as compared to control cells (Fig 3A and B). Treatment with with mDivi1 reduced the migration rates of mtDNAdepleted cells (Fig. 3C). The mtDNA depleted cells exhibited unorganized trajectory, typical of cells with high metastatic potential. These results suggest the contribution of higher mitochondrial fission towards cellular migration and therefore tumor progression. (41)This suggests that mitochondrial dysfunction induces alterations in mitochondrial fusion-fission dynamics, which by a feedback loop further modulates mitochondrial integrity and functions, exacerbating the effects of mitochondrial dysfunction and promoting oncogenesis.
Figure 2. Altered mitochondrial fission and fusion dynamics in mtDNA depleted cells.

(A) Immunofluorescence images showing OPA1 (mitochondrial fusion marker in red) and DAPI (nuclei in blue) staining pattern in control, mtDNA depleted (Tfam sh) and CcOIVi1kd C2C12 cells. (B) Primary esophageal epithelial cells derived from Tfam fl/fl mice expressing either Adeno GFP (control) or AdenoL2- Cre (Tfam KO) stained with OPA1 (mitochondrial fusion marker in red) and DAPI (nuclei in blue). (C) Primary esophageal epithelial cells derived from Tfam fl/fl mice expressing either Adeno GFP (control) or AdenoL2- Cre (Tfam KO) stained with DRP1 (mitochondrial fission marker in red) and DAPI (nuclei in blue). (D) Primary esophageal epithelial cells derived from Tfam fl/fl mice expressing either Adeno GFP (control) or AdenoL2- Cre (Tfam KO) treated with or without mDivi1 (10μM, 48h) stained with Texas-Red Phalloidin (red, actin) and DAPI (blue, nuclei). Images are captured under 40x objective of a NIKON E600 microscope.
Figure 3. Acquired Migratory pattern of C2C12 cells.

(A) Windrose plot showing the directionality of migration of Control, mtDNA depleted and mtDNA depleted cells treated with mDivi1as indicated in the figure. Individual cells tracked are indicated by different colors. (B) Individual cells in each category were tracked using the Volocity software (Perkin Elmer) to estimate the maximum distance covered during the 10h migration. Movie Legends: Time lapse recordings for “scratch wound healing” migration assay: Control (Video1), mtDNA- depleted (Video 2) and mtDNA-depleted + mDivi1 treated (Video 3) C2C12 cells were grown to confluence and wounded with a pipette tip. Wound healing as a measure of cell motility and images were captured every 5 mins and followed for 10 hours.
V. Retrograde signaling by dysfunctional mitochondria
Functional interaction between mitochondria and nucleus is maintained by signaling network that controls both the biogenesis and functioning of mitochondria. The anterograde signals from nucleus to mitochondria control import of proteins, availability of substrates, mtDNA maintenance and gene expression in accordance with the energy and growth requirements of the cell (42–45). In a healthy cell, under physiological conditions, retrograde feedback from mitochondria to nucleus is likely to be normal mitochondrial output of ATP, metabolic intermediates and basal reactive oxygen radicals, and serve to fine-tune the metabolic flux to meet cellular energy demands. On the other hand, mitochondrial retrograde signaling (MtRS), which is activated by mitochondria under stress, has received considerable attention in recent years due to its ability to bring about global changes in nuclear gene expression and phenotypic changes in cells (5;46). Of the multiple retrograde signaling pathways, at least three are triggered directly by mitochondrial stress (47–50). Interestingly all three pathways suggest important role for retrograde stress signaling in cellular transformation and cancer cell survival implicating a role of mitochondrial dysfunction in cancer development(47–50).
V.1 Reactive oxygen species initiated signaling
Reactive oxygen species (ROS) in mitochondria are result of premature oxidation of electrons in the mitochondrial electron transport chain. It is estimated that 2–5% of oxygen consumed by mitochondria is converted to ROS. In healthy mitochondria, ROS is rapidly detoxified by antioxidant defenses (51). Excess ROS produced under hypoxia or by increased inflammatory cytokines, on the other hand, activates MtRS leading to cellular adaptations (52–54). ROS produced by complex III under hypoxia was shown to activate AMPK and inhibit mTOR to conserve energy at the same time increase ATP production (55). Similarly, TNF alpha induced mitochondrial ROS production and triggered cell death by activation of JNK pathway (56). In RAW 264.7 macrophages, hypoxia alone or synergistically with RANKL, produces mitochondrial ROS, triggering retrograde signaling and differentiating the cells into bone resorbing osteoclasts (57;58). In the case of macrophages however, we observed that the initial ROS signal resulted in the activation of calcium-calcineurin mediated MtRS signaling pathway discussed in detail below (57;58). Some reports showed that mitochondrial ROS generated under hypoxia stabilize HIF1alpha and facilitate cellular transformation (48). Similarly, high ROS levels in cancers characterized by loss of tumor suppressor mutations could activate proliferation, angiogenic and antiapoptotic pathways aiding in cancer progression (59–62).
V.2 Unfolded protein response (mtUPR) initiated signaling
Mitochondrial stress and dysfunction affects protein homeostasis. Oxidatively damaged proteins and unfolded or mis-folded proteins are degraded by mitochondrial proteases for quality control (63–65). In mammalian cells mitochondrial ability to communicate the matrix unfolded protein response to the nucleus involve activation of CHOP and CREB (66;67). Experimental targeting of misfolded ornithine transcarbomoylase to mitochondrial matrix resulted in upregulation of several heat shock proteins like HSP60, HSP10 and chaperones without affecting cytosolic or ER stress response (67). Similarly, presence of mutant EndoG in intermembrane space resulted in Akt phosphorylation mediated activation of estrogen receptor alpha and induction of quality control proteases localized to IMS (47). Direct evidence for retrograde signaling by unfolded protein response has been shown in C. elegans, in which excessive accumulation of unfolded proteins resulted in their degradation and efflux of peptides (68). The peptides activate a transcriptional response mediated by ATFS-1 that localizes to nucleus and along with two other factors DVE-1 and Ubl-5 induce expression of genes involved in mitochondrial quality control and cellular metabolism (68–71). Although there is no direct evidence, it is likely that the retrograde signaling by unfolded protein response may play a major role in cancer cell survival. Aberrant growth and exposure to adverse conditions like hypoxia are common feature of cancer cells and likely result in very high rates of oxidatively damaged and misfolded proteins inside mitochondria, conditions that could initiate unfolded protein response. In this context, it is interesting that expression of HSP60 and HSP90 related protein TRAF1 is elevated in many tumors. It is however unclear if this is an effect of retrograde signaling by UPR.
V.3 Calcium-calcineurin mediated retrograde signaling
Calcium-calcineurin mediated signaling is activated in response to mtDNA depletion, loss of electron transport chain proteins and disruption of mitochondrial membrane potential (24;49;50;72;73). Mitochondria play an important role in Ca2+ homeostasis by transiently removing Ca2+ released by intracellular stores or from outside the cell and releasing back to the cytosol to regulate calcium dependent (74–76) signaling. Mitochondrial dysfunction due to loss of membrane potential from either mtDNA defects or exposure to xenobiotic toxins impairs mitochondrial calcium uptake (49;73;77). Interestingly, in models of mtDNA depletion by ethidium bromide treatment and membrane potential disruption by CCCP treatment, the cells exhibited elevated expression of Ryanodine receptor (RYR1, 2 or 3), which is a calcium leak channel in the endoplasmic reticulum membrane (24;49;73). Collectively, inability of mitochondria to take up calcium, excessive calcium leak through upregulated RYR channels and likely reduced activity of the ATP dependent Endoplasmic Reticulum calcium pump together result in sustained elevation of [Ca2+]c. Increased steady state [Ca2+]c activates calcineurin, a cytosolic phosphatase, which in turn activates NFAT and a unique IkB dependent NFκB pathway (29;49;78). Calcium dependent kinases PKC, CAMKIV, JNK and MAPK are also activated, which leads to activation of transcription factors CEBP/δ and CREB (5). An important factor activated by this pathway is the heterogeneous nuclear ribonuclear protein hnRNPA2, which acts as a transcriptional co-activator (79;80). It assembles the stress induced transcription factors in enhanceosome complexes at promoter sequences leading to synergistic activation of several stress response genes (46;79–81).
Recently we reported that hnRNPA2 is a novel mitochondrial stress activated protein lysine acetyltransferase that acetylates histone H4 at lysine K8 on target gene promoters. This H4K8 acetylation is essential for the transcriptional activation of the mitochondrial stress-responsive target genes (82). In ethidium bromide treated C2C12 cells, mtDNA depletion results in induced expression of more than 120 genes that bring about phenotypic changes like altered morphology, increased invasiveness, metabolic shift to glycolysis and resistance to apoptosis (50;83–85). Similar activation of Calcium-calcineurin mediated MtRS signaling was seen in mtDNA depleted immortalized RAW264.7 and MCF10A cells as well as transformed cells such as A549, MCF7 and HCT116 (29;73;81;86).
VI. Mitochondrial defects in cancer
Altered mitochondrial function is a hallmark of many cancers although the nature of functional modification depends on the type of cancer (87–91). Nearly 80 years ago Warburg noted that cancer cells have damaged respiration and increased lactate production, a phenomenon referred to as aerobic glycolysis or Warburg Effect (92). Since then, several studies have shown reduced mitochondrial respiration in a wide range of cancers (93). Supporting evidence for the defective mitochondria in cancer tissue has been shown by both genetic screening for mtDNA defects and by providing experimental support by using cybrids (91;94;95). Low copy number of mtDNA has been observed in many cancers including breast, colon, hepatocellular carcinomas, astrocytomas and prostate cancers (6;8;11;96;97). Experimentally induced mtDNA depletion in colorectal and prostate cancer cells promotes aggressive cancers (98;99). Similarly, increased tumor growth is seen in the intestine of mice with adenomatous polyposis coli intestinal neoplasia when crossed with mice heterozygous for the mitochondrial transcription factor A (TFAM) with reduced mtDNA copy number (100). Mutations in mitochondrial DNA has been reported in several cancers including breast, renal adenocarcinoma, thyroid tumors, colon cancer, head and neck cancer and prostate cancer (95;97;101;102). Although the causal role of mitochondrial DNA defect in tumorigenesis has not been clearly established, cybrid cell lines generated by fusing cytoplasts and nuclei from different cell lines provide a means for testing the role of WT and mutant mtDNA under the same nuclear genetic background. Mutations in complex I subunit ND6 increased the metastatic potential by producing excessive ROS, whereas an ND5 mutation promoted tumorigenesis by oxidative stress and Akt activation (94;95).
Mitochondrial function is affected by changes in nuclear coded proteins. Mutations in SDH subunits and FH genes have been observed in paragangliomas, pheochromocytomas, multiple cutaneous and uterine leiomyomatas and aggressive forms of renal cell cancer (103–107). In both these instances, loss of function causes accumulation of substrates succinate and fumarate, which have been shown to activate specific stress pathways with roles in tumor development (103;107–109). Heterozygous missense mutations in IDH have been shown in gliomas, chondromas and astrocytomas (110–112). Heterodimers of mutant IDH1 and IDH3 have been shown to increase accumulation of 2-hydroxyglutarate that has been shown to affect methylation and other epigenetic modifications (110;111;113;114).
In addition to mutations, levels of mitochondrial proteins can also be affected by environmental changes. Long term hypoxia as commonly seen in solid tumors has been shown to specifically degrade subunits IV and Vb of cytochrome oxidase (115;116). Using stable cell lines expressing shRNA to subunits IV and Vb, we showed that mitochondrial dysfunction results in activation of several genes that have role in tumor development (29). The causal role of this loss of CcO subunits in the observed phenotype was further confirmed by rescue of CcO activity WT subunit cDNAs, which reverted most of the phenotypic changes (29).
Mitochondrial dysfunction is also shown to play important role in metastasis. Depletion of mtDNA resulting in mitochondrial dysfunction induced an epithelial to mesenchymal transition in multiple cell lines including MCF 10A breast cancer cells (81). Mutation in complex I subunit ND6 was shown to increase the metastatic potential of a mouse lung carcinoma cell line (95). In human cervix squamous cell carcinoma and murine melanoma cell lines, clonally selected cells with high metastatic potential produced elevated levels of superoxide (117). Furthermore, experimental inhibition of electron transport chain functions seemed to attenuate the metastatic potential of these cells suggesting the causal role of superoxide in conferring metastatic potential. Superoxide was shown to promote tumor cell migration by activating Pyk2, the focal adhesion kinase in Src mediated pathway. Moreover, partial inhibition of ETC resulted in prometastatic phenotype, which was reversed by treatment with mitochondria targeted antioxidants (117) clonal selection of high metastatic cells over successive generations could have attributed to the phenotypic drift commonly reported in transformed cell lines (118–120)(119–121). It is also likely that different mitochondrial outputs are needed for cells at different stages of metastatic progression.
A more recent focus of research in the field has been on the contribution of mitochondrial dynamics towards tumor initiation and progression. Although the exact mechanism and the signaling of defective mitochondrial dynamics in cancer development is not known, it has been observed that excessive fission and reduced fusion is a feature of many tumors (121–123). Interestingly most of the available literature shows dysregulated Drp1 action as responsible for excessive fission (124–126). In human pancreatic cancer, expression of oncogenic Ras or activation of MAPK pathway leads to Erk2 mediated phosphorylation of Drp1 on Ser 616 that leads to increased mitochondrial fragmentation (126). Moreover, inhibition of this phosphorylation in xenografts is sufficient to block tumor growth (126). Similarly, both A549 lung adenocarcinoma cell lines and human tissues exhibit higher expression of Drp1 with increased phosphorylation (121). Interestingly this was accompanied by reduced Mfn2 expression. Increasing mitochondrial networking by Mdivi1 treatment resulted in higher apoptosis and reduced proliferation in A549 cells and reduced tumor growth in xenografts expressing Mfn2 delivered by adenoviral vector (121). The trigger and the mechanism resulting in Drp1 overexpression and phosphorylation are not well understood. In high stage neuroblastoma, Survivin, an antiapoptotic protein recruits Drp1 to mitochondria and induces mitochondrial fragmentation (127). Survivin overexpressing cells were shown to have reduced complex I and IV activities with a shift to aerobic glycolysis (127).
As mentioned earlier, mtDNA depletion in C2C12 cells induces mitochondrial retrograde signaling that transforms them to tumorigenic cells. We recently observed in these cells that mitochondrial dysfunction caused by mtDNA depletion leads to increased Drp1 mitochondrial localization and reduced OPA1 expression, accompanied with fragmented mitochondria. As shown in Fig 1 and 2, mtDNA depletion (either by EtBr or knock down of Tfam mRNA) induced higher levels of mitochondrial fission. The mtDNA-depleted cells also showed remarkably altered cytoskeleton and pseudopodia like structures, characteristic of tumor cells (Fig. 2). Interestingly, the treatment with Mdivi1 (128), a specific inhibitor of fission protein DRP1 attenuated these tumorigenic morphological changes suggesting a connecting link between mtDNA depletion, mitochondrial dynamics, altered morphology and tumor-like phenotype. However, the mechanistic link between increased mitochondrial fragmentation and phenotypic transformation remains to be investigated.
VII. Alterations in Cellular Metabolism effected by mitochondrial dysfunction
As the hub of metabolic integration, mitochondrial metabolome consists of hundreds of intermediates generated in metabolic pathways like TCA cycle, fatty acid metabolism, amino acid oxidation and oxidative phosphorylation. Dysfunctional mitochondria, either due to specific defects in mitochondrial enzymes or general effects like hypoxia, loss of membrane potential or mtDNA depletion exhibit characteristic accumulation of metabolites. Aberrant levels of metabolites are not only signatures of pathologies, but also responsible for signaling and disease phenotype. For example, high fat diet induced mitochondrial dysfunction in metabolic syndrome and inflammation is characterized by presence of medium and long chain acyl carnitines (129). One of the metabolites, lauryl carnitine was shown to increase expression of pro inflammatory cytokines in bone marrow derived macrophages (129). Similarly, in mouse models of Alzheimer s disease, differential levels of intermediates of nucleotide metabolism, TCA cycle and amino acid metabolic pathways accumulate (130). These data provide evidence of causal effect of defective energy metabolism. As detailed in an earlier section, altered mitochondrial function is a feature of several cancers. Mutations in enzymes of TCA cycle, components of the electron transport chain and mtDNA defects affect mitochondrial metabolism and alter the metabolome. Metabolic profiling of hereditary paraganglioma and phenochromocytoma harboring mutations in succinate dehydrogenase showed increased levels of succinate, which has been shown to inhibit prolyl hydroxylases (PHD) and stabilize HIF1 (107;109;112). Heriditary leiomyomatosis and renal cell cancers that have a high frequency of mutation in Fumarate hydratase similarly accumulate millimolar levels of fumarate, which has been shown to also inhibit PHDs and histone demethylases (108;109;112).
Fumarate has also been shown to react with the thiol group of glutathione by a process called succination and increase oxidative stress (131). Isocitrate dehydrogenase is another enzyme of the TCA cycle that is mutated in many human cancers like colon cancer, gliomas, AML and osteosarcoma. Mutations in IDH generates a neomorphic enzyme that converts isocitrate to 2-hydroxyglutarate (2-HG) instead of oxo-glutarate (110;114). 2-HG has been shown to be oncogenic through its action on PHDs, and can bring about epigenetic changes (110;114). mtDNA depletion mediated mitochondrial dysfunction induces stress signaling that transforms non-tumorigenic cells to acquire tumorigenic phenotype.
To understand the metabolic shift by partial mtDNA depletion, we carried out a metabolic profile analysis on control and mtDNA depleted C2C12 cells (Fig 4). The depleted cells showed between 3–4 fold higher lactate levels indicating a metabolic shift to glycolysis. Interestingly mtDNA depleted cells also showed higher levels of 2-HG and Fumarate, although their roles in cancer promotion and signaling need to be investigated.
Figure 4. Metabolic changes in response to mtDNA depletion.

Intracellular lactate, fumarate and 2-hydroxyglutarate were quantified by reverse phase LC coupled to an Orbitrap Mass Spectrometer. Graphs show fold change in indicated metabolites between control and mtDNA depleted C2C12 cells. Values normalized to control cells.
VIII. Mitochondrial dysfunction modulates cellular morphological changes
It is well documented that mitochondrial dysfunction influences pathological phenotypes and morphological changes has been postulated to be an adaptive cellular response to the mitochondrial stress Reports from our laboratory and others have shown that immortalized mammary cells harboring mitochondrial defects undergo cellular morphological reprograming similar to an epithelial-mesenchymal transition (81;132;133). Actin microfilaments are critical components of cellular cytoskeleton. It is known that actin filaments acquire conformational changes in response to changes in nucleotide binding or cellular stressors (134–136). The interaction between actin filaments and various actin-binding proteins are critical for the functional differentiation of actin filaments in vivo (134;135;137). Cell polarity is dynamic and dependent on various physiological processes such as cell division, migration, and morphogenesis. Only recently, the critical role of cell polarity in regulating cancer phenotype has gained interest (138–140). We observed mtDNA-depleted C2C12 and HEK293T cells have extensive actin reorganization with long stretched F-actin filaments shown by phalloidin staining (Fig 5A, B). These are characteristics of a highly invading tumor cell.
Figure 5. MtDNA depletion induced cytoskeletal reorganization in immortalized cells.

Control and mtDNA depleted C2C12 (A) and HEK293T (B) stained with Texas-Red® conjugated Phalloidin (Molecular Probes) for gamma Actin (red) and DAPI for nuclei (blue). Phalloidin staining was performed according to manufacturer s suggested protocol. Cells were imaged under Nikon E600 microscope 40x Objective. Scale Bar: 20μm.
In primary esophageal cells derived from Tfamfl/fl mice, we observed, cells transduced ex vivo with adenoviral Cre contained >95% reduced Tfam mRNA and also with reduced mtDNA content compared to cells transduced with the adenoviral vector expressing GFP (Fig 6A,B). We observed that mtDNA depleted primary esophageal cells (Tfamfl/fl/+Cre) were ~20% larger in size (Fig 6C). Similar to immortalized cells, primary esophageal mtDNA depleted cells also exhibit higher levels of F-actin and stretched F-actin filaments (Fig. 6D) suggesting that mitochondrial stress signaling regulate cell polarity. Turnover of actin filament and dynamic regulation of actin structures are critical for cell migration, cell adhesion and protrusion, important processes during oncogenesis (141–143). Moreover, cell polarity regulating proteins also modulate microtubule dynamics, which is significant because dysregulation of microtubule dynamics is used in cancer therapy (144). Therefore, the changes in actin reorganization we observed in response to mitochondrial stress are of relevance to tumor cell physiology, morphology and cell growth characteristics.
Figure 6. Alterations in cellular morphology in Tfamfl/fl/Cre-primary esophageal cells.

Real Time PCR quantitation of relative Tfam mRNA levels (A) and mtDNA content (B) in primary esophageal epithelial cells derived from Tfam fl/fl mice expressing either Adeno GFP (control) or AdenoL2- Cre (Tfam KO). (C) Cell Size distribution in primary esophageal epithelial cells derived from Tfam fl/fl mice expressing either Adeno GFP (control) or AdenoL2- Cre (Tfam KO) assessed on Nexcelom Vision CBA. (D) Primary esophageal epithelial cells derived from Tfam fl/fl mice expressing either Adeno GFP (control) or AdenoL2- Cre (Tfam KO) stained with Texas-Red Phalloidin (red, actin) and DAPI (blue, nuclei) imaged under 40x objective of a NIKON E600 microscope.
The 3D organoid cultures of esophageal epithelial cells (EEC) are called “mini organs” because of their close similarity to the esophageal tissue in terms of cell types, overall organization and function (145). These organoids therefore provide an excellent ex vivo model for studying the pathophysiology of esophagi. Using 3D organoid cultures of primary EEC we demonstrate that mitochondrial functions are necessary for normal organoid development (Fig 7). Organoids derived from Tfam knockout Tfamfl/fl/+Cre murine primary esophageal cells as described in the above section, were fewer in numbers and showed reduced basal and parabasal cells. Additionally these organoids also showed higher keratinization as indicative of terminal differentiation, supporting the role of mitochondria in regulating cellular development and morphology.
Figure 7. Three dimensional organoids in Tfamfl/fl/Cre-primary esophageal cells.

Bright Field images of Hematoxylin-Eosin stained sections of 3D organoids from primary esophageal epithelial cells derived from Tfamfl/fl mice expressing either Adeno GFP (control) or AdenoL2-Cre (Tfam KO.
Summary and Conclusions
The Warburg hypothesis proposed nearly 80 years ago still remains an enigma in cancer biology and tumor development. The first part of the Warburg hypothesis on markedly increased glycolysis and lactate production by fast growing tumors in culture or in vivo in tumors is universally accepted. The second part of the hypothesis suggesting a role for defective mitochondrial function as a possible cause of cancer initiation or progression remains contentious, despite intense investigations in many laboratories. Although clinical studies show strong correlation between mitochondrial dysfunction, including respiratory defects, mtDNA mutations and low mtDNA contents in a variety of human cancers, the cause or effect relationship remains unclear. Many studies including ours have shown that partial depletion of mtDNA, heteroplasmic mtDNA mutations, or disruption of Complex I and complex IV induce tumorigenic phenotypes in immortalized epithelial cells. However, in vivo models of tumor production specifically induced by mitochondrial dysfunction are currently limited, and more research efforts and more models would be needed to address this important question.
It is widely known that anapleurotic metabolic processes including the activity of TCA cycle and metabolism of carbon skeletons of Gln and Asn that are used as preferred fuel sources by tumor cells are essential for the production of biomass (146;147). This has led to the concept that a full mitochondrial function is essential for tumor development. Some studies show that metastasizing tumors and those resistant to chemotherapeutic drugs, show more robust respiration and TCA cycle activity. It should be noted that mitochondrial dysfunction induced by partial mtDNA depletion or disruption of ETC complexes retain nearly full ability to metabolize amino acids and generate citrate as their citrate synthase activities are equal to or even higher than control cells (148). However, the metabolic status in Rho zero cells used in some studies (149) is quite different than that we have observed in partial mtDNA depleted cells. Furthermore, the role of mitochondrial dysfunction in early stages of tumor initiation and progression observed in our and others studies are likely to be different from that reported in tumor metastasis and drug resistance (150) where cellular signals from the microenvironment might play more critical roles. The details of these differences need to be investigated to gain a firm understanding of the second part of Warburg hypothesis. Our observations over the years clearly show that mitochondrial dysfunction and associated mtRS plays a critical role in inducing oncogenic phenotypes in immortalized cells.
It is becoming increasingly clear that mitochondrial fission and fusion play a critical role in quality control and mitochondrial damage repair (151). Our results show extensive mitochondrial fission and reduced fusion by partial mtDNA depletion and shRNA mediated knock down of CcO subunits. Thus, altered fusion and fission are closely linked to mitochondrial dysfunction and mtRS activation. The Drp1 inhibitor Mdivi-1 inhibits colony formation, MtRS and other tumorigenic characteristics. A recent study also showed that Mdivi-1 treatment inhibited tumor growth in a KRAS induced mouse tumor model (126). It is therefore likely that altered quality control associated with mitochondrial dysfunction plays a role in tumor induction and progression, while these processes are partly or wholly repaired in tumor metastasis and drug resistance.
Methods
Electron Microscopy
Cell Pellets for electron microscopic examination were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer, pH7.4, overnight at 4oC. After subsequent buffer washes, the samples were post-fixed in 2.0% osmium tetroxide for 1 hour at room temperature, and then washed again in buffer followed by DH2O. After dehydration through a graded ethanol series, the tissue was infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image capture software.
Esophageal Three Dimensional Organoid Cultures
Three dimensional organoid cultures were grown as described in Tanaka et al (152). Briefly, 1x 103 cell suspension in 50 μl Matrigel were seeded per well in 24 well plates. Cells were grown in organoid growth medium supplemented with 1X B27, 0.1 mM N-acetyl-L-cysteine (Sigma-Aldrich), mouse recombinant epidermal growth factor (R&D Systems, Minneapolis, MN), 2.0% Noggin/R-Spondin-conditioned media and 10 μM Y27632 (Tocris Biosciences, Bristol, UK). For H&E sections, organoids were recovered by digesting Matrigel® (BD Biosciences, San Jose, CA) with Dispase I (BD Biosciences, San Jose, CA; 1 U/ml) and fixed overnight in 4.0% paraformaldehyde. Samples were embedded in 2.0% Bacto-Agar: 2.5% gelatin prior to paraffin embedding.
Cell Migration (wound-healing) Assay
Migration assay was performed as described before (81). Confluent monolayer of C2C12 cells were scratched using a pipette tip, and cells migrating into this area were observed at 5-min intervals for 20 h under an inverted bright-field microscope. For quantitative analysis, individual cells were tracked and their direction of movement, velocity and distance covered in the direction of the wound were measured using Volocity software (Perkin Elmer, Waltham, MA, USA).
Metabolite analysis
For metabolomic analysis, metabolites were extracted in 80% methanol in water solution. Samples were analysed using an Orbitrap mass spectrometer coupled to reverse phase ion pairing chromatography (153). Data was normalized to the median and fold change were calculated using control C2C12 cells as reference.
Supplementary Material
Highlights.
Mitochondrial defects affecting Δμm induce stress signaling
MtDNA depletion induces reorganization of actin cytoskeleton and regulates cell polarity
MtDNA depletion induces mitochondrial fission and alters dynamics
Inhibition of mitochondrial fission by Mdivi1 abrogates the acquired migratory potential of mtDNA depleted cells
MtDNA depletion affects 3D organoid formation by primary esophageal epithelial cells
Acknowledgments
We thank Gordon Ruthel for assistance with the imaging studies. This work was funded by NCI grant CA-22762, an Endowment from Harriet Ellison Woodward Foundation to NGA and a Breast Cancer Alliance Young Investigator Grant to MG.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lane N, Martin W. The energetics of genome complexity. Nature. 2010 Oct 21;467(7318):929–34. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondria, bioenergetics, and the epigenome in eukaryotic and human evolution. Cold Spring Harb Symp Quant Biol. 2009;74:383–93. doi: 10.1101/sqb.2009.74.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016 Jan 4;44(D1):D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012 Oct;12(10):685–98. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004 Apr 9;14(1):1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 6.Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, Lee LM, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer. 2006 Jul;45(7):629–38. doi: 10.1002/gcc.20326. [DOI] [PubMed] [Google Scholar]
- 7.Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005 Mar 1;65(5):1655–63. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- 8.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005 Jan 18;102(3):719–24. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JS, Sharma LK, Li H, Xiang R, Holstein D, Wu J, et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet. 2009 May 1;18(9):1578–89. doi: 10.1093/hmg/ddp069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, Takenaga K, et al. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS One. 2011;6(8):e23401. doi: 10.1371/journal.pone.0023401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton TM, Petros JA, Heddi A, Shoffner J, Kaufman AE, Graham SD, Jr, et al. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosomes Cancer. 1996 Feb;15(2):95–101. doi: 10.1002/(SICI)1098-2264(199602)15:2<95::AID-GCC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 12.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005 Jul 15;309(5733):481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 13.Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012 Jan;226(2):274–86. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 14.Mandavilli BS, Santos JH, Van HB. Mitochondrial DNA repair and aging. Mutat Res. 2002 Nov 30;509(1–2):127–51. doi: 10.1016/s0027-5107(02)00220-8. [DOI] [PubMed] [Google Scholar]
- 15.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010 Jan;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci U S A. 2014 Sep 23;111(38):E4033–E4042. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011 Jun;11(6):393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 18.Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999 Dec;66(6):889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 19.Niranjan BG, Bhat NK, Avadhani NG. Preferential attack of mitochondrial DNA by aflatoxin B1 during hepatocarcinogenesis. Science. 1982 Jan 1;215(4528):73–5. doi: 10.1126/science.6797067. [DOI] [PubMed] [Google Scholar]
- 20.Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene. 1999 Nov 18;18(48):6641–6. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- 21.Srinivasan S, Avadhani NG. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic Biol Med. 2012 Sep 15;53(6):1252–63. doi: 10.1016/j.freeradbiomed.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashley N, Poulton J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem Biophys Res Commun. 2009 Jan 16;378(3):450–5. doi: 10.1016/j.bbrc.2008.11.059. [DOI] [PubMed] [Google Scholar]
- 23.Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J. 2009 Feb 18;96(4):1388–98. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas G, Srinivasan S, Anandatheerthavarada HK, Avadhani NG. Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc Natl Acad Sci U S A. 2008 Jan 8;105(1):186–91. doi: 10.1073/pnas.0706183104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bansal S, Leu AN, Gonzalez FJ, Guengerich FP, Chowdhury AR, Anandatheerthavarada HK, et al. Mitochondrial targeting of cytochrome P450 (CYP) 1B1 and its role in polycyclic aromatic hydrocarbon-induced mitochondrial dysfunction. J Biol Chem. 2014 Apr 4;289(14):9936–51. doi: 10.1074/jbc.M113.525659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benbrik E, Chariot P, Bonavaud S, Ammi-Said M, Frisdal E, Rey C, et al. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI) and zalcitabine (ddC) on cultured human muscle cells. J Neurol Sci. 1997 Jul;149(1):19–25. doi: 10.1016/s0022-510x(97)05376-8. [DOI] [PubMed] [Google Scholar]
- 27.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011 Apr;278(6):941–54. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Hajnoczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011 Oct;18(10):1561–72. doi: 10.1038/cdd.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srinivasan S, Guha M, Dong DW, Whelan KA, Ruthel G, Uchikado Y, et al. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene. 2016 Mar 24;35(12):1585–95. doi: 10.1038/onc.2015.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. 2013 Mar 15;304(6):R393–R406. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–80. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- 32.Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003 Sep;134(3):333–44. doi: 10.1093/jb/mvg150. [DOI] [PubMed] [Google Scholar]
- 33.Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002 Dec;13(12):4343–54. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, et al. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Cell Physiol. 2006 Jul;291(1):C176–C184. doi: 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- 35.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003 Mar 7;278(10):7743–6. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 36.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001 Aug;12(8):2245–56. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007 Jul 27;282(30):21583–7. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 38.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008 Jun;19(6):2402–12. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003 Aug;23(15):5409–20. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003 Sep 19;278(38):36373–9. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 41.Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol. 2013;3:292. doi: 10.3389/fonc.2013.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He J, Cooper HM, Reyes A, Di RM, Sembongi H, Litwin TR, et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012 Jul;40(13):6109–21. doi: 10.1093/nar/gks266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bestwick ML, Shadel GS. Accessorizing the human mitochondrial transcription machinery. Trends Biochem Sci. 2013 Jun;38(6):283–91. doi: 10.1016/j.tibs.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008 Jul 11;134(1):112–23. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garesse R, Vallejo CG. Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene. 2001 Jan 24;263(1–2):1–16. doi: 10.1016/s0378-1119(00)00582-5. [DOI] [PubMed] [Google Scholar]
- 46.Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion. 2013 Nov;13(6):577–91. doi: 10.1016/j.mito.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013 Feb;1833(2):410–6. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000 Aug 18;275(33):25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 49.Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, et al. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999 Feb 1;18(3):522–33. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol. 2003 May 12;161(3):507–19. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem. 1997 Jul 25;272(30):18515–7. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 52.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011 Jul 11;194(1):7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006 Jun 30;312(5782):1882–3. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 54.Collins Y, Chouchani ET, James AM, Menger KE, Cocheme HM, Murphy MP. Mitochondrial redox signalling at a glance. J Cell Sci. 2012 Feb 15;125(Pt 4):801–6. doi: 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- 55.Emerling BM, Weinberg F, Snyder C, Burgess Z, Mutlu GM, Viollet B, et al. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009 May 15;46(10):1386–91. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J Immunol. 2000 Jul 15;165(2):1013–21. doi: 10.4049/jimmunol.165.2.1013. [DOI] [PubMed] [Google Scholar]
- 57.Srinivasan S, Avadhani NG. Hypoxia-mediated mitochondrial stress in RAW264.7 cells induces osteoclast-like TRAP-positive cells. Ann N Y Acad Sci. 2007 Nov;1117:51–61. doi: 10.1196/annals.1402.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srinivasan S, Koenigstein A, Joseph J, Sun L, Kalyanaraman B, Zaidi M, et al. Role of mitochondrial reactive oxygen species in osteoclast differentiation. Ann N Y Acad Sci. 2010 Mar;1192:245–52. doi: 10.1111/j.1749-6632.2009.05377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007 Sep;12(3):230–8. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010 May 11;107(19):8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993 Sep 1;90(17):7915–22. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991 Feb 1;51(3):794–8. [PubMed] [Google Scholar]
- 63.Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans. 2011 Oct;39(5):1509–13. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- 64.Baker MJ, Palmer CS, Stojanovski D. Mitochondrial protein quality control in health and disease. Br J Pharmacol. 2014 Apr;171(8):1870–89. doi: 10.1111/bph.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benischke AS, Hemion C, Flammer J, Neutzner A. Proteasome-mediated quality control of S-nitrosylated mitochondrial proteins. Mitochondrion. 2014 Jul;17:182–6. doi: 10.1016/j.mito.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Arnould T, Vankoningsloo S, Renard P, Houbion A, Ninane N, Demazy C, et al. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002 Jan 15;21(1–2):53–63. doi: 10.1093/emboj/21.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002 Sep 2;21(17):4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010 Feb 26;37(4):529–40. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt) Mol Cell. 2015 Apr 2;58(1):123–33. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006 Sep;174(1):229–39. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012 Aug 3;337(6094):587–90. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001 Apr 17;20(8):1910–20. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002 Nov 7;21(51):7839–49. doi: 10.1038/sj.onc.1205983. [DOI] [PubMed] [Google Scholar]
- 74.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003 Jul;4(7):517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 75.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995 Aug 11;82(3):415–24. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 76.Cobbold PH, Cuthbertson KS. Calcium oscillations: phenomena, mechanisms and significance. Semin Cell Biol. 1990 Aug;1(4):311–21. [PubMed] [Google Scholar]
- 77.Luo Y, Bond JD, Ingram VM. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc Natl Acad Sci U S A. 1997 Sep 2;94(18):9705–10. doi: 10.1073/pnas.94.18.9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang W, Chowdhury AR, Guha M, Huang L, Van WT, Rustgi AK, et al. Silencing of IkBbeta mRNA causes disruption of mitochondrial retrograde signaling and suppression of tumor growth in vivo. Carcinogenesis. 2012 Sep;33(9):1762–8. doi: 10.1093/carcin/bgs190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guha M, Pan H, Fang JK, Avadhani NG. Heterogeneous nuclear ribonucleoprotein A2 is a common transcriptional coactivator in the nuclear transcription response to mitochondrial respiratory stress. Mol Biol Cell. 2009 Sep;20(18):4107–19. doi: 10.1091/mbc.E09-04-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guha M, Tang W, Sondheimer N, Avadhani NG. Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets. Biochim Biophys Acta. 2010 Jun;1797(6–7):1055–65. doi: 10.1016/j.bbabio.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guha M, Srinivasan S, Ruthel G, Kashina AK, Carstens RP, Mendoza A, et al. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene. 2013 Nov 4; doi: 10.1038/onc.2013.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guha M, Srinivasan S, Guja K, Mejia E, Garcia-Diaz M, Johnson B, et al. HnRNPA2 is a novel histone acetyltransferase which mediates mitochondrial stress induced nuclear gene expression. Cell Discovery. 2016 doi: 10.1038/celldisc.2016.45. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Biswas G, Guha M, Avadhani NG. Mitochondria-to-nucleus stress signaling in mammalian cells: nature of nuclear gene targets, transcription regulation, and induced resistance to apoptosis. Gene. 2005 Jul 18;354:132–9. doi: 10.1016/j.gene.2005.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Biswas G, Anandatheerthavarada HK, Avadhani NG. Mechanism of mitochondrial stress-induced resistance to apoptosis in mitochondrial DNA-depleted C2C12 myocytes. Cell Death Differ. 2005 Mar;12(3):266–78. doi: 10.1038/sj.cdd.4401553. [DOI] [PubMed] [Google Scholar]
- 85.Biswas G, Tang W, Sondheimer N, Guha M, Bansal S, Avadhani NG. A distinctive physiological role for Ikappa Bbeta in the propagation of mitochondrial respiratory stress signaling. J Biol Chem. 2008 Feb 13; doi: 10.1074/jbc.M710481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chowdhury AR, Long A, Fuchs SY, Rustgi A, Avadhani NG. Mitochondrial stress-induced p53 attenuates HIF-1alpha activity by physical association and enhanced ubiquitination. Oncogene. 2016 Jun 27; doi: 10.1038/onc.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gallardo ME, Moreno-Loshuertos R, Lopez C, Casqueiro M, Silva J, Bonilla F, et al. m.6267G>A: a recurrent mutation in the human mitochondrial DNA that reduces cytochrome c oxidase activity and is associated with tumors. Hum Mutat. 2006 Jun;27(6):575–82. doi: 10.1002/humu.20338. [DOI] [PubMed] [Google Scholar]
- 88.Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008 Feb 1;68(3):700–6. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- 89.Dakubo GD, Parr RL, Costello LC, Franklin RB, Thayer RE. Altered metabolism and mitochondrial genome in prostate cancer. J Clin Pathol. 2006 Jan;59(1):10–6. doi: 10.1136/jcp.2005.027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006 Aug 7;25(34):4647–62. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 91.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006 Aug 7;25(34):4663–74. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 92.Warburg O. On the origin of cancer cells. Science. 1956 Feb 24;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 93.Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006 Dec 18;175(6):913–23. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet. 2011 Dec 1;20(23):4605–16. doi: 10.1093/hmg/ddr395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008 May 2;320(5876):661–4. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 96.Correia RL, Oba-Shinjo SM, Uno M, Huang N, Marie SK. Mitochondrial DNA depletion and its correlation with TFAM, TFB1M, TFB2M and POLG in human diffusely infiltrating astrocytomas. Mitochondrion. 2011 Jan;11(1):48–53. doi: 10.1016/j.mito.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 97.Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, Wu CW, et al. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci. 2005 May;1042:109–22. doi: 10.1196/annals.1338.011. [DOI] [PubMed] [Google Scholar]
- 98.Guo J, Zheng L, Liu W, Wang X, Wang Z, Wang Z, et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011 Apr 15;71(8):2978–87. doi: 10.1158/0008-5472.CAN-10-3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moro L, Arbini AA, Yao JL, di Sant’Agnese PA, Marra E, Greco M. Mitochondrial DNA depletion in prostate epithelial cells promotes anoikis resistance and invasion through activation of PI3K/Akt2. Cell Death Differ. 2009 Apr;16(4):571–83. doi: 10.1038/cdd.2008.178. [DOI] [PubMed] [Google Scholar]
- 100.Woo DK, Green PD, Santos JH, D’Souza AD, Walther Z, Martin WD, et al. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am J Pathol. 2012 Jan;180(1):24–31. doi: 10.1016/j.ajpath.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Coller HA, Khrapko K, Bodyak ND, Nekhaeva E, Herrero-Jimenez P, Thilly WG. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet. 2001 Jun;28(2):147–50. doi: 10.1038/88859. [DOI] [PubMed] [Google Scholar]
- 102.Fan W, Lin CS, Potluri P, Procaccio V, Wallace DC. mtDNA lineage analysis of mouse L-cell lines reveals the accumulation of multiple mtDNA mutants and intermolecular recombination. Genes Dev. 2012 Feb 15;26(4):384–94. doi: 10.1101/gad.175802.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Picaud S, Kavanagh KL, Yue WW, Lee WH, Muller-Knapp S, Gileadi O, et al. Structural basis of fumarate hydratase deficiency. J Inherit Metab Dis. 2011 Jun;34(3):671–6. doi: 10.1007/s10545-011-9294-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001 Jul;69(1):49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000 Nov;26(3):268–70. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 106.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000 Feb 4;287(5454):848–51. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 107.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011 Nov;1807(11):1432–43. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 108.Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011 Sep 8;477(7363):225–8. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 109.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012 Jun 15;26(12):1326–38. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009 Dec 10;462(7274):739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010 Mar 16;17(3):225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 2009 Feb 19;360(8):813–5. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012 Mar 22;483(7390):474–8. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, et al. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012 May 10;31(19):2491–8. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007 Apr 6;129(1):111–22. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 116.Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem. 2006 Jan 27;281(4):2061–70. doi: 10.1074/jbc.M507741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014 Aug 7;8(3):754–66. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 118.Barnes LM, Moy N, Dickson AJ. Phenotypic variation during cloning procedures: analysis of the growth behavior of clonal cell lines. Biotechnol Bioeng. 2006 Jun 20;94(3):530–7. doi: 10.1002/bit.20856. [DOI] [PubMed] [Google Scholar]
- 119.Masramon L, Vendrell E, Tarafa G, Capella G, Miro R, Ribas M, et al. Genetic instability and divergence of clonal populations in colon cancer cells in vitro. J Cell Sci. 2006 Apr 15;119(Pt 8):1477–82. doi: 10.1242/jcs.02871. [DOI] [PubMed] [Google Scholar]
- 120.Torsvik A, Stieber D, Enger PO, Golebiewska A, Molven A, Svendsen A, et al. U-251 revisited: genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014 Aug;3(4):812–24. doi: 10.1002/cam4.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012 May;26(5):2175–86. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Inoue-Yamauchi A, Oda H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem Biophys Res Commun. 2012 Apr 27;421(1):81–5. doi: 10.1016/j.bbrc.2012.03.118. [DOI] [PubMed] [Google Scholar]
- 123.Arismendi-Morillo G. Electron microscopy morphology of the mitochondrial network in human cancer. Int J Biochem Cell Biol. 2009 Oct;41(10):2062–8. doi: 10.1016/j.biocel.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 124.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013 Oct;32(40):4814–24. doi: 10.1038/onc.2012.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, et al. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One. 2015;10(3):e0122308. doi: 10.1371/journal.pone.0122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015 Feb 5;57(3):537–51. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013 Oct;32(40):4748–57. doi: 10.1038/onc.2012.500. [DOI] [PubMed] [Google Scholar]
- 128.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008 Feb;14(2):193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sampey BP, Freemerman AJ, Zhang J, Kuan PF, Galanko JA, O’Connell TM, et al. Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS One. 2012;7(6):e38812. doi: 10.1371/journal.pone.0038812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Paglia G, Stocchero M, Cacciatore S, Lai S, Angel P, Alam MT, et al. Unbiased Metabolomic Investigation of Alzheimer’s Disease Brain Points to Dysregulation of Mitochondrial Aspartate Metabolism. J Proteome Res. 2016 Feb 5;15(2):608–18. doi: 10.1021/acs.jproteome.5b01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006 Aug 7;25(34):4675–82. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 132.Magda D, Lecane P, Prescott J, Thiemann P, Ma X, Dranchak PK, et al. mtDNA depletion confers specific gene expression profiles in human cells grown in culture and in xenograft. BMC Genomics. 2008;9:521. doi: 10.1186/1471-2164-9-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Naito A, Cook CC, Mizumachi T, Wang M, Xie CH, Evans TT, et al. Progressive tumor features accompany epithelial-mesenchymal transition induced in mitochondrial DNA-depleted cells. Cancer Sci. 2008 Aug;99(8):1584–8. doi: 10.1111/j.1349-7006.2008.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Oda T, Maeda Y. Multiple Conformations of F-actin. Structure. 2010 Jul 14;18(7):761–7. doi: 10.1016/j.str.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 135.Hild G, Bugyi B, Nyitrai M. Conformational dynamics of actin: effectors and implications for biological function. Cytoskeleton (Hoboken) 2010 Oct;67(10):609–29. doi: 10.1002/cm.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Uyeda TQ, Iwadate Y, Umeki N, Nagasaki A, Yumura S. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS One. 2011;6(10):e26200. doi: 10.1371/journal.pone.0026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McGough A, Pope B, Chiu W, Weeds A. Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol. 1997 Aug 25;138(4):771–81. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011 Sep;18(9):1470–7. doi: 10.1038/cdd.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lobert VH, Stenmark H. Cell polarity and migration: emerging role for the endosomal sorting machinery. Physiology (Bethesda) 2011 Jun;26(3):171–80. doi: 10.1152/physiol.00054.2010. [DOI] [PubMed] [Google Scholar]
- 140.Wodarz A, Nathke I. Cell polarity in development and cancer. Nat Cell Biol. 2007 Sep;9(9):1016–24. doi: 10.1038/ncb433. [DOI] [PubMed] [Google Scholar]
- 141.van RJ, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci. 2009 Feb 1;122(Pt 3):305–11. doi: 10.1242/jcs.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007 May;1773(5):642–52. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lorente G, Syriani E, Morales M. Actin filaments at the leading edge of cancer cells are characterized by a high mobile fraction and turnover regulation by profilin I. PLoS One. 2014;9(1):e85817. doi: 10.1371/journal.pone.0085817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014;4:153. doi: 10.3389/fonc.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, et al. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc. 2012 Feb;7(2):235–46. doi: 10.1038/nprot.2011.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dang CV. Links between metabolism and cancer. Genes Dev. 2012 May 1;26(9):877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, Vander Heiden MG, Stephanopoulos G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016;4:16. doi: 10.1186/s40170-016-0156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guantes R, Rastrojo A, Neves R, Lima A, Aguado B, Iborra FJ. Global variability in gene expression and alternative splicing is modulated by mitochondrial content. Genome Res. 2015 May;25(5):633–44. doi: 10.1101/gr.178426.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell. 2016 Jan 21;61(2):199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016 May 2;126(5):1834–56. doi: 10.1172/JCI82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012 Aug 31;337(6098):1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tanaka K, Whelan KA, Chandramouleeswaran PM, Kagawa S, Rustgi SL, Noguchi C, et al. ALDH2 modulates autophagy flux to regulate acetaldehyde-mediated toxicity thresholds. Am J Cancer Res. 2016;6(4):781–96. [PMC free article] [PubMed] [Google Scholar]
- 153.Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem. 2010 Apr 15;82(8):3212–21. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.