

Abstract
Current advances in systems biology suggest a new change of paradigm reinforcing the holistic nature of the drug discovery process. According to the principles of systems biology, a simple drug perturbing a network of targets can trigger complex reactions. Therefore, it is possible to connect initial events with final outcomes and consequently prioritize those events, leading to a desired effect. Here, we introduce a new concept, ‘Systemic Chemogenomics/Quantitative Structure—Activity Relationship (QSAR)’. To elaborate on the concept, relevant information surrounding it is addressed. The concept is challenged by implementing a systemic QSAR approach for phenotypic virtual screening (VS) of candidate ligands acting as neuroprotective agents in Parkinson’s disease (PD). The results support the suitability of the approach for the phenotypic prioritization of drug candidates.
‘It has been said that something so small as the flutter of a butterfly’s wing can ultimately cause a typhoon halfway around the world.’ The Butterfly Effect, 2004
Keywords: butterfly effect, drug discovery, Parkinson’s disease, phenotypic virtual screening, systemic chemogenomics/QSAR
Introduction
There have been several parallelisms surrounding the different paradigms of drug discovery. All these parallelisms capture in some way the essence of the underlying phenomenon that guides the drug discovery process. The underlying principle of every paradigm today is the ‘lock-and-key’ model proposed by Fisher [1], which explains the way a ligand (small molecule) binds to, and modulates, a target (protein enzyme or receptor). This concept has been further refined in terms of the flexibility of the ligand—target complex with the theory of ‘induced fit’ [2].
Since its inception, particularly when there was (apparently) no ‘rational’ approach, the drug discovery process has been compared to finding ‘a needle in a haystack’, a parallelism that unfortunately remains valid. Initially, phenotypes obtained after the administration of several candidate compounds in animal models were used to establish a SAR that ultimately determined the course of further trial-and-error experiments. The eminently random nature of such a systems-based chemocentric drug discovery approach [3], which did not result in outstanding productivity, is not significantly different from that used today [4]. Historically, the transition from a well-established drug discovery paradigm to a novel one has been guided mainly by productivity or efficiency criteria, with the hope of increasing them by rethinking the way a drug ‘really’ works.
The accelerated progress of molecular biology and genetics research made it possible to have a mechanistic view of drug action, leading to Paul Ehrlich’s ‘magic bullet’: drugs that go straight to their intended cell structural targets [5]. The hope for improved productivity based on a mechanistic understanding of a highly specific targeted drug action resulted in this target-based paradigm [3] dominating the drug discovery process until very recently; however, after decades of drug discovery, no significant improvements in productivity have been seen. On the contrary, the pharmaceutical industry is currently in a deep productivity crisis [4,6]. All of this is a complicated way of saying something simple: we still lack a complete understanding of biological systems (and probably never will, because of their dynamic nature), and most of the low-hanging fruits have already been picked.
Current advances in systems biology suggest the adoption of a paradigm that incorporates the multiparametric and holistic nature of the drug discovery process [4]. Several concepts have recently been proposed to adapt the current knowledge to a new paradigm that, as with its predecessors, will have to be challenged, hopefully to prevail. Focusing on the importance of dealing with all the determinant parameters of a drug (such as potency, bioavailability, and safety) in a parallel manner rather than using the traditional sequential approach, Lusher et al. [7] compared the drug discovery process to ‘solving a Rubik’s cube’. Here, each face represents a required parameter and changing one face will affect another face, perhaps detrimentally. Therefore, solving the puzzle might require sacrificing a completed face; that is, taking a local step back, to make a global step forward. In addition, Medina-Franco recently conceptualized the need for a multitarget approach as finding ‘master keys’, referring to compounds that favorably interact with multiple targets (i.e., operate a set of desired locks to gain access to the expected clinical effects), but not with ‘any’ target (that would probably lead to adverse effects) [8].
The principles of systems biology also indicate that an apparently simple drug acting on a specific target (or a set of targets) can trigger complex reactions somewhere else. Such sensitivity to small events is widely known as the ‘butterfly effect’, which is an integral feature of all complex, interconnected systems. This term was coined by Lorenz in 1972 (based on a previous simple insight extracted from a rather fortuitous experiment on a computer program to simulate weather) to describe how small events can resonate through complex networks and systems to cause major effects [9,10]. This concept can be adapted to an emerging drug discovery paradigm relying heavily on the network behavior of biological systems, if the main conclusion implicit in the original research leading to the concept (‘when small imprecisions matter greatly, the world is radically unpredictable’) does not discourage us.
Chemogenomics, emerging as a discipline to systematically study the relations between biological and chemical space [11], has a key role in this endeavor. Much progress has been made in chemogenomics, but there are still major challenges [12,13]. Accordingly, if we focus our efforts on at least roughly understanding which elements are perturbed after the initial event until the final major effect and how they are related or interconnected, there is a possibility of connecting initial events with final outcomes and consequently establishing a system to prioritize those events leading to a desired effect. Here, we introduce a new concept that can be parallelized with the aforementioned ‘butterfly effect’: Systemic Chemogenomics/QSAR. Terms such as ‘systems approach’, ‘network approach’, or ‘system-wide approach’ are well established in the drug discovery arena. However, here we resort to ‘systemic’, which is a less common term, but one that better reflects in medical terms an effect on the whole body.
To introduce the concept, the relevant information and knowledge surrounding it is addressed. Next, the concept is challenged by the introduction of a systemic QSAR approach for phenotypic VS. As a case study, the approach is applied to the prioritization of candidate ligands acting as neuroprotective agents in PD.
On the need for a systemic approach in drug discovery
If we aim to induce an effect on a whole system (a systemic effect), targeting a single, specific element of that system, regardless of the other elements and their interrelationships, is not an option [14]. This is the essence of the proven inefficiency of drug discovery approaches based on the magic bullet paradigm, with its own reductionist nature.
The plethora of evidence collected today indicates that most pathologies, especially complex diseases, have a polygenic nature involving the deregulation of complex networks of proteins [15]. By contrast, the inherent redundancy and robustness of biological networks and pathways [16] imply that targeting a single ‘disease-modifying’ gene might not be enough to produce the desired therapeutic effect [17,18]. Instead, the strategy should be to perturb disease-associated network states rather than individual proteins alone [19,20].
Therefore, we have two facts that appear to lead to a dead end: (i) no matter how much we know about a single specific disease-related target, it will not necessarily translate into a desired phenotype if targeted; and (ii) our currently immature chemogenomics knowledge does not allow us to fully understand the network of interconnections present in complex biological systems. Faced with the situation of making a choice between two opposite extreme solutions (a fully understood molecular biology centric reductionist approach versus a poorly understood chemogenomic centric holistic approach), we might be forced to find a solution that leverages a situation half way from each extreme. Such an intermediate solution to the standing challenge would be to encode what is happening in the whole system rather than fully understand it. Several key elements need to be considered to find such a solution.
Multitarget drugs: from systems biology to network pharmacology
The first element to be aware of is the need to consider an approach based on the identification of multitargeted drugs to increase the probabilities of inducing the desired phenotype or therapeutic effect [21]. Multitarget drugs are compounds that are prospectively and intentionally designed to specifically interact with multiple targets involved in a disease state [22]. These drugs can act on targets in a single common pathway as well as on targets involved in different pathways. Here, we refer to multitarget drugs in the broader sense, as acting in any of the two forms described. Among the many facts supporting multitarget drugs, the most obvious is the underlying complexity of multifactorial diseases, which shows that simple drugs do not cure complex diseases [23]. Consequently, the application of a multitarget approach to the discovery of drug candidates for complex diseases, such as cancer or central nervous system pathologies, has experienced exponential growth over the past few years [24]. However, although the rational design of such compounds is conceptually attractive, in practice it is a challenging task.
The rationale behind the efficiency of targeting multiple targets in drug discovery relies on the network-like nature of complex systems [25]. System biology analyses have shown little effect on disease networks after the deletion of individual protein nodes, suggesting that targeting multiple proteins is required to perturb robust disease phenotypes [25,26]. This fact is reflected in systems that are not only resilient to the random deletion of any one protein (node), but also critically dependent on a few highly connected hubs (highly connected essential protein nodes). This resilience is biologically explained by alternative compensatory signaling routes that allow a reorganization of the disease network, compensating a single initial perturbation [27]. These observations indicate that the inhibition of a single protein target in complex diseases might not be sufficient to induce lasting therapeutic effects because compensatory pathways might become activated to keep the initial disease phenotype.
Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops clearly illustrates this phenomenon [28]. For example, the inhibition of kinases by targeted drugs is evaded by compensatory changes in the signaling pathways of treated cancer cells [29]. Here, the signaling pathway activity of cancer cells inhibited by the targeted drug is restored or activated by alternative signaling components that elicit the same phenotypic consequences as the original pathway. Other illustrative examples of signaling crosstalk and drug-mediated activation of compensatory pathways are provided in [29].
These findings constitute the foundation of the network pharmacology [30] and polypharmacology [15] paradigms in drug discovery. Strictly speaking, polypharmacology refers to molecules that bind to different molecular targets. Although it is mostly associated with positive outcomes, the affinity towards multiple targets also implies serious safety concerns because of possible off-target effects. Polypharmacy or the combination of drugs is a concept related to polypharmacology. It refers to prescribing either many drugs (appropriately) or too many drugs (inappropriately). Combinations of drugs could improve the clinical efficacy and/or prevent resistance issues associated with treatments based on single agents. However, similar to polypharmacology, this approach can often lead to increased risks and adverse effects from medication [31].
This dual face of polypharmacology and polypharmacy highlights the need for ‘master key compounds’ (as defined above) rather than simply looking for multitarget ligands [32]. In this sense, the application of safety panels can have a key role in segregating off-target effects and consequently in prioritizing successful master key compounds. Such safety panels are designed to detect unintended interactions of candidate molecules with molecular targets associated with serious adverse drug reactions, which are known as ‘antitargets’. The minimal panels of common antitargets used by four major pharmaceutical companies (AstraZeneca, GlaxoSmithKline, Novartis, and Pfizer) are presented and illustrated in [33], with examples of their impact on the drug discovery process.
From phenotypic screening and back again
In general terms, phenotypic screening is a semiempirical, system-based drug discovery approach relying only on understanding the biology related to the disease. Contrary to the target-based discovery approach, it does not rely on a clear understanding of the mechanism of action [3]. The eminent trial-and-error approach of the early years of drug discovery was flawed by its random nature. However, this phenotype centric approach granted a productivity that was not very different from the target-based approach that has dominated the drug discovery scene for decades [34]. This could explain the resurgence of phenotypic screening in drug discovery [35].
Although the target-based discovery approach has led to the generation of new molecular entities, phenotypic screening has had a significant role in the discovery of first-in-class drugs [3,34]. Several reasons support the current and future key role of phenotypic screening [3]: (i) it has guided the discovery of drug leads and clinical candidates that are more likely to have relevant mechanisms of action [34]; (ii) it has the potential to accelerate the development of important pharmacological tools to study new biology [36,37]; and (iii) it holds the promise to uncover new therapeutic principles and molecular pathways of currently untreatable diseases [38,39].
By contrast, the integration of chemogenomics data and tools for the analysis of phenotypic screening data has recently been highlighted as an effective approach to uncovering new insights into small-molecule modes of action. The work of Bender and colleagues [40,41] is representative of current efforts in this direction. They propose the integration of information pertaining to the chemical structure, biological targets, and pathways to obtain phenotypically relevant bioactivity profiles. From these profiles, further biological insights into the system modulated or the differences in the compound mode of action that explain different phenotypes can be obtained. Previously, Jenkins et al. [42] proposed the integration of high-content screening data with ligand—target prediction for similar purposes.
However, all these facts supporting phenotypic screening should not be understood as a call to move back to the classical, system-based chemocentric approach to drug discovery. In the current context, phenotypic screening should be considered as a logical evolution of the current target-based approaches. Consequently, it is important to recognize it as a new discipline that needs new technologies and methods [3].
A fundamental challenge for phenotypic screening will be the prioritization of promising compounds from current large chemical libraries, typically contaminated with unselective or toxic compounds, substances with unwanted mechanisms of action, or false positives [3]. Contrary to the target-based approach, no hypothesis drives the screening of such chemical libraries and, consequently, the randomness of the screening process hampers the effective selection of interesting compounds. Therefore, it is important to assume that new methodologies and approaches will be required to improve the success rate of phenotypic screens.
The take-home message is simple: we need to go back to phenotypic screening but incorporate all the current knowledge to eliminate as much as possible the randomness inherent to the screening process. This process could imply screening phenotypically in an efficient and effective manner in combination with target-based approaches.
Phenotypic VS
At this point of the discussion on the need for a systemic approach in drug discovery, a fundamental limitation needs to be addressed: the inherent random nature of biological screening approaches. In this sense, computers and computer scientists have provided the solution to overcome such a bottleneck. Specifically, chemoinformatics has been a major contributor fueling the development of VS approaches and methodologies. However, to date (as far as we know), no computer-aided approach or methodology connects the two core concepts of a systemic approach and VS.
The problem connecting the two concepts finds its roots in the nature (target-based) of the approach dominating the drug discovery process at the time when the chemoinformatics approaches that laid the basis for the development of VS tools emerged. That is, QSAR and related chemoinformatics approaches with predictive capabilities arose as promising techniques to accelerate a drug discovery process guided by a target-based approach. Consequently, in the context of drug discovery, computational approaches were thought of as virtual (informatics) surrogates of target-based assays conducted in the wet lab. Additionally, the VS methodologies generated are usually fed and benchmarked with data from target-based assays [43]. Under these conditions, it is unlikely to depart from a target-based paradigm.
However, the current landscape is enriched with new and valuable systemic information stored in massive and publicly available ‘omics databases (i.e., genomic, proteomic, and chemogenomic) [44] and web servers [45], which favor the emergence of holistic or systemic computational approaches supporting drug discovery. In this context, phenotypic VS can be defined in the same terms as current VS [46,47], as a prioritization tool, but focused on the expected therapeutic response (the phenotype) of the candidate ligand instead of its binding or inhibitory capacity over a therapeutic target.
The elements essential to integrating a systemic approach for phenotypic VS rely on current advances in computational system chemical biology (CSCB) [48]. CSCB is a concept introduced by Oprea that refers to the use of computational resources to address the information available at the interface between chemical biology and systems biology [49]. According to Oprea, through CSCB, ‘scientists will be able to start addressing, in an integrated simulation environment, questions that make the best use of our ever-growing chemical and biological data repositories at the system-wide level’. It allows us to develop ‘an integrated in silico pharmacology/systems biology continuum that embeds drug—target—clinical outcome triplets’, which is essentially what is needed to develop a systemic chemogenomics/QSAR approach for phenotypic VS.
Systemic chemogenomics: the butterfly effect behind phenotypic VS
The concept we are introducing here departs from the classical target-based approach but does not necessarily neglect it. We base our proposal on the established fact that small molecules acting on a single or multiple targets are likely to produce direct and/or indirect cascading effects throughout the physiological system. Such cascading effects come from the fact that the body comprises a meta-network of targets interacting with each other to regulate complex biological processes [50]. Therefore, systemic chemogenomics or systemic QSAR can be understood as an approach directed to prioritize ‘multiple magic bullets’ [5] inducing a desired phenotype. Such a concept, although system based, is in accordance with Ehrlich’s magic bullet concept.
Let us elaborate on this using sunitinib as an example; this is a selective multikinase inhibitor that acts as an antiangiogenic agent for the treatment of solid tumors. Attending to the multifactorial nature of cancer and the role of angiogenesis in tumor development, multitargeted inhibitors, such as sunitinib, have been designed to act over several receptor tyrosine kinases involved in neovascularization [51,52]. It is known that, after the natural ligand of receptor tyrosine kinases binds to the extracellular domains, the intracellular subunits undergo transautophosphorylation, which leads to receptor activation and the phosphorylation of downstream substrates. Multikinase inhibitors, such as sunitinib, are ATP-competitive inhibitors that block the ATP-binding site within the kinase domain, thereby preventing phosphorylation of the receptor and its downstream targets [53,54]. Therefore, the initial inhibition of multiple targets induces a downstream cascading effect ending with the desired phenotype comprising the impairment of angiogenesis, an essential prerequisite for the sufficient supply of oxygen and nutrients, and, thus, for the growth of all solid tumors. In addition, in this context, the case of imatinib was recently discussed by Maggiora as a prototypical example of targeted drug therapy [55].
This is a simplified illustration of a phenomenon that Lorenz introduced as the ‘butterfly effect’ [10]. In his seminal work, Lorenz noted that the innumerable interconnections of nature mean that the flap of the wings of a butterfly could cause a tornado elsewhere in the world or, for all we know, prevent one. The work set the basis for the analogy giving rise to the concept proposed in this work. We need to consider that there are many butterflies, but not just any one could cause a tornado, just like not any molecule in the vast chemical space [56] can induce a desired phenotype or therapeutic response. Additionally, the current biological knowledge falls short in fully explaining the complex and entangled meta-network of targets interacting with each other to regulate complex biological processes in the whole body (i.e., the ‘innumerable interconnections of nature’ noted by Lorenz). Accordingly, under current conditions, it is more feasible to apply the current system chemical biology knowledge and tools to codify such interconnections between initial states (the flap of the wings of a butterfly or the target binding of a ligand) and final outcomes (tornado or desired phenotype) rather than trying to fully understand it (Figure 1).
Figure 1.
Schematic representation of systemic chemogenomics as the butterfly effect behind phenotypic virtual screening in drug discovery.
This concept has been simplified and adapted to drug discovery: by trying to encode what is going on in the whole system between the first binding(s) of a ligand to its target(s) and the final phenotype (expected therapeutic response) in terms of biological processes or any other biologically relevant information source, it is possible to use such information for the prioritization of those ligands inducing the desired phenotype. Therefore, in light of the network behavior of complex biological systems and in terms of the butterfly effect, this new drug discovery paradigm can be compared with chasing butterflies. However, not any butterfly, but only those whose flutter can ultimately cause a typhoon halfway around the world, as the original idea suggested [10].
Overall outline of the systemic chemogenomics/QSAR approach
A systemic chemogenomics/QSAR approach for phenotypic VS is outlined by the following sequence of integrated steps (schematized in Figure S1 of the supplementary information online).
Targeting the disease and a measurable therapeutic phenotype. The first step to establishing a systemic QSAR methodology for the phenotypic prioritization of candidate ligands is to define both the target disease and a measurable phenotype of the intended therapeutic effect. In this work, for example, we addressed PD by targeting neuroprotection as the disease-modifying measurable phenotype.
Collecting ligands representative of the intended phenotype. A proper design of the experiment is then required to ensure the efficacy and reliability of the final result. For this, the first element to consider is the source of information used to collect the ligands representative of the intended phenotype. For PD, for example, those compounds successfully evaluated in a clinical trial for neuroprotective action in PD in Phase II or higher are appropriate starting points. In this way, we grant that the selected compounds induce the desired phenotype (neuroprotection in PD) because the main goal of Phase II clinical trials is to evaluate the therapeutic efficacy of the candidate [57].
Setting the disease-relevant chemogenomic space. Once the ligands representative of the targeted phenotype have been selected, we take advantage of currently available target-based assays. We do so by collecting information on the gene or protein targets reported to be involved in the therapeutic mode of action (i.e., the molecular mechanism of neuroprotection) of each ligand. The set of collected genes or proteins targets constitutes a potentially neuroprotective target space that will be used to collect the associated potentially neuroprotective ligand space. The latter can be achieved by mining publicly available chemogenomics databases [44], such as ChEMBL [58] or BindingBD [59]. The significance of the collected ligand space will rely heavily on the appropriate application of several criteria that describe the ligand—target interaction. Among these criteria are the type of activity measure (i.e., IC50, Ki, etc.), the type of binding assay (i.e., agonistic or antagonistic action) or the cutoff used to consider a ligand—target interaction as significant. In this way, we collect a chemogenomic space that is relevant to the targeted disease phenotype and that comprises information on the ligand—target interactions associated with it. The collected chemogenomic space is further enriched with ligands bound to the key gene or protein targets involved in the molecular interactions and reaction networks included in the known disease pathways, as reported in databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) [60] or REACTOME [61]. The joint result is a disease-relevant chemogenomic space.
Target fishing of the ligands included in the disease-relevant chemogenomic space. This step is crucial because it is responsible for the addition of systemic information to the disease-relevant chemogenomic space. If we intended to capture the systemic effect of the ligands included in the disease-relevant chemogenomic space, the more realistic approach would be to predict all possible gene or protein targets to which each ligand binds. Although our currently insufficient chemogenomic knowledge prevents us from fully understanding every possible ligand—target interaction, it is sufficient to know that ligand—target interactions induce direct and/or indirect cascading effects throughout the physiological system [50]. Therefore, we can predict ligand—target interactions to identify the set of targets potentially involved in the biological processes perturbed by a ligand at a system level, although we cannot be 100% certain about these interactions. Several approaches for the prediction of ligand—target interactions or target fishing have been reported [62–64]. The foundations, advantages, and limitations of representative target-fishing approaches are reviewed in [65]. Regardless of the theory behind each approach, when choosing a target fishing tool, the priority must be granted to its predictive capability. After efficient target fishing over the ligands in the disease-relevant chemogenomic space, we can say that we have a systemic picture of this space. Therefore, at this point, we have set the conditions to encode the systemic effect of the ligands in a disease-relevant chemogenomic space. The problem now is how to encode such information.
Defining the ligand gene ontology systemic fingerprint (LGOSFP). There are many possible sources of information to encode the systemic effect of ligands. Such sources range from well-established chemical descriptors [66] to emerging descriptions based on biological information [67]. Given their biological relevance, we propose the use of gene ontology (GO) terms [68,69]. The problem then is how to connect a ligand to a set of GO terms. For that purpose, we can take advantage of the disease-relevant chemogenomic space, which includes the association of each ligand with experimentally determined and/or predicted gene targets. By codifying each ligand through the GO terms associated with their interacting targets, it is possible to access a systemic ‘picture’ or ‘fingerprint’ of the biological process (BP), molecular functions (MF), and cellular components (CC) perturbed by the ligand through its interaction with the corresponding network of gene or protein targets. In this way, what we call a ‘LGOSFP’ is obtained, which is used to describe the systemic effect of ligands in a disease-relevant chemogenomic space.
Defining the phenotype-positive and phenotype-negative classes. After codifying our disease-relevant chemogenomic space by using a LGOSFP, it is time to design a classification problem to extract the LGOSFP terms that allow significant discrimination of ligands with a high probability to induce the desired phenotype. For this, we are restricted to the chemogenomic space relevant to the targeted disease phenotype (only ligands with confirmed interactions with targets associated with the disease phenotype are considered in this step). By using this subset of the disease-relevant chemogenomic space, an appropriate cutoff based on the binding-affinity measures reported for the ligand—target interaction (i.e., IC50, Ki, etc.) needs to be applied to split it into two classes. The first class comprises all ligands that significantly interact with at least one of the gene or protein targets involved in the therapeutic mode of action associated with the desired phenotype (the phenotype-positive class). The second class comprises the rest of compounds with no significant interaction with any of the targets associated with the desired phenotype (the phenotype-negative class).
Phenotypic VS by using machine learning-based QSAR models. Once we have established a significant classification problem in the terms described above, the remaining steps can be resumed in a classical QSAR-based VS methodology [70–72]. Here, machine learning is central in providing biologically relevant and predictive classification models, which will be used as VS tools [73]. For this, we need to set a VS benchmark set comprising a few ligands confirmed to induce the desired therapeutic phenotype (i.e., neuroprotection in PD) dispersed in a significantly larger set of ligands with no significant interaction with any of the targets associated with the desired phenotype. In terms of VS, the first small set can be labeled as the phenotypic ligand set and must comprise those initial candidate drugs successfully evaluated in Phase II (or higher) clinical trials, which should never be used in model training. The remaining larger set can be labeled as the phenotypic decoys set and must comprise those compounds from the phenotype-negative class never used for model training. The phenotypic decoy set can also be generated by standard ligand decoy generation tools [74,75], but we recommend extracting it from the phenotype-negative class, which is closer to a confirmed inactive set [46], whenever a sufficiently large set can be collected from this source. Even so, the risk of false negative contamination is significant. The appropriate selection of the cut-off to set the classes will also be directly related to this risk.
Phenotypic VS validation. Finally, standard metrics used in the QSAR and VS arena can be used to evaluate both the generalization ability of the classifiers obtained [70] and their overall VS performance [47], especially the early recognition ability. The VS performance of the methodology will be the ultimate measure of its success in prioritizing candidate ligands with a desired therapeutic phenotype.
Proof of concept
We challenge the potential of the approach we are proposing for phenotypic VS by applying it to the prioritization of candidate ligands acting as neuroprotective agents in PD. It is important to highlight that, because we are conducting a proof-of-concept experiment, the most basic approaches will be used to provide what should be expected from the approach under the worst-case scenario. In this way, if more advanced approaches are used, improved results can be expected.
First, our target disease is PD, a multifactorial and multigenic disease of unknown etiology, which constitutes a perfect fit to a complex disease [76] that can benefit from a systemic approach [77]. The selected phenotype was neuroprotection because it is the most realistic and studied disease-modifying therapeutic mode of action in PD [77]. We inspected the literature to locate a reliable source that could provide ligands fitting the selected disease and phenotype. Consequently, from the drug candidates reported in [57], we collected a set of 12 of those successfully evaluated in Phase II and Phase III clinical trials as neuroprotective agents in PD. This set constitutes the starting point of our proof-of-concept exercise. Tables 1 and 2 provide information on the 12 drug candidates selected and the set of therapeutic targets associated with their mechanism of neuroprotective action, respectively. The corresponding molecular structure representations were used as provided in [57].
Table 1.
Neuroprotective candidate drugs
| Compound IDa | Clinical Trial IDb |
|---|---|
| GM1 ganglioside (sygen) | NCT00037830 |
| AFQ056 (mavoglurant) | NCT01092065 |
| BIIB014 | NCT00442780 |
| SR57667B (paliroden) | NCT00220272 |
| EMD 1195686 (safinamide) | NCT00865579 |
| ADS-5102 (nurelin) | NCT01397422 |
| IPX066 (carbidopa) | NCT00880620 |
| SCH 420814 (preladenant) | NCT01215227 |
| Caffeine | NCT01738178 |
| Zonisamide | NCT01766128 |
| E2007 (perampanel) | NCT00360308 |
| Rasagiline | NCT01187888 |
From [57].
Table 2.
Therapeutic targets associated with the neuroprotective candidate drugs
| Gene symbol | Gene name | Gene ID | ChEMBL ID | Ligand mode of action |
|---|---|---|---|---|
| ACHE | Acetylcholinesterase | 43 | CHEMBL220 | Inhibitor |
| ADORA2A | Adenosine A2a Receptor | 135 | CHEMBL251 | Antagonist |
| DRD2 | Dopamine Receptor D2 | 1813 | CHEMBL217 | Agonist |
| DRD3 | Dopamine Receptor D3 | 1814 | CHEMBL234 | Agonist |
| GRM5 | Glutamate Metabotropic Receptor 5 | 2915 | CHEMBL3227 | Antagonist |
| MAOB | Monoamine Oxidase B | 4129 | CHEMBL2039 | Inhibitor |
| PPARG | Peroxisome Proliferator Activated Receptor Gamma | 5468 | CHEMBL235 | Binder |
| CACNA | Calcium Voltage-Gated Channel | CHEMBL4478; CHEMBL1940; CHEMBL2095229; CHEMBL4641; CHEMBL1859 |
Blocker | |
| CACNA1B | Calcium Voltage-Gated Channel Subunit Alpha1 B | 774 | ||
| CACNA1C | Calcium Voltage-Gated Channel Subunit Alpha1 C | 775 | ||
| CACNA1F | Calcium Voltage-Gated Channel Subunit Alpha1 F | 778 | ||
| CACNA1G | Calcium Voltage-Gated Channel Subunit Alpha1 G | 8913 | ||
| CACNA1H | Calcium Voltage-Gated Channel Subunit Alpha1 H | 8912 | ||
| GRIA | Glutamate Ionotropic Receptor AMPA Type | CHEMBL2009; CHEMBL4016; CHEMBL2096670; CHEMBL3190 |
Antagonist | |
| GRIA1 | Glutamate Ionotropic Receptor AMPA Type Subunit 1 | 2890 | ||
| GRIA2 | Glutamate Ionotropic Receptor AMPA Type Subunit 2 | 2891 | ||
| GRIA3 | Glutamate Ionotropic Receptor AMPA Type Subunit 3 | 2892 | ||
| GRIA4 | Glutamate Ionotropic Receptor AMPA Type Subunit 4 | 2893 | ||
| GRIN | Glutamate Ionotropic Receptor NMDA Type | CHEMBL2094124; CHEMBL1907603; CHEMBL1907604 |
Antagonist | |
| GRIN1 | Glutamate Ionotropic Receptor NMDA Type Subunit 1 | 2902 | ||
| GRIN2A | Glutamate Ionotropic Receptor NMDA Type Subunit 2A | 2903 | ||
| GRIN2B | Glutamate Ionotropic Receptor NMDA Type Subunit 2B | 2904 | ||
| GRIN2C | Glutamate Ionotropic Receptor NMDA Type Subunit 2C | 2905 | ||
| GRIN2D | Glutamate Ionotropic Receptor NMDA Type Subunit 2D | 2906 | ||
| GRIN3A | Glutamate Ionotropic Receptor NMDA Type Subunit 3A | 116443 | ||
| GRIN3B | Glutamate Ionotropic Receptor NMDA Type Subunit 3B | 116444 |
As described above, the potentially neuroprotective target space shown in Table 2 is used to set the potentially neuroprotective ligand space that will later be used to set the classification QSAR problem. The ligands associated with each of the targets reported in Table 2 were extracted from ChEMBL (https://www.ebi.ac.uk/chembl/). Only ligands with reported IC50, EC50, Ki, or Kd binding data and the corresponding mode of action (agonist, antagonist, inhibitor, or binder) were considered. When the same ligand was reported with more than one assay, the geometric mean was used as binding data. Duplicates were then removed by using the EdiSDF program (http://edisdf.software.informer.com/5.0/). The collected data comprised 16 867 ligands assayed for at least one of the targets reported in Table 2. Ligands bound to at least one of the targets at a concentration lower than or equal to 1 MM comprised the phenotype-positive class (Class_1: 9094 potentially neuroprotective ligands), and the rest were labeled as the phenotype-negative class (Class_0: 7773 ligands with no neuroprotective potential). The full set of ligands comprising the potentially neuroprotective ligand space is provided in the supplementary information online. The same procedure was applied to an additional set of gene or protein targets involved in the PD pathway reported in KEGG (http://www.genome.jp/dbget-bin/www_bget?pathway1hsa05012). The list of additional targets is provided in Table S1 of the supplementary information online. A set of 11 280 unique ligands comprising the PD pathway ligand space was collected and added to the previously collected potentially neuroprotective ligand space to finally conform our PD-relevant chemogenomics space of 28 147 ligands and their respective associations to neuroprotective and PD pathway-associated targets.
A target-fishing process over the 28 147 ligands collected in the PD-relevant chemogenomics space was then conducted. For this, version 19 of the ChEMBL database was filtered to obtain a curated subset comprising only ligands with at least a weak interaction with the corresponding target. Recall that, when considering a multitarget approach, weak interactions matter [22]. As before, when the same ligand was reported in more than one assay, the geometric mean was used. Only human targets (Homo sapiens) with ten or more ligands binding at a concentration ≤ 10 MM were considered. Duplicates were removed by inspecting the respective canonical SMILES. No salts or disconnected structures were considered. In these cases, only the largest fragment was used as the input structure representation. The final chemogenomic space used for target fishing comprised 1386 human targets and 317 008 ligands.
For target fishing, we implemented the approach proposed by Liu et al. [62]. Although more sophisticated approaches are available that can be used for target fishing, we used this one because of its simplicity and easy implementation. This is an approach based on ligand similarity, which assumes the similarity principle as the rationale to infer new ligand—target interactions (structurally similar ligands should bind to the same target). The chemical structure was encoded by using the extended chemical fingerprints implemented in the rcdk package [78] for R [79] using the Tanimoto coefficient (Tc) as the molecular similarity metric. In our implementation, a query ligand was considered to bind to a database target if its average molecular similarity (based on Tc) with the top three closest ligands reported in the database for that target was >0.55 or >0.85 for the closest ligand. In a tenfold cross-validation experiment, the prediction performance of our implementation was similar to the performance reported in [62]. In this way, we were able to obtain a predicted binding profile for the 28 147 ligands in our PD-relevant chemogenomics space. The information about their interactions with neuroprotective and PD pathway-related targets was extended with predicted binding information of an additional set of 1386 human targets. This provided a systemic picture of the effect of these ligands in a network of 1386 human targets.
Once the predicted binding profile of the 28 147 ligands in the PD-relevant chemogenomics space was obtained, it was possible to derive the corresponding LGOSFPs. For this, we resorted to the GO.db package for R [80], which was used to process the GO database (http://geneontology.org/). The package is used to connect every GO term with it ancestors to avoid redundancy in the final set of GO terms collected. For this, the GO.db package was first used to extract the full set of unique GO terms (BPs, MFs, and CCs) present in the GO database. Next, the corresponding protein/gene—GO terms associations were set by using the mapping file for H. sapiens from the UNIPROT database [81]. Each protein/gene reported for H. sapiens in the UNIPROT database was then associated with a binary vector of length 11 412 (BP, 9313; MF, 1399; and CC, 700), codifying for the presence (1) or absence (0) of each GO term. From this file, a subset was extracted comprising only the GO term vectors associated with the 1386 human targets previously generated from ChEMBL. This new file was then used to connect every ligand in the collected PD-relevant chemogenomic space with the set of predicted interacting targets resulting from the target-fishing process and the corresponding vector of GO terms. An in-house R code was used to generate the final LGOSFPs. The simplest form of LGOSFP for a given ligand only codifies for the presence or absence of the 11 412 GO terms by considering a GO term as present if at least one of the targets predicted for the ligand is associated with it. This is the simplest form of LGOSFP and was the one used in our proof of concept.
As described above, the 16 867 ligands comprising the potentially neuroprotective ligand space, now described through LGOSFPs, were used to solve a traditional QSAR classification problem. After a simple random partition of training, test, and external evaluation sets (Training Class_1/Class_0, 5468/4657; Test Class_1/Class_0,: 1814/1565; External Evaluation Class_1/Class_0, 1812/1551), multiple least square support vector machine (LSSVM) base classifiers were trained to obtain the ensemble classifier that was ultimately used as a VS tool. In-house MatLab codes were used for feature selection, modeling, and genetic algorithm optimization. Given that this stage is based on standard procedures already reported [72,82,83], the details are provided in the supplementary information online.
The best ensemble classifier obtained that was later used as a VS tool was based on 12 base classifiers comprising a total of 224 unique GO terms. The accuracy, sensitivity, and specificity of the external evaluation set of this ensemble classifier were 0.70, 0.66, and 0.74, respectively. This was a significantly good performance [70] considering the complexity of the classification problem and the simplicity of the LGOSFP description used. The classification performance on the training and test sets are also provided in Table 3. Details of the classification and VS performance of the 12 base classifiers are provided in Tables S2 and S3 in the supplementary information online.
Table 3.
Classification performance of the LSSVM ensemble classifier used as a VS toola
| Acc | Se | Sp |
|---|---|---|
| Training set | ||
| 0.7510 | 0.6979 | 0.7963 |
| Test set | ||
| 0.7521 | 0.6773 | 0.7663 |
| External evaluation set | ||
| 0.7032 | 0.6634 | 0.7373 |
Abbreviatons: Acc, accuracy (overall correct classification rate); Se, sensitivity (correct classification rate for the phenotype-positve Class_1); Sp, specificity (correct classification rate for the phenotype-negative Class_0).
To evaluate the ability of the systemic QSAR approach to prioritize drug candidates with a desired therapeutic phenotype, a retrospective phenotypic VS experiment was designed. For this, we assembled a VS set including the 12 neuroprotective drug candidates collected (Table 1) dispersed in a subset of 3116 compounds of the phenotype-negative class included in the test and external evaluation sets, which were never used for training. This screening set was used to evaluate the ability of the ensemble classifier to prioritize those 12 neuroprotective drug candidates among more than 3000 compounds confirmed not to be binders of any of the neuroprotective targets reported in Table 2. For this, the aggregated scores generated by the ensemble classifier were used as ranking criterion. These scores were obtained through the arithmetic mean of the normalized rankings provided by the 12 base LSSVM classifiers.
The ordered list generated by using the aggregated scores from the LSSVM ensemble classifier was evaluated through established VS performance metrics [47]. These metrics provide a quantitative estimate of the ability of a VS tool to place the desired ligands at the top positions of the generated ordered list. To evaluate the overall VS performance, we used the area under the accumulation curve (AUAC). The local VS performance at the relevant top fractions was evaluated through the enrichment factor (EF). The most important feature of a VS tool, the early recognition ability, was evaluated through the Boltzmann-enhanced discrimination of the Receiver Operating Characteristic curve (BEDROC). The definition and practical utility of these metrics are provided in the supplementary information online.
The aggregated scores used as the ranking criterion are not unique for each compound in the screening set. This implies that the same score can be assigned to fractions of the data set where ties can happen. Although the largest possible fraction with equal scores including at least one of the 12 active ligands was not higher than 0.8 % of the whole data set, it is important to account for this small randomness when computing the VS performance metrics. For this, the tie regions, including at least one active ligand, were identified in the ordered list generated by the LSSVM ensemble classifier. Detailed information on these tie regions is provided in Table S4 in the supplementary information online. Each tie region was then subject to a random resampling of 1 000 000 repetitions, so that each repetition generated an ordered list that was used to compute the VS metrics (AUAC, EF, and BEDROC). In this way, the resulting average values are considered instead of using those from a single run of the ensemble classifier. The results of this experiment are provided in Table 4.
Table 4.
VS performance of the LSSVM ensemble classifier used as a VS tool
| Overall enrichment (AUAC) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Min. | 0.6389 | ||||||||
| Mean | 0.6395 | ||||||||
| Max. | 0.6402 | ||||||||
| Std. Dev. | 0.0003 | ||||||||
| Local enrichment (EF) | |||||||||
| Top 1 % | Top 2% | Top 3% | Top 4% | Top 5% | Top 8% | Top 10% | Top 15% | Top 20% | |
| Min. | 16.29 | 8.28 | 5.55 | 4.14 | 3.32 | 2.08 | 2.50 | 1.66 | 2.08 |
| Mean | 16.29 | 8.28 | 5.55 | 4.14 | 3.32 | 2.08 | 2.50 | 1.66 | 2.08 |
| Max. | 16.29 | 8.28 | 5.55 | 4.14 | 3.32 | 2.08 | 2.50 | 1.66 | 2.08 |
| Std. Dev. | ,0.0000000001 | ||||||||
| Early recognition (BEDROC) | |||||||||
| Top 1 % | Top 2% | Top 3% | Top 4% | Top 5% | Top 8% | Top 10% | Top 15% | Top 20% | |
| Min. | 0.0587 | 0.0993 | 0.1187 | 0.1309 | 0.1404 | 0.1652 | 0.1810 | 0.2196 | 0.2562 |
| Mean | 0.1134 | 0.1357 | 0.1455 | 0.1520 | 0.1579 | 0.1767 | 0.1903 | 0.2259 | 0.2611 |
| Max. | 0.1915 | 0.1794 | 0.1757 | 0.1752 | 0.1766 | 0.1886 | 0.1999 | 0.2324 | 0.2660 |
| Std. Dev. | 0.0287 | 0.0174 | 0.0124 | 0.0096 | 0.0079 | 0.0051 | 0.0041 | 0.0028 | 0.0021 |
The data in Table 4 show that the overall enrichment performance attained can be considered as acceptable. According to the AUAC values, the probability of ranking a drug candidate inducing a neuroprotective phenotype earlier than an inactive drug candidate was approximately 0.64, which is acceptable, but not impressive. However, the enrichment ability shown by the proposed approach was significantly better at very early fractions (top 1%) of the screened data. From the EF values, we can infer that the top 1% of the screened data would be enriched with active ligands 16 more times than what would be expected from a random selection. In this case, approximately 16% of the neuroprotective drug candidates could be identified by just filtering the top 1% of the data. From the BEDROC values, it is possible to assert that, in the worst-case scenario (minimum BEDROC values), drug candidates inducing a neuroprotective phenotype can be found in the top 1% fraction of the filtered data.
As mentioned above, the randomness of small data fractions could challenge the reliability of the inferences made from VS metrics. The standard deviation associated with each VS metric in Table 4 shows that the variability was focused mainly in the top 1% fraction and affected only BEDROC values. The best, worst, and mean accumulation curves based on the minimum, maximum, and mean rank positions obtained from the 1 000 000 repetitions are shown in Figure 2. Here, specifically in the zoom of the top 5% fraction of the data, it is possible to confirm that randomness had no significant effect, except before the top 1% fraction of the screened data. Even so, in the worst-case scenario, significant enrichment was obtained at very early fractions. The best, worst, and mean enrichment cumulative curves (based on maximum, minimum, and mean EF values, respectively) shown in Figure 3 also suggest that the highest possible screening efficiency can be attained before the top 1% of the screened data.
Figure 2.
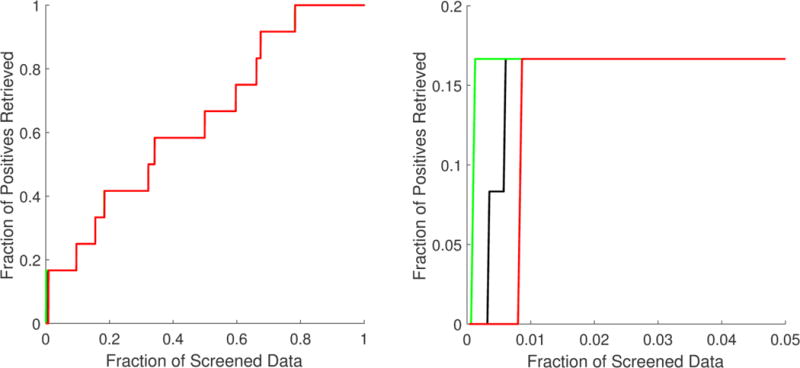
Best (green), worst (red), and mean (black) accumulation curves for the ordered list generated from the multiple least square support vector machine (LSSVM) ensemble classifier used as a virtual screening tool. A zoom of the top 5% fraction (B) is provided in addition to the curve for the full set (A).
Figure 3.
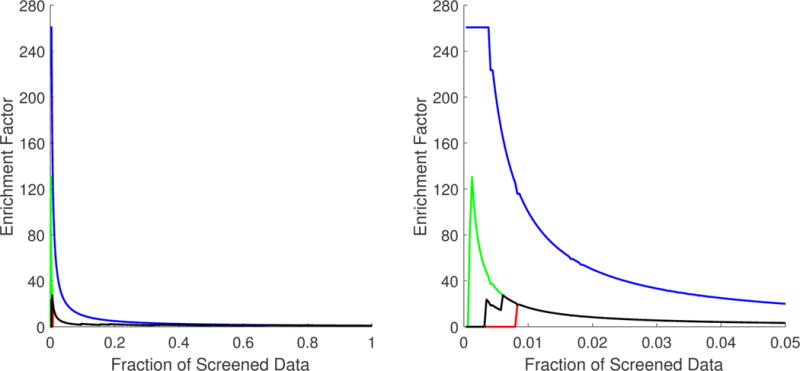
Best (green), worst (red), and mean (black) enrichment cumulative curve for the ordered list generated from the multiple least square support vector machine (LSSVM) ensemble classifier used as a virtual screening tool. A zoom of the top 5% fraction (B) is provided in addition to the curve for the full set (A). The optimal enrichment curve is provided (in blue) for comparison purposes
Even after the ghost of randomness vanished, the results obtained can be a matter of chance unless evidence of biological relevance is provided. For this, we analyzed the set of GO terms used in the LSSVM ensemble classifier to check whether this information was significantly associated with PD. A consensus score quantifying the relevance of the GO terms according to their role in the ensemble LSSVM classifier was used for this task (see the definition in the supplementary information online). The consensus scores associated with the list of 224 GO terms were then used to summarize and visualize them as a tree map by means of the Web tool REVIGO [84]. The full list of GO terms with the corresponding consensus scores is provided in the Table S5 in the supplementary information online. According to this analysis, the 224 GO terms used by the LSSVM ensemble classifier could be summarized in eight major biological processes. The tree map representation obtained is shown in Figure 4.
Figure 4.
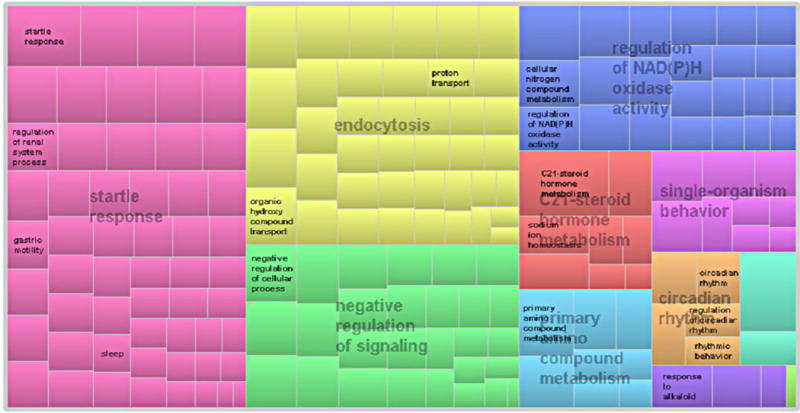
Tree map [scaled with the consensus scores associated to the Gene Ontology (GO) terms] summarizing the biological process corresponding with the GO terms used by the multiple least square support vector machine (LSSVM) ensemble classifier.
All these biological processes have been previously associated with PD to a higher or lesser extent: (i) startle response: patients with PD have attenuated habituation to startling stimuli [85], which was found to be a sensitive measure in detecting the disease [86]; (ii) endocytosis: synucleins, including a-synuclein, preferentially regulate synaptic vesicles endocytosis [87]. Strong genetic evidence links SNCA, the gene encoding α-synuclein, to PD [88]; (iii) negative regulation of signaling: the modulating role of purinergic signaling in dopaminergic neurotransmission was recently reviewed [89]. Oxidative stress-induced signaling pathways are also implicated in the pathogenesis of PD [90], supporting the neuroprotective role of antioxidant agents in PD; (iv) the regulation of NAD(P)H oxidase activity: It is known that NAD(P)H oxidase has a crucial role in neurodegeneration [91] by modulating the behavior of α-synuclein expression and aggregation in dopaminergic neurons [92]; (v) C21-steroid hormone metabolism: the sex difference in PD, with a higher susceptibility in men, suggests a modulatory effect of sex steroids in the brain. It is known that hormonal effects on enzymes in the metabolic pathway of dopamine modulate its levels. Numerous studies highlight that sex steroids, such as progesterone (a C21-steroid hormone), have neuroprotective properties against various brain injuries [93,5]; (vi) single-organism behavior: a behavioral-cognitive decline is a prominent feature of PD [94,95]; (vii) primary amino compound metabolism: neurotransmitters, such as serotonin, catecholamine, and especially dopamine, are primary amino compounds whose metabolism is largely associated with PD [96]; and (viii) circadian rhythm: a recent study suggested that the loss of the midbrain dopaminergic neurons leads to impairments of the circadian control of rest—activity rhythms [97]. This finding could explain why sleep dysfunction seen in early PD can be the result of a more fundamental pathology in the molecular clock underlying circadian rhythms [98].
Considering the process applied to associate a ligand with its corresponding LGOSFP, the data presented suggest that the biological processes controlled by the network of targets perturbed by the ligands in the PD-relevant collected ligand space are a biologically relevant systemic description that can be used to efficiently prioritize neuroprotective drug candidates. Finally, the results obtained in the experiments conducted in our proof of concept allow us to assert that systemic QSAR is a suitable approach for the prioritization of drug candidates inducing a neuroprotective phenotype.
Potential, limitations, and future directions
As noted above, the proof of concept was conducted by using the most basic approach, which implies that, at every stage of the approach, other alternatives exist with the potential of improving its performance. Among the elements with more room for improvement is the approach used to describe the ligand, which in terms of QSAR are known as ‘molecular descriptors’ [66]. The description used determines the similarity relations between ligands and constitutes the core of the QSAR paradigm. The first improvement to this approach is the use of biologically relevant information instead of the chemical information traditionally used in QSAR. The advantages of rethinking molecular similarities on the basis of biological activity have been highlighted by Petrone et al. [67]. First, because similarity relations are not established based on a description of the chemical structure, the scaffold-hopping ability (i.e., the ability to retrieve structurally diverse active ligands) is improved. The most important advantage relies on the systemic information encoded by LGOSFPs, which perfectly fits with multitarget or network-based drug discovery paradigms. In addition to GO terms, other biologically relevant descriptions can be used within the systemic QSAR approach. In this sense, the BioAssay Ontology (BAO) [99,100] proposed by Schürer et al., is an attractive alternative with the potential to provide an improved phenotypic VS performance.
Instead of only using the presence of GO terms, the information content of the LGOSFP can be improved by incorporating frequency and/or binding-affinity information. A frequency LGOSFP comprises elements defined as the ratio of targets associated with the GO term and the total number of predicted targets for a given ligand. A binding-affinity LGOSFP comprises elements resulting from weighting the presence of a GO term with the binding affinity reported for the ligand—target complex. Finally, a binding affinity-weighted frequency LGOSFP results from the product of both variants.
The content of the information of the different variants of LGOSFP is inversely related to their degree of redundancy. The latter decreases from the simplest presence LGOSFP to the binding affinity-weighted frequency version, and has a direct influence on QSAR modeling. Such redundant information contaminates the modeling data with instances with similar descriptions (LGOSFP vector) and opposite labels (opposite classes). These types of data exception are known as instances that should be misclassified (ISM) [101] in machine-learning terms or activity cliffs [102] in QSAR and drug discovery terms. The presence of such noisy instances is recognized as an important limitation for the predictive performance of similarity-based approaches [103]. Consequently, considering the presence of these types of data exception at the QSAR modeling stage will improve the final performance of the systemic QSAR approach.
Another element that can be addressed to improve the efficiency of the approach is the modeling tool applied. Given that information completeness is a distinctive mark of the system under study, gray box modeling [104] might be a suitable alternative for dealing with some of the issues associated with incomplete information. In gray box models, the input, output, and some but not all internal mechanistic information is available. These models are in sharp contrast to black box models, where the internal workings are completely unknown and only the inputs and their corresponding outputs are known, and to white box models, where the inputs, outputs, and internal working are all completely known.
The most significant drawback of the approach is its own associative nature. The reader has probably noted a recurrent use of the adverb ‘potentially’. This is because the initial set of neuroprotective drug candidates is the only space that can be confirmed to show the referred property. In the rest of the resultant spaces (neuroprotective targets or ligands), the referred property (neuroprotective) is inferred from associations established from the information stored in chemogenomics databases. It constitutes a significant source of noise, which hampers the predictive ability of the machine learning-based QSAR models derived from such space. The dependence on the availability of drugs known to treat the phenotype as well as the use of predicted ligand—target interactions to derive the LGOSFP are also major limitations of the approach. Consequently, these stages must be conducted with great caution.
The most important clue supporting the systemic QSAR approach is that a significant fraction of the neuroprotective drug candidates used as starting point could be recovered at very early fractions of the screened data after several steps of data associations (from a few neuroprotective drug candidates to a few neuroprotective targets to thousands of ligands potentially neuroprotective or not). Additionally, the fact that most (if not all) of the GO terms used in the QSAR problem for the classification of neuroprotective ligands are associated with PD not only reinforces the validity of the systemic QSAR approach, but also highlights the key role of a careful experimental design. Consider that the biological processes encoded by the GO terms used for classification are ultimately retrieved from only 12 neuroprotective drug candidates. The extended chain of complex associations applied to finally obtain the LGOSFP description would not likely end in an efficient prioritization of neuroprotective drug candidates based on a description relying on a biological process significantly associated with PD, unless a well-conceived design of the experiment was applied. This is bad news for those using informatics resources to generate tools providing automatic solutions. Systemic QSAR relies heavily on informatics and computer sciences, but to be successful, it needs to be applied in the old-fashioned way, where expertise and multidisciplinary work are central.
Finally, although the systemic chemogenomics/QSAR approach is proposed mainly for complex diseases, it is theoretically applicable to any therapeutic condition targeted in drug discovery. Its worth as a phenotypic VS tool was challenged by a proof-of-concept experiment designed to use the most basic settings. The results obtained grant further efforts in optimizing or evolving new forms of the approach. We are aware that this approach is far from being the definite solution, but we trust that these weak flutters can resonate enough to finally produce the tornado of efficiency that current drug discovery needs.
Supplementary Material
Research Highlights.
We introduce a new concept, the systemic chemogenomics/QSAR
We address all the relevant information and knowledge surrounding the concept
We propose an approach for phenotypic virtual screening
We conduct a proof-of-concept experiment providing evidences on the validity of the approach
We discuss the potential and limitations of the systemic chemogenomics/QSAR approach
Acknowledgments
This project was supported by Foundation for Science and Technology (FCT), FEDER/COMPETE (Grants UID/ QUI/00081/2013, POCI-01-0145-FEDER-006980, NORTE-01-0145-FEDER-000028), and the NCI through the NIH Common Fund. F.B. also thanks the COST action CA15135 (Multi-Target Paradigm for Innovative Ligand Identification in the Drug Discovery Process, MuTaLig) for the support. M.C-M. (Grant SFRH/BPD/90673/2012) was also supported by FCT and FEDER/COMPETE funds. S.S. acknowledges support from the grants U54CA189205 (Illuminating the Druggable Genome Knowledge Management Center, IDG-KMC) and U54HL127624 (Data Coordination and Integration Center for BD2K-LINCS, BD2K-LINCS DCIC). J.L.M-F. acknowledges support from the Universidad Nacional Autónoma de México (UNAM) through the grant Programa de Apoyo a la Investigación y el Posgrado (PAIP) 5000-9163.
Biographies

Maykel Cruz-Monteagudo
Maykel Cruz-Monteagudo is currently a postdoctoral researcher at CIQUP in the Department of Chemistry and Biochemistry, Faculty of Sciences of the University of Porto. He received his BSc in pharmaceutical sciences from the Central University of Las Villas, Cuba, in 2003; and was awarded his PhD in pharmaceutical sciences from the Faculty of Pharmacy, University of Porto, in 2010. His current research is devoted to the development and application of chemoinformatics approaches to drug discovery, focusing on the application of system chemical biology concepts to multitarget/multiobjective drug discovery. He has authored more than 40 publications published in peer-reviewed journals and two book chapters.

Stephan Schürer
Stephan Schürer is the director of drug discovery at the Center for Computational Science and associate professor in the Department of Pharmacology at the University of Miami. He was awarded his PhD in synthetic organic chemistry from the Technical University of Berlin and studied chemistry at Humboldt University-Berlin and the University of California, Berkeley. The research focus of his group is in systems drug discovery. The group integrates and models small molecule—protein interactions, systems biology ‘omics’, and chemistry data to improve the translation of disease models into novel functional small molecules. Dr Schürer is a principal investigator in two national Consortia, the Library of Integrated Network-based Cellular Signatures (LINCS), which is also part of the Big Data to Knowledge (BD2K) program, and the Illuminating the Druggable Genome (IDG) project. He has authored more than 80 publications in peer-reviewed journals, six book chapters, and several patents.

Fernanda Borges
Fernanda Borges is an associate professor in the Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, and a senior researcher in CIQUP. She received her MSc and PhD in pharmaceutical sciences from the Faculty of Pharmacy, University of Porto, Portugal. Her current research is focused on medicinal chemistry, namely in the design and development of drugs to be used in the prevention and/or therapy of neurodegenerative diseases. She has authored more than 240 publications in peer-reviewed journals, 21 book chapters, and several patents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Teaser: Systemic chemogenomics/QSAR is a new concept that forms the basis for a phenotypic virtual screening approach in drug discovery.
References
- 1.Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber Dtsch Chem Ges. 1894;27:2985–2993. [Google Scholar]
- 2.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci U S A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eder J, et al. The discovery of first-in-class drugs: origins and evolution. Nat Rev DrugDiscov. 2014;13:577–587. doi: 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- 4.Mignani S, et al. Why and how have drug discovery strategies in pharma changed? What are the new mindsets? Drug Discov Today. 2016;21:239–249. doi: 10.1016/j.drudis.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 6.FitzGerald GA. Perestroika in pharma: evolution or revolution in drug development? Mt Sinai J Med. 2010;77:327–332. doi: 10.1002/msj.20198. [DOI] [PubMed] [Google Scholar]
- 7.Lusher SJ, et al. A molecular informatics view on best practice in multi-parameter compound optimization. Drug Discov Today. 2011;16:555–568. doi: 10.1016/j.drudis.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Medina-Franco JL, et al. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18:495–501. doi: 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorenz EN. Deterministic nonperiodic flow. J Atm Sci. 1963;20:130–141. [Google Scholar]
- 10.Lorenz EN. Predictability: Does the Flap of a Butterfly’s Wings in Brazil Set Off a Tornado in Texas? AAAS 1972 [Google Scholar]
- 11.Marechal E. Chemogenomics: a discipline at the crossroad of high throughput technologies, biomarker research, combinatorial chemistry, genomics, cheminformatics, bioinformatics and artificial intelligence. Comb Chem High Throughput Screen. 2008;11:583–586. doi: 10.2174/138620708785739961. [DOI] [PubMed] [Google Scholar]
- 12.Jacoby E. Chemogenomics: drug discovery’s panacea? Mol Biosyst. 2006;2:218–220. doi: 10.1039/b603004c. [DOI] [PubMed] [Google Scholar]
- 13.Medina-Franco JL, et al. The interplay between molecular modeling and chemoinformatics to characterize protein-ligand and proteinprotein interactions landscapes for drug discovery. Adv Protein Chem Struct Biol. 2014;96:1–37. doi: 10.1016/bs.apcsb.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Bruggeman FJ, et al. Introduction to systems biology. In: Baginsky S, Fernie AR, editors. Plant Systems Biology. Birkhäuser; Basel: 2007. pp. 1–19. [Google Scholar]
- 15.Anighoro A, et al. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 16.Jeong H, et al. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 19.Morphy R, Rankovic Z. Fragments, network biology and designing multiple ligands. Drug Discov Today. 2007;12:156–160. doi: 10.1016/j.drudis.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Hellerstein MK. A critique of the molecular target-based drug discovery paradigm based on principles of metabolic control: advantages of pathway-based discovery. Metab Eng. 2008;10:1–9. doi: 10.1016/j.ymben.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Morphy JR, Harris CJ. Designing Multi-Target Drugs. The Royal Society of Chemistry; 2012. [Google Scholar]
- 22.Morphy R, et al. From magic bullets to designed multiple ligands. Drug Discov Today. 2004;9:641–651. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- 23.Hornberg JJ. Simple drugs do not cure complex diseases: the need for multi-targeted drugs. In: Morphy JR, Harris CJ, editors. Designing Multi-Target Drugs. The Royal Society of Chemistry; 2012. pp. 1–13. [Google Scholar]
- 24.Bansal Y, Silakari O. Multifunctional compounds: smart molecules for multifactorial diseases. Eur J Med Chem. 2014;76:31–42. doi: 10.1016/j.ejmech.2014.01.060. [DOI] [PubMed] [Google Scholar]
- 25.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 26.Korcsmáros T, et al. How to design multi-target drugs. Expert Opin Drug Discovery. 2007;2:799–808. doi: 10.1517/17460441.2.6.799. [DOI] [PubMed] [Google Scholar]
- 27.Kitano H. Towards a theory of biological robustness. Mol Syst Biol. 2007;3:137. doi: 10.1038/msb4100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azmi AS. Network pharmacology for cancer drug discovery: are we there yet? Future Med Chem. 2012;4:939–941. doi: 10.4155/fmc.12.44. [DOI] [PubMed] [Google Scholar]
- 29.von Manstein V, et al. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr Signal Transduct Ther. 2013;8:193–202. doi: 10.2174/1574362409666140206221931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 31.Aronson JK. In defence of polypharmacy. Br J Clin Pharmacol. 2004;57:119–120. doi: 10.1111/j.1365-2125.2004.02067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Méndez-Lucio O, et al. Review. One drug for multiple targets: a computational perspective. J Mex Chem Soc. 2016;60:168–181. [Google Scholar]
- 33.Bowes J, et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov. 2012;11:909–922. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- 34.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 35.Zheng W, et al. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. 2013;18:1067–1073. doi: 10.1016/j.drudis.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall SE. Chemoproteomics-driven drug discovery: addressing high attrition rates. Drug Discov Today. 2006;11:495–502. doi: 10.1016/j.drudis.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Pruss RM. Phenotypic screening strategies for neurodegenerative diseases: a pathway to discover novel drug candidates and potential disease targets or mechanisms. CNS Neurol Disord Drug Targets. 2010;9:693–700. doi: 10.2174/187152710793237377. [DOI] [PubMed] [Google Scholar]
- 38.Fishman MC, Porter JA. Pharmaceuticals: a new grammar for drug discovery. Nature. 2005;437:491–493. doi: 10.1038/437491a. [DOI] [PubMed] [Google Scholar]
- 39.Gao M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liggi S, et al. Extending in silico mechanism-of-action analysis by annotating targets with pathways: application to cellular cytotoxicity readouts. Future Med Chem. 2014;6:2029–2056. doi: 10.4155/fmc.14.137. [DOI] [PubMed] [Google Scholar]
- 41.Liggi S, et al. Extensions to in silico bioactivity predictions using pathway annotations and differential pharmacology analysis: application to Xenopus laevis phenotypic readouts. Mol Inform. 2013;32:1009–1024. doi: 10.1002/minf.201300102. [DOI] [PubMed] [Google Scholar]
- 42.Young DW, et al. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat Chem Biol. 2008;4:59–68. doi: 10.1038/nchembio.2007.53. [DOI] [PubMed] [Google Scholar]
- 43.Lagarde N, et al. Benchmarking data sets for the evaluation of virtual ligand screening methods: review and perspectives. J Chem Inf Model. 2015;55:1297–1307. doi: 10.1021/acs.jcim.5b00090. [DOI] [PubMed] [Google Scholar]
- 44.Rigden DJ, et al. The 2016 database issue of Nucleic Acids Research and an updated molecular biology database collection. Nucleic Acids Res. 2016;44:D1–D6. doi: 10.1093/nar/gkv1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benson G. Editorial: Nucleic Acids Research annual Web Server Issue in 2015. Nucleic Acids Res. 2015;43:W1–W2. doi: 10.1093/nar/gkv581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scior T, et al. Recognizing pitfalls in virtual screening: a critical review. J Chem Inf Model. 2012;52:867–881. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 47.Truchon JF, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the ‘early recognition’ problem. J Chem Inf Model. 2007;47:488–508. doi: 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]
- 48.Oprea TI, et al. Computational systems chemical biology. Methods Mol Biol. 2011;672:459–488. doi: 10.1007/978-1-60761-839-3_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oprea TI, et al. Systems chemical biology. Nat Chem Biol. 2007;3:447–450. doi: 10.1038/nchembio0807-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pujol A, et al. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol Sci. 2010;31:115–123. doi: 10.1016/j.tips.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 51.Fong TA, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- 52.Jubb AM, et al. Predicting benefit from anti-angiogenic agents in malignancy. Nat Rev Cancer. 2006;6:626–635. doi: 10.1038/nrc1946. [DOI] [PubMed] [Google Scholar]
- 53.Millauer B, et al. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 54.Faivre S, et al. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 55.Maggiora G. Is imatinib a prototypical example of targeted drug therapy? Future Med Chem. 2016;8:1907–1911. doi: 10.4155/fmc-2016-0145. [DOI] [PubMed] [Google Scholar]
- 56.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 57.Harikrishna Reddy D, et al. Advances in drug development for Parkinson’s disease: present status. Pharmacology. 2014;93:260–271. doi: 10.1159/000362419. [DOI] [PubMed] [Google Scholar]
- 58.Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu T, et al. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanehisa M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fabregat A, et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44:D481–D487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu X, et al. In Silico target fishing: addressing a ‘Big Data’ problem by ligand-based similarity rankings with data fusion. J Cheminform. 2014;6:33. doi: 10.1186/1758-2946-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim H, et al. Large-scale off-target identification using fast and accurate dual regularized one-class collaborative filtering and its application to drug repurposing. PLoS Comput Biol. 2016;12:e1005135. doi: 10.1371/journal.pcbi.1005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 65.Cereto-Massague A, et al. Tools for in silico target fishing. Methods. 2015;71:98–103. doi: 10.1016/j.ymeth.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 66.Todeschini R, Consonni V. Molecular Descriptors for Chemoinformatics. Wiley-VCH; 2009. [Google Scholar]
- 67.Petrone PM, et al. Rethinking molecular similarity: comparing compounds on the basis of biological activity. ACS Chem Biol. 2012;7:1399–1409. doi: 10.1021/cb3001028. [DOI] [PubMed] [Google Scholar]
- 68.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huntley RP, et al. The GOA database: Gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015;43:D1057–D1063. doi: 10.1093/nar/gku1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol Inf. 2010;29:476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 71.Castillo-Gonzalez D, et al. Harmonization of QSAR best practices and molecular docking provides an efficient virtual screening tool for discovering new G-quadruplex ligands. J Chem Inf Model. 2015;55:2094–2110. doi: 10.1021/acs.jcim.5b00415. [DOI] [PubMed] [Google Scholar]
- 72.Perez-Castillo Y, et al. Toward the computer-aided discovery of FabH inhibitors. Do predictive QSAR models ensure high quality virtual screening performance? Mol Divers. 2014;18:637–654. doi: 10.1007/s11030-014-9513-y. [DOI] [PubMed] [Google Scholar]
- 73.Varnek A, Baskin I. Machine learning methods for property prediction in chemoinformatics: Quo Vadis? J Chem Inf Model. 2012;52:1413–1437. doi: 10.1021/ci200409x. [DOI] [PubMed] [Google Scholar]
- 74.Cereto-Massague A, et al. DecoyFinder: an easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics. 2012;28:1661–1662. doi: 10.1093/bioinformatics/bts249. [DOI] [PubMed] [Google Scholar]
- 75.Mysinger MM, et al. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perez-Tur J. Parkinson’s disease genetics: a complex disease comes to the clinic. Lancet Neurol. 2006;5:896–897. doi: 10.1016/S1474-4422(06)70580-4. [DOI] [PubMed] [Google Scholar]
- 77.Van der Schyf CJ. Rational drug discovery design approaches for treating Parkinson’s disease. Expert Opin Drug Discov. 2015;10:713–741. doi: 10.1517/17460441.2015.1041495. [DOI] [PubMed] [Google Scholar]
- 78.Guha R. Chemical informatics functionality in R. J Stat Softw. 2007;18:16. [Google Scholar]
- 79.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2016. [Google Scholar]
- 80.Carlson M. GO.db: A set of annotation maps describing the entire Gene Ontology. Bioconductor 2016 [Google Scholar]
- 81.UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perez-Castillo Y, et al. GA(M)E-QSAR: a novel, fully automatic genetic-algorithm-(meta)-ensembles approach for binary classification in ligand-based drug design. J Chem Inf Model. 2012;52:2366–2386. doi: 10.1021/ci300146h. [DOI] [PubMed] [Google Scholar]
- 83.Helguera AM, et al. Ligand-based virtual screening using tailored ensembles: a prioritization tool for dual A2Aadenosine receptor antagonists/monoamine oxidase B inhibitors. Curr Pharm Des. 2016;22:3082–3096. doi: 10.2174/1381612822666160302103542. [DOI] [PubMed] [Google Scholar]
- 84.Supek F, et al. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen KH, et al. Startle habituation and midfrontal theta activity in Parkinson disease. J Cogn Neurosci. 2016;28:1923–1932. doi: 10.1162/jocn_a_01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nieuwenhuijzen PH, et al. Startle responses in Parkinson patients during human gait. Exp Brain Res. 2006;171:215–224. doi: 10.1007/s00221-005-0270-0. [DOI] [PubMed] [Google Scholar]
- 87.Vargas KJ, et al. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34:9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Devine MJ, et al. Parkinson’s disease and alpha-synuclein expression. Mov Disord. 2011;26:2160–2168. doi: 10.1002/mds.23948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Navarro G, et al. Purinergic signaling in Parkinson’s disease. Relevance for treatment Neuropharmacology. 2016;104:161–168. doi: 10.1016/j.neuropharm.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 90.Gaki GS, Papavassiliou AG. Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. Neuromolecular Med. 2014;16:217–230. doi: 10.1007/s12017-014-8294-x. [DOI] [PubMed] [Google Scholar]
- 91.Hernandes MS, Britto LR. NADPH oxidase and neurodegeneration. Curr Neuropharmacol. 2012;10:321–327. doi: 10.2174/157015912804143540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cristóvão AC, et al. NADPH oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J Neurosci. 2012;32:14465–14477. doi: 10.1523/JNEUROSCI.2246-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bourque M, et al. Neuroprotective actions of sex steroids in Parkinson’s disease. Front Neuroendocrinol. 2009;30:142–157. doi: 10.1016/j.yfrne.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 94.Girotti F, Soliveri P. Cognitive and behavioral disturbances in Parkinson’s disease. Neurol Sci. 2003;24(Suppl. 1):S30–S31. doi: 10.1007/s100720300034. [DOI] [PubMed] [Google Scholar]
- 95.Anderson KE. Behavioral disturbances in Parkinson’s disease. Dialogues Clin Neurosci. 2004;6:323–332. doi: 10.31887/DCNS.2004.6.3/kanderson. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goodall M, Alton H. Dopamine (3-hydroxytyramine) metabolism in Parkinsonism. J Clin Invest. 1969;48:2300–2308. doi: 10.1172/JCI106196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fifel K, Cooper HM. Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol Dis. 2014;71:359–369. doi: 10.1016/j.nbd.2014.08.024. [DOI] [PubMed] [Google Scholar]
- 98.Breen DP, et al. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014;71:589–595. doi: 10.1001/jamaneurol.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Visser U, et al. BioAssay Ontology (BAO): a semantic description of bioassays and high—throughput screening results. BMC Bioinformatics. 2011;12:257. doi: 10.1186/1471-2105-12-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abeyruwan S, et al. Evolving BioAssay Ontology (BAO): modularization, integration and applications. J Biomed Semantics. 2014;5(Suppl. 1):S5. doi: 10.1186/2041-1480-5-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smith MR, et al. An instance level analysis of data complexity. Mach Learn. 2014;95:225–256. [Google Scholar]
- 102.Maggiora GM. On outliers and activity cliffs — why QSAR often disappoints. J Chem Inf Model. 2006;46:1535–1535. doi: 10.1021/ci060117s. [DOI] [PubMed] [Google Scholar]
- 103.Cruz-Monteagudo M, et al. Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde? Drug Discov Today. 2014;19:1069–1080. doi: 10.1016/j.drudis.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 104.Kroll A. Grey-box models: concepts and application. In: Mohammadian M, editor. New Frontiers in Computational Intelligence and its Applications. IOS Press; 2000. pp. 42–51. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.