

ABSTRACT
Pyrazinamide (PZA), an indispensable component of modern tuberculosis treatment, acts as a key sterilizing drug. While the mechanism of activation of this prodrug into pyrazinoic acid (POA) by Mycobacterium tuberculosis has been extensively studied, not all molecular determinants that confer resistance to this mysterious drug have been identified. Here, we report how a new PZA resistance determinant, the Asp67Asn substitution in Rv2783, confers M. tuberculosis resistance to PZA. Expression of the mutant allele but not the wild-type allele in M. tuberculosis recapitulates the PZA resistance observed in clinical isolates. In addition to catalyzing the metabolism of RNA and single-stranded DNA, Rv2783 also metabolized ppGpp, an important signal transducer involved in the stringent response in bacteria. All catalytic activities of the wild-type Rv2783 but not the mutant were significantly inhibited by POA. These results, which indicate that Rv2783 is a target of PZA, provide new insight into the molecular mechanism of the sterilizing activity of this drug and a basis for improving the molecular diagnosis of PZA resistance and developing evolved PZA derivatives to enhance its antituberculosis activity.
KEYWORDS: PNPase, pyrazinoic acid, antibiotic resistance, drug target, ppGpp, pyrazinamide
INTRODUCTION
In its most recent report on global tuberculosis (TB), the World Health Organization declared that TB killed 1.8 million people in 2015 (1). The report also reiterated an urgent need for new drugs with sterilizing activity that can prevent relapse and the development of drug resistance. Pyrazinamide (PZA) is considered a sterilizing drug in TB treatment regimens owing to its unique ability to act on Mycobacterium tuberculosis persisters in the acidic environments of macrophages and inflammatory foci (2). It is a vital component of the modern short-course TB treatment regimen, as its inclusion shortens the treatment duration from 9 to 12 months to 6 months (3). Because the activity of PZA is synergistic with that of several new drugs and drug candidates presently in clinical development, it is considered a part of future regimens in combination with bedaquiline, pretomanid, and moxifloxacin (4). These new drug combinations are expected to further shorten the period of treatment for drug-susceptible as well as drug-resistant TB. Thus, PZA has proven to be a unique enhancer of a variety of current and emerging TB therapeutic regimens.
PZA is a prodrug that requires conversion into its active form, pyrazinoic acid (POA). The conversion is catalyzed by pyrazinamidase (PZase), an enzyme encoded by pncA of M. tuberculosis (5). Although the mechanisms of PZA activation by PZase and PZA resistance mediated by mutations of pncA and its flanking regions have been extensively studied, a complete picture of the molecular target(s) and cellular functions inhibited by POA that together prove lethal to M. tuberculosis has not been elucidated. The fatty acid synthase I (FAS I), encoded by fasI, has been reported to be one of the targets of PZA (6). The eukaryote-like fasI from M. avium, M. bovis bacillus Calmette-Guérin (BCG), or M. tuberculosis was found to confer resistance to the PZA analogue 5-chloro-pyrazinamide (5-Cl-PZA), when present in multicopy vectors in M. smegmatis (6). However, no mutations in fasI have been found in PZA-resistant M. tuberculosis strains, and FAS I was shown to be the target of 5-Cl-PZA but not of PZA (7). The ribosomal protein S1 (RpsA), a vital protein involved in the ribosome-rescuing process of trans-translation, has also been reported to be a target of POA (8). This followed an earlier identification of a low level of PZA resistance in M. tuberculosis clinical isolate DHM444 which had no mutations in pncA but which harbored a deletion of alanine at position 438 in RpsA (RpsAΔA438) (8). Overexpression of RpsA rendered M. tuberculosis resistant to PZA, and unlike wild-type RpsA, RpsAΔA438 did not bind POA (8). However, a strain defective for trans-translation was fully susceptible to PZA (9), suggesting that altered trans-translation does not fully account for PZA resistance. Recently, aspartate decarboxylase, encoded by panD and involved in the synthesis of the β-alanine precursor for pantothenate and coenzyme A biosynthesis, was also suggested to be an additional target of PZA in M. tuberculosis (10). However, an M. tuberculosis strain with panC (encoding pantothenate synthetase) and panD deleted was cultivated with pantetheine at a concentration that did not antagonize PZA activity (11). This finding uncouples pantothenate synthesis and the action of PZA, suggesting that pantothenate-mediated antagonism occurs via a novel mechanism that is independent of PanD. An ATP-dependent ATPase (ClpC1) involved in protein degradation was very recently reported to be associated with PZA resistance in M. tuberculosis (12).
In summary, the molecular targets and mechanisms described so far do not fully account for the activity of PZA, and additional mechanisms that are yet unknown exist. Herein, for the first time, we demonstrate that a mutation in Rv2783, a bifunctional enzyme, confers resistance to PZA and further explore the properties of this enzyme to develop insights into the mechanism that leads to PZA resistance.
RESULTS
M. tuberculosis Rv2783c is associated with PZA resistance.
We hypothesized that an unknown mechanism conferred PZA resistance in four M. tuberculosis clinical isolates that we recently described (13), as they lacked mutations in previously described resistance determinants, namely, pncA and rpsA. We further hypothesized that molecular factors that bind POA or its metabolites but whose potential involvement in PZA resistance has not been identified could yield valuable leads in pursuing the resistance determinants. Therefore, we selected proteins that have been demonstrated to bind to a POA derivative, 5-hydroxyl-2-pyrazinecarboxylic acid (8), namely, those encoded by Rv2731, Rv2783c, Rv3169, and Rv3601c. Analysis of the sequences of these genes identified the same G199A mutation in Rv2783c in two clinical strains. Simultaneously, we amplified pncA and its flanking regions again and verified the pyrazinamidase (PZase) activities of the two strains. The PZase activity in the two strains was similar to that in wild-type M. tuberculosis (see Fig. S1 in the supplemental material), and the sequences of pncA and its flanking regions were identical to those in the wild type.
Expression of Rv2783 with the Asp67Asn mutation (Rv2783Asp67Asn) in M. tuberculosis H37Rv causes PZA resistance.
To further explore the potential role of Rv2783c in PZA resistance, we engineered a recombinant M. tuberculosis strain to express Rv2783c with the G199A mutation (Rv2783cG199A) and compared its PZA susceptibility to that of wild-type M. tuberculosis (Table 1). Expression of the Rv2783cG199A mutant in M. tuberculosis H37Rv using the p60luxN vector (14) containing the strong hsp60 promoter caused a 5-fold increase in the MIC of PZA (MIC = 500 μg/ml) compared with the MICs for the hsp60 vector control and the parental M. tuberculosis H37Rv strain (MICs = 100 μg/ml) at pH 5.5. On the other hand, the MIC of PZA against the recombinant M. tuberculosis strain containing additional wild-type Rv2783c in the same vector was 150 μg/ml. In addition, both the wild-type Rv2783c and mutant Rv2783cG199A transformants were positive in the PZase activity assay, as were the parental M. tuberculosis strain and the M. tuberculosis empty vector control (Fig. S1).
TABLE 1.
PZA susceptibility testing of M. tuberculosis Rv2783c recombinant strainsa
| M. tuberculosis recombinant strain | In vitro PZA susceptibility | MIC (μg/ml) |
|---|---|---|
| Wild-type Rv2783c | Susceptible | 150 |
| Mutant Rv2783cD67N | Resistant | 500 |
| Hsp60RvC (vector control) | Susceptible | 100 |
| Parental M. tuberculosis | Susceptible | 100 |
The PZA susceptibility test, which was performed with the Bactec MGIT 960 system, was repeated 4 times. Expression of mutant Rv2783cD67N and wild-type Rv2783c in M. tuberculosis H37Rv was done using the extrachromosomal p60luxN vector (14) containing the strong mycobacterial hsp60 promoter. The PZase assay result was positive for all strains.
Rv2783 binds to POA but not to PZA.
To determine if POA binds to Rv2783 and whether the Asp67Asn substitution affects binding to POA, we used isothermal titration calorimetry (ITC) to measure the binding interactions of the purified wild-type Rv2783, the mutant Rv2783Asp67Asn, and the M. smegmatis polynucleotide phosphorylase (PNPase) with POA or PZA. Wild-type Rv2783 bound to POA via a sequential binding sites model (n = 2), with dissociation constant 1 (Kd1)) being equal to 1.05 mM and Kd2 being equal to 3.17 mM (Fig. 1A), but did not bind to the prodrug PZA even at higher concentrations (Fig. 1B). On the contrary, the Rv2783Asp67Asn mutant and PNPase from naturally PZA-resistant M. smegmatis failed to bind to POA or PZA even at higher concentrations (Fig. S2). We observed, however, that POA bound to wild-type Rv2783 at relatively higher concentrations and only weakly at lower concentrations.
FIG 1.
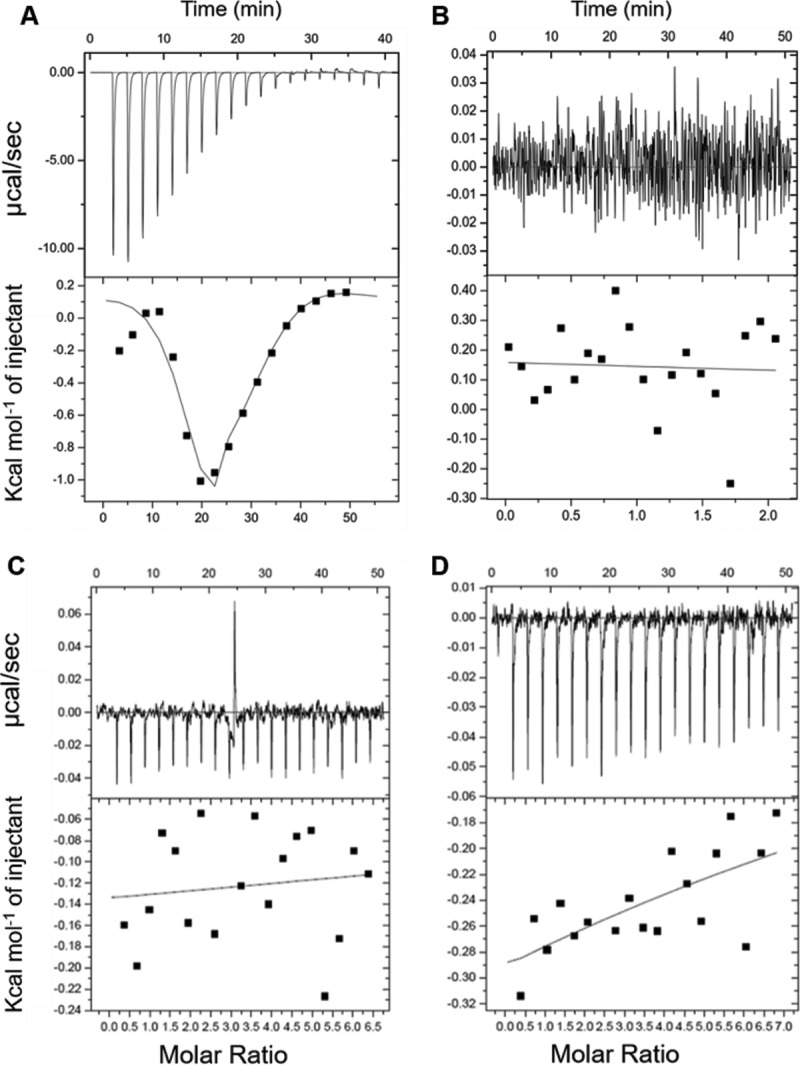
Interaction of POA with wild-type Rv2783, mutant Rv2783Asp67Asn, and M. smegmatis PNPase. (A) Representative results of an isothermal titration calorimetry (ITC) binding study indicate that POA binds to wild-type Rv2783 at high concentrations. (Top) Raw data. The data on the y axis indicate the amount of heat released per second during wild-type Rv2783 and POA binding. (Bottom) Integrated heat in each injection of POA together with the fit. The data on the y axis are expressed as the amount of heat released per mole in each injection. The association constants were obtained from fits of POA binding with wild-type Rv2783. (B) Representative results of an ITC binding study indicate that PZA did not bind to the wild-type Rv2783 protein. (Top) No heat was released during the wild-type Rv2783 and PZA interaction; (bottom) there was no integrated heat following each injection of PZA, and thus, there was no curve of the fit. (C and D) Representative results of an ITC binding study indicate that POA did not bind to the mutant Rv2783Asp67Asn protein (C) or to the M. smegmatis PNPase protein (D). (Top) Raw data indicating that no heat was released during the interaction between POA and the two proteins; (bottom) there was no integrated heat following each injection of POA, and thus, there was no curve of the fit.
ssDNA polymerization and phosphorolysis activities of Rv2783.
Rv2783 is predicted to function as a PNPase enzyme, which catalyzes synthetic and degradative activities in RNA metabolism (15). Exemplary bacterial PNPase enzymes have been reported to catalyze DNA activities in vitro when manganese or iron is provided in lieu of magnesium as the metal cofactor (16). Therefore, it was of considerable relevance to determine whether Rv2783 could catalyze single-stranded DNA (ssDNA) polymerization and phosphorolysis activities. Both the Rv2783 and Rv2783Asp67Asn proteins catalyzed the elongation of the ssDNA of a 5′ 6-carboxyfluorescein (FAM)-labeled 36-mer ssDNA substrate in the presence of Mn2+ and dADP when incubated at 37°C for 60 min, yielding a bimodal distribution of fluorescently labeled end products ranging from 45-mer to 93-mer in size. We observed that the ssDNA polymerization activity of wild-type Rv2783 but not that of Rv2783Asp67Asn was significantly inhibited by POA, while PZA inhibited neither of them (Fig. 2). This implies that the mutant Rv2783Asp67Asn protein but not the wild-type Rv2783 protein can withstand the inhibitory effects of POA. However, there was no significant difference in the POA-free ssDNA polymerization activities of the wild-type Rv2783 and mutant Rv2783Asp67Asn proteins. On the other hand, upon substitution of dADP with inorganic phosphate, both proteins were able to degrade the 3′ end of a 5′ FAM-labeled 36-mer ssDNA substrate, resulting in the appearance of fluorescently 5′ FAM-labeled decay products shorter than the input 36-mer ssDNA. However, unlike for wild-type Rv2783, where the inhibitory effect of POA on ssDNA phosphorolysis was significantly higher (P < 0.0001), the inhibitory effect of POA on the ssDNA phosphorolytic activity of Rv2783Asp67Asn was comparatively lower (P = 0.0399). Moreover, there was a significant difference (P = 0.0074) in the POA-free ssDNA phosphorolysis activities of the two proteins (Fig. 3).
FIG 2.

Capillary electrograms and graphical presentations of the ssDNA polymerization activities of Rv2783. (A to C) Representative capillary electrograms showing the uninhibited ssDNA polymerization activity of wild-type Rv2783 (A) and inhibition of the ssDNA polymerization activity of wild-type Rv2783 by PZA (B) and POA (C). (D) Comparison of the effects of POA and PZA on the ssDNA polymerization activity of the wild-type Rv2783 protein. (E to G) Representative capillary electrograms showing the uninhibited ssDNA polymerization activity of mutant Rv2783Asp67Asn (E) and inhibition of the ssDNA polymerization activity of mutant Rv2783Asp67Asn by PZA (F) and POA (G). (H) Comparison of the effects of POA and PZA on the ssDNA polymerization activity of the mutant Rv2783Asp67Asn protein. For panels D and H, data are presented as means ± SEMs. Student's t test was used for the statistical analysis. ns, not significant; RFU, relative fluorescence units.
FIG 3.

Capillary electrograms and graphical presentations of the ssDNA phosphorolysis activities of Rv2783. (A to C) Representative capillary electrograms showing the uninhibited ssDNA phosphorolysis activity of wild-type Rv2783 (A) and inhibition of the ssDNA phosphorolysis activity of wild-type Rv2783 by PZA (B) and POA (C). (D) Comparison of the effects of POA and PZA on the ssDNA phosphorolysis activity of the wild-type Rv2783 protein. (E to G) Representative capillary electrograms showing the uninhibited ssDNA phosphorolysis activity of mutant Rv2783Asp67Asn (E) and inhibition of the ssDNA phosphorolysis activity of mutant Rv2783Asp67Asn by PZA (F) and POA (G). (H) Comparison of the effects of POA and PZA on the ssDNA phosphorolysis activity of the mutant Rv2783Asp67Asn protein. For panels D and H, data are presented as means ± SEMs. Student's t test was used for the statistical analysis.
RNA polymerization and phosphorolysis activities of M. tuberculosis Rv2783.
We studied the RNA polymerization and phosphorolysis activities of both wild-type Rv2783 and Rv2783Asp67Asn. When reacted with a 5′ FAM-labeled 24-mer single-stranded RNA (ssRNA) substrate in the presence of Mg2+ and ADP (in lieu of Mn2+ and dADP), the proteins catalyzed the extension of the input ssRNA into a polynucleotide tail. On the other hand, when the 5′ FAM-labeled 24-mer ssRNA template was reacted with the proteins in the presence of Mg2+ and phosphate (in lieu of ADP), most of the input ssRNA was degraded into a mixture of shorter nucleotide end products. Comparatively, as observed with the ssDNA activities, the ssRNA polymerization and phosphorolysis activities of wild-type Rv2783 but not those of the mutant protein were significantly inhibited by POA, while PZA did not significantly inhibit either activity (Fig. 4). In addition, there was no significant difference in the POA-free ssRNA metabolism activities between the wild-type and Rv2783Asp67Asn proteins. Taken together, these findings suggest the direct relevance of the Rv2783 protein in RNA metabolism.
FIG 4.

Capillary electrograms and graphical presentations of RNA metabolism activities of Rv2783. (A and B) Representative capillary electrograms showing the uninhibited RNA polymerization activity of wild-type Rv2783 (A) and inhibition of RNA polymerization activity of wild-type Rv2783 by POA (B). (C) Comparison of the effects of POA and PZA on the RNA polymerization activity of the wild-type Rv2783 protein. (D and E) Representative capillary electrograms showing the uninhibited RNA polymerization activity of mutant Rv2783Asp67Asn (D) and inhibition of the RNA polymerization activity of mutant Rv2783Asp67Asn by POA (E). (F) Comparison of the effects of POA and PZA on the RNA polymerization activity of the mutant Rv2783Asp67Asn protein. (G and H) Representative capillary electrograms showing the uninhibited RNA phosphorolysis activity of wild-type Rv2783 (G) and inhibition of the RNA phosphorolysis activity of wild-type Rv2783 by POA (H). (I) Comparison of the effects of POA and PZA on the RNA phosphorolysis activity of the wild-type Rv2783 protein. For panels C, F, and I, the data are presented as means ± SEMs. Student's t test was used for the statistical analysis.
ppGpp synthetase and hydrolase activities of Rv2783c.
On the basis of sequence homology, Rv2783 has been predicted to be a probable guanosine pentaphosphate synthetase (GpsI) that is involved in the synthesis and hydrolysis of (p)ppGpp (17) and the stringent response in many bacteria (18). Therefore, it was of considerable interest to determine whether the protein possessed ppGpp synthesis and ppGpp hydrolysis activities, as have been previously demonstrated for the M. tuberculosis Rel (RelMtb) protein (19). When reacted with various concentrations of ATP (phosphate donor) and GDP (substrate) in the presence of various concentrations of Mg2+ and Na+ at various pH values (5.5 to 8.0), both the wild-type Rv2783 and the Rv2783Asp67Asn proteins demonstrated only weak ppGpp synthesis activities, while RelMtb demonstrated strong ppGpp synthesis activity under similar reaction conditions (Fig. 5). On the other hand, when the same concentrations of the wild-type Rv2783 and Rv2783Asp67Asn proteins were reacted with 0.1 mM ppGpp in the presence of various concentrations of Mn2+ and Na+, ppGpp was strongly hydrolyzed into GDP and ATP. However, while the wild-type Rv2783 protein exhibited relatively stronger ppGpp hydrolysis activity than Rv2783Asp67Asn, its activity, unlike that of the mutant, was significantly inhibited by POA (Fig. 6).
FIG 5.
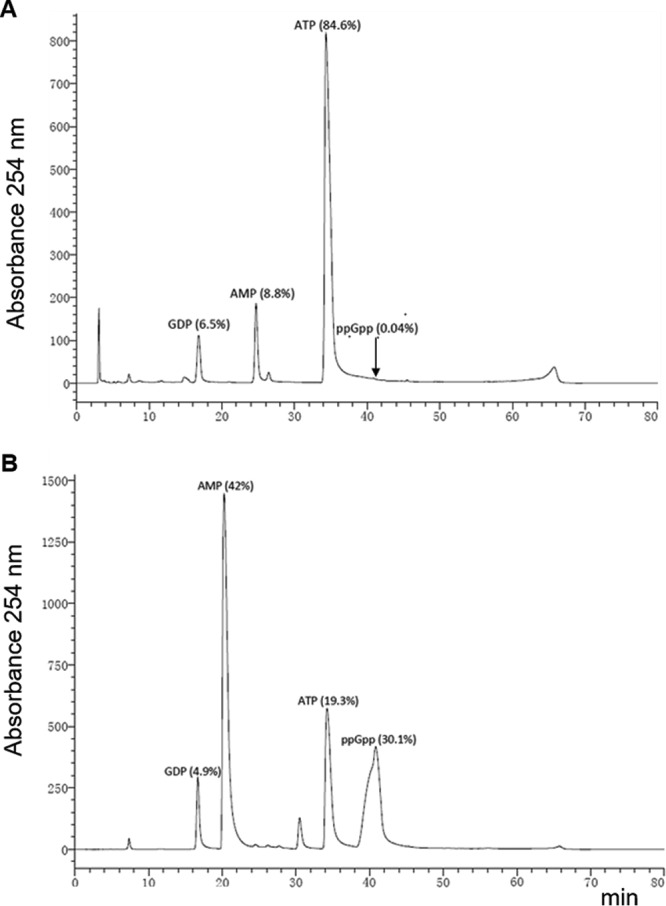
ppGpp synthetase assays. Representative HPLC separation of ppGpp synthesis reaction products catalyzed by the wild-type Rv2783 protein (A) and the wild-type M. tuberculosis Rel protein (positive control) (B).
FIG 6.

ppGpp hydrolase assays. Representative HPLC separation of ppGpp hydrolysis reaction products catalyzed by the wild-type Rv2783 protein in the absence (A) and presence (B) of POA and the mutant Rv2783Asp67Asn protein in the absence (C) and presence (D) of POA.
DISCUSSION
Herein, we demonstrated the involvement of the M. tuberculosis gene Rv2783c in PZA resistance. In our previous study (13), we identified four clinical strains that lacked mutations in genes reported to confer PZA resistance. This indicated the potential presence of an additional molecular mechanism that could confer PZA resistance. We sequenced the genes of M. tuberculosis proteins that were previously reported to bind with POA (8) as leads to seek a target(s) that conferred PZA resistance in these clinical strains. Analysis of the sequences of these genes indicated that two out of the four strains harbored the same G199A mutation in Rv2783c. The two PZA-resistant strains were found to be for positive PZase activity, and sequence analysis of their pncA genes, including the flanking regions, indicated no mutation. This allowed us to rule out the possibility of alterations in the potential regulatory regions of pncA that could result in a lack of PZase activity as a possible cause of the PZA resistance (13). Expression of Rv2783Asp67Asn increased the MIC of PZA in wild-type M. tuberculosis, providing direct evidence of the involvement of Rv2783 in PZA resistance. The finding that both the wild-type Rv2783c and Rv2783cG199A transformants were positive in the PZase activity assay ruled out the possibility of a mutation in pncA as a possible cause of the PZA resistance in the transformant strain expressing Rv2783Asp67Asn. An increase in the level of transcription of a target gene, leading to its higher level of expression, is a well-known drug resistance mechanism in bacteria (20). In this study, expression of Rv2783Asp67Asn in wild-type M. tuberculosis H37Rv was therefore possibly responsible for the observed PZA resistance in vitro. However, introducing wild-type Rv2783c under the control of a strong promoter on a multicopy extrachromosomal plasmid into the PZA-sensitive M. tuberculosis H37Rv strain only weakly increased the strain's resistance to PZA. It is possible that the increased number of copies of wild-type Rv2783c was not sufficient to withstand the inhibitory effects of the high concentrations of PZA. In addition, we observed a similar phenomenon in our recent study (14) in which the introduction of the wild-type target gene (rplC) did not increase the level of resistance to linezolid, as was the case after introduction of a wild-type potential prothionamide target gene (unpublished data). Therefore, these findings illustrate that the Rv2783Asp67Asn mutation confers resistance to POA.
While wild-type Rv2783 bound to POA, it failed to bind to the prodrug PZA; this suggests that Rv2783 is a potential target of POA and confirms that, indeed, it is POA and not the prodrug PZA that has the anti-TB activity. We found, however, that POA bound to wild-type M. tuberculosis Rv2783 at a relatively higher concentration and bound only weakly at lower concentrations. This finding is concordant with an earlier observation (8) in which Rv2783 was also shown to bind only weakly to a POA derivative, 5-hydroxyl-2-pyrazinecarboxylic acid. This could probably explain why PZA (with a lower molecular weight of 123.11) must be administered at a higher dosage than other anti-TB drugs. On the other hand, the failure by Rv2783Asp67Asn and PNPase from naturally PZA-resistant M. smegmatis to bind to POA or PZA, even at higher concentrations, further supports the suggestion that Rv2783 is a target of POA and that the Asp67Asn mutation affects its ability to bind to POA. We cannot exclude the possibility that the mutant Rv2783 is more stable or expressed at a higher level, but it did affect the susceptibility of M. tuberculosis to PZA. These findings suggest that POA may bind to Rv2783 and, in particular, to residue Asp67, as it was the altered site found in the two PZA-resistant strains and thus influences the PZA susceptibility of M. tuberculosis. PZA is a mysterious drug which is active only at acidic pH in vitro and has weak activity only at a very high dose in vivo, despite its lower molecular weight. In our unpublished works in vitro, even at 1 mg/ml, PZA exhibited no activity against M. tuberculosis at normal pH. In that case, the behavior of PZA in the biochemical assays and in vivo against M. tuberculosis can be very different. On the other hand, the binding of POA to wild-type Rv2783 is sequential (n = 2 binding sites). The two binding sites are nonindependent, meaning that the binding by the first POA molecule is required to facilitate access and binding by the second POA molecule. These could probably explain POA's poor Kd (dissociation constant).
Our findings on the ssDNA polymerase and phosphorylase catalytic activities of Rv2783 contribute to an emerging picture of mycobacterial PNPases as catalysts of DNA metabolism, in addition to their synthetic and degradative roles in RNA metabolism (16). As suggested, Rv2783 is likely involved in conserving DNA integrity through DNA repair and mutagenesis in vivo. A similar PNPase-associated DNA metabolism in Bacillus subtilis has also been described (21). The study provided genetic evidence that B. subtilis PNPase participates in the homologous recombination and nonhomologous end-joining pathways of double-strand break repair in response to damage by hydrogen peroxide (21). Considering that mycobacteria possess three distinct pathways for the repair of DNA double-strand breaks (homologous recombination, nonhomologous end joining, and single-strand annealing), a similar participation by PNPase is highly possible. Alonso and colleagues suggested that PNPase reacts with broken DNA ends, either converting them from dirty breaks that cannot be ligated to clean ends that can be sealed by DNA ligase or adding nontemplated single-stranded 3′ tails that can then influence the repair pathway choice (21, 22). The differential inhibitory effects of POA on the ssDNA polymerization and phosphorolysis activities of the wild-type and mutant Rv2783 proteins could help explain the difference in the PZA susceptibilities of their respective transformants. Taken together, it appears that the Asp67Asn substitution in Rv2783 alters the binding of POA, resulting in reduced susceptibility to POA. In such a case, the Rv2783Asp67Asn mutant strain would catalyze DNA activities (such as DNA repair) unimpeded under conditions that impact PZA susceptibility, especially during dormancy.
The RNA degradosome, a multienzyme complex comprised of proteins such as RNase E and PNPase, is involved in RNA metabolism and posttranscriptional control of gene expression in many bacteria (23–25). Although the mechanism of action of the RNA degradosome in Escherichia coli is relatively well understood, very little is known about the mechanisms involved in RNA metabolism in other bacteria (26, 27), including mycobacteria. Given the central role of the RNA degradosome in RNA metabolism in E. coli, it is conceivable that the RNA degradosome-dependent regulation of RNA stability in M. tuberculosis might also be a very important mechanism associated with cellular metabolism. During dormancy, M. avium exhibits a decline in susceptibility to antibiotics and an increase in RNase E levels (28). Likewise, having demonstrated the involvement of Rv2783 in RNA metabolism, we hypothesize an increased level of expression of Rv2783Asp67Asn and its subsequent involvement in DNA/RNA metabolism during dormancy, thereby influencing PZA susceptibility. This is likely true, as Rv2783Asp67Asn was able to catalyze RNA metabolism even in the presence of POA and could, in addition, contribute toward sustaining RNA stability during dormancy. On the other hand, the ability of Rv2783 to catalyze RNA polymerization in a template-independent manner would possibly result in the production of inaccurate mRNAs, rRNAs, and tRNAs and further mistranslation. New protein variants with an adaptive function could be synthesized, producing some new bacillary phenotypes that could tolerate or resist various stresses and antibiotics (29). Specific mistranslation of the mycobacterial RNA polymerase, the target of rifampin (RIF), was demonstrated to be necessary and sufficient for phenotypic RIF resistance (30). In addition, we speculate that the potential diverse small RNAs synthesized by Rv2783 may regulate the gene expression of M. tuberculosis bacilli in different ways, enabling some of them to survive during persistence without a genome mutation.
The long-term survival of nonreplicating M. tuberculosis bacilli has been associated with RelMtb-mediated (p)ppGpp synthesis, which triggers the stringent response (19). The enzyme transfers a pyrophosphate from ATP to GDP or GTP to synthesize ppGpp and pppGpp, respectively, and also hydrolyzes (p)ppGpp into pyrophosphate and GDP or GTP (19). However, the strong ppGpp hydrolysis and weak ppGpp synthesis activities of Rv2783 demonstrated herein also strongly suggest its involvement in the regulation of intracellular concentrations of ppGpp in the bacilli under stressful conditions during dormancy. These catalytic activities of Rv2783 are similar to those of the E. coli SpoT protein, which is a bifunctional enzyme with strong (p)ppGpp hydrolase and only weak synthetase activities, which are activated under starvation conditions (31). PZA exhibits a preferential sterilizing activity against nonreplicating persisting bacilli during dormancy/persistence (2). Therefore, our findings that expression of Rv2783Asp67Asn caused PZA resistance and that the mutant protein retained the strong ppGpp hydrolysis and weak ppGpp synthesis activities even in the presence of POA strongly suggest the protein's vital role in the regulation of intracellular (p)ppGpp and, hence, the stringent response. The synthesis of (p)ppGpp without its hydrolysis directly impacts cellular pathways that are detrimental for the bacteria (32). The expression of a hydrolase-null RelMtb mutant incapable of hydrolyzing (p)ppGpp but still able to synthesize (p)ppGpp decreased the growth rate of M. tuberculosis and changed the colony morphology of the bacteria. Moreover, the expression of the RelMtb mutant during acute or chronic M. tuberculosis infection in mice was lethal to the infecting bacteria (32). Taken together, our findings further highlight the distinct importance of (p)ppGpp hydrolysis or regulation of its content that is essential for M. tuberculosis pathogenesis. We propose that Rv2783 plays an important role in the general homeostasis of (p)ppGpp during dormancy, and while this function is inhibited by POA in the wild type, the Asp67Asn mutation helps circumvent the effects of POA.
The Rv2783 protein plays many important roles in M. tuberculosis, so it could be essential. Rv2783c is located within a cluster of co-oriented open reading frames (see Fig. S3 in the supplemental material). The upstream lppU gene (encoding a lipoprotein), the downstream pepR gene (encoding a zinc protease), and the Rv2781c gene are deemed nonessential for M. tuberculosis growth, as was illustrated by the recovery of viable bacteria with a highly efficient transposon recognizing and inserting the binucleotide sequence TA within these respective open reading frames (33). In contrast, no transposon insertion within Rv2783c was recovered in several independent saturated M. tuberculosis mutation libraries using the same transposon combined with whole-genome sequencing (34, 35), suggesting the essentiality of Rv2783c for M. tuberculosis growth.
In conclusion, it is likely that Asp67 of Rv2783 is flexible and dispensable for enzymatic activity but is required for POA binding. POA binds to the Rv2783 protein, and the ensuing conformational changes inhibit its catalytic activities. The Asp67Asn mutation obstructs POA binding to the protein, and hence, the mutant protein proceeds with its catalytic activities. However, further studies by crystal structure analysis and functional analysis of some specific amino acid residues of Rv2783c are needed to confirm this hypothesis and delineate their roles. The limitation of this study is that the binding experiment and enzyme activity assays carried out in vitro may not represent the native conditions in M. tuberculosis.
MATERIALS AND METHODS
General DNA techniques.
M. tuberculosis genomic DNA was extracted using the cetyltrimethylammonium bromide method as previously described (36) and used for subsequent PCRs. PCR amplification reactions were performed with Pfu DNA polymerase (TaKaRa, China), and 5% dimethyl sulfoxide was added due to the high G+C content of the mycobacterial genomes. The PCR-amplified products were analyzed by electrophoresis in agarose gels and purified using a DNA gel extraction kit (Bioflux). Plasmids were also extracted and purified using kits from the same company. Purified PCR products and recombinant plasmids were sequenced at The Beijing Genomics Institute, Shenzhen, China.
PCR and DNA sequencing of new genes associated with PZA resistance.
To identify possible new genes associated with PZA resistance in four previously reported (13) PZA-resistant clinical strains lacking pncA and rpsA mutations, we amplified three genes (Rv2731, Rv2783c, and Rv3169), which encode proteins previously reported to bind a POA derivative, 5-hydroxyl-2-pyrazinecarboxylic acid (8), plus their flanking regions (FR), using primer pairs Rv2731F and Rv2731R, Rv2783cF and Rv2783cR, and Rv3169F and Rv3169R, respectively (Table 2). We also amplified panD (Rv3601c), another gene implicated in PZA resistance (10), plus its FR using primers panDF and panDR (Table 2). We amplified pncA plus its FR using primers pncAF and pncAR (Table 2) to verify the absence of any mutation. The PCR-amplified DNA products were then sequenced, and the gene sequences from the four PZA-resistant strains were compared against the respective wild-type gene sequences of M. tuberculosis H37Rv to identify potential mutations.
TABLE 2.
DNA primers used in this study
| Primer pair | Nucleotide sequence (5′-3′)a |
|---|---|
| Rv2731F/Rv2731R | ATCGGCAAATATCGCG/GCACCATCGTGCAGCT |
| Rv2783cF/Rv2783cR | GGGAATTCCATATGTCTGCCGCTGAAAT/CCCAAGCTTATTCGGTGACCACTCG |
| Rv3169F/Rv3169R | AACTTGGTGACCTGCA/CCGCATTTCGGCGGTT |
| pncAF/pncAR | ATTTGTCGCTCACTACA/ATGCCCCACCTGCGGCT |
| panDF/panDR | TCGACTACCTGGAGCT/TGACTTCGGATTCGGT |
| Ms2656F/Ms2656R1 | GGGAATTCCATATGTCTGTAGTCGAACT/AAAAGTACTCGGACCTGCGACGTTTC |
| HygF/HygR | AGAGCACCAACCCCGTACTG/GTGAAGTCGACGATCCCGGT |
| Ms2656F/Ms2656R2 | GGGAATTCCATATGTCTGTAGTCGAACT/ATAAGAATGCGGCCGCCGGACCTGCGACGTTTC |
| RelF/RelR | GGGAATTCCATATGGTGGCCGAGGACCAG/CCCAAGCTTAAATCAGCCCGCCCAAT |
Restriction enzyme sites are underlined.
Determination of PZase activities of the two PZA-resistant strains.
The PZase test of Wayne (37) was performed as previously modified (38). Briefly, two to three discrete colonies from the same batch were picked and transferred into 1 ml Middlebrook 7H9 medium supplemented with albumin-dextrose-catalase medium containing PZA (100 μg/ml and 200 μg/ml) in a 1.5-ml tube. The cells were incubated overnight at 37°C while shaking. Fifteen microliters of 2% Fe2+ was added to the cells, and the cells were incubated at 4°C for 2 h for detection of a color reaction. Wild-type M. tuberculosis H37Rv was used as a positive control (PZA sensitive), while Mycobacterium bovis bacillus Calmette-Guérin (BCG) Tice and a clinical strain with a pncA mutation were used as two negative controls (both strains were PZA resistant). PZA in the presence of the PZase enzyme from M. tuberculosis is converted to POA, which then reacts with ferrous ion to produce a brown compound, which can be detected as an indication of positivity for PZase activity.
Rv2783 overexpression in M. tuberculosis.
The wild-type Rv2783c and the Rv2783cG199A mutant genes were amplified from M. tuberculosis H37Rv and the PZA-resistant mutant strains, respectively, using primer pair Rv2783cF and Rv2783cR (Table 2). The PCR fragments were digested with the same enzymes, ligated to the extrachromosomal plasmid p60luxN (14) downstream of the strong hsp60 promoter, and transformed into E. coli DH5α cells. An empty p60luxN vector was included as a control. Competent wild-type M. tuberculosis H37Rv cells were first incubated at 37°C for 10 min before electroporation, and transformation was performed at room temperature as previously described (39). Verification of the transformed M. tuberculosis cells was done by amplification of the 529-bp hyg marker gene open reading frame using the primer pair HygF and HygR (Table 2) and testing for PZase enzyme activity. Verified transformants were plated on 7H11 agar plates containing hygromycin (50 μg/ml) and used for in vitro PZA susceptibility testing using the Bactec MGIT 960 system (BD, Franklin Lakes, NJ, USA).
In vitro PZA susceptibility testing of M. tuberculosis transformants.
PZA susceptibility tests were carried out in MGIT PZA medium (pH 5.5; BD, Franklin Lakes, NJ, USA) according to the manufacturer's instructions. Briefly, a 0.5 McFarland suspension was diluted 1:5 and 1:50 in sterile distilled water. Dilutions of 1:50 were inoculated into MGIT PZA medium without drug, while 1:5 dilutions were inoculated into MGIT PZA medium supplemented with 25 to 500 μg/ml PZA, and the dilutions were incubated in the MGIT instrument at 37°C. Results were read automatically within 14 and 21 days after inoculation of the medium. The M. tuberculosis H37Rv parental strain, which is susceptible to PZA, was used as a positive control, while BCG Tice, which is naturally resistant to PZA, was used as a negative control.
Construction of protein expression plasmids.
The wild-type and mutant M. tuberculosis Rv2783c DNA fragments were PCR amplified from the genomic DNA of M. tuberculosis H37Rv and the clinical PZA-resistant mutant, respectively, using primer pair Rv2783cF and Rv2783cR (Table 2). The M. smegmatis PNPase protein, encoded by the M.smeg_2656 gene (2,292 bp), was expressed to serve as a positive control for the PNPase assays with the M. tuberculosis Rv2783 protein. Genomic DNA from wild-type M. smegmatis was used to amplify the M.smeg_2656 gene using primer pair Ms2656F and Ms2656R2 (Table 2). The M. tuberculosis Rel protein, encoded by the Rv2583c gene (2,373 bp), was also expressed to serve as a positive control for the ppGpp synthetase and hydrolysis assays with the Rv2783 protein. Genomic DNA from wild-type M. tuberculosis H37Rv was used to amplify the Rv2583c gene using primer pair RelF and RelR (Table 2). The PCR-amplified DNA fragments were digested with the appropriate enzymes and ligated to the pET-28a(+) plasmid vector that had been digested with the same enzymes to yield recombinant plasmids.
Protein expression and purification of the mycobacterial proteins.
The mycobacterial proteins were overexpressed in E. coli strain BL21(λDE3) containing the recombinant plasmids described above after induction with isopropyl 1-thiogalactopyranoside (IPTG), which was added to a final concentration of 0.5 mM, at 16°C overnight. The proteins, expressed as His-tagged derivatives, were purified from soluble bacterial extracts by sequential nickel-affinity, anion-exchange, and gel filtration chromatography steps.
The bacterial cells were harvested by centrifugation at 4°C, and all subsequent procedures were performed at 4°C. The supernatants containing the recombinant proteins were purified on Ni2+-nitrilotriacetic acid agarose (Qiagen, Hilden, Germany) that had been equilibrated with buffer A (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10 mM imidazole). The columns were washed with buffer A, and protein was eluted with buffer B (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 300 mM imidazole). The protein compositions of the fractions were monitored by SDS-PAGE. The appropriate protein fractions were pooled and loaded onto a Q-Sepharose column (180 ml) equilibrated with buffer C (20 mM Tris-HCl [pH 80], 5 mM NaCl) and eluted with buffer D (20 mM Tris-HCl [pH 8.0], 1 M NaCl) with a linear gradient (0 to 100%). The protein fractions were again monitored by SDS-PAGE, pooled, and gel filtered through a Superdex-200 size exclusion column equilibrated with buffer E (20 mM Tris-HCl [pH 8.0], 150 mM NaCl). The peak fractions were pooled, concentrated by centrifugal ultrafiltration, and stored at −80°C. The protein concentrations were determined using the Bio-Rad dye reagent with bovine serum albumin as the standard.
Isothermal titration calorimetry (ITC) binding study.
Titration of the mycobacterial recombinant proteins with POA or PZA was performed in a VP-ITC 200 microcalorimeter (MicroCal, LLC, USA) at room temperature. The VP-ITC system was used to determine whether any interaction existed between Rv2783 and POA or PZA. Initially, protein solutions consisting of 10 μM wild-type Rv2783 or mutant Rv2783Asp67Asn and M. smegmatis PNPase in 10 mM phosphate-buffered saline (pH 7.5) were titrated with 100 μM POA or PZA (molar concentration ratio, 1:10). Blank titration of the drug solution in the same buffer in the absence of the proteins was performed. Further, the proteins were titrated with saturated POA or PZA (up to a 1:200 molar concentration ratio) at pH 5.5 and pH 6.5. The binding constants were estimated from the isotherms obtained using the calorimetric analysis origin software. Typically, 20 injections with 2 μl per injection were made at 300-s intervals, and the reaction temperature was 25°C. The heat of the reaction per injection (microcalories per second) was determined by integration of the peak areas.
ssDNA and ssRNA catalytic activities of M. tuberculosis Rv2783.
The DNA/RNA polymerization and phosphorolytic activities of Rv2783 were determined according to previously published work on M. smegmatis PNPase (16), which served as a positive control, with some modifications.
ssDNA polymerization assay.
Reaction mixtures (10 μl) containing 20 mM Tris-HCl (pH 7.5), 5 or 10 mM MnCl2, 2 mM dADP, 0.1 μM (1 pmol) 36-mer ssDNA substrate (5′-FAM-GCCCTGCTGCCGACCAACGAAGGTAAAAAAAAAAAA-3′), and different concentrations of the wild-type and mutant Rv2783 and M.smeg_2656 (positive-control) proteins were incubated for 30 min at 37°C. The reactions were quenched by adding 10 μl of 90% formamide, 50 mM EDTA buffer. The samples were heated for 5 min at 100°C and then analyzed by capillary electrophoresis at BGI, Wuhan, China.
ssDNA 3′-phosphorylase assay.
Reaction mixtures (10 μl) containing 20 mM Tris-HCl (pH 7.5), 5 mM MnCl2, 30 μM (NH4)3PO4, 0.1 μM (1 pmol) 36-mer ssDNA substrate (5′-FAM-GCCCTGCTGCCGACCAACGAAGGTAAAAAAAAAAAA-3′), and different concentrations of the wild-type and mutant Rv2783 and M.smeg_2656 (positive-control) proteins were incubated for 60 min at 37°C. The reactions were quenched by adding 10 μl of 90% formamide, 50 mM EDTA buffer. The samples were heated for 5 min at 100°C and then analyzed by capillary electrophoresis at BGI, Wuhan, China.
RNA polymerization assay.
Reaction mixtures (10 μl) containing 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 2 mM ADP, 0.1 μM (1 pmol) 24-mer ssRNA substrate (5′-FAM-GGGUCGCAAUUGAUUCCGAUAGUG-3′), and different concentrations of the wild-type and mutant Rv2783 and Msmeg_2656 (positive-control) proteins were incubated at 37°C for 15 min. The reactions were quenched by adding 10 μl of 90% formamide, 50 mM EDTA buffer. The samples were heated for 5 min at 100°C and then analyzed by capillary electrophoresis at Sangon Biotech, Shanghai, China.
RNA 3′-phosphorylase assay.
RNA phosphorylase assay reaction mixtures (10 μl) containing 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.5 mM (NH4)3PO4, 0.1 μM (1 pmol) 24-mer ssRNA substrate (5′-FAM-GGGUCGCAAUUGAUUCCGAUAGUG-3′), and different concentrations of the wild-type and mutant Rv2783 and M.smeg_2656 (positive-control) proteins were incubated for 15 min at 37°C. The reactions were quenched by adding 10 μl of 90% formamide, 50 mM EDTA buffer. The samples were heated for 5 min at 100°C and then analyzed by capillary electrophoresis at Sangon Biotech, Shanghai, China.
Inhibition of Rv2783 ssDNA and RNA catalytic activities by POA or PZA.
Both ssDNA and ssRNA polymerization and phosphorylation assays were repeated as described above but in the presence of POA or PZA (100 μg/ml).
ppGpp synthetase and hydrolysis assays of the Rv2783 protein.
ppGpp synthetase and hydrolysis assays of the Rv2783 protein were done as described by Avarbock and colleagues on the ribosome-independent (p)ppGpp synthetase and hydrolase activities of Rel from M. tuberculosis (40) with some modifications.
ppGpp synthetase assays.
Reaction mixtures (25 μl) containing 50 mM HEPES (pH 8.0), 150 mM NaCl, 1 mM dithiothreitol (DTT), various concentrations of GDP (Sigma-Aldrich), ATP (Sigma-Aldrich), MgCl2, and 5 μM the wild-type and mutant Rv2783 proteins were incubated at 30°C for 1 h. The M. tuberculosis Rel protein was used as the positive control at a similar concentration. Three negative-control reactions in which the reaction mixtures included no wild-type or mutant Rv2783c protein, no GDP, and no ATP, respectively, were also run. The reactions were quenched by adding 1 μl of formic acid. Additionally, the reactions were repeated in the presence of POA (100 μg/ml) at different pHs to evaluate the inhibitory effects of POA. After 5 min at room temperature, the reaction tubes were centrifuged, their contents were filtered to remove the precipitated proteins, and the proteins were analyzed by high-pressure liquid chromatography (HPLC) using ppGpp, ATP, GTP, GDP, and AMP substrates as standards for both reactions.
ppGpp hydrolysis assays.
Hydrolysis reaction mixtures containing 50 mM HEPES (pH 8.0), 150 mM NaCl, 1 mM DTT, 0.1 μM ppGpp (TriLink Biotechnologies), various concentrations of MnCl2, and 5 μM the wild-type and mutant Rv2783 proteins were incubated at 30°C for 20 min. The M. tuberculosis Rel protein was used as the positive control. The reactions were quenched by adding 1 μl of formic acid. Additionally, the reactions were repeated in the presence of different concentrations of POA or PZA at different pHs to evaluate the inhibitory effects of POA and PZA. After 5 min at room temperature, the reaction tubes were centrifuged, their contents were filtered to remove the precipitated proteins, and the proteins were analyzed by HPLC as mentioned above.
HPLC analysis of the ppGpp synthetase and hydrolysis products.
The chromatographic system consisted of a Waters in-line degasser, a Waters 600 controller connected to a Waters 600 pump, a Waters 486 tunable absorbance detector, and a Waters Millennium workstation (version 3.05) chromatography manager. The nucleotides were separated by reversed-phase chromatography using a Waters Symmetry C18 3.5-μm (150- by 4.6-mm) column equipped with a NovaPak C18 Sentry guard column (Waters), adapting a prior method used for the separation of nucleotides (41). Separation was done using a method that started with a 70:30 concentration of buffer A-buffer B at a flow rate of 0.8 ml/min, which was changed in a linear gradient to 40:60 buffer A-buffer B after 30 min. From 30 to 60 min, the gradient was changed linearly from 40:60 buffer A-buffer B to 0:70:30 buffer A-buffer B-buffer C. The injection volume was 50 μl, and detection was by UV at 254 nm. Buffer A consisted of 5 mM t-butyl ammonium phosphate (PicA reagent), 10 mM KH2PO4, and 0.25% methanol adjusted to pH 6.9. Buffer B consisted of 5 mM t-butyl ammonium phosphate, 50 mM KH2PO4, and 30% methanol (pH 7.0). Buffer C was acetonitrile. Buffers A and B were degassed, passed through a 0.45-μm-pore-size filter to avoid the risk of microbial contamination, prepared fresh, and stored in the dark at 4°C prior to use. The Millennium workstation (version 3.05) chromatographic manager was used to pilot the HPLC instrument and to process the data throughout the method validation and sample analysis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Xiantao Zhang at Guangzhou Eggbio Co. Ltd. for helping us with the HPLC analysis of the ppGpp synthetase and hydrolase assay products in this study.
This work was supported by the National Natural Science Foundation of China (81572037), by Chinese Academy of Sciences grants (154144KYSB20150045, KFZD-SW-207), by University of Chinese Academy of Sciences scholarships (to M.N., J.M., H.M.A.H.), by the Ph.D. Start-up Fund of the Natural Science Foundation of Guangdong Province, China (2016A030310123 to J.G.), and a Key Project grant (SKLRD2016ZJ003) from the State Key Lab of Respiratory Disease, Guangzhou Institute of Respiratory Diseases, First Affiliated Hospital of Guangzhou Medical University, and was partially supported by the Guangzhou Municipal Industry and Research Collaborative Innovation Program (201508020248, 201604020019), by the National Mega-project of China for Innovative Drugs (2017ZX09201001-003-003), and by the Guangzhou Municipal Clinical Medical Center Program (155700012).
M. Njire, Jinsong Liu, W. W. Yew, E. Nuermberger, G. Lamichhane, and T. Zhang conceived and designed the experiments. M. Njire, N. Wang, B. Wang, J. Mugweru, Y. Tan, X. Cai, Y. Liu, J. Guo, and T. Zhang performed the experiments. M. Njire, N. Wang, Jinsong Liu, J. Mugweru, Y. Tan, J. Guo, B. Wang, S. Tan, Jianxiong Liu, W. W. Yew, E. Nuermberger, G. Lamichhane, and T. Zhang analyzed the data. Y. Tan, Jinsong Liu, S. Tan, Jianxiong Liu, and T. Zhang contributed reagents, materials, or analysis tools. M. Njire, Jinsong Liu, W. W. Yew, H. M. A. Hameed, E. Nuermberger, G. Lamichhane, and T. Zhang wrote the manuscript.
The Rv2783c gene has been filed as a patent in China (201610564946.X) for the molecular diagnosis of PZA-resistant Mycobacterium tuberculosis.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00070-17.
REFERENCES
- 1.World Health Organization. 2016. Global tuberculosis report 2016. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Heifets L, Lindholm-Levy P. 1992. Pyrazinamide sterilizing activity in vitro against semidormant Mycobacterium tuberculosis bacterial populations. Am Rev Respir Dis 145:1223–1225. doi: 10.1164/ajrccm/145.5.1223. [DOI] [PubMed] [Google Scholar]
- 3.Mitchison D. 1985. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 66:219–225. doi: 10.1016/0041-3879(85)90040-6. [DOI] [PubMed] [Google Scholar]
- 4.Dawson R, Diacon AH, Everitt D, van Niekerk C, Donald PR, Burger DA, Schall R, Spigelman M, Conradie A, Eisenach K, Venter A, Ive P, Page-Shipp L, Variava E, Reither K, Ntinginya NE, Pym A, von Groote-Bidlingmaier F, Mendel CM.. 2015. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: a phase 2b, open-label, partly randomized trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 385:1738–1747. doi: 10.1016/S0140-6736(14)62002-X. [DOI] [PubMed] [Google Scholar]
- 5.Scorpio A, Zhang Y. 1996. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med 2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 6.Zimhony O, Cox JS, Welch Vilcheze JTC, Jacobs WR. 2000. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat Med 6:1043–1047. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- 7.Boshoff HI, Mizrahi V, Barry CE. 2002. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J Bacteriol 184:2167–2172. doi: 10.1128/JB.184.8.2167-2172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Personne Y, Parish T. 2014. Mycobacterium tuberculosis possesses an unusual tmRNA rescue system. Tuberculosis 94:34–42. doi: 10.1016/j.tube.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Shi W, Chen J, Feng J, Cui P, Zhang S, Weng X, Zhang W, Zhang Y. 2014. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg Microbes Infect 3:e58. doi: 10.1038/emi.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dillon NA, Peterson ND, Rosen BC, Baughn AD. 2014. Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob Agents Chemother 58:7258–7263. doi: 10.1128/AAC.04028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, Chen J, Shi W, Cui P, Zhang J, Cho S, Zhang W, Zhang Y. 2017. Mutation in clpC1 encoding an ATP-dependent ATPase involved in protein degradation is associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg Microbes Infect 6:e8. doi: 10.1038/emi.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan Y, Hu Z, Zhang T, Cai X, Kuang H, Liu Y, Chen J, Yang F, Zhang K, Tan S, Zhao Y. 2014. Role of pncA and rpsA gene sequencing in detection of pyrazinamide resistance in Mycobacterium tuberculosis isolates from southern China. J Clin Microbiol 52:291–297. doi: 10.1128/JCM.01903-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makafe GG, Cao Y, Tan Y, Julius M, Liu Z, Wang C, Njire MM, Cai X, Liu T, Wang B, Pang W, Tan S, Zhang B, Yew WW, Lamichhane G, Guo J, Zhang T. 2016. Oxazolidinone resistance in Mycobacterium tuberculosis: what is the role of Cys154Arg mutation in the ribosomal protein L3? Antimicrob Agents Chemother 60:3202–3206. doi: 10.1128/AAC.00152-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole S, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 16.Unciuleac MC, Shuman S. 2013. Distinctive effects of domain deletions on the manganese-dependent DNA polymerase and DNA phosphorylase activities of Mycobacterium smegmatis polynucleotide phosphorylase. Biochemistry 52:2967–2981. doi: 10.1021/bi400281w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones GH, Bibb MJ. 1996. Guanosine pentaphosphate synthetase from Streptomyces antibioticus is also a polynucleotide phosphorylase. J Bacteriol 178:4281–4288. doi: 10.1128/jb.178.14.4281-4288.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cashel M, Rudd KE. 1987. The stringent response, p 1458–1496. In Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella typhimurium: cellular and molecular biology, vol 2 American Society for Microbiology, Washington, DC. [Google Scholar]
- 19.Avarbock D, Salem J, Li L-S, Wang Z-M, Rubin H. 1999. Cloning and characterization of a bifunctional RelA/SpoT homologue from Mycobacterium tuberculosis. Gene 233:261–269. doi: 10.1016/S0378-1119(99)00114-6. [DOI] [PubMed] [Google Scholar]
- 20.Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P. 2007. Modes and modulations of antibiotic resistance gene expression. Clin Microbiol Rev 20:79–114. doi: 10.1128/CMR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardenas PP, Carrasco B, Sanchez H, Deikus G, Bechhofer DH, Alonso JC. 2009. Bacillus subtilis polynucleotide phosphorylase 3′-to-5′ DNase activity is involved in DNA repair. Nucleic Acids Res 37:4157–4169. doi: 10.1093/nar/gkp314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardenas PP, Carzaniga T, Zangrossi S, Briani F, Garcia-Tirado E, Dehò G, Alonso JC. 2011. Polynucleotide phosphorylase exonuclease and polymerase activities on single-stranded DNA ends are modulated by RecN, SsbA and RecA proteins. Nucleic Acids Res 39:9250–9261. doi: 10.1093/nar/gkr635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carpousis AJ, van Houwe G, Ehretsmann C, Krisch HM. 1994. Copurification of E. coli RNAse E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 24.Miczak A, Kaberdin VR, Wei C-L, Lin-Chao S. 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci U S A 93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 26.Condon C. 2003. RNA processing and degradation in Bacillus subtilis. Microbiol Mol Biol Rev 67:157–174. doi: 10.1128/MMBR.67.2.157-174.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Condon C, Putzer H. 2002. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res 30:5339–5346. doi: 10.1093/nar/gkf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Archuleta RJ, Hoppes PY, Primm TP. 2005. Mycobacterium avium enters a state of metabolic dormancy in response to starvation. Tuberculosis 85:147–158. doi: 10.1016/j.tube.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino-Gelo F, Leibler S, McKinney JD. 2013. Dynamic persistence of antibiotic-stressed mycobacteria. Science 339:91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 30.Javid B, Sorrentino F, Toosky M, Zheng W, Pinkham JT, Jain N, Pan M, Deighan P, Rubin EJ. 2014. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc Natl Acad Sci U S A 111:1132–1137. doi: 10.1073/pnas.1317580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das B, Pal RR, Bag S, Bhadra RK. 2009. Stringent response in Vibrio cholerae: genetic analysis of spoT gene function and identification of a novel (p)ppGpp synthetase gene. Mol Microbiol 72:380–398. doi: 10.1111/j.1365-2958.2009.06653.x. [DOI] [PubMed] [Google Scholar]
- 32.Weiss LA, Stallings CL. 2013. Essential roles for Mycobacterium tuberculosis Rel beyond the production of (p)ppGpp. J Bacteriol 195:5629–5638. doi: 10.1128/JB.00759-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 34.Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ. 2012. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog 8:e1002946. doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Helden PD, Victor TC, Warren RM, van Helden EG. 2001. Isolation of DNA from Mycobacterium tuberculosis, p 19–30. In Mycobacterium tuberculosis protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 37.Wayne LG. 1974. Simple pyrazinamidase and urease tests for routine identification of mycobacteria. Am Rev Respir Dis 109:147–151. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Chen J, Shi W, Liu W, Zhang W, Zhang Y. 2013. Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg Microbes Infect 2:e34. doi: 10.1038/emi.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang T, Bishai WR, Grosset JH, Nuermberger EL. 2010. Rapid assessment of antibacterial activity against Mycobacterium ulcerans by using recombinant luminescent strains. Antimicrob Agents Chemother 54:2806–2813. doi: 10.1128/AAC.00400-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avarbock D, Avarbock A, Rubin H. 2000. Differential regulation of opposing RelMtb activities by the aminoacylation state of a tRNA · ribosome · mRNA · RelMtb complex. Biochemistry 39:11640–11648. doi: 10.1021/bi001256k. [DOI] [PubMed] [Google Scholar]
- 41.Huang D, Zhang Y, Chen X. 2003. Analysis of intracellular nucleoside triphosphate levels in normal and tumor cell lines by high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 784:101–109. doi: 10.1016/S1570-0232(02)00780-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.