

Abstract
Nucleic acid directed bioorthogonal reactions offer the fascinating opportunity to unveil and redirect a plethora of intracellular mechanisms. Nano‐ to picomolar amounts of specific RNA molecules serve as templates and catalyze the selective formation of molecules that 1) exert biological effects, or 2) provide measurable signals for RNA detection. Turnover of reactants on the template is a valuable asset when concentrations of RNA templates are low. The idea is to use RNA‐templated reactions to fully control the biodistribution of drugs and to push the detection limits of DNA or RNA analytes to extraordinary sensitivities. Herein we review recent and instructive examples of conditional synthesis or release of compounds for in cellulo protein interference and intracellular nucleic acid imaging.
Keywords: in situ drug release, in situ drug synthesis, nucleic acid encoded reactions, nucleic acid sensing
1. Introduction
Given the wide range of highly functionalized molecules present in cells, the design of target‐specific agents is the single most important challenge in medicinal chemistry and chemical biology. Most drugs and inhibitor molecules act on proteins. Small and mid‐sized molecules typically interact with more than one protein target which may lead to undesirable side effects. This problem is most pressing in cancer therapy, owing to the small differences between the molecular composition of cancer cells and that of healthy cells. Larger molecules such as oligonucleotides or antibodies show higher specificity. In a very actively pursued research field, small cytotoxic molecules are attached to antibodies.1 The antibody–drug conjugates are designed for targeted therapy, which is based on a selective enrichment of the cytotoxic drug. According to a perhaps more futuristic approach, oligonucleotide conjugates may provide an alternative means to target cytotoxic molecules to diseased cells. In fact, cancer cells can be recognized by their RNA expression, because endogenous mRNA molecules also encode for the proteins that are deregulated in cancer. The idea is to design oligonucleotide conjugates in a way that allows enrichment of the cytotoxic payload within the targeted cell. Disease‐relevant mRNA molecules may serve as templates that instruct the assembly, formation, or release of cytotoxic molecules. It is envisioned that the approach would provide a solution to the problem of unspecific biodistribution, as the bioactive compound is generated only in response to the presence of a specific genetic fingerprint of a cell. In principle, the simple and robust Watson and Crick base‐pairing rules provide target specificity.
RNA‐templated processes that involve the formation or cleavage of chemical bonds should proceed with high chemoselectivity to avoid chemical reactions with target‐unrelated cellular components. The chemoselective biocompatible chemistries developed to date have enabled site‐specific modifications of proteins, nucleic acids, lipids, and glycans, thus opening new possibilities for the investigation and modulation of biological processes.2 Similarly, biorthogonal reactions have been successfully applied to the formation and release of active drugs from prodrug species.3 Herein we describe biocompatible reactions applied in an RNA‐templated fashion. In this approach, reactive oligonucleotide conjugates are designed to undergo proximity‐driven chemical reactions upon adjacent hybridization with the disease‐encoding RNA instructor.
The templated hydrolysis of a nitrophenyl ester was the first example of a reaction applied to designing a selective nucleic acid triggered drug release.4 To date, many nucleic acid encoded chemistries have been developed and improved, and a broad set of biorthogonal reactions is available, including templated native chemical ligation (NCL), SN2/SNAr, Staudinger reaction, tetrazine‐mediated transfers, carbon–carbon bond formation reactions, amongst others.4, 5 Processes that turn on bioactivity bear resemblance to processes that turn on fluorescence upon chemical reactions on RNA targets, and often activation of fluorescence is used to demonstrate the feasibility of newly developed templated chemistries. As described later in this review, reaction systems for fluorescence activation are potentially useful for RNA imaging.
Nucleic acid templated chemical reactions may proceed with turnover in template. As a result, the concentration of the molecules formed by the template reaction may exceed the concentration of the RNA instructor by 100‐ to 1000‐fold. This amplification is a valuable asset when the concentration of the targeted RNA template is low.
Herein we focus on some illustrative examples of nucleic acid templated reactions for the in situ synthesis and uncaging of bioactive molecules, and nucleic acid sensing. Given the potential relevance for weakly expressed RNA targets, emphasis is placed on approaches that allow turnover in template (Figure 1). Reaction systems applied in DNA‐directed chemistry for compound discovery from synthetic libraries constitute a very broad area that is not discussed here (see ref. 6 for recent reviews on this topic).
Figure 1.
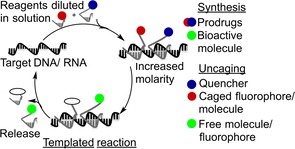
Nucleic acid triggered reactions proceeding with turnover.
2. In situ protein interference
RNA molecules overexpressed in diseased cells are regarded simultaneously as the recognition target and the instructor for conveniently designed conjugates triggering the: 1) synthesis, 2) arrangement or assembly, or 3) release of bioactive compounds. Representative examples of these nucleic acid instructed systems are described in the following paragraphs.
2.1. Nucleic acid programmed in situ synthesis
Erben et al.7 conceived a nucleic acid directed peptidyl transfer reaction for the synthesis of bioactive peptides proceeding by native chemical ligation (NCL; Figure 2). A thioester‐linked peptide–PNA conjugate (donor) and a cysteinyl peptide–PNA conjugate (acceptor) were designed so that the PNAs annealing to a targeted DNA triggered the ligation. The increase in effective concentration lowered the energy barrier for the transthioesterification, followed by irreversible S‐ to N‐acyl shift, leading to the formation of the full‐length bioactive peptide.
Figure 2.

DNA/mRNA‐triggered peptidyl transfer by NCL proceeds with turnover at nanomolar amounts of template, enabling the formation of full‐length bioactive peptides. Transferred peptidyl units are shown in red.
The reaction system was designed to enable the transfer of an alanine residue, which furnished the peptide ACPIAQK. This sequence mimics the N‐terminal fragment of the second mitochondria‐derived activator of caspase (Smac) protein. The Smac‐like peptide activates apoptosis by antagonizing the inhibitory interaction between caspase‐9, a protease involved in apoptosis, and the BIR3 domain of the X‐linked inhibitor of apoptosis protein (XIAP). Only trace amounts of the peptide product were formed when donor and acceptor conjugates were mixed at 1.5 and 0.75 μm, respectively. The presence of stoichiometric DNA template conferred a dramatic rate acceleration, affording 79 % ACPIAQK peptide within 1 h. The reaction proceeded with turnover, yielding over 60 % product yield after 5 h with only 75 nm DNA template. When performed in HEK293 cell lysate, the DNA‐templated transfer rescued the activity of BIR3‐inhibited caspase‐9.7
Vázquez and Seitz8 used templated transfer reactions as a means to place the formation of cytotoxic [KLAKLAK]2 peptide–PNA conjugates under the control of an RNA template (Figure 2). The aim was to design a reaction system that provides biological proof for turnover. Both donor and acceptor conjugates were decorated with octaarginine, a cell‐penetrating peptide (CPP). The PNA oligomers were designed to hybridize with a fragment of the XIAP RNA. The transfer product is oligocationic and will therefore be scavenged by the RNA template unless turnover provides excess product. Indeed, the RNA‐driven synthesis proceeded with turnover, and after 180 min 0.5 μm RNA template induced 3.75 μm product (75 % yield, acceptor 5 μm). This was sufficient to inhibit HeLa cell proliferation by over 50 %. Both the 14‐mer [KLAKLAK]2 peptide and the CPP were necessary for the conjugate to exert its biological effect. The formation of the full‐length [KLAKLAK]2 peptide was sequence specific. The presence of an RNA segment encoding for glyceraldehyde 3‐phosphate dehydrogenase (GADPH) did not trigger cytotoxic effects.8
Typically, mRNA templates are expressed at low‐nanomolar concentrations or below. In a very recent study, we scrutinized RNA‐templated peptidyl transfer chemistry in greater detail.9 The aim was to maximize the output of reactions on decreased amounts of template. We studied the RNA‐encoded formation of a 16‐mer peptide, which has been reported to inhibit the anti‐apoptotic protein Bcl‐xL and restore apoptosis in cancer cells. Surprisingly, we found that the length of the transferred peptide has no dramatic influence on the rate enhancements provided by the RNA template. Instead, we observed remarkable differences in the reaction rates of various aminoacyl thioesters. At sub‐stoichiometric loads of RNA template, alanyl and glycyl thioesters afforded the highest reactivity over background. Bulky/β‐branched amino acids such as isoleucine were unreactive. The distance between the annealing sites on the target template turned out to be an important factor as well. In this reaction system, two unpaired uridines juxtaposing the ligation site tripled the yields. This and the use of PNA oligomers that allow dynamic strand exchange under the reaction conditions provided a reaction system that gave 102 nm product with nanomolar amounts of RNA (1–10 nm).9
2.2. Nucleic acid programmed arrangement and rearrangement
In the previous section, we discussed the conditional in situ formation of bioactive molecules. The interaction with specific DNA/RNAs can also be used to trigger a conformational change that conditionally activates a bioactive compound. Self‐hybridizing peptide–PNA conjugates (also known as chimera nucleotide molecules), peptide–PNA/DNA or RNA duplexes and peptide–aptamer hybrids have been conceived to exploit the base pairing of nucleic acids as a means to induce a stapled conformation of peptides and/or tune their interaction with target proteins.10
Choi et al.11 pioneered the idea of modulating signal transduction pathways by controlling protein conformation and activity through mechanical forces. The mechanical stress is a consequence of the interaction between protein–DNA constructs (also termed chimeras) with single‐stranded DNAs (ssDNAs). In an initial experiment, they accomplished DNA‐dependent allosteric control of the maltose binding protein (MBP). The hybridization of a 60‐base‐long DNA component of the chimera with an ssDNA resulted in increased stiffness and mechanical stress of the overall system, affecting MBP affinity for its substrate (60 % lower maltose binding affinity).11 They later used this approach to modulate the activity of enzymes such as protein kinase A (PKA), guanylate kinase (GK), and Renilla luciferase (RLuc).12 PKA activity increased by a factor of 1.5 upon hybridization of the 60‐base DNA, covalently bound to the regulatory subunit of the enzyme, with complementary ssDNAs. In contrast, mechanical stress significantly decreased the enzymatic activities of GK and RLuc.12b,12c
Margulies and co‐workers13 devised a multivalent chimeric signal transducer. This construct was composed of: 1) a platelet‐derived growth factor (PDGF)‐binding aptamer, 2) a bivalent bis‐ethacrynic amide (EA) inhibitor of glutathione S‐transferase (GST), and 3) a fluorophore/quencher system for fluorescence monitoring (Figure 3, inset). An on/off reversible circuit of GST‐mediated catalysis was envisaged. The PDGF–aptamer–EA conjugate was found to inhibit the catalytic activity of GST, as a consequence of simultaneous binding of the EA units to GST. Binding of the PDGF aptamer to PDGF or formation of stable DNA–aptamer duplexes by strand displacement hampered binding to the bivalent EA inhibitor and restored GST activity.
Figure 3.

Logic circuit leading to the release of nitric oxide. Inset: multivalent chimeric signal transducer.
They validated the utility of this molecular actuator by testing the PDGF‐aptamer‐dependent activation of JS‐K, an anticancer prodrug. JS‐K is cleaved by GST, and the reaction produces toxic nitric oxide (NO) as output (Figure 3). To a solution containing JS‐K (45 μm), PDGF aptamer (750 nm) and glutathione (750 μm), were added in different combinations: GST (10 nm), PDGF (2 μm), and displacing DNA (2 μm). Significant amounts of NO (∼14 μm) were obtained only if GST was combined with PDGF and/or displacing DNA.13
Systems responsive to DNA, RNA, and protein inputs have found application in signal amplification, targeted disease diagnosis, signal thresholding, consensus gating, and feedback control.5a, 14 Hybridization with DNA/RNA has been used to modulate the binding of a phosphopeptide–PNA conjugate to the SH2 domain of Src kinase (Src‐SH2).15 The hybridization of non‐complementary PNA strands, flanking from the QpYEEI peptide ends, with an ssDNA target can induce increases or decreases in the affinity for Src‐SH2 (Figure 4 a). Notably, the DNA‐dependent affinity switching was reversible. A “DNA scavenger” was fully complementary to the “DNA activator” which offered toehold sequences to facilitate the release of non‐hybridized peptide–PNA by strand displacement (Figure 4 a, left).15a
Figure 4.

A) Interaction of peptide–PNA probe with single‐strand DNA modulates peptide binding to Src‐SH2. Left: extended peptide conformation displays increased affinity for Src‐SH2. Further interaction with scavenger DNA fully complementary to the activator DNA disassembles the chimera, releasing the unstructured peptide–PNA complex. Right: seamless hybridization leads to constrained peptides with low affinity. B) Activation of Src kinase by a PNA–phosphopeptide chimera. The inactive‐closed kinase conformation is stabilized by the interaction between pTyr527 and the SH2 domain. Addition of activator DNA/RNA induces strand exchange to form an activated chimera complex with high affinity for SH2, thereby displacing pTyr527 from the SH2 binding pocket. This triggers the activation of Src kinase. C) A hairpin‐structured peptide–PNA conjugate that signals the presence of the Src‐SH2 protein. Left: red labels are pyrene units.
In another experiment, Röglin used the PNA–peptide chimera for an RNA‐triggered activation of the Src kinase.15b Phosphorylation at Tyr527 stabilized a closed, inactive state of the kinase. The hairpin‐like constrained PNA–peptide chimera was found to have low affinity for the Src‐SH2 domain, and the closed state of Src‐kinase prevailed. However, addition of the “DNA activator” increased SH2 affinity. The activated PNA–peptide chimera was found to bind to the Src‐SH2 domain, thereby pushing the equilibrium to the opened activated state of Src kinase (Figure 4 b).15b When the EPQpYEEIP sequence was armed with complementary PNA strands, the conjugate adopted a stem–loop structure (Figure 4 c). Binding to Src‐SH2 favors stem opening, which disrupts the proximity between two terminally appended chromophores. PNA labelling with two pyrene units afforded so‐called hairpin peptide beacons (HPBs), which signaled the presence of Src‐SH2 by up to 11‐fold increases of the pyrene monomer emission.15c
A similar system was reported by Plaxco and co‐workers. They developed stem–loop peptide–PNA conjugates for the detection of anti‐HIV‐1 p17 IgG antibodies. A 6‐mer epitope from the HIV p17 protein served as recognition element. In the presence of the target IgGs, the hairpin assumed an extended conformation, which allowed BODIPY fluorescence emission, otherwise quenched by a proximal tryptophan residue via photoinduced electron transfer.16 Lim et al.17 adopted the use of peptide–oligonucleotide beacon for the detection of influenza A subtype H1N1 viruses (H1N1). Peptide interaction with H1N1 caused hairpin opening and, in turn, a 23‐fold increased fluorescence emission due to the separation of a trimethine cyanine fluorophore (Cy3) from a black hole quencher‐2.17
Ranallo et al.18 designed a versatile hybrid hairpin that can accommodate different target molecules for mono‐ and bivalent protein sensing (Figure 5). A DNA hairpin was hybridized “head‐to‐head” with two oligonucleotide strands bearing the recognition elements for the targeted protein. In the absence of the protein target, the DNA stem–loop is closed and the fluorescence quenched. The binding of mono‐ or bivalent macromolecules opens the stem and triggers fluorescence emission. This system showed no cross‐reactivity and fast responsiveness (<10 min). Anti‐DNP (dinitrophenol), anti‐FLAG (8‐mer FLAG peptide), anti‐HIV (13‐mer epitope from HIV p17 matrix protein) IgG antibodies, as well as monovalent targets such as anti‐Dig Fab fragment (fragment antigen‐binding digoxigenin) and TBP (transcription factor TATA binding protein) were detected at nanomolar concentrations in serum.18
Figure 5.

DNA hairpin system for sensing of mono‐ and bivalent proteins.
Fischbach et al.19 described a hairpin‐constrained peptide that provided pyrene excimer emission upon cleavage by proteases such as matrix metalloprotease 7 (MMP‐7), which is a biomarker in human blood serum. The RPLALWRS peptide substrate was equipped with two complementary PNA arms. One PNA arm contained two pyrene units within the stem‐forming PNA segment. The other arm featured an anthraquinone unit. In the closed hairpin conformation, the anthraquinone quenched long‐lived pyrene excimer signaling. MMP‐7 cleavage of the RPLALWRS peptide prompted hairpin disruption and induced a 50‐fold increase in pyrene excimer emission. This was sufficient to detect the cancer biomarker MMP‐7 in human blood serum.19
The Winssinger research group recently showed that hairpin‐constrained self‐hybridization by PNA conferred phosphatase activity to a hexapeptide containing His, Ser, Asp, and Lys (commonly found in hydrolytic enzymes).20 When folded as a hairpin, this peptide hydrolyzed a phosphate ester yielding a quinazolinone‐based fluorescent precipitate. The hydrolytic efficiency was allosterically controlled by the extent of overlap of the flanking PNA monomers, and toehold strand displacement compromised hybridization‐induced peptide folding. This responsiveness of the catalytic activity to the presence of competing oligonucleotides strands can be exploited for protein interference as well as nucleic acid sensing.20
Chu et al.22 conceived an miRNA‐triggered activation of a DNA–small‐molecule chimera for protein inhibition (Figure 6). The interaction of the DNA conjugate duplex with miRNA, through a fully complementary toehold, triggered the release of the ssDNA–small‐molecule conjugate and its binding to the target protein (allosteric control of the activity). They proved the effectiveness of their system by designing a conjugate that offered a toehold for interaction with miR‐21 (overexpressed in various cancers),21 and a small molecule that is a strong binder of human carbonic anhydrase II (hCA‐II). The activated single‐stranded conjugate interacted with the hCA‐II active site with a K i value of 3.12 μm upon interaction with miR‐21.22
Figure 6.

Allosteric control of binding to human carbonic anhydrase II (hCA‐II) through strand displacement.
In 2014, Abendroth and Seitz developed a peptide–phosphorothioate antisense oligonucleotide (PSAO) hybrid to combine the advantages of protein interference and antisense.23 A strain‐promoted [2+3] cycloaddition in buffer allowed the conjugation of up to four peptidomimetics with the phosphorothioate‐modified oligonucleotides. This method was used to decorate an antisense oligonucleotide downregulating c‐Flip (inhibitor of the extrinsic apoptotic pathway) with a Smac‐peptidomimetic XIAP inhibitor (protein upregulated in cancer cells), as shown in Figure 7. The two moieties acted synergistically, and the peptide–PSAO conjugates were able to decrease the viability of cancer cells to 20 %.23
Figure 7.

Strain‐promoted cycloaddition yields peptide–PSAO conjugates. The conjugates comprise an antisense oligophosphorothiate for downregulation of c‐flip mRNA (master regulator of the extrinsic pathway of apoptosis) and a Smac peptidomimetic for antagonizing the inhibitor of apoptosis proteins (IAPs; master regulators of the intrinsic pathway of apoptosis).
2.3. Triggered release of bioactive molecules
Bioorthogonal reactions catalyzed by spatial proximity24 and photoinduction5d can be used to trigger the release of prodrugs upon interaction with unique or overexpressed RNA or DNA sequences. Winssinger and co‐workers developed a para‐azidobenzyl‐based immolative linker for the uncaging of functional molecules (Figure 8 a). A so‐called pro‐functional molecule and a reducing agent were covalently bound to two PNAs annealing with a three‐base gap on a DNA template. The spatial proximity triggered cleavage upon azide reduction, which resulted in the release of estradiol, rhodamine, or doxorubicin (with the templated release of estradiol reaching 85 % yield after 30 min).25
Figure 8.
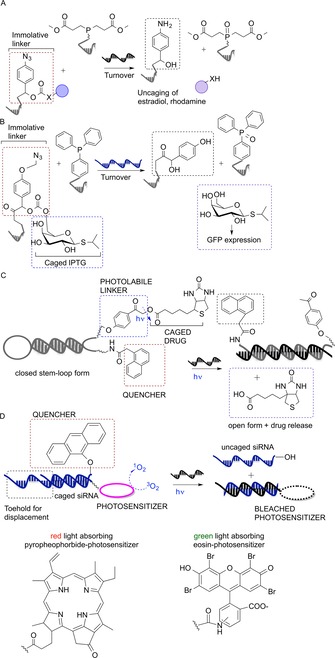
A) p‐Azidobenzyl‐based immolative linker for the uncaging of functional molecules. B) 23S rRNA‐dependent IPTG release system in bacterial cells triggers GFP expression. C) Photoinduced cleavage of a phenacyl ester linker triggers the release of biotin. D) Uncaging of siRNA to modulate gene expression in HeLa cells. Red/green light‐responsive photosensitizer catalyzes 1O2 formation, which cleaves the 9‐alkoxyanthracenyl‐based quencher, thereby releasing active siRNA.
Abe and colleagues devised a 23S rRNA‐directed isopropyl‐β‐d‐thiogalactopyranoside (IPTG) release system in a bacterial cell which relied on a Staudinger reaction followed by 1,6‐elimination (Figure 8 b). Two DNA probes were designed to anneal to a specific fragment of the E. coli 23S rRNA, with one unpaired nucleotide as optimum spacer. One probe carried triphenylphosphine as reducing agent, while the other strand presented a caged IPTG. Probes were introduced in E. coli by incubation of bacterial cells in buffer containing 0.1 % sodium dodecyl sulfate. The sequence‐dependent liberation of IPTG induced the expression of GFP in E. coli. In an important control experiment, cells were charged with scrambled sequence conjugates. Fluorescence remained low, which provides evidence for the target specificity of the method.26
Conditional phototriggered drug release has garnered attention as a convenient strategy to improve the control of release. However, to be applicable to live species, photodeprotection of caged bioactive molecules must proceed at non‐harmful wavelengths and with high quantum yield.27
Saito and co‐workers were the first to propose a molecular beacon strategy for the conditional photorelease of molecules (Figure 8 c). Their stem‐like probe carried two photoactive groups at the 3′ and 5′ ends: a photocleavable phenacyl ester moiety and a substituted naphthalene moiety (quencher), spatially close to each other in the probe‐closed conformation. Upon interaction with a DNA target, the stem opened up, and the para‐hydroxyphenacyl ester could be cleaved via a triplet excited state by UV irradiation (312 nm), thus releasing biotin.28 Tanabe et al. improved this strategy by developing a similar system, but with irradiation at 365 nm.29 Röthlingshöfer et al.30 reported the templated photocleavage of a nitrobenzyl‐type linker occurring at 405 nm. A light‐responding thioxanthenone sensitizer, hanging from a 7‐mer PNA strand, mediated the energy transfer to a complementary PNA, resulting in cleavage of the appended linker and release of rhodamine. The templated reaction showed a 20‐fold rate enhancement over background.30
The research groups of Mokhir and Gothelf developed gene‐specific photosensitizer‐containing probes for the release of singlet oxygen (1O2) as cytotoxic drug. Pyropheophorbide or eosin‐based photosensitizers were appended at the 5′ end of a nucleic acid sequence and silenced by a quencher appended at the 5′ end of a complementary strand. The interaction of the latter with a nucleic acid target and exposure to red or green light (pyropheophorbide and eosin, respectively) triggered generation of cytotoxic 1O2 in vitro and in live cells. Pyropheophorbide photosensitizer reactions proceeded with remarkable turnover and high sequence specificity. A turnover number of 377 was reached within 40 min, and the presence of a single mismatch generated a background‐like signal.31 Eosin‐based photosensitizers allowed facile detection due to the generation of a reacted fluorescent probe, but the number of turnovers achieved in the same time was roughly one order of magnitude smaller. No binding occurred to templates with a single mismatch.32
More recently, Meyer and Mokhir showed another application of conditional phototriggered reaction: the photoactivation of a caged siRNA, by using nontoxic wavelengths (red and green), with the aim to modulate gene expression (Figure 8 d). The 9‐alkoxyanthracenyl moiety at the 5′ end of an siRNA functioned as 1O2‐responsive fragment caging the siRNA activity in the quiescent state. The red/green‐light‐responding photosensitizer at the 3′ end of the complementary strand catalyzed 1O2 production from 3O2 upon irradiation, triggering the uncaging. In contrast to the previously developed 9,10‐dialkoxyanthracene,33 the cleavage of the 9‐alkoxyanthracenyl moiety proceeded with no need for additional electrons, and the photocatalyst was quickly bleached after irradiation, thus stopping the unwanted production of extra singlet oxygen molecules.34 Free siRNAs, delivered inside HeLa cells with Lipofectamine, were able to target two designed genes, and their photorelease resulted in the inhibition of gene expression. A system based on the uncaging of reactive 1O2 from its prodrug form (3O2) was exploited for selective photosensitizer‐based therapies, as well as for the detection of nucleic acids.35
3. Uncaging of smart probes for nucleic acid imaging
Nucleic acid detection is a frequently explored testing ground for the development of new templated chemistries, and the sequence‐specific release of fluorescent molecules has enabled the selective detection of nucleic acid sequences within living systems.5f The typical nucleic acid templated reaction relies on adjacent annealing of two comparatively short probes, which proceeds with higher specificity than hybridization of one long probe. Therefore, nucleic acid templated reactions can discriminate a single base mismatch in the DNA or RNA analyte. Furthermore, turnover may provide for signal amplification, which increases the sensitivity of nucleic acid detection. On the other hand, turnover should be avoided in RNA imaging because signaling molecules would no longer remain localized to the target. In this section, we review examples of templated bioorthogonal chemistries applied to nucleic acid sensing. Templated Staudinger reaction, photocatalytic reduction of azides, tetrazine transfers, native chemical ligation, Wittig olefination, and aldol condensation reactions have elicited robust fluorescence enhancements upon target interaction.
3.1. Staudinger‐triggered release of fluorescence
Templated Staudinger reactions have attracted attention for the development of robust fluorescence activation systems that overcome the limitations of previously developed systems suffering from low reaction rates and partial bioorthogonality (i.e., fluorescence uncaging by SN2 displacement).36 Application of the Staudinger reduction of azides to a templated format has enabled the detection and imaging of RNAs in mammalian and bacterial cells.
Franzini and Kool introduced a versatile fluorescence uncaging system based on templated Staudinger reduction of so‐called Q‐STAR probes (quenched‐Staudinger triggered α‐azidoether release), which responded to a specific RNA target expressed in E. coli and S. enterica. They designed a Q‐STAR DNA probe, modified at the 5′ end with a fluorophore and a quencher covalently bound to the DNA strand by a α‐azidoether linker, and a TPP‐DNA probe carrying a triphenylphosphine (TPP) at the 3′ position (Figure 9 a). When these two probes hybridize adjacently with the bacterial RNA target, the TPP reduces the azide, triggering cleavage of the linker with subsequent strong fluorescence enhancement. Notable sequence fidelity and significant signal amplification under isothermal conditions enabled the discrimination of closely related RNA sequences both in vitro and in the aforementioned organisms.37 This concept was further elaborated in the design of 2‐STAR probes containing two quencher groups tethered again by azide‐based linkers to a DNA strand. In this case, the fluorescence was released only after a two‐step mechanism, which was demonstrated to be effective for decreasing auto‐fluorescence background, but also compromised sequence discrimination.38
Figure 9.

A) Templated Staudinger reduction of azides to unleash fluorescence signal. B) Introduction of an immolative linker to trigger fluorescence signal. C),D) Tetrazine‐mediated transfer (TMT) to uncage a fluorescence signal. E) Catalyzed transfer of a reporter group by templated acyl transfer. Red labels: quenchers; blue labels: quenched fluorophores; purple labels: free fluorophores; white circles: reacted/transferred quencher.
Very recently, Velema and Kool developed another two‐step mechanism based on a quenched autoligation (QUAL)36a, 39/QSTAR‐templated reaction cascade. Upon recognition of a target, the phosphorothioate hanging from a QUAL probe attacks the electrophilic carbon atom of a butyl linker conveniently modified with a leaving group, yielding the ligated oligonucleotides tethered by a butyl linker. The newly ligated strand spontaneously dissociates from the former target and acts as template itself, instructing the Staudinger reaction between two QSTAR probes. One of these QSTAR probes carries a triphenylphosphine, the other bears a quenched fluorophore/α‐azidoether‐linked quencher system. Fluorescence unquenching allowed the detection of specific targets down to 10 picomolar concentrations in buffer and a single point mutation of 16S rRNA in H. pylori lysate.40
The idea of doubling the reactive probes was also applied by Winnsinger who developed the templated reduction/activation of bis‐rhodamine PNA/guanidinium modified PNA (GPNA) probes for the identification of mi‐RNA‐21. GPNA monomers confer cell‐penetrating properties to the conjugates due to similarity with oligoarginine‐based CPPs.41 The amplified miR‐21‐templated phosphine reduction of a bis‐rhodamine–PNA/GPA allowed miR‐21 identification and imaging, both in vitro and in breast cancer cell lines.42 More recently, Abe and co‐workers exploited the Staudinger reduction to trigger luminescence of a lanthanide‐based DNA probe in an RNA‐directed fashion. The activation of a phenanthridinone‐based antenna upon templated azide reduction produced a long‐lived luminescence. This was due to autofluorescence of the uncaged lanthanide complex (Eu3+ or Tb3+) upon recognition of target 23S rRNA in E. coli.43
3.2. Immolative linkers to unleash fluorophores
Phosphines, used as reducing agents in the Staudinger reaction, are prone to oxidation. Thus, stoichiometric amounts of ascorbic acid or excess phosphine probe must be added to the reaction media, which can decrease cellular viability and increase non‐templated reaction (background). Other strategies have been devised to overcome these limitations.
Liu and co‐workers discovered the photocatalytic reduction of azides with catalytic amounts of [Ru(bpy)3]2+ and stoichiometric amounts of ascorbate or NADPH.44 The Winssinger group successfully applied this reaction to a nucleic acid template arming two γ‐serine‐modified PNA strands, one with a [Ru(bpy)3]2+ analogue, and one with a pro‐fluorescent rhodamine through an immolative linker (Figure 9 b). Photoexcitation (455 nm) triggered fluorescence emission with only 2 % of Ru‐derivatized probe if a matching DNA was present.45 The RuII‐catalyzed photoreduction of the proximal azide to aniline led to the decomposition of the immolative linker, resulting in rhodamine uncaging. The reaction proceeded with 4000 turnovers (over 24 h), with picomolar concentrations of template, in buffer. This enabled the selective imaging of miR‐21 and miR‐31 in human breast BT474 and cervical HeLa cancer cells, respectively. The reactive probes were introduced within BT474 or HeLa cells by using a pore‐forming peptide (streptolysin‐O) or transfection.46 This system was tested for the specific recognition and imaging of mature miRNAs in vivo. Ruthenium and rhodamine‐azide‐modified PNA probes were injected in wild‐type zebrafish embryos raised in the dark. After irradiation at 450 nm and rhodamine uncaging, miR‐9, miR‐196, and miR‐206 were imaged in zebrafish with striking specificity, almost no background, and in a tissue dependent‐manner (probes imaging miR‐9 yielded distinct localization in brain).47
Very recently, the same group reported an improvement of the templated photocatalyst by introducing a pyridinium‐based immolative linker in place of the azido moiety. The templated photocatalytic reaction proceeded with enzyme‐like efficiency (10−5 m −1 s−1) and stunning turnover efficiency (10 h−1) when the two counterparts were appended to two 5‐mer PNAs (short PNAs with low T m values facilitate a dynamic strand exchange).9 The 5‐mer PNA was inserted in a beacon architecture composed of an 8‐mer l‐γ‐PNA for DNA recognition and a 5‐mer d‐γ‐PNA to ensure sufficient specificity for nucleic acid sensing. The interaction with sub‐stoichiometric DNA template and light irradiation at 455 nm induced hairpin opening and triggered photocatalytic reduction of the pyridinium linker.48
3.3. Tetrazine‐based fluorogenic reactions in live cells
The research group of Devaraj has reported several examples of the application of highly fluorogenic tetrazine cycloadditions for the detection of oligonucleotide target sequences in live cells.49 In 2014, they reported a successful example of tetrazine‐mediated transfer (TMT) for the detection of endogenous miR‐21 in two cancer cell lines. A tetrazine moiety, responsible for the fluorescence quenching of a BODIPY dye (by through‐bond energy transfer, TBET), reacted with a 7‐azabenzonorbornadiene (dienophile) through a retro Diels–Alder reaction to form a dihydropirazine (Figure 9 c). This species spontaneously aromatized to yield a pyridazine as the final product. The release of this highly fluorescent TMT product allowed the detection of miR‐21 with remarkable sensitivity (down to low picomolar amounts), displaying single‐base mismatch recognition.49c More recently, the authors described a novel bioorthogonal way to unmask the fluorescence of a near‐infrared‐emitting fluorophore caged with vinyl ether (Figure 9 d). Live cell detection by fluorogenic tetrazine uncaging in transiently transfected Chinese hamster ovary (CHO) cells showed high fluorescence in the presence of target mRNA versus control.49e
3.4. Native chemical ligation based detection
The development of nucleic acid sensing by template‐instructed native chemical ligation (NCL) has been extensively pursued in our group. This reaction system relies on the interaction between two conveniently modified oligonucleotides (donor thioester‐ON and acceptor cysteinyl‐ON), which, by hybridizing with a target DNA or mRNA template, trigger NCL and generate a measurable signal.50
Grossmann and Seitz were the first to develop the DNA‐directed transfer of a reporter group from a thioester‐linked donor probe to an isocysteinyl‐acceptor probe (Figure 9 e). The reporter was a Dabsyl group, quenching either the FAM donor or TAMRA acceptor probes. The fluorescence‐based readout of this reaction allowed the identification of single mismatches with high accuracy and pronounced signal amplification, enabling the detection of RasT DNA.50d
In 2014, it was demonstrated that templated NCL also occurs in heterogeneous systems, i.e., on the surface of streptavidin‐modified quantum dots (QD). QD‐bound 5′‐cysteinyl‐modified acceptors armed with a biotin at the 3′ end hybridized with the DNA analyte, which, in turn, recruited a thioester‐linked cyanine dye placed at the 3′ end of a DNA/PNA strand. The thioester‐linked fluorophore was transferred via NCL to the acceptor once spatial proximity between donor and acceptor was reached, triggering significant fluorescence resonance energy transfer (FRET) signal enhancements. This method is an easily applicable detection methodology with, in principle, no restriction concerning the choice of the fluorophores.50j
Roloff and Seitz pushed the limits of the templated NCL, affording PNA ligation during polymerase chain reaction (PCR). The use of attomolar amounts of DNA template represented the main advantage of such development. The overall optimization of the process required the adaptation to challenging requirements such as: 1) stability of reactants at high temperatures (up to 72 °C during PCR), 2) extremely rapid reaction kinetics (1 min time elapsed between annealing and primer extension), and 3) the affinity of ligated PNA for the DNA template must be tuned to avoid hampering DNA polymerase activity. Internal labeling of the PNA strands with rhodamine and fluorescein‐modified monomers enabled FRET reaction monitoring. β‐Alanine thioesters and isocysteine pairs displayed remarkable stability against hydrolysis, low background, and fast NCL kinetics.50h,50m
3.5. Templated formation of double bonds
Templated Wittig olefination and aldol‐type chemistry constitute two examples of biocompatible reactions affording fluorogenic compounds by carbon–carbon bond formation.51 Chen et al.52 devised a sensing system that relies on templated Wittig reaction and host–guest complex formation. A DNA template catalyzed the transfer of a benzylidene moiety from a phosphonium–DNA donor conjugate to a benzaldehyde–DNA acceptor yielding a fluorescent stilbene unit. The newly formed stilbene was efficiently encapsulated in the hydrophobic α‐cyclodextrin binding pocket. α‐Cyclodextrin hosted stilbene and increased fluorescence emission (Figure 10 a). A 100‐fold fluorescence enhancement at 380 nm was obtained after 24 h with sub‐stoichiometric amounts of DNA template (0.1 equiv); 10−4 equivalents of DNA yielded a turnover of 84 after 24 h, which allowed the specific detection of template at 40 pm.52
Figure 10.
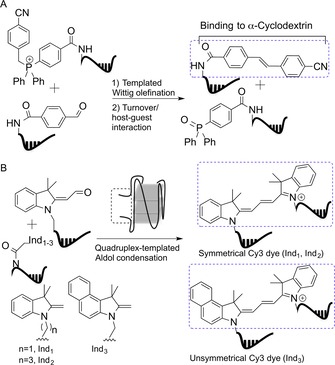
A) Templated fluorophore synthesis and host–guest interaction lead to fluorescence exacerbation. B) Quadruplex‐templated aldol condensation yields fluorogenic Cy3 dyes.
The research group of Ladame used aldol condensation for the in situ synthesis of fluorescent probes detecting G‐quadruplexes and DNAs.53 Symmetrical or unsymmetrical Cy3 dyes were formed upon reaction of two non‐fluorescent precursors: 2‐methylene indoline and aldehyde derivative‐modified PNAs, at physiological pH (Figure 10 b). The PNA strands were designed in a way that the fluorogenic reaction occurred only if a quadruplex was formed. This feature enabled the specific detection of ckit21T quadruplex, a promoter region of c‐kit gene, down to 500 nm concentration.53a The introduction of a modified aldehyde derivative led to the formation of a pentamethine cyanine dye (Cy5) in a similar fashion. Cy3 and Cy5 were used for simultaneous sensing of DNA hairpins and quadruplexes (fluorescence emission recorded at 540 and 660 nm, respectively).53b
Ladame and colleagues developed a sensing approach for the quantification of circulating miRs as biomarkers for prostate cancer that could compete with real‐time quantitative PCR (RT‐qPCR; the gold standard technique). The method relied on the templated synthesis of fluorescent coumarin–PNA conjugates from a quenched coumarin–PNA and a PNA modified with a butanethiol group at the C terminus. The quenched coumarin probe was obtained by aldol condensation of coumarin 334 and 3‐formylbenzoic acid. Upon hybridization of the two PNAs to complementary miRNAs, Michael addition of the thiol group to the α,β‐unsaturated ketone of the coumarin unit triggered the release of fluorescence. This system allowed the in vitro sequence‐specific detection of miR‐141 and miR‐375 in a concentration‐dependent manner and a two‐digit nanomolar detection limit.54
4. Conclusions
Nucleic acid instructed reactions are powerful and versatile tools that are increasingly applied in drug discovery. Herein we have described nucleic acid encoded chemical reactions developed to explore options for gene‐expression‐specific synthesis inside cells. The idea is to use products formed upon RNA instructed synthesis as agents that interfere with deregulated proteins. Such an endeavor involves many challenges. The chemical reactions must proceed with very high chemoselectivity to avoid perturbations of bystander molecules. Furthermore, RNA is expressed at low concentrations. Therefore, RNA‐templated reactions should proceed with turnover to trigger enough product for protein interference. A key challenge is delivery of the nucleic acid conjugates to cells. At first glance, the hurdles seem insurmountable. However, the idea of a targeted therapy based on RNA‐templated synthesis offers the promise to overcome unspecific biodistribution and to decrease side effects in cancer therapy. Although the idea still sounds like science fiction, recent progress described in this review has moved the field closer to real‐world applications. Considering the wealth of new conjugation chemistries, the biocompatibility of chemical reactions no longer appears to be an issue. The templated reactions in current use proceed with very high chemoselectivity, which almost routinely allows reactions in complex environments such as cells, cell lysates, and human serum. Many reactions provide for turnover, which has been used in the detection of minute (picomolar) amounts of template RNA outside and within cells. Cellular delivery is still the most significant challenge. However, the vast literature precedence for systemic activity of antisense oligonucleotides55 and the recent application of RNA‐templated chemistry inside live zebrafish47 show that this challenge can be overcome.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Margherita Di Pisa graduated from the University of Florence (Italy) in 2009, where she also obtained a PhD in Chemistry in 2013. The focus of her research was the design and setup of a peptide‐based diagnostic for autoimmune diseases. She then moved to the Laboratory of Biomolecules at the University Pierre and Marie Curie in Paris to work as a postdoctoral researcher, studying the translocation processes of cell‐penetrating peptides. In September 2015 she joined the Seitz research group at the Humboldt University of Berlin. Her current research interests include oligonucleotide‐encoded reactions for the in situ synthesis of bioactive molecules.

Biographical Information
Oliver Seitz studied chemistry at Johannes Gutenberg University Mainz (Germany), where he obtained his PhD in organic chemistry. After postdoctoral research at The Scripps Research Institute in La Jolla, California (USA), he moved to the Technical University Karlsruhe. He became group leader at the Max Planck Institute for Molecular Physiology and obtained the venia legendi in Organic Chemistry from the Technical University Dortmund. In 2003 he was appointed Full Professor at the Humboldt University of Berlin. He is broadly interested in fashioning nucleic acid and protein molecules to provide tools for life sciences.

Acknowledgements
The authors gratefully acknowledge the Alexander von Humboldt Foundation for a grant to M.D.P. and the European Research Commission for the ERC Advanced Grant to O.S..
M. Di Pisa, O. Seitz, ChemMedChem 2017, 12, 872.
Contributor Information
Dr. Margherita Di Pisa, Email: margherita.dipisa@chemie.hu-berlin.de
Prof. Dr. Oliver Seitz, Email: oliver.seitz@chemie.hu-berlin.de
References
- 1.
- 1a. Agarwal P., Bertozzi C. R., Bioconjugate Chem. 2015, 26, 176–192; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Chari R. V. J., Miller M. L., Widdison W. C., Angew. Chem. Int. Ed. 2014, 53, 3796–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3872–3904; [Google Scholar]
- 1c. Polakis P., Pharmacol. Rev. 2016, 68, 3–19. [DOI] [PubMed] [Google Scholar]
- 2. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133. [Google Scholar]
- 3.
- 3a. Li J., Chen P. R., Nat. Chem. Biol. 2016, 12, 129–137; [DOI] [PubMed] [Google Scholar]
- 3b. Patterson D. M., Nazarova L. A., Prescher J. A., ACS Chem. Biol. 2014, 9, 592–605; [DOI] [PubMed] [Google Scholar]
- 3c. Row R. D., Prescher J. A., ACS Cent. Sci. 2016, 2, 493–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma Z., Taylor J.-S., Proc. Natl. Acad. Sci. USA 2000, 97, 11159–11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Battle C., Chu X., Jayawickramarajah J., Supramol. Chem. 2013, 25, 848–862; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Diezmann F., Seitz O., Chem. Soc. Rev. 2011, 40, 5789–5801; [DOI] [PubMed] [Google Scholar]
- 5c. Li X., Liu D. R., Angew. Chem. Int. Ed. 2004, 43, 4848–4870; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4956–4979; [Google Scholar]
- 5d. Jacobsen M. F., Cló E., Mokhir A., Gothelf K. V., ChemMedChem 2007, 2, 793–799; [DOI] [PubMed] [Google Scholar]
- 5e. Gorska K., Winssinger N., Angew. Chem. Int. Ed. 2013, 52, 6820–6843; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6956–6980; [Google Scholar]
- 5f. Shibata A., Abe H., Ito Y., Molecules 2012, 17, 2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Brenner S., Lerner R. A., Proc. Natl. Acad. Sci. USA 1992, 89, 5381–5383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. R. A. Goodnow, Jr. , Dumelin C. E., Keefe A. D., Nat. Rev. Drug Discovery 2017, 16, 131–147; [DOI] [PubMed] [Google Scholar]
- 6c. Yuen L. H., Franzini R. M., ChemBioChem 2017, 18, 829–836; [DOI] [PubMed] [Google Scholar]
- 6d. Zambaldo C., Barluenga S., Winssinger N., Curr. Opin. Chem. Biol. 2015, 26, 8–15; [DOI] [PubMed] [Google Scholar]
- 6e. Zimmermann G., Neri D., Drug Discovery Today 2016, 21, 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erben A., Grossmann T. N., Seitz O., Angew. Chem. Int. Ed. 2011, 50, 2828–2832; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2880–2884. [Google Scholar]
- 8. Vázquez O., Seitz O., Chem. Sci. 2014, 5, 2850–2854. [Google Scholar]
- 9. Di Pisa M., Hauser A., Seitz O., ChemBioChem 2017, 18, 872–879. [DOI] [PubMed] [Google Scholar]
- 10. Röglin L., Seitz O., Org. Biomol. Chem. 2008, 6, 3881–3887. [DOI] [PubMed] [Google Scholar]
- 11. Choi B., Zocchi G., Canale S., Wu Y., Chan S., Perry L. J., Phys. Rev. Lett. 2005, 94, 038103. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Choi B., Zocchi G., J. Am. Chem. Soc. 2006, 128, 8541–8548; [DOI] [PubMed] [Google Scholar]
- 12b. Wang A., Zocchi G., Biophys. J. 2009, 96, 2344–2352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Tseng C.-Y., Zocchi G., J. Am. Chem. Soc. 2013, 135, 11879–11886. [DOI] [PubMed] [Google Scholar]
- 13. Peri-Naor R., Ilani T., Motiei L., Margulies D., J. Am. Chem. Soc. 2015, 137, 9507–9510. [DOI] [PubMed] [Google Scholar]
- 14. Han D., Kang H., Zhang T., Wu C., Zhou C., You M., Chen Z., Zhang X., Tan W., Chem. Eur. J. 2014, 20, 5866–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Röglin L., Ahmadian M. R., Seitz O., Angew. Chem. Int. Ed. 2007, 46, 2704–2707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2759–2763; [Google Scholar]
- 15b. Röglin L., Altenbrunn F., Seitz O., ChemBioChem 2009, 10, 758–765; [DOI] [PubMed] [Google Scholar]
- 15c. Thurley S., Röglin L., Seitz O., J. Am. Chem. Soc. 2007, 129, 12693–12695. [DOI] [PubMed] [Google Scholar]
- 16. Oh K. J., Cash K. J., Lubin A. A., Plaxco K. W., Chem. Commun. 2007, 4869–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lim E.-K., Guk K., Kim H., Chung B.-H., Jung J., Chem. Commun. 2016, 52, 175–178. [DOI] [PubMed] [Google Scholar]
- 18. Ranallo S., Rossetti M., Plaxco K. W., Vallée‐Bélisle A., Ricci F., Angew. Chem. Int. Ed. 2015, 54, 13214–13218; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13412–13416. [Google Scholar]
- 19. Fischbach M., Resch‐Genger U., Seitz O., Angew. Chem. Int. Ed. 2014, 53, 11955–11959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12149–12153. [Google Scholar]
- 20. Machida T., Dutt S., Winssinger N., Angew. Chem. Int. Ed. 2016, 55, 8595–8598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8737–8740. [Google Scholar]
- 21. Ambros V., Nature 2004, 431, 350–355. [DOI] [PubMed] [Google Scholar]
- 22. Chu X., Battle C. H., Zhang N., Aryal G. H., Mottamal M., Jayawickramarajah J., Bioconjugate Chem. 2015, 26, 1606–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abendroth F., Seitz O., Angew. Chem. Int. Ed. 2014, 53, 10504–10509; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10672–10677. [Google Scholar]
- 24. Ma Z., Taylor J.-S., Bioconjugate Chem. 2003, 14, 679–683. [DOI] [PubMed] [Google Scholar]
- 25. Gorska K., Manicardi A., Barluenga S., Winssinger N., Chem. Commun. 2011, 47, 4364–4366. [DOI] [PubMed] [Google Scholar]
- 26. Shibata A., Ito Y., Abe H., Chem. Commun. 2013, 49, 270–272. [DOI] [PubMed] [Google Scholar]
- 27. Mayer G., Heckel A., Angew. Chem. Int. Ed. 2006, 45, 4900–4921; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5020–5042. [Google Scholar]
- 28. Okamoto A., Tanabe K., Inasaki T., Saito I., Angew. Chem. Int. Ed. 2003, 42, 2502–2504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 2606–2608. [Google Scholar]
- 29. Tanabe K., Nakata H., Mukai S., Nishimoto S.-i., Org. Biomol. Chem. 2005, 3, 3893–3897. [DOI] [PubMed] [Google Scholar]
- 30. Röthlingshöfer M., Gorska K., Winssinger N., J. Am. Chem. Soc. 2011, 133, 18110–18113. [DOI] [PubMed] [Google Scholar]
- 31. Arian D., Cló E., Gothelf K. V., Mokhir A., Chem. Eur. J. 2010, 16, 288–295. [DOI] [PubMed] [Google Scholar]
- 32. Dutta S., Mokhir A., Chem. Commun. 2011, 47, 1243–1245. [DOI] [PubMed] [Google Scholar]
- 33. Arian D., Kovbasyuk L., Mokhir A., J. Am. Chem. Soc. 2011, 133, 3972–3980. [DOI] [PubMed] [Google Scholar]
- 34. Meyer A., Mokhir A., Angew. Chem. Int. Ed. 2014, 53, 12840–12843; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13054–13057. [Google Scholar]
- 35. Schikora M., Mokhir A., Inorg. Chim. Acta 2016, 452, 118–124. [Google Scholar]
- 36.
- 36a. Sando S., Kool E. T., J. Am. Chem. Soc. 2002, 124, 9686–9687; [DOI] [PubMed] [Google Scholar]
- 36b. Abe H., Kool E. T., Proc. Natl. Acad. Sci. USA 2006, 103, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franzini R. M., Kool E. T., J. Am. Chem. Soc. 2009, 131, 16021–16023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franzini R. M., Kool E. T., Chem. Eur. J. 2011, 17, 2168–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silverman A. P., Kool E. T., Nucleic Acids Res. 2005, 33, 4978–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Velema W. A., Kool E. T., J. Am. Chem. Soc. 2017, 139, 5405–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dragulescu-Andrasi A., Zhou P., He G., Ly D. H., Chem. Commun. 2005, 244–246. [DOI] [PubMed] [Google Scholar]
- 42. Gorska K., Keklikoglou I., Tschulena U., Winssinger N., Chem. Sci. 2011, 2, 1969–1975. [Google Scholar]
- 43. Saneyoshi H., Ito Y., Abe H., J. Am. Chem. Soc. 2013, 135, 13632–13635. [DOI] [PubMed] [Google Scholar]
- 44. Chen Y., Kamlet A. S., Steinman J. B., Liu D. R., Nat. Chem. 2011, 3, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Röthlingshöfer M., Gorska K., Winssinger N., Org. Lett. 2012, 14, 482–485. [DOI] [PubMed] [Google Scholar]
- 46. Sadhu K. K., Winssinger N., Chem. Eur. J. 2013, 19, 8182–8189. [DOI] [PubMed] [Google Scholar]
- 47. Holtzer L., Oleinich I., Anzola M., Lindberg E., Sadhu K. K., Gonzalez-Gaitan M., Winssinger N., ACS Cent. Sci. 2016, 2, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang D., Lindberg E., Winssinger N., J. Am. Chem. Soc. 2017, 139, 1444–1447. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Devaraj N. K., Hilderbrand S., Upadhyay R., Mazitschek R., Weissleder R., Angew. Chem. Int. Ed. 2010, 49, 2869–2872; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2931–2934; [Google Scholar]
- 49b. Han H.-S., Devaraj N. K., Lee J., Hilderbrand S. A., Weissleder R., Bawendi M. G., J. Am. Chem. Soc. 2010, 132, 7838–7839; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49c. Wu H., Cisneros B. T., Cole C. M., Devaraj N. K., J. Am. Chem. Soc. 2014, 136, 17942–17945; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49d. Wu H., Yang J., Šečkutė J., Devaraj N. K., Angew. Chem. Int. Ed. 2014, 53, 5805–5809; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5915–5919; [Google Scholar]
- 49e. Wu H., Alexander S. C., Jin S., Devaraj N. K., J. Am. Chem. Soc. 2016, 138, 11429–11432. [DOI] [PubMed] [Google Scholar]
- 50.
- 50a. Ficht S., Mattes A., Seitz O., J. Am. Chem. Soc. 2004, 126, 9970–9981; [DOI] [PubMed] [Google Scholar]
- 50b. Ficht S., Dose C., Seitz O., ChemBioChem 2005, 6, 2098–2103; [DOI] [PubMed] [Google Scholar]
- 50c. Dose C., Ficht S., Seitz O., Angew. Chem. Int. Ed. 2006, 45, 5369–5373; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5495–5499; [Google Scholar]
- 50d. Grossmann T. N., Seitz O., J. Am. Chem. Soc. 2006, 128, 15596–15597; [DOI] [PubMed] [Google Scholar]
- 50e. Dose C., Seitz O., Bioorg. Med. Chem. 2008, 16, 65–77; [DOI] [PubMed] [Google Scholar]
- 50f. Grossmann T. N., Röglin L., Seitz O., Angew. Chem. Int. Ed. 2008, 47, 7119–7122; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7228–7231; [Google Scholar]
- 50g. Grossmann T. N., Seitz O., Chem. Eur. J. 2009, 15, 6723–6730; [DOI] [PubMed] [Google Scholar]
- 50h. Roloff A., Seitz O., Chem. Sci. 2013, 4, 432–436; [Google Scholar]
- 50i. Roloff A., Seitz O., ChemBioChem 2013, 14, 2322–2328; [DOI] [PubMed] [Google Scholar]
- 50j. Michaelis J., van Noort G. J. V., Seitz O., Bioconjugate Chem. 2014, 25, 18–23; [DOI] [PubMed] [Google Scholar]
- 50k. Michaelis J., Maruyama A., Seitz O., Chem. Commun. 2013, 49, 618–620; [DOI] [PubMed] [Google Scholar]
- 50l. Kern A., Seitz O., Chem. Sci. 2015, 6, 724–728; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50m. Roloff A., Seitz O., Bioorg. Med. Chem. 2013, 21, 3458–3464. [DOI] [PubMed] [Google Scholar]
- 51.
- 51a. Gartner Z. J., Kanan M. W., Liu D. R., Angew. Chem. Int. Ed. 2002, 41, 1796–1800; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1874–1878; [Google Scholar]
- 51b. Huang Y., Coull J. M., J. Am. Chem. Soc. 2008, 130, 3238–3239. [DOI] [PubMed] [Google Scholar]
- 52. Chen X. H., Roloff A., Seitz O., Angew. Chem. Int. Ed. 2012, 51, 4479–4483; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4556–4561. [Google Scholar]
- 53.
- 53a. Meguellati K., Koripelly G., Ladame S., Angew. Chem. Int. Ed. 2010, 49, 2738–2742; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2798–2802; [Google Scholar]
- 53b. Koripelly G., Meguellati K., Ladame S., Bioconjugate Chem. 2010, 21, 2103–2109. [DOI] [PubMed] [Google Scholar]
- 54. Metcalf G. A. D., Shibakawa A., Patel H., Sita-Lumsden A., Zivi A., Rama N., Bevan C. L., Ladame S., Anal. Chem. 2016, 88, 8091–8098. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. DeVos S. L., Miller T. M., Neurotherapeutics 2013, 10, 486–497; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55b. Sharma V. K., Sharma R. K., Singh S. K., MedChemComm 2014, 5, 1454–1471; [Google Scholar]
- 55c. Evers M. M., Toonen L. J. A., van Roon-Mom W. M. C., Adv. Drug Delivery Rev. 2015, 87, 90–103; [DOI] [PubMed] [Google Scholar]
- 55d. Lundin K. E., Gissberg O., Smith C. I. E., Human Gene Ther. 2015, 26, 475–485; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55e. Havens M. A., Hastings M. L., Nucleic Acids Res. 2016, 44, 6549–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]