

Abstract
Our understanding of the events taking place within the blood following severe injury with hemorrhagic shock is quickly evolving. Traditional concepts have given way to a detailed and nuanced understanding of coagulopathy, bleeding, and shock at the cellular and biochemical levels. In doing so, the tremendous complexity of events taking place within the blood have been illuminated and present an additional challenge. In this review, we seek to understand shock, endotheliopathy, and coagulopathy not as isolated events, but rather as the result of changes taking place within a single dynamic organ system. This review will highlight the key linkages existing between blood and endothelium and how these processes are perturbed by hemorrhagic shock to produce a syndrome that we call “hemorrhagic blood failure.” From this perspective, it may be regarded that the blood organ system fails in providing its vital functions predictably after injury. We review how accumulation of oxygen debt during shock leads to endotheliopathy and coagulopathy, and how current transfusion strategies may impact the syndrome of hemorrhagic blood failure.
Keywords: Trauma, coagulopathy, endotheliopathy, oxygen debt, shock, hemorrhage, blood failure
The burden of traumatic bleeding
Trauma is responsible for 1 in 10 deaths worldwide and is the leading cause of death of people less than 45 years old (1). The two most common causes of death in trauma victims are brain injury and hemorrhage (2). Hemorrhage accounts for the preponderance (~60%) of deaths in patients with potentially salvageable injuries (~50% of those injured), amounting to about one third of trauma deaths (1). Over 90% of potentially-survivable US military combat deaths are attributed to hemorrhage (3). The majority of all victims who die from hemorrhage will die within the first several hours following injury, indicating that a great number of them could be rescued if hemorrhage could be stopped quickly (4,5).
Impaired clot formation, or coagulopathy, that increases bleeding is also present in 20–30% of severely-injured trauma patients immediately after injury and is associated with increased incidence of multi-organ failure, intensive care utilization, and death (6). Traumatic coagulopathy is a distinct and multilayered biochemical response to tissue injury and hemorrhagic shock (7). When present on arrival to the Emergency Department, coagulopathy is associated with 4–6-fold increased mortality (8). The pathophysiology of traumatic coagulopathy is now recognized to be multifactorial and networked in nature, consisting of contrasting coagulation states that produce an emergent syndrome that contributes to failure of the vital functions of the blood.
Blood as an organ system
Body organs are composed of a variety of distinct tissues that work together to perform one or more coordinated functions. Of utmost importance is the circulatory system. The main function of the circulatory system is to provide oxygen and nutrients, clear waste products, and provide a conduit to connect organs that are separated physically from one another. In doing so, the circulatory system links all organ systems and dynamically-regulates physiological homeostasis.
Using this perspective, it can be realized that the many components of the cardiovascular system must interact intimately and that many subsystems exist therein. None are more complex nor dynamic than the blood-endothelial system. The intimate coordination required of these two separate tissues to provide a coordinated physiological function meets the criteria for classification as a separate organ system. In fact, they may be considered the largest organ system in the body. Such separations are artificial at best, and must be made if only in the hope of improving overall understanding. Nonetheless, it is useful to consider the liquid blood and its components as existing and functioning in concert with the endothelium which it bathes, and also derives so much close regulation. The blood-endothelial unit, which we will refer to simply as “blood” in this review, will be approached as a single organ system in the sense that these individual components are inextricably linked and closely support each other’s function.
Definition of hemorrhagic blood failure
We define hemorrhagic blood failure as an emergent state of blood that arises during accumulation of a critical level of oxygen debt (shock). Critically-low tissue oxygen delivery, endotheliopathy, platelet dysfunction, and coagulopathy should all be present to meet our definition of blood failure. (Fig. 1) These processes are currently recognized to be present individually. However, it is useful to consider them together given their interconnectedness. Some changes brought about by blood failure may help to maintain blood fluidity or protect by activating coagulation during limited blood loss or hypotension (9). However, in the particular case of combined tissue injury with hemorrhagic shock where blood can be lost from wounds, emergence of blood failure is associated with dramatically-increased mortality (8).
Figure 1.
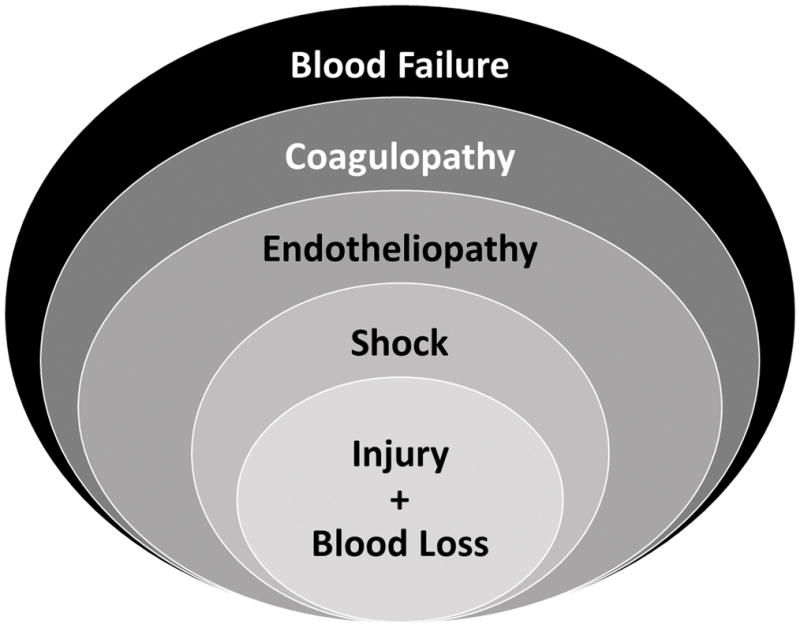
Schematic representing the components of hemorrhagic blood failure.
Who gets hemorrhagic blood failure?
Evidence suggests that trauma victims having both tissue injury and hemorrhagic shock are susceptible to intrinsic hemorrhagic blood failure, which is further modified by plasma dilution, hypothermia, and acidosis, some of which can be iatrogencially produced. Approximately 25–35% of severely injured victims presenting with both severe anatomical injuries defined by an injury severity score > 15 and hemorrhagic shock defined by base deficit < 4 meq/L will also have coagulopathy which includes platelet dysfunction, partly meeting our definition of hemorrhagic blood failure (7,8). Significant tissue injury may not be absolutely required to induce blood failure because coagulopathy can arise after blood loss with minimal tissue injury such as in the case of rupture abdominal aortic aneurysms (10). However, tissue injury likely contributes to coagulopathy and may speed its onset with less blood lost through processes that exacerbate tissue hypoxia. Those experiencing polytrauma or high-energy mechanisms of injury, such as blast injuries or high-velocity gunshot wounds seen with military trauma, typically have an increased incidence of coagulopathy (11). Specific types of tissue injury, such as traumatic brain injury, may generate especially strong perturbations in blood homeostasis, contributing to the overall syndrome of blood failure when shock is also present (12). However, in trauma patients it is often difficult or impossible to distinguish between the independent contributions of shock and tissue injury. When stratified in a non-biased way using hierarchical clustering analysis of multiple coagulation parameters, overt coagulopathy typical of that seen as a component of blood failure was present in approximately 11% of Emergency Department trauma patients (13). These subjects were bluntly injured (78%), were significantly injured (median ISS = 41), and were in shock (median base deficit = 8.1 meq/L). It is clear that blood failure can emerge as a result of hemorrhagic shock, but is also modulated and exacerbated by tissue injury. Clinical suspicion should remain highest in polytrauma patients having significant tissue injury that also have evidence of blood loss.
Effect of blood failure on clot formation
Of paramount importance are the effects of hemorrhagic blood failure on hemostatic clot formation. Measurements obtained in the field at the site of injury prior to hospital transport indicate that coagulopathy contributing to blood failure can manifest almost immediately after injury (14,15). Abnormal thrombelastography (TEG) measurements and Protein C activity measurements taken in the field tend to persist or worsen over time (14,15). Clot phenotypes change in a predictable way as shock severity increases when measured using viscoelastic techniques such as TEG or rotational thromboelastometery (ROTEM). Davenport et al. found that a decrease of clot amplitude (or strength) was the first significant viscoelastic change noted in the Emergency Department when comparing trauma patients grouped according to a mild increase of prothrombin ratio > 1.2 (16). Notably, platelet-induced clot contraction, which is a major contributor to clot amplitude, was the first major abnormality noted to be present in blood obtained prehospital (14). Over the ensuing minutes and hours clot amplitude tends to decrease further and clot onset times become prolonged (16). Increased clot lysis then often becomes apparent, with the intensity of lysis becoming strongly predictive of increased mortality (17). Once terminal stages are reached, i.e. after traumatic cardiac arrest, the viscoelastic tracing approximates a diamond shape with shortened onset time and a rapid, but short lived, amplitude that decreases to zero within 30 minutes of clot formation time (18). Termed the “Death Diamond” by Chapman et al, this clot formation phenotype signals a terminal state of blood failure. Interestingly, asphyxia alone can produce similar clot formation phenotypes to that seen in the most severe trauma patients. In drowning victims, Schwameis et al. found that ROTEM clot formation was severely diminished or abolished, and positively correlated with plasma tPA concentration. Clot formation could be rescued in these drowning victims using fibrinogen supplementation, antifibrinolytics, and heparinase (19). These results support a strong linkage between injury, tissue hypoperfusion, and coagulopathy manifested by immediate platelet dysfunction that progresses to rapid anticoagulation within moments, and overwhelming hyperfibrinolysis within hours of injury.
Blood failure is initiated by shock
Blood failure is strongly-linked to the presence of shock. This linkage is supported by both preclinical animal studies of hemorrhagic shock and observational human clinical data. The evidence suggests that hemorrhagic blood failure is initiated and propagated by blood loss that decreases systemic oxygen delivery to critically low levels incapable of supporting aerobic metabolism. Once the lower threshold for critical oxygen delivery is surpassed, an instantaneous oxygen deficit is incurred, oxygen becomes maximally extracted from the blood by the tissues, and anaerobic metabolism increases. Oxygen debt is the accumulation of oxygen deficit over time, and represents both the severity of shock and time spent in the shock state (20). The burden of accumulated oxygen debt must be repaid by increasing oxygen delivery above baseline levels in a timely fashion in order to restore metabolic function and prevent ongoing organ injury at the cellular level (21,22). Therefore, simply returning oxygen delivery to normal basal levels after accumulation of oxygen debt is not sufficient to prevent subsequent organ injury. Instead, a substantial amount of oxygen debt must be repaid to prevent significant or permanent organ injury because metabolic activity continues during cellular ischemia, leading to altered cellular and subcellular membrane potentials, accumulation of byproducts of anaerobic metabolism, and activation of neurovascular compensatory mechanisms (23). A period of increased aerobic metabolism is required to reverse these changes, replenish oxygen and the phosphagen, glycogen-lactic acid system, and return to physiological homeostasis. Therefore, to understand how shock induces blood failure, it is important to understand the cellular and metabolic changes that take place during shock.
Oxygen participates directly in aerobic metabolism as a terminal electron acceptor (or oxidant). More specifically, oxygen interacts with mitochondrial cytochrome c oxidase (Complex IV) which transfers membrane electrons produced by upstream cytochromes to oxygen, creating water, and pumping the hydrogen protons required to drive ATP synthase (Complex V), producing ATP. When oxygen delivery is critically-low, oxygen becomes limiting as the terminal electron acceptor and ATP production decreases. This results in a shift towards anaerobic metabolism, and an increase of mitochondrial membrane electron burden which alters and destabilizes mitochondrial membrane electrochemical potentials (23). Electrons begin to leak from the mitochondrial membrane, and what oxygen is available is pathologically reduced to form reactive oxygen species such as superoxide radical. Superoxide can subsequently react with nitric oxide to form peroxynitrite, a highly toxic radical which nitrosylates tyrosine moieties and alters protein function. In addition, superoxide dismutase catalyzes formation of hydrogen peroxide from superoxide. Peroxides can form extremely-damaging hydroxyl radicals when antioxidant systems based on catalase and glutathione peroxidase become overwhelmed (24,25). As oxygen debt accumulates, ongoing production of these oxidants can induce increasing and irreversible cellular damage in the form of protein nitrosylation, lipid peroxidation, and DNA damage (24). Oxidants can also play a role in inflammatory signaling as cytokine-induced second messengers (25,26).
During shock, aerobic mitochondrial metabolism subsequently shifts to anaerobic pathways within the cytosol and lactate assumes the alternative role of the terminal electron acceptor from NADH by the action of lactate dehydrogenase on pyruvate (27). Intracellular lactate is shuttled from the cytosol to the extracellular space resulting in rapid accumulation of extracellular lactate. Importantly, adrenergic activation and catecholamine release induced by neurovascular compensation also stimulates lactate production by activation of the Na+-K+-ATPase so that lactate is enriched in the blood to be used as an alternative fuel for vital organs (28). Concomitant vasoconstriction occurring in part due to increase adrenergic output further decreases nutritive flow to many organ systems thus increasing tissue hypoxia. These changes affect the blood by causing altered blood redox potential, lactic acidosis, and an increase in adrenergic mediators such as epinephrine (29,30). The resulting effects on endothelial cells, plasma proteins, and blood cells induce the endotheliopathy of trauma that includes the coagulopathy that represents blood failure.
The role of endothelium
The vascular endothelium is an organ in itself that is heterogeneous and communicates systemically throughout the body (31). In severe hemorrhagic shock and trauma, the endothelium is damaged and this leads to the development of an endotheliopathy of trauma characterized by three main components: 1) endothelial compromise and paracellular permeability, 2) dysfunctional coagulation, and 3) inflammation; all components which potentially contribute to blood failure (32, 33). Endothelial cells regulate the integrity of the blood-organ barrier and clot formation including both pro-coagulant and anticoagulant functions. Induction of the endotheliopathy of trauma by hemorrhage and trauma involves hypoxia, endothelial cell surface receptor activation by inflammatory mediators and growth factors, binding of platelets, red blood cells and leukocytes to activated endothelial cells, and production of coagulation pathway intermediates. An understanding of the role of the endothelium in initiation and propagation of the endotheliopathy of trauma and blood failure is primarily based upon pre-clinical work in vitro and in vivo in animal models of hemorrhagic shock and trauma. Human clinical trials have also provided some observational insight (34,35). Under normal physiological conditions, the endothelial surface maintains blood fluidity and regulates flow by multiple anticoagulant mechanisms. The surface-linked protein Thrombomodulin binds thrombin converting it from a potent procoagulant to an anticoagulant by increasing its affinity for protein C above that of fibrinogen (36). Activation of Protein C can further reduce Factor V and VIII levels. Activated protein C also interacts with endothelial cells to activate cell survival responses and maintain the endothelial barrier (37). Local synthesis of prostacylin (PGI2) and metabolism of ADP to adenine nucleotides, also inhibits platelet activation, adhesion, and aggregation. Endothelial cells also regulate vasoreactivity and vascular patency by synthesizing nitric oxide, a potent vasodilator, and tPA which activates plasminogen to plasmin, the primary proteolytic enzyme of fibrin (31,37).
Paracellular permeability leading to organ edema and organ failure is caused by breakdown of endothelial cell-cell tight and adherens junctions that regulate the endothelial blood-organ barrier in various tissues. This barrier is tissue specific and is maintained by structural components (i.e. adherens junctions), cellular components (i.e. smooth muscle cells and pericytes), and extracellular matrix proteins working in unison to prevent vascular permeability. These components form the tethers that hold the endothelium together. Cell surface receptor based signaling is a key component that also regulates the blood-organ barrier. Antagonistic signaling of angiopoietins 1 and 2 signal via the Tie-2 receptor to tighten (Ang-1) or loosen (Ang-2) the barrier that separates the strongly procoagulant subendothelial tissues from blood (38). An additional layer of protection is afforded by a thick surface matrix made up of membrane-bound glycoproteins and proteoglycans having heparin-like activity called the glycocalyx. The glycocalyx is a protective border on endothelial cells that regulates endothelial permeability, shear stress, limits interactions with circulating blood cells, and inhibits local thrombin activity (39). However, with traumatic injury and shock, injured endothelium can promote coagulation and barrier disruption when activated by releasing Ang-2, tissue factor, von Willebrand factor (VWF), platelet activating factor, and PAI-1 (39). Therefore, the endothelium can exist in a quiescent anticoagulant or activated procoagulant phenotype depending upon local and systemic blood and tissue conditions.
Hemorrhagic shock with cellular ischemia affects endothelium by activating both procoagulant and anticoagulant responses to various degrees. Local acidosis can induce a decrease in nitric oxide and PIG2 production promoting vasoconstriction and platelet adhesion (40). Amongst the first events are shedding of the glycocalyx, which releases syndecans with antithrombotic properties into the blood stream (34,35,41,42). The extent to which these shed products are capable of measurably anticoagulating the blood remains unknown. However, clot formation can be improved in blood from coagulopathic trauma patients in the presence of heparinase, suggesting a role for autoheparinization of the blood as a component of coagulopathy (43). Circulating epinephrine and high circulating syndecan-1, a marker of endothelial glycocalyx degradation, has been positively correlated with the degree of glycocalyx shedding and mortality in trauma patients (35). The catecholamine surge correlated to patient mortality also includes biomarkers of endothelial cell damage, degradation of the glycocalyx, hyperfibrinolysis and coagulopathy that occurs after trauma, all components of the endotheliopathy of trauma that potentially lead to blood failure.
Endothelial cell-blood cell adhesion also leads to vascular endothelial compromise in hemorrhagic shock. An important consequence of glycocalyx shedding is the enabling of direct interactions between inflammatory blood cells and their mediators and the endothelial surface. Endothelial exposure may activate platelets as indicated by increased plasma soluble CD40 ligand (sCD40L), a platelet-derived inflammatory mediator that is associated with sympathoadrenal activation, immune system activation, and increased mortality (44). Activated neutrophils migrate to the endothelial surface, contribute to oxidation, and activate coagulation locally by release of neutrophil extracellular traps (44). Local endothelial oxidation is also capable of directly promoting red blood cell adhesion and inducing local thrombosis as demonstrated experimentally by local application of Ferric Chloride (46).
Oxidative stress from activated neutrophils may also directly affect important coagulation factors. Oxidation of a single key methionine in Aα-C domain of fibrinogen to methionine sulfoxide by hypochlorous acid, produced by activated neutrophils, can disrupt fibrin polymerization in vitro (47). Methionine sulfoxide content was also found to be significantly increased at the same position in coagulopathic trauma patients (48). Taken together, the complex processes regulated on the surface of the vascular endothelium are critical to homeostasis and repair in both health and disease. Elucidation of the complex components and mechanisms of action of the endotheliopathy of trauma and its contributions to blood failure is vital to understanding relevant therapeutic targets in treating the disease.
Coagulopathy
Many observational human clinical studies support coagulopathy as a major component of blood dysfunction after trauma. During shock the endothelium responds to maintain blood fluidity through the action of multiple anticoagulant responses. A primary response to hypotension and central hypovolemia is release of tPA to activate fibrinolysis (9). In addition, soluble thrombomodulin and activated Protein C are increased in the blood (49). However, levels of activated Protein C reported in trauma patients are insufficient to anticoagulate normal plasma alone (50). Platelets can also provide sufficient activated FV to support hemostasis and overcome even super-physiologic concentrations of activated Protein C (50). In addition, procoagulant activity and thrombin generation is generally accepted to be increased above healthy control levels in the blood of trauma patients, even those with coagulopathy (51–53). This suggests that trauma patient blood, even in the setting of prolonged PT/INR may not be truly anti-coagulated. Abnormalities in clot-based assays like PT/INR that might suggest anticoagulation likely reflect complex interactions of fibrinogen depletion, fibrin degradation product interference with fibrin mesh formation, dysregulated thrombin generation, and excess plasmin activity manifested most obviously in fibrinolysis.
Perhaps more important to the pathophysiology of coagulopathy during blood failure is the increase of proteolytic activity within the blood. Multiple proteolytic enzymes are increased in blood after trauma, including neutrophil elastase and plasmin (54). Plasmin is a serine protease having wide proteolytic activity against a multitude of coagulation proteins, membrane proteins, and integrins. (Table 1.) Plasmin proteolysis can inactivate FV, FVIII, FXIIIa, and can activate FXII, thus linking its activation to complement, inflammation, and immunity by direct generation of bradykinin from high molecular weight kininogen (55–63). However, the most direct effect of plasmin on coagulation is its activity against both fibrinogen and fibrin which contributes to rapid fibrinogen consumption and fibrinolysis after trauma (64,65). The degree of fibrinolysis is positively associated with mortality and likely proportional to the degree of shock, even when it is not sufficient to induce whole blood clot lysis (66). Overall, severe trauma and shock appear to increase both thrombin generation and plasmin activation, with an early increase in thrombin and a gradual increase in relative plasmin activity over time. Interestingly, some degree of physiological fibrinolysis may be beneficial after trauma. Moore et al, have demonstrated in a multicenter cohort that both increased fibrinolysis and a lack of fibrinolysis or fibrinolytic resistance are both associated with increased mortality after trauma (67). Therefore, some degree of fibrinolysis may support vascular patency during low flow states, but may also exacerbate blood loss when significantly increased or when bleeding wounds are present. Randomized controlled human data suggests that using an antifibrinolytic agent to inhibit plasmin-induced fibrinolysis confers a significant survival advantage for trauma patients. The CRASH-2 trial demonstrated a clear mortality benefit for the use of tranexamic acid, an antifibrinolytic lysine analogue, if administered within 3 hours of injury (68). Questions remain regarding appropriate patient selection for antifibrinolytic therapy, however, it is clear that shock-induced blood proteolysis is an important component of coagulopathy and a driver of blood failure. Inhibition of plasmin activation with tranexamic acid in the setting of trauma and shock also improves epithelial and endothelial barrier function and reduces tissue edema and injury, likely in part due to reduced bradykinin generation (69). This effect underscores the interconnectedness of blood and the endothelium as an integrated organ system.
Table 1.
Proteolytic targets of Plasmin. Superscripted numbers represent the corresponding reference numbers used to support the claim.
Another important contributor to coagulopathy and hemorrhagic blood failure is platelet dysfunction. Circulating platelets act to initiate clot formation at wounds by adhesion and aggregation during primary hemostasis. They also provide a local environment that supports thrombin generation and they forcefully contract fibrin to stabilize clots. Platelet dysfunction after trauma, measured by decreased impedance aggregation has been strongly associated with increased mortality (70). The mechanism of platelet dysfunction remains unclear, given that the surface of dysfunctional platelets also appear to be paradoxically activated (71). Platelet-induced clot contraction is a determinant of clot strength after injury and partly explains the variability in clot strength seen in Emergency Department trauma patients (72).
Shock appears to induce a spectrum of endothelial-driven coagulation responses that initially promote a procoagulant phenotype that rapidly transitions to anticoagulation as oxygen debt accumulates and shock worsens. These responses are coordinated at the blood-endothelial interface and are modulated by circulating catecholamines, anaerobic metabolites, inflammation, oxidation, proteolysis, and cellular dysfunction. (Fig. 2)
Figure 2.

Schematic of key linkages between oxygen debt, cellular dysfunction, and coagulopathy during hemorrhagic blood failure.
Blood transfusion to treat blood failure
To adequately treat hemorrhagic blood failure, we must consider holistic treatments that simultaneously address its multiple components. Repayment of oxygen debt must be accomplished in addition to simultaneous treatment of both endothelial injury and coagulopathy. The role of using oxygen carriers to increase tissue oxygen delivery as well as metabolic therapies which reduce tissue oxygen consumption should be considered and researched to counter the primary effects of tissue hypoxia on blood failure. Further insight into how blood failure may treated can be gained by identifying the contributions and limitations of individual blood product components currently in use. (Fig. 3)
Figure 3.

Schematic summarizing the effects of individual blood products on the three components of hemorrhagic blood failure. PRBC= packed red blood cells, Cryo= cryoprecipitate
Packed red blood cells (PRBC’s) are transfused to primarily address oxygen debt by increasing oxygen carrying capacity (oxygen content), cardiac output, and thus oxygen delivery. The role of red blood cell age remains questionable as the ability to offload oxygen and support hypoxic vasodilation decreases with increased storage length (73). It seems reasonable to transfuse the freshest red blood cells to the sickest patient population. However, data to support the use of fresh red blood cells preferentially for critically-ill patients is lacking (74). Hematocrit also contributes to platelet margination to the vessel wall under flow and low hematocrit is associated with increased bleeding time (75,76).
Plasma transfusion supports cardiac output through intravascular volume expansion and provides coagulation factors to support hemostasis. All coagulation factors are present in plasma in roughly mg/ml concentrations, providing for balanced coagulation factor replacement. However, their volume of distribution may limit the hemostatic efficacy of plasma when given in large volumes alone (77). Plasma may also provide the additional benefit of providing endothelial protection by replacing important protective enzymes that can contribute to sealing the endothelial barrier, speed recovery of the glycocalyx, and rebalance thrombin generation by the provision of antithrombin (78–80). However, plasma has also been independently linked to multi-organ failure and lacks the cellular elements required to fully restore clot formation (81).
Early platelet transfusion is associated with improved outcome in the acutely bleeding trauma patients. Platelet concentrates clearly contribute to hemostasis by increasing thrombin formation, increasing clot stiffness, and increasing resistance to clot lysis (82). Platelets are also a rich source of proteins which may confer a degree of fibrinolytic protection by inhibition of tPA (83). However, platelets are the most precious blood product by way of having the most limited shelf life partly due to the requirement for room-temperature storage. Interestingly, shelf life limitations have a regional variation from 3 days in Japan to 5 days in the US and 7 days in Europe. The historical paper by Murphy and Gardner published in 1969 that defined the essentials of platelet storage (room temperature storage under constant agitation) has been used for the last 45 years (84). In addition, the current model for platelet transfusion is based on prophylactic treatment of hypoproliferative thrombocytopenia (i.e. during chemotherapy) neglecting other patient populations requiring platelet transfusion for bleeding. More recent data has suggested that platelets stored at 4°C retain better hemostatic properties and remain energetic and viable up to 15 days of storage (85,86). Even though refrigerated platelets may also remain for a shorter period of time in the circulation, they remain long enough to contribute to hemostasis in the most crucial period of acute traumatic injury, and may also decrease endothelial permeability compared to 22°C-stored platelets (87). A recent intravital microscopy study in a rat hemorrhagic shock model has documented that cold platelets can also contribute to clot formation without excess thrombogenicity (88).
Cryoprecipitate provides a rich source of fibrinogen, Factor VIII, and von Willebrand factor and is primarily used to enhance hemostasis while also likely offering benefit to endothelium. Early and aggressive fibrinogen supplementation can recover viscoelastic clot strength when given in concentrated doses (89) and cryoprecipitate provides grams of fibrinogen per dose. Retrospective data supports an association between the combined use of cryoprecipitate and tranexamic acid and reduced mortality after battlefield injury (90). However logistical limitations and lack of evidence have prevented the widespread use of cryoprecipitate in civilian trauma centers. While the early administration of cryoprecipitate to trauma patients has been shown to be feasible, a mortality benefit has yet to be realized (91). One reason may be that cryoprecipitate can only restore traumatic hypofibrinogenemia and improve clot strength when given in high concentrations, but it is often given in limited quantities and provided late during resuscitation (92). A review of cryoprecipitate use in major trauma centers in the United States revealed that its median time from admission to first cryoprecipitate unit was 2.7 hours, and that more than 70% of those who died from hemorrhage in the first six hours did not receive any cryoprecipitate (93).
Fresh whole blood and cold stored whole blood have reemerged as an approach to providing holistic resuscitation that can simultaneously address the three components of blood failure; oxygen debt, endotheliopathy, and coagulopathy. The Early Whole Blood Investigators reported a comparison of coagulation status after administering modified whole blood (platelet and leukodepleted) or components (PRBC’s + Plasma) as the initial trauma resuscitation fluid to trauma patients (94). They found that the type and timing of transfusion were both important to coagulation status and that there was an improvement in platelet function in the whole blood group. The same group also reported previously from the same cohort that those receiving whole blood received less total transfusions at 24 hours after subjects with traumatic brain injury were excluded (95). There is also evidence that fresh whole blood transfusion is associated with increased survival for combat-related injuries (96).
Due to logistical considerations and the need for far-forward resuscitation of blood failure, there has been increased interest from militaries in using fresh whole blood via “walking blood banks” for resuscitation during prehospital trauma care (97). The “walking blood bank” concept is advantageous because it appears to be technically feasible and reduces the need for prolonged blood transport and storage in austere settings (98). In addition, cold stored leukoreduced whole blood was found to retain its hemostatic function after 14 days of austere storage aboard a naval vessel comparably to components containing 5-day warm stored platelets (99). Blood donation of 450ml also appeared to be safe and had minimal detrimental effects on the physical performance of donors who were highly trained Special Forces personnel and are expected to return to vigorous combat activity after donation (100). Fresh or cold-stored whole blood appears to be not only logistically advantageous, but also feasible for use during resuscitation of traumatic hemorrhagic shock. However, more direct study of the effects of whole blood transfusion as a treatment of hemorrhagic blood failure is needed. In particular, the dose required to restore the vital functions of the blood after it has failed.
Conclusion
Blood and endothelium interact to provide coordinated physiological functions, thus encouraging us to view blood and the vascular system as a single dynamic organ system. This interconnectedness is highlighted by the effect of accumulating oxygen debt after traumatic blood loss that eventually leads to a multifactorial pathological syndrome that we call hemorrhagic blood failure. Examining blood as an organ during traumatic hemorrhagic shock not only provides a useful perspective on the pathophysiology of traumatic hemorrhagic shock and coagulopathy, but also suggests potential treatment strategies that may be useful to rescue those afflicted. Successful treatment would simultaneously counteract key pathological processes involved in blood failure, including endotheliopathy and proteolysis, and also provide holistic replacement therapies. Moving forward, future trials should view blood as an organ system and begin to combine targeted therapies with more holistic replacement therapies, such as fresh whole blood.
Acknowledgments
Support: N.J. White, was supported by Grant Number KL2 TR000421 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). These contents are solely the responsibility of the authors and do not necessarily represent the official view of NCATS or NIH.
Footnotes
Presented at the 6th Annual Remote Damage Control Resuscitation Symposium of the Trauma Hemostasis and Oxygenation Research Network, June 20–22, 2016, in Os, Norway.
Conflicts of Interest Statement: N.J.W holds intellectual property regarding detection methods for oxidative modifications of fibrinogen. K.R. Ward has a number of invention disclosures and patents pending for coagulation and oxidative stress monitoring technologies through the University of Michigan and no direct conflicts with the information contained in this manuscript. S. Pati, G. Strandenes, and A. Cap report no direct conflicts of interest with the information contained in this manuscript. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the US Department of the Army or the US Department of Defense.
Authorship Statement
N.J.W., K.R.W., S.P., G.S., and A.C. drafted the manuscript, provided critical revisions, and approve the final version to be published.
Contributor Information
Nathan J. White, University of Washington Division of Emergency Medicine and Harborview Medical Center.
Kevin R. Ward, Michigan Center for Integrative Research in Critical Care and University of Michigan Department of Emergency Medicine, Ann Arbor, MI USA.
Shibani Pati, Blood Systems Research Institute and the University of California, San Francisco, CA USA.
Geir Strandenes, Norwegian Naval Special Operations Command and Department of Immunology and Transfusion Medicine, Haukeland University Hospital, Bergen, Norway.
Andrew P. Cap, Coagulation and Blood Research, US Army Institute of Surgical Research, JBSA Fort Sam Houston, TX USA.
References
- 1.Murray CL, Lopez AD. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet. 1997 May;349:1269–1276. doi: 10.1016/S0140-6736(96)07493-4. [DOI] [PubMed] [Google Scholar]
- 2.Shackford SR, Mackersie RC, Davis JW, Wolf PL, Hoyt DB. Epidemiology and pathology of traumatic deaths occurring at a level-I trauma center in a regionalized system - the importance of secondary brain injury. J Trauma Acute Care Surg. 1989 Oct;29(2):1392–1397. doi: 10.1097/00005373-198910000-00018. [DOI] [PubMed] [Google Scholar]
- 3.Eastridge BJ, Mabry RL, Seguin P, Cantrell J, Tops T, Uribe P, Mallett O, Zubko T, Oetjen-Gerdes L, Rasmussen TE, et al. Death on the battlefield (2001–2011): implications for the future of combat casualty care. J Trauma Acute Care Surg. 2012 Dec;73(6 Suppl 5):S431–7. doi: 10.1097/TA.0b013e3182755dcc. [DOI] [PubMed] [Google Scholar]
- 4.Baker SP, Whitfield RA, O’Neill B. Geographic variations in mortality from motor vehicle crashes. N Engl J Med. 1987 May;316:1384–1387. doi: 10.1056/NEJM198705283162206. [DOI] [PubMed] [Google Scholar]
- 5.Scope A, Farkash U, Lynn M, Abargel A, Eldad A. Mortality epidemiology in low-intensity warfare: Israel Defense Forces’ experience. Injury. 2001 Jan;32(1):1–3. doi: 10.1016/s0020-1383(00)00101-7. [DOI] [PubMed] [Google Scholar]
- 6.Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. J Trauma Acute Care Surg. 2003 Jun;54(6):1127–30. doi: 10.1097/01.TA.0000069184.82147.06. [DOI] [PubMed] [Google Scholar]
- 7.Hess JR, Brohi K, Dutton RP, Hauser CJ, Holcomb JB, Kluger Y, Mackway-Jones K, Parr MJ, Rizoli SB, Yukioka T, et al. The Coagulopathy of Trauma: A Review of Mechanisms. J Trauma Acute Care Surg. 2008 Oct;65(4):748–54. doi: 10.1097/TA.0b013e3181877a9c. [DOI] [PubMed] [Google Scholar]
- 8.Maegele M, Lefering R, Yucel N, Tjardes T, Rixen D, Paffrath T, Simanski C, Neugebauer E, Bouillon BAG Polytrauma of the German Trauma Society (DGU) Early coagulopathy in multiple injury: An analysis from the German Trauma Registry on 8724 patients. Injury. 2007 Mar;38(3):298–304. doi: 10.1016/j.injury.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 9.van Helmond N, Johnson BD, Curry TB, Cap AP, Convertino VA, Joyner MJ. Coagulation changes during lower body negative pressure and blood loss in humans. Am J Physiol Heart Circ Physiol. 2015 Nov;309(9):H1591–7. doi: 10.1152/ajpheart.00435.2015. [DOI] [PubMed] [Google Scholar]
- 10.Reed MJ, Burfield LC. Initial emergency department coagulation profile does not predict survival in ruptured abdominal aortic aneurysm. Eur J Emerg Med. 2013 Dec;20(6):397–401. doi: 10.1097/MEJ.0b013e32835d92c5. [DOI] [PubMed] [Google Scholar]
- 11.Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) Study. Arch Surg. 2012 Feb;147(2):113–9. doi: 10.1001/archsurg.2011.287. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Z, Wang M, Tian Y, Hilton T, Salsbery B, Zhou EZ, Wu X, Thiagarajan P, Boilard E, Li M, et al. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood. 2016 Jun 2;127(22):2763–72. doi: 10.1182/blood-2015-12-688838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White NJ, Contaifer D, Jr, Martin EJ, Newton JC, Mohammed BM, Bostic JL, Brophy GM, Spiess BD, Pusateri AE, Ward KR, et al. Early Hemostatic Responses to Trauma Identified Using Hierarchical Clustering Analysis. J Thromb Haemost. 2015 Jun;13(6):978–88. doi: 10.1111/jth.12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll RC, Craft RM, Langdon RJ, Clanton CR, Snider CC, Wellons DD, Dakin PA, Lawson CM, Enderson BL, Kurek SJ. Early evaluation of acute traumatic coagulopathy by thrombelastography. Transl Res. 2009 Jul;154(1):34–9. doi: 10.1016/j.trsl.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Floccard B, Rugeri L, Faure A, Saint Denis M, Boyle EM, Peguet O, Levrat A, Guillaume C, Marcotte G, Vulliez A, et al. Early coagulopathy in trauma patients: an on-scene and hospital admission study. Injury. 2012 Jan;43(1):26–32. doi: 10.1016/j.injury.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Davenport R, Manson J, De’Ath H, Platton S, Coates A, Allard S, Hart D, Pearse R, Pasi KJ, MacCallum, et al. Functional definition and characterization of acute traumatic coagulopathy. Crit Care Med. 2011 Dec;39(12):2652–8. doi: 10.1097/CCM.0b013e3182281af5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schöchl H, Frietsch T, Pavelka M, Jámbor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma Acute Care Surg. 2009 Jul;67(1):125–31. doi: 10.1097/TA.0b013e31818b2483. [DOI] [PubMed] [Google Scholar]
- 18.Chapman MP, Moore EE, Moore HB, Gonzalez E, Morton AP, Chandler J, Fleming CD, Ghasabyan A, Silliman CC, Banerjee A, et al. The “Death Diamond”: Rapid thrombelastography identifies lethal hyperfibrinolysis. J Trauma Acute Care Surg. 2015 Dec;79(6):925–9. doi: 10.1097/TA.0000000000000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwameis M, Schober A, Schörgenhofer C, Sperr WR, Schöchl H, Janata-Schwatczek K, Kürkciyan EI, Sterz F, Jilma B. Asphyxia by Drowning Induces Massive Bleeding Due To Hyperfibrinolytic Disseminated Intravascular Coagulation. Crit Care Med. 2015 Nov;43(11):2394–402. doi: 10.1097/CCM.0000000000001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rixen D, Siegel JH. Bench-to-bedside review: oxygen debt and its metabolic correlates as quantifiers of the severity of hemorrhagic and post-traumatic shock. Crit Care. 2005 Oct;9:441–53. doi: 10.1186/cc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbee RW, Reynolds PS, Ward KR. Assessing shock resuscitation strategies by oxygen debt repayment. Shock. 2010 Feb;33:113–22. doi: 10.1097/SHK.0b013e3181b8569d. [DOI] [PubMed] [Google Scholar]
- 22.Siegel JH, Fabian M, Smith JA, Kingston EP, Steele KA, Wells MR. Oxygen debt criteria quantify the effectiveness of early partial resuscitation after hypovolemic hemorrhagic shock. J Trauma Acute Care Surg. 2003 May;54:862–880. doi: 10.1097/01.TA.0000066186.97206.39. [DOI] [PubMed] [Google Scholar]
- 23.Chaudry IH, Ohkawa M, Clemens MG, Baue AE. Alterations in electron transport and cellular metabolism with shock and trauma. Prog Clin Biol Res. 1983;111:67–88. [PubMed] [Google Scholar]
- 24.Szabó C, Módis K. Pathophysiological roles of peroxynitrite in circulatory shock. Shock. 2010 Sep;34(Suppl 1):4–14. doi: 10.1097/SHK.0b013e3181e7e9ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weidinger A, Kozlov AV. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules. 2015 Apr 15;5(2):472–84. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007 Aug;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Chaudry IH, Clemens MG, Baue AE. Alterations in cell function with ischemia and shock and their correction. Arch Surg. 1981 Oct;116(10):1309–17. doi: 10.1001/archsurg.1981.01380220053009. [DOI] [PubMed] [Google Scholar]
- 28.James JH, Luchette FA, McCarter FD, Fischer JE. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 1999 Aug 7;354(9177):505–8. doi: 10.1016/S0140-6736(98)91132-1. [DOI] [PubMed] [Google Scholar]
- 29.Rael LT, Bar-Or R, Salottolo K, Mains CW, Slone DS, Offner PJ, Bar-Or D. Injury severity and serum amyloid A correlate with plasma oxidation-reduction potential in multi-trauma patients: a retrospective analysis. Scand J Trauma Resusc Emerg Med. 2009 Nov 19;17:57. doi: 10.1186/1757-7241-17-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansson PI, Ostrowski SR. Acute coagulopathy of trauma: balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulation. Med Hypotheses. 2010 Dec;75(6):564–7. doi: 10.1016/j.mehy.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 31.Aird WC. Endothelium and haemostasis. Hämostaseologie. 2015;35(1):11–16. doi: 10.5482/HAMO-14-11-0075. [DOI] [PubMed] [Google Scholar]
- 32.Holcomb JB, Pati S. Optimal trauma resuscitation with plasma as the primary resuscitative fluid: the surgeon’s perspective. Hematology Am Soc Hematol Educ Program. 2013:656–9. doi: 10.1182/asheducation-2013.1.656. [DOI] [PubMed] [Google Scholar]
- 33.Watson JJ, Pati S, Schreiber MA. Plasma Transfusion: History, Current Realities, and Novel Improvements. Shock. 2016 Nov;46(5):468–479. doi: 10.1097/SHK.0000000000000663. [DOI] [PubMed] [Google Scholar]
- 34.Rahbar E, Cardenas JC, Baimukanova G, Usadi B, Bruhn R, Pati S, Ostrowski SR, Johansson PI, Holcomb JB, Wade CE. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J Transl Med. 2015 Apr;13:117. doi: 10.1186/s12967-015-0481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansson PI, Henriksen HH, Stensballe J, Gybel-Brask M, Cardenas JC, Baer LA, Cotton BA, Holcomb JB, Wade CE, Ostrowski SR. Traumatic Endotheliopathy: A Prospective Observational Study of 424 Severely Injured Patients. Ann Surg. doi: 10.1097/SLA.0000000000001751. Epub 2016 May 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spronk HM, Borissoff JI, ten Cate H. New insights into modulation of thrombin formation. Curr Atheroscler Rep. 2013 Nov;15(11):363. doi: 10.1007/s11883-013-0363-3. [DOI] [PubMed] [Google Scholar]
- 37.Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006 Apr;32(Suppl 1):49–60. doi: 10.1055/s-2006-939554. [DOI] [PubMed] [Google Scholar]
- 38.Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000 Apr;6(4):460–3. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 39.Tuma M, Canestrini S, Alwahab Z, Marshall J. Trauma and Endothelial Glycocalyx: The Microcirculation Helmet? Shock. 2016 Oct;46(4):352–7. doi: 10.1097/SHK.0000000000000635. [DOI] [PubMed] [Google Scholar]
- 40.Crimi E, Taccone FS, Infante T, Scolletta S, Crudele V, Napoli C. Effects of intracellular acidosis on endothelial function: an overview. J Crit Care. 2012 Apr;27(2):108–18. doi: 10.1016/j.jcrc.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Haywood-Watson RJ, Holcomb JB, Gonzalez EA, Peng Z, Pati S, Park PW, Wang WW, Zaske AM, Menge T, Kozar RA. Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS ONE. 2011 Aug;6(8):e23530. doi: 10.1371/journal.pone.0023530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozar RA, Pati S. Syndecan-1 restitution by plasma after hemorrhagic shock. J Trauma Acute Care Surg. 2015 Jun;78(6 Suppl 1):S83–6. doi: 10.1097/TA.0000000000000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrowski SR, Johansson PI. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J Trauma Acute Care Surg. 2012 Jul;73(1):60–6. doi: 10.1097/TA.0b013e31825b5c10. [DOI] [PubMed] [Google Scholar]
- 44.Johansson PI, Sørensen AM, Perner A, Welling KL, Wanscher M, Larsen CF, Ostrowski SR. High sCD40L levels early after trauma are associated with enhanced shock, sympathoadrenal activation, tissue and endothelial damage, coagulopathy and mortality. J Thromb Haemost. 2012 Feb;10(2):207–16. doi: 10.1111/j.1538-7836.2011.04589.x. [DOI] [PubMed] [Google Scholar]
- 45.Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, Sandler N, Grimm MJ, Segal BH, Otterbein LE, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS One. 2015 Mar 16;10(3):e0120549. doi: 10.1371/journal.pone.0120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013 May 2;121(18):3733–41. doi: 10.1182/blood-2012-11-468983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weigandt KM, White NJ, Chung D, Ellingson E, Wang Y, Fu X, Pozzo DC. Alterations in Fibrin Clot Structure and Mechanics Associated with Specific Oxidation of Methionine Residues in Fibrinogen. Biophys J. 2012 Dec 5;103(11):2399–407. doi: 10.1016/j.bpj.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White NJ, Wang, Fu X, Cardenas JC, Martin EJ, Brophy DF, Wade CE, Wang X, St John AE, Lim EB, Stern SA, et al. Post-translational oxidative modification of fibrinogen is associated with coagulopathy after traumatic injury. Free Radic Biol Med. 2016 Jul;96:181–189. doi: 10.1016/j.freeradbiomed.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, Pittet JF. Critical role of activated Protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Annals of Surgery. 2012 Feb;255:379–85. doi: 10.1097/SLA.0b013e318235d9e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campbell JE, Meledeo MA, Cap AP. Comparative response of platelet fV and plasma fV to activated protein C and relevance to a model of acute traumatic coagulopathy. PLoS ONE. 2014 Jun;9:e99181. 32. doi: 10.1371/journal.pone.0099181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandler WL. Procoagulant activity in trauma patients. Am J Clin Pathol. 2010 Jul;134(1):90–6. doi: 10.1309/AJCP3WPOYSKK6BFE. [DOI] [PubMed] [Google Scholar]
- 52.Dunbar NM, Chandler WL. Thrombin generation in trauma patients. Transfusion. 2009 Dec;49(12):2652–60. doi: 10.1111/j.1537-2995.2009.02335.x. [DOI] [PubMed] [Google Scholar]
- 53.Cardenas JC, Rahbar E, Pommerening MJ, Baer LA, Matijevic N, Cotton BA, Holcomb JB, Wade CE. Measuring thrombin generation as a tool for predicting hemostatic potential and transfusion requirements following trauma. J Trauma Acute Care Surg. 2014 Dec;77(6):839–45. doi: 10.1097/TA.0000000000000348. [DOI] [PubMed] [Google Scholar]
- 54.Hayakawa M, Sawamura A, Gando S, Kubota N, Uegaki S, Shimojima H, Sugano M, Ieko M. Disseminated intravascular coagulation at an early phase of trauma is associated with consumption coagulopathy and excessive fibrinolysis both by plasmin and neutrophil elastase. Surgery. 2011 Feb;149(2):221–30. doi: 10.1016/j.surg.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 55.Omar MN, Mann KG. Inactivation of factor Va by plasmin. J Biol Chem. 1987 Jan;262:9759–5. [PubMed] [Google Scholar]
- 56.Nogami K, Shima M, Matsumoto T, Nishiya K, Tanaka I, Yoshioka A. Mechanisms of plasmin-catalyzed inactivation of factor VIII: a crucial role for proteolytic cleavage at Arg336 responsible for plasmin-catalyzed factor VIII inactivation. J Biol Chem. 2007 Feb 23;282(8):5287–95. doi: 10.1074/jbc.M607816200. [DOI] [PubMed] [Google Scholar]
- 57.Hur WS, Mazinani N, Lu XJ, Britton HM, Byrnes JR, Wolberg AS, Kastrup CJ. Coagulation factor XIIIa is inactivated by plasmin. Blood. 2015 Nov 12;126(20):2329–37. doi: 10.1182/blood-2015-07-650713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaplan AP, Ghebrehiwet B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol. 2010 Aug;47(13):161–9. doi: 10.1016/j.molimm.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 59.Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A, Frigout Y, Hommet Y, Lebouvier L, Montaner J, et al. Hyperfibrinolysis increases blood brain barrier permeability by a plasmin and bradykinin-dependent mechanism. Blood. 2016 doi: 10.1182/blood-2016-03-705384. Epub Aug 16. [DOI] [PubMed] [Google Scholar]
- 60.Sane DC, Moser TL, Greenberg CS. Limited proteolysis of vitronectin by plasmin destroys heparin binding activity. Thromb Haemost. 1991 Sep;66(3):310–4. [PubMed] [Google Scholar]
- 61.Hermel M, Dailey W, Hartzer MK. Efficacy of plasmin, microplasmin, and streptokinase-plasmin complex for the in vitro degradation of fibronectin and laminin- implications for vitreoretinal surgery. Curr Eye Res. 2010 May;35(5):419–24. doi: 10.3109/02713680903572517. [DOI] [PubMed] [Google Scholar]
- 62.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000 May;98(4):323–32. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 63.Loew D, Perrault C, Morales M, Moog S, Ravanat C, Schuhler S, Arcone R, Pietropaolo C, Cazenave JP, van Dorsselaer A, et al. Proteolysis of the exodomain of recombinant protease-activated receptors: prediction of receptor activation or inactivation by MALDI mass spectrometry. Biochemistry. 2000 Sep;39(35):10812–22. doi: 10.1021/bi0003341. [DOI] [PubMed] [Google Scholar]
- 64.Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, Mitra S, Ghasabyan A, Chin TL, Sauaia A, et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J Trauma Acute Care Surg. 2016 Jan;80(1):16–23. doi: 10.1097/TA.0000000000000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardenas JC, Matijevic N, Baer LA, Holcomb JB, Cotton BA, Wade CE. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock. 2014 Jun;41(6):514–21. doi: 10.1097/SHK.0000000000000161. [DOI] [PubMed] [Google Scholar]
- 66.Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, Khan S, De’Ath HD, Allard S, Hart DP, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013 Feb;11(2):307–14. doi: 10.1111/jth.12078. [DOI] [PubMed] [Google Scholar]
- 67.Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, Sauaia A, Cotton BA. Acute Fibrinolysis Shutdown after Injury Occurs Frequently and Increases Mortality: A Multicenter Evaluation of 2,540 Severely Injured Patients. J Am Coll Surg. 2016 Apr;222(4):347–55. doi: 10.1016/j.jamcollsurg.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shakur H, Roberts I, Bautista R, Caballero J, Coats T, Dewan Y, El-Sayed H, Gogichaishvili T, Gupta S, Herrera J, et al. CRASH-2 trial collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010 Jul;376(9734):23–32. doi: 10.1016/S0140-6736(10)60835-5. [DOI] [PubMed] [Google Scholar]
- 69.Cap AP. Plasmin: a driver of hemovascular dysfunction. Blood. 2016 Nov;128(20):2375–2376. doi: 10.1182/blood-2016-09-735720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012 Jul;73(1):13–9. doi: 10.1097/TA.0b013e318256deab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma. 2001 Oct;51(4):639–47. doi: 10.1097/00005373-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 72.White NJ, Newton JC, Martin EJ, Mohammed BM, Contaifer D, Jr, Bostic JL, Brophy GM, Spiess BD, Pusateri AE, Ward KR, et al. Clot Formation Is Associated with Fibrinogen and Platelet Forces in a Cohort of Severely Injured Emergency Department Trauma Patients. Shock. 2015 Aug;44(Suppl 1):39–44. doi: 10.1097/SHK.0000000000000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spinella PC, Doctor A. Role of transfused red blood cells for shock and coagulopathy within remote damage control resuscitation. Shock. 2014 May;41(Suppl 1):30–4. doi: 10.1097/SHK.0000000000000089. [DOI] [PubMed] [Google Scholar]
- 74.Lacroix J, Hébert PC, Fergusson DA, Tinmouth A, Cook DJ, Marshall JC, Clayton L, McIntyre L, Callum J, Turgeon AF, et al. ABLE Investigators; Canadian Critical Care Trials Group. Age of transfused blood in critically ill adults. N Engl J Med. 2015 Apr;372(15):1410–8. doi: 10.1056/NEJMoa1500704. [DOI] [PubMed] [Google Scholar]
- 75.Spann AP, Campbell JE, Fitzgibbon SR, Rodriguez A, Cap AP, Blackbourne LH, Shaqfeh ES. The Effect of Hematocrit on Platelet Adhesion: Experiments and Simulations. Biophys J. 2016 Aug 9;111(3):577–88. doi: 10.1016/j.bpj.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hellem AJ, Borchgrevink CF, Ames SB. The role of red cells in haemostasis: the relation between haematocrit, bleeding time and platelet adhesiveness. Br J Haematol. 1961 Jan;7:42–50. doi: 10.1111/j.1365-2141.1961.tb00318.x. [DOI] [PubMed] [Google Scholar]
- 77.Emerson CP, Ebert RV. A Study of Shock in Battle Casualties: Measurements of the Blood Volume Changes Occurring in Response to Therapy. Ann Surg. 1945 Nov;122(5):745–72. doi: 10.1097/00000658-194511000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kozar RA, Peng Z, Zhang R, Holcomb JB, Pati S, Park P, Ko TC, Paredes A. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011 Jun;112(6):1289–95. doi: 10.1213/ANE.0b013e318210385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pati S, Wataha K, Menge T, Deng X, Bode A, Holcomb JB, Potter D, Kozar R, Spinella RC, Pati S. Sprayed dried plasma and fresh frozen plasma modulate permeability and inflammation in vitro in vascular endothelial cells. Transfusion. 2013 Jan;53(Suppl 1):80S–90S. doi: 10.1111/trf.12040. [DOI] [PubMed] [Google Scholar]
- 80.Cardenas JC, Cap AP, Swartz MD, Huby MP, Baer LA, Matijevic N, Cotton BA, Holcomb JB, Wade CE. Plasma Resuscitation Promotes Coagulation Homeostasis Following Shock-Induced Hypercoagulability. Shock. 2016 Feb;45(2):166–73. doi: 10.1097/SHK.0000000000000504. [DOI] [PubMed] [Google Scholar]
- 81.Johnson JL, Moore EE, Kashuk JL, Banerjee A, Cothren CC, Biffl WL, Sauaia A. Effect of blood products transfusion on the development of postinjury multiple organ failure. Arch Surg. 2010 Oct;145(10):973–7. doi: 10.1001/archsurg.2010.216. [DOI] [PubMed] [Google Scholar]
- 82.Moore HB, Moore EE, Chapman MP, Gonzalez E, Slaughter AL, Morton AP, D’Alessandro A, Hansen KC, Sauaia A, Banerjee A, et al. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. J Thromb Haemost. 2015 Oct;13(10):1878–87. doi: 10.1111/jth.13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moore HB, Moore EE, Gonzalez E, Hansen KC, Dzieciatkowska M, Chapman MP, Sauaia A, West B, Banerjee A, Silliman CC. Hemolysis exacerbates hyperfibrinolysis, whereas platelolysis shuts down fibrinolysis: evolving concepts of the spectrum of fibrinolysis in response to severe injury. Shock. 2015 Jan;43(1):39–46. doi: 10.1097/SHK.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murphy S, Gardner FH. Effect of storage temperature on maintenance of platelet viability - deleterious effect of refrigerated storage. N Engl J Med. 1969 May;(280):1094–1098. doi: 10.1056/NEJM196905152802004. [DOI] [PubMed] [Google Scholar]
- 85.Valeri CR. Circulation and hemostatic effectiveness of platelets stored at 4 C or 22 C: studies in aspirin-treated normal volunteers. Transfusion. 1976 Jan-Feb;16(1):20–3. doi: 10.1046/j.1537-2995.1976.16176130832.x. [DOI] [PubMed] [Google Scholar]
- 86.Bynum JA, Meledeo MA, Getz TM, Rodriguez AC, Aden JK, Cap AP, Pidcoke HF. Bioenergetic profiling of platelet mitochondria during storage: 4°C storage extends platelet mitochondrial function and viability. Transfusion. 2016 Mar;56(Suppl 1):S76–84. doi: 10.1111/trf.13337. [DOI] [PubMed] [Google Scholar]
- 87.Baimukanova G, Miyazawa B, Potter DR, Gibb SL, Keating S, Danesh A, Beyer A, Dayter Y, Bruhn R, Muench MO, et al. The effects of 22°C and 4°C storage of platelets on vascular endothelial integrity and function. Transfusion. 2016 Mar;56(Suppl 1):S52–64. doi: 10.1111/trf.13455. [DOI] [PubMed] [Google Scholar]
- 88.Torres Filho IP, Torres LN, Valdez C, Salgado C, Cap AP, Dubick MA. Refrigerated platelets stored in whole blood up to 5 days adhere to thrombi formed during hemorrhagic hypotension in rats. J Thromb Haemost. 2017 Jan;15(1):163–175. doi: 10.1111/jth.13556. [DOI] [PubMed] [Google Scholar]
- 89.Fries D, Martini WZ. Role of fibrinogen in trauma-induced coagulopathy. Br J Anaesth. 2010 Aug;105(2):116–21. doi: 10.1093/bja/aeq161. [DOI] [PubMed] [Google Scholar]
- 90.Morrison JJ, Ross JD, Dubose JJ, Jansen JO, Midwinter MJ, Rasmussen TE. Association of cryoprecipitate and tranexamic acid with improved survival following wartime injury: findings from the MATTERs II Study. JAMA Surg. 2013 Mar;148(3):218–25. doi: 10.1001/jamasurg.2013.764. [DOI] [PubMed] [Google Scholar]
- 91.Curry N, Rourke C, Davenport R, Beer S, Pankhurst L, Deary A, Thomas H, Llewelyn C, Green L, Doughty H, et al. Early cryoprecipitate for major haemorrhage in trauma: a randomised controlled feasibility trial. Br J Anaesth. 2015 Jul;115(1):76–83. doi: 10.1093/bja/aev134. [DOI] [PubMed] [Google Scholar]
- 92.Rourke C, Curry N, Khan S, Taylor R, Raza I, Davenport R, Stanworth S, Brohi K. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J Thromb Haemost. 2012 Jul;10(7):1342–51. doi: 10.1111/j.1538-7836.2012.04752.x. [DOI] [PubMed] [Google Scholar]
- 93.Holcomb JB, Fox EE, Zhang X, White N, Wade CE, Cotton BA, del Junco DJ, Bulger EM, Cohen MJ, Schreiber MA, et al. the PROMMTT Study Group. Cryoprecipitate use in the PROMMTT study. J Trauma Acute Care Surg. 2013 Jul;75(1 Suppl 1):S31–9. doi: 10.1097/TA.0b013e31828fa3ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rahbar E, Cardenas JC, Matijevic N, Del Junco D, Podbielski J, Cohen MJ, Cotton BA, Holcomb JB, Wade CE the Early Whole Blood Investigators. Trauma, Time, and Transfusions: A Longitudinal Analysis of Coagulation Markers in Severely Injured Trauma Patients Receiving Modified Whole Blood or Component Blood Products. Shock. 2015 Nov;44(5):417–25. doi: 10.1097/SHK.0000000000000449. [DOI] [PubMed] [Google Scholar]
- 95.Cotton BA, Podbielski J, Camp E, Welch T, del Junco D, Bai Y, Hobbs R, Scroggins J, Hartwell B, Kozar RA, et al. Early Whole Blood Investigators. A randomized controlled pilot trial of modified whole blood versus component therapy in severely injured patients requiring large volume transfusions. Ann Surg. 2013 Oct;258(4):527–32. doi: 10.1097/SLA.0b013e3182a4ffa0. [DOI] [PubMed] [Google Scholar]
- 96.Spinella PC, Perkins JG, Grathwohl KW, Beekley AC, Holcomb JB. Warm fresh whole blood is independently associated with improved survival for patients with combat-related traumatic injuries. J Trauma Acute Care Surg. 2009 Apr;66(4 Suppl):S69–76. doi: 10.1097/TA.0b013e31819d85fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hooper TJ, Nadler R, Badloe J, Butler FK, Glassberg E. Implementation and execution of military forward resuscitation programs. Shock. 2014 May;41(Suppl 1):90–7. doi: 10.1097/SHK.0000000000000081. [DOI] [PubMed] [Google Scholar]
- 98.Strandenes G, De Pasquale M, Cap AP, Hervig TA, Kristoffersen EK, Hickey M, Cordova C, Berseus O, Eliassen HS, Fisher L, et al. Emergency whole-blood use in the field: a simplified protocol for collection and transfusion. Shock. 2014 May;41(Suppl 1):76–83. doi: 10.1097/SHK.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 99.Strandenes G, Austlid I, Apelseth TO, Hervig TA, Sommerfelt-Pettersen J, Herzig MC, Cap AP, Pidcoke HF, Kristoffersen EK. Coagulation function of stored whole blood is preserved for 14 days in austere conditions: A ROTEM feasibility study during a Norwegian antipiracy mission and comparison to equal ratio reconstituted blood. J Trauma Acute Care Surg. 2015 Jun;78(6 Suppl 1):S31–8. doi: 10.1097/TA.0000000000000628. [DOI] [PubMed] [Google Scholar]
- 100.Eliassen HS, Aandstad A, Bjerkvig C, Fosse T, Hervig TA, Pidcoke HF, Strandenes G. Making whole blood available in austere medical environments: donor performance and safety. Transfusion. 2016 Apr;56(Suppl 2):S166–72. doi: 10.1111/trf.13510. [DOI] [PubMed] [Google Scholar]