

Abstract
Photo-triggered α-helix formation of a 16-residue peptide featuring a built-in conformational photoswitch is monitored by time-resolved IR spectroscopy. An experimental approach with 2-ps time resolution and a scanning range up to 30 μs is used to cover all time scales of the peptide dynamics. Experiments are carried out at different temperatures between 281 and 322 K. We observe single-exponential kinetics of the amide I′ band at 322 K on a time scale comparable to a recent temperature-jump folding experiment. When lowering the temperature, the kinetics become slower and nonexponential. The transition is strongly activated. Spectrally dispersed IR measurements provide multiple spectroscopic probes simultaneously in one experiment by resolving the amide I′ band, isotope-labeled amino acid residues, and side chains. We find differing relaxation dynamics at different spectral positions.
Keywords: α-helix folding, femtosecond IR spectroscopy, protein folding
Conformational dynamics of peptides and proteins range from subpicosecond fluctuations of backbone dihedral angles (1) to collective motions of large regions of the molecule, extending to milliseconds and longer (2). Attempts to model dynamics of peptides and proteins often adopt a hierarchical view, which implies a separation of time scales generating classes of events that can be treated separately. The lower, faster hierarchical levels are typically handled in a collective statistical fashion, leading to a transitionstate-theory-like picture (3). This picture is justified if the coupling of the processes, occurring on different time and length scales, allows the selection of a reaction coordinate with a well defined barrier for a simplified description (3, 4). However, the overlap of time scales often brings about questions of the applicability of hierarchical models and leads to controversies, such as those about the interplay between hydrophobic collapse, secondary structure formation, and tertiary structure formation in protein folding (5, 6). In this article, we report on stretched kinetics, overlapping dynamics of different spectroscopic probes, and oscillations of the transient absorption during α-helix formation. These results indicate that even such simple phenomena as the formation of a single stretch of secondary structure are governed by multiple processes, and a separation of their time scales is not given.
Except in a few cases (7–9), the kinetics of helix folding has been inferred indirectly from thermal unfolding experiments (10–17) that require a number of assumptions. If one aims to enter a regime in which these approximations are likely to break down, it is clearly preferable to start from a largely unfolded ensemble and observe the relaxation into a helical state. Helix formation has been achieved previously by temperature (T)-jumping a cold denatured peptide (7). Also, photoexcitation of a ruthenium complex attached to a peptide chain has been used as a trigger (8). Although the complex is electronically excited, its large dipole moment promotes helix formation. A drawback of this approach is the perturbation of the peptide dynamics by electronic relaxation. Vibrational stark effects make the interpretation of spectral shifts difficult. A nonreversible approach for triggering is photocleavage of a disulfide cross-linking group that prevents helix formation in its bridged form (9). However, most of the sulfide biradicals recombine before secondary structure formation can take place.
Our present approach features the following three main ingredients to provide a detailed picture of folding dynamics.
To trigger folding, we use an azobenzene-based ultrafast reversible photoswitch that is covalently linked to the peptide and can be switched back and forth by using light of different wavelengths (18–23) (Fig. 1).
To cover the whole range of dynamics, we use a setup based on two synchronized femtosecond laser systems (24). In the present version, the delay can be continuously scanned up to 30 μs with a time resolution of 2 ps.
The importance of using multiple spectroscopic probes to discriminate different relaxation mechanisms has been stressed (4, 25). Spectrally dispersed multichannel detection of broadband femtosecond IR pulses provides multiple spectroscopic probes in a single experiment: The amide I′ band reports on the dynamics of the whole backbone. Isotope labeling reveals the dynamics at specific backbone sites (13). In the present work, we also resolve the dynamics of amino acid side chains. If folding was a simple two-state process, all observables must follow the same dynamics, yielding the same single-exponential rate constant; we will show that this is not the case here.
Fig. 1.

The photoswitchable FK11X peptide. (A) Cross-linker in cis (Left) and trans (Right) conformation. (B) FK11X sequence, showing the positions of the 13C labels (underlined residues) and the attached cross-linker. (C) Schematic models of cis-FK11X (Left) and trans-FK11X (Right), illustrating the conformational transition induced by the photoswitchable linker. Hydrogens and side chains are omitted for clarity.
We designed FK11X for strong helix propensity, starting from the general sequence Ac-(EAAAK)3-A-NH2 (26, 27), which is stabilized by interactions between the Glu and Lys side chains of opposite charge. In FK11X, Lys has been replaced by Arg to further increase helicity (28), and a Gln residue is used for C-terminal capping (29). Capping has been observed not only to stabilize the helix but also to speed up folding (30). Two alanines at positions 3 and 14 have been replaced by cysteine to allow for the attachment of the photoswitchable linker. Switching the linker between cis and trans modifies the energy landscape of the peptide and allows for larger changes in helicity than typically achieved in T-jump experiments. Three helical turns are required to form an α-helix out of the 12 residues bridged by the linker. These three turns do not fit well into the distance spanned by the linker in the cis form; thus, helicity is greatly reduced when the linker is in this conformation (CD yields 34% helicity at 5°C as described below). Only after stretching the linker by switching it to trans can the α-helix fold properly (93% helicity at 5°C). Therefore, ultrafast cis → trans isomerization of the linker projects the unfolded ensemble onto an energy landscape that favors the folded state, and folding can be observed as the dominant kinetic contribution. For isotope labeling, we chose two residues with an i, i + 3 spacing, because vibrational coupling between their amide I′ modes should change considerably after transition from extended to helical structure (31).
The complementary process of trans → cis isomerization was investigated recently by Chen et al. (32) using time-resolved optical rotary dispersion with a time resolution of 16 ns. They determined a time constant of 55 ns for the relaxation that is dominated by unfolding. Note that, although the same molecule has been investigated by Chen et al., the results cannot be compared directly, because trans → cis isomerization is followed by equilibration on the cis energy landscape, whereas we investigate dynamics on the trans landscape.
Materials and Methods
Peptide Synthesis. The 16-residue peptide Ac-EACAREAAAREAACRQ-NH2 [underlined residues are 13C-labeled in FK11X(iso)] was prepared by using standard fluorenylmethoxycarbonyl-based solid-phase peptide-synthesis methods (19). The two cysteine residues were cross-linked with the photoisomerizable linker (Fig. 1A) (18, 19) to obtain the isotope-labeled and -unlabeled photoswitchable peptides FK11X(iso) and FK11X (Fig. 1B).
Time-Resolved IR Spectroscopy. Most time-resolved measurements were performed on FK11X(iso). FK11X(iso) (700 μg) was dissolved in 260 μl of 10 mM deuterated phosphate buffer (pD 7.0). The sample was circulated through a closed-cycle CaF2 flow cell (pathlength, 50 μm) (33). The closed cycle was adjusted with a thermostat to ±1°C.
The dark-adapted FK11X(iso) is in the trans-azo conformation (ccis < 1%) (19), which favors helical conformation of the peptide (Fig. 1). To monitor folding, the initial cis (unfolded) state was prepared by continuous UV irradiation of the trans π → π* band of the linker with an Ar-ion laser (Innova 100, 363 nm, 200 mW; Coherent Radiation, Palo Alto, CA). The transition from cis (unfolded) to trans (helical) was initiated by a short (700-fs) 425-nm laser pulse.
The evolution of the peptide after photoswitching the linker was monitored by time-resolved IR spectroscopy. To investigate the whole range of the dynamics with 2-ps time resolution up to 30 μs, a setup of two synchronized 1-kHz Ti:sapphire-oscillator/regenerative amplifier femtosecond laser systems was used (24). System 1 was frequency-doubled to generate pulses at 425 nm for switching the linker. System 2 pumped an optical parametric amplifier with a difference-frequency mixing stage to obtain IR probe pulses (100 fs; center frequency, 1,620 cm-1; bandwidth, 240 cm-1 full width at half maximum) (34). The delay of the pulses was controlled electronically. The IR beam was split into a probe and a reference beam that were focused into the sample with a spot size of 80 μm. The probe beam was centered in the 425-nm pump spot (140 μm), and the reference beam passed the flow cell 1 mm upstream. Probe and reference beams were frequency-dispersed in a spectrometer and imaged onto a 2 × 32-pixel HgCdTe detector array.
CD Measurements. CD measurements were performed on a Jasco (Easton, MD) model J-710 spectropolarimeter. Helix content was calculated by using [θ]222 of FK11X dissolved in 50% trifluoroethanol at 5°C as 100% helical (27).
Experimental Results and Assignments
Steady-State IR Spectroscopy. The IR absorption spectrum of trans-FK11X shows a broad amide I′ band centered at 1,638 cm (Fig. 2 Upper), as commonly observed for small α-helical peptides in 2H2O (35, 36). Between 1,520 cm and 1,620 cm-1, we find several bands that exist in both FK11X(iso) and FK11X. They belong to the side chains of Glu [1,563 cm-1, antisymmetric carboxylate (37)] and Arg [1,583 and 1,600 cm-1, symmetric and antisymmetric CN3H5+ (37)]. A ring mode of azobenzene causes a tiny absorption at 1,601 cm-1, which can be better seen in the transient spectra at picosecond delays (see below). The band of the two isotope labels is not clearly resolved, because it is obscured by the absorption of the six side chains. It leads to an increased absorption around 1,590 cm-1 in FK11X(iso), which is the position expected for a doubly labeled helix, with the labeled amino acids separated by two unlabeled residues (35).
Fig. 2.

IR spectra. (Upper) Fourier transform IR (FTIR) absorption spectra of trans-FK11X(iso) and FK11X. (Lower) Transient difference spectra of FK11X(iso) at different delays and magic angle polarization. Steady-state Fourier transform IR difference spectra of FK11X(iso) and FK11X. All measurements were taken at 20°C. See Experimental Results and Assignments for discussion of numbered peaks.
Steady-state Fourier transform IR difference spectra were obtained by subtracting the cis (unfolded) from the trans (helical) spectrum (Fig. 2, lowest plot). We observe a red shift of the amide I′ band with a decreased absorbance at 1,665 cm [“random-coil” frequency (10, 14, 36)] and an increase at 1,633 cm [position 9; solvent-exposed α-helix in 2H2O (14, 36)]. At 1,590 cm (position 10) we find that FK11X(iso) differs from FK11X as expected for an isotope-labeled α-helix (35). Side-chain absorption at 1,560 cm is also slightly changed by helix formation.
Overview of the Transient Spectral Features. The time-resolved measurements discussed here were carried out for FK11X(iso). At early times (1 ps), the signal is dominated by the response of the linker (Fig. 2), which is heated by absorption of the UV photon and subsequent ultrafast electronic relaxation of the azobenzene moiety (20, 23, 38). The local heating leads to a red shift and broadening of the vibrational bands of the linker (38). By comparison with other molecules (20), we can assign the signals at 1 ps to N=N stretching of azobenzene (Fig. 2, position 1), a ring mode of azobenzene (position 2), and the C=O stretching of the amide groups of the linker that are conjugated with the azobenzene moiety (position 3; see Fig. 1 for the linker structure). The heat signal decays within a few picoseconds as the local excess energy is rapidly dissipated (20).
Accordingly, the heat signature is greatly reduced in the 11-ps spectrum. Already on this very short time scale we observe a bleach at the position of the amide I′ band (position 4), as well as at the position of the Glu side chains (position 5), indicating a perturbation of the peptide backbone by the stretching of the photoswitch.
The amide I′ bleach is growing and shifting slightly until ≈100 ps, reflecting small changes in the backbone at this early time. The heat is fully dissipated into the buffer after ≈20 ps, inducing a temperature rise of ≈10 mK, which causes a small background signal of the deuterated phosphate buffer (Fig. 2, dashed line at 100-ps delay, measured by exciting the 1,550-cm band of 2H2O and scaled accordingly) two to three orders of magnitude smaller than in typical T-jump measurements. This background stays constant for longer delays. It is essentially flat, and thus, the structure in the signal clearly originates from peptide dynamics.
Between 100 and 7 ns, only minor changes of the signal are observed at the position of the isotope labels (position 6). Comparison of FK11X(iso), unlabeled FK11X (Fig. 2, dotted line), and the buffer background shows that the difference signal around 1,590 cm (position 6) in FK11X(iso) is due to the isotope-labeled residues. The details of the dynamics in this spectral range are discussed below.
The spectrum after 420 ns shows the signature of helix formation. The red shift of the amide I′ band is now clearly visible (position 7), as is the rising absorption at the Glu side-chain position (position 8). Folding is largely finished after ≈10 μs at 20°C (2 μs at 49°C; see Fig. 3), and the transient spectrum then closely resembles the steady-state difference spectrum.
Fig. 3.

Time-dependent amplitude of the amide I′ difference signal at different temperatures and exponential and stretched exponential fits. (Inset) Dots, Arrhenius plot of the rates (taken from exponential fits); solid line, fit using Eq. 2 assuming non-Arrhenius (quadratic in 1/T, where T is temperature) behavior. Within the small temperature interval we cannot distinguish an Arrhenius (linear in 1/T) from a quadratic dependence on 1/T.
Amide I′ Dynamics and Their Temperature Dependence. The main signal reporting on helix formation is the amide I′ difference signal. Fig. 3 shows the dynamics of the unlabeled amide I′ band at different temperatures. The amplitude [ΔA(1,635 cm-1) - ΔA(1,662 cm-1)] is plotted, and its maximum is normalized to 1 for better comparison. Exponential (e-t/τ) and stretched exponential exp[-(t/τ)β] (39) fits are shown. Lowering the temperature from 49° to 8°C slows down equilibration by one order of magnitude. Moreover, with lower temperature the kinetics become more and more stretched, which is measured by the exponent β varying from β = 1.00 at 49°C to β = 0.69 at 8°C (see Table 1). We cannot completely rule out a small contribution to the signal from the reverse trans → cis process. However, we estimate it to be 5% at most, which is too small to explain the observed values of β.
Table 1.
Helicities in the trans state and parameters for the fits in Fig. 3
|
e-t/τ
|
e-(t/τ)β
|
|||
|---|---|---|---|---|
| T, °C | Helicity, % | τ, μs | τ, μs | β |
| 8 | 93 | 2.2 | 2.4 | 0.69 |
| 20 | 86 | 1.2 | 1.2 | 0.82 |
| 39 | 71 | 0.44 | 0.44 | 0.91 |
| 49 | 64 | 0.27 | 0.27 | 1.00 |
Dynamics at Selected Spectral Positions. During the first nanoseconds, we observe spectral changes mainly at the position of the labeled residues (Fig. 2, position 6). This is highlighted in Fig. 4 A and B, showing selected transient spectra, and in Fig. 4C, showing the dynamics at selected spectral positions. After the first 20 ps that are dominated by heat dissipation and deflection of the backbone by the linker, the absorption at the position of the isotope labels around 1,590 cm first rises until ≈0.5 ns (see Fig. 4C, isotope-labeled transient). It subsequently decreases from 0.5 to 18 ns, whereas there is no comparable change in the amide I′ band of the unlabeled residues at 1,638 cm-1 (Fig. 4A). Fig. 4 B and C show how the decrease at the labeled position continues up to 100 ns. However, after 18 ns, the signal of the unlabeled amide I′ band now grows in, leading to a rising absorption at 1,638 cm-1. At the isotope position, the absorption begins to rise toward its final value only after 100 ns. The oscillating transient absorption at the position of the isotope labels and its shift against the unlabeled amide I′ band clearly show that different processes overlap in time during relaxation.
Fig. 4.

Dynamics of selected spectral positions in the early nanosecond range at 20°C. (A) Decreasing absorption at the isotope position. (B) Decrease at the isotope position continues until 100 ns, by which time build up at the amide I′ and Glu position has begun already. Only after 100 ns does the absorption at the isotope position rise. (C) Dynamics at selected spectral positions. The solid lines are fits with stretched exponentials.
The side-chain signal (Fig. 2, position 8) around 1,555 cm shows a similar time dependence as the amide I′ band at 1,638 cm (position 4): at short times, we observe a bleach (position 5), and at longer times, we observe a positive signal (position 8) rising together with the amide I′ band (position 7), as shown in greater detail in Fig. 4B. The transients in Fig. 4C indicate that the side-chain dynamics may be slightly faster than the rise of the amide I′ band. Fits with stretched exponentials yield time constants of 810 versus 1,150 ns, respectively.
Discussion
To understand the action of the photoswitch on the peptide backbone, it is instructive to compare the results on FK11X with our previous work on a cyclic octapeptide, in which we observed dynamics to be faster by several orders of magnitude (20). The small, strained, cyclic peptide features a photoisomerizable azobenzene-based ω-amino acid as a rigidly linked part of its backbone. Thus, stretching of this switch exerts strong forces on the rest of the backbone. Already after 20 ps, we observed a transient signal closely resembling the steady-state difference spectrum, showing that the major part of the conformational transition takes place on this ultrafast time scale. This result is in strong contrast to FK11X, for which the time scale of linker isomerization in the range of 1 ps (40) is very much separated from the major changes of amide I′ absorption that report on α-helix formation. Therefore, we conclude that the bias imposed by the photoswitch in FK11X is soft and changes the energy landscape globally without introducing strong gradients that drive the system toward the final state. Molecular mechanics calculations show that the linker in the cis state prefers distances between the cysteine sulfur atoms of 6.2–14.6 Å, whereas in the trans state, the favored distances are 17.0–18.6 Å (41), better matching the distance expected for an α-helical conformation. This change between cis and trans is small compared with the length of the peptide chain bridged by the linker, which would measure ≈40 Å in extended conformation. Thus, switching the linker leads to a comparably slight deflection of the backbone that is reflected by the small bleach in the amide I′ band, occurring on the time scale of tens of picoseconds (see Fig. 2). Furthermore, the four flexible single bonds on each side of the linker provide a soft coupling between the backbone and the more rigid conjugated part of the azobenzene moiety. Unlike in the cyclic octapeptide, the linker does not force the molecule into a certain secondary structure. A more adequate picture of the switching in FK11X is that stretching the linker opens up a new part of conformational space for the backbone. The system is transferred from the cis to the trans energy landscape, where it folds in a diffusion-controlled way on time scales that are comparable with those obtained by a recent T-jump α-helix folding experiment (7).
A striking observation in FK11X is the stretching of amide I′ kinetics at lower temperatures (Fig. 3). The high time resolution reveals these kinetics without being affected by the instrument response. It is well known from chemical kinetics that if the system has to surmount a single high barrier (large compared with kBT), the kinetics is single-exponential. In this case, the system is effectively a two-state system. The observed nonexponentiality implies that a single pronounced barrier does not exist and that the free-energy surface is relatively flat (because, in addition, the equilibrium constants do not deviate much from 1). A large body of theoretical work and simulations predict that the folding kinetics of polypeptides should become nonexponential at lower temperature (4, 42–46). Despite a systematic search for this effect (47), experimental support for this kind of temperature dependence is sparse (48), and it has been observed only for a synthetic (phenylacetylene) helical oligomer (49). Sabelko et al. (50) investigated refolding of two cold-denatured proteins with kinetics that, in contrast, became more exponential as the temperature was lowered. However, one has to keep in mind that in the cold-denaturation regime the stability of the native state as well as the surface roughness is reduced with lower temperature (50). If the reason for nonexponentiality is the stability of the folded relative to the unfolded state, our observations in fact do comply with those of Sabelko et al. (50). With increasing stability of the folded state, any rate-limiting dominant barrier might be flattened out, and folding becomes a downhill process with kinetics governed by surface roughness (51). This conclusion has been confirmed by simple lattice dynamics models (44, 45) that revealed stretching factors β < 1 with decreasing temperature. The kinetic effect of helix stabilization was investigated by Wang et al. (30). For 19-mer peptides (length comparable with FK11X), the use of stabilizing capping residues resulted in acceleration and stretching of the kinetics. For proteins, it has been shown that reducing the folding barrier by mutagenesis may result in nonexponential kinetics as well (52).
We find helix folding to be a strongly thermally activated process (Fig. 3 Inset), in agreement with the T-jump experiment by Werner et al. (7). This finding is corroborated by more indirect conclusions from unfolding experiments (12, 14, 30, 53). Previously, the strong temperature dependence was often interpreted in terms of a two-state model in which unfolded and folded states are separated by a large enthalpic barrier. Fitting the temperature dependence of the rates by an Arrhenius law,
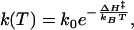 |
[1] |
would yield an apparent activation enthalpy of ΔH‡ = 38.9 kJ/mol and a large preexponential factor of k0 = 6.9 × 1012 s-1. However, a single rate-limiting barrier much larger than kBT would lead to strictly single-exponential kinetics, which is inconsistent with the observation of stretched exponentials (unless the large activation enthalpy is compensated by a large activation entropy resulting in a relatively flat free-energy barrier). Instead of relating the observed thermal activation to a single dominant barrier, it can also be attributed to multiple local barriers between misfolded microstates or, in other words, to an activated diffusion process on a rugged energy landscape. It has been shown that for a random distribution of barrier heights, the diffusion constant depends more strongly on temperature (54, 55),
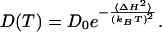 |
[2] |
Within the small temperature range of Fig. 3, fits based on Eqs. 1 and 2 both work equally well; thus, we cannot distinguish Arrhenius (linear in 1/T) from non-Arrhenius (quadratic in 1/T) behavior. Yet, Eq. 2 yields a much more realistic number for the average barrier heights (roughness) (ΔH = 7.1 kJ/mol), which agrees well with what is expected for hydrogen-bond breaking (56). The preexponential factor is 3 × 109 s-1 in this case.
Indeed, there is increasing evidence from both experimental work and simulations that the so-called unfolded state of proteins and even of small peptides is by no means random, as assumed in statistical models of helix–coil relaxation (57–65). Only in recent years has it become possible to simulate peptide dynamics in atomic detail by using force fields that allow for a realistic description of native and nonnative interactions (61, 62). Simulations show that the unfolded state of peptides comprises vastly less conformations than one would expect based on the number of conformational degrees of freedom present, and that those states are stabilized by more or less specific nonnative contacts (61–66). Significant enthalpic barriers in the folding process arise from the breaking of nonnative contacts such as nonlocal hydrogen bonds of the backbone and of the side chains as well as interactions between hydrophobic side chains (64, 65, 67, 68), rendering the energy landscape rugged. Chowdhury et al. (68) found rapid nucleation of the helix within 1 ns in their simulations of a hexadecapeptide, in agreement with the simulations of Hummer et al. (65). However, instead of subsequent rapid helix propagation driven by the formation of backbone hydrogen bonds, as envisioned in the statistical “kinetic zipper model” of helix–coil relaxation (11), they found that folding is hampered by nonnative contacts, the breaking of which determines the rate of helix formation. They concluded that helix nucleation does not trigger quick formation of the entire helix. Similar results are obtained by Bertsch et al. (67), who observe the frequent inhibition of helix propagation by nonnative hydrogen bonds.
Fig. 5 shows the sketch of a rugged energy-landscape model inspired by the work of Bicout and Szabo (69). It can qualitatively account for all our experimental observations (i.e., the temperature dependence of the overall rate, equilibrium constant, and stretching factor). It contains a distinguished folded state, a largely flat, but rugged conformational space of unfolded states, and a relatively small barrier separating both regions. The barrier is entropic, reflecting the narrowing conformational space in the vicinity of the folded state (69). If the overall rate keq connecting unfolded states was much faster than the rate kfold toward the folded state, the folding kinetics would be exponential, because all unfolded states would be in equilibrium among each other at any time. In this case, one could view them as one unified ensemble of unfolded states, and a two-state picture would be appropriate. However, if both rates are of the same order of magnitude (or if kfold > keq), nonexponential folding kinetics is expected. In this case, we indeed find that β≈ 0.7 in simple kinetic simulations. Hence, the diffusion on the rugged energy surface becomes rate limiting and might even dominate the barrier connecting to the folded state. To obtain a situation in which the nonexponentiality increases with lowering temperature, keq should decrease quicker with temperature than kfold. Diffusion on a rugged energy surface supports a large variation of keq with temperature (Eq. 2), whereas an entropic barrier will render kfold temperature independent. An additional negative activation enthalpy for this barrier may further assist the large variation of the stretching factor β within a relatively small temperature range, as observed in this work.
Fig. 5.

A simple model explaining the experimental observations. The solid line is a free-energy surface, and the dotted line represents the enthalpic contribution only. The roughness of the unfolded manifolded is characterized by ΔH.
The model shown in Fig. 5, although certainly not unique, explains all the experimental observations in a physically reasonable manner (69). Several alternative models that we tested in simulations, including initiation–propagation models, produce stretching factors β > 1 (contrary to what we observe) when starting as far away from equilibrium as in the present case. A discussion of this finding would go beyond the scope of the present article and will be published elsewhere.
The time-resolved amide I′ spectra of FK11X(iso) provide no hints for the existence of distinct intermediates resulting from traps or wells on the rugged landscape. However, the amide I′ absorption measures the peptide conformation averaged over all residues and therefore is relatively insensitive to the underlying heterogeneity. The isotope labels in FK11X(iso) avoid this averaging and allow measurement of dynamics at a specific backbone site. The high time resolution reveals oscillations of the isotope signal on a logarithmic time scale (Fig. 4C). Such logarithmic oscillations have been found to be indicative for relaxation processes on a rugged energy landscape (39) and clearly discard a simple two-state folding model. It is very likely that other residues would show a similar behavior if labeled and that the smooth but nonexponential kinetics of the unlabeled amide I′ band is just the average of local processes of similar complexity. However, extended labeling studies are needed to obtain a more detailed picture.
Uncertainty exists in the literature as to the origins of the so-called instantaneous component, a fast component beyond the time resolution (≥10 ns) of previous approaches, which has been observed in most T-jump experiments on helical systems (10, 13–17, 30). It often has the same or even larger amplitude than the slower component attributed to the actual α-helix folding/unfolding dynamics. Two origins have been discussed: an instantaneous spectral shift caused by changes in solvation due to the T-jump (10, 15, 17, 53) or a very fast conformational change (13, 15, 53). We observe a rapid bleach of the amide I′ band during the first 20 ps that changes until ≈100 ps and then stays constant to the early nanosecond range. With the buffer background subtracted, its amplitude is only 15% of the final amplitude of the difference signal, much less than the instantaneous component typically observed in T-jump experiments. In our case, the T-jump and its influence on the peptide spectrum is negligible, and we attribute this rapid bleach to a deflection of the backbone by the linker, as described above. Arguing along the lines of the kinetic zipper model it has been presumed that the instantaneous component is caused by the fast redistribution of helix lengths (“zipping”). However, in our spectrally resolved measurements, the phase up to 10 ns does not show any spectral signature of the helical state. In light of the present observations, it seems likely that the instantaneous component in T-jump experiments is caused, to a large extent, by a change in solvation.
Conclusions
The fast isomerization of the azobenzene photoswitch on the time scale of 1 ps is decoupled from the backbone dynamics during folding. Our folding rates compare well to those obtained from T-jump experiments, confirming that triggering with a built-in photoswitch can be designed for essentially unperturbed folding dynamics. The high time resolution of the experiment enables discussion of deviations from exponential behavior without ambiguities introduced by overlap with the instrument response. We observed increasingly stretched kinetics of α-helix formation with lower temperature. Spectrally dispersed multichannel detection revealed kinetic heterogeneity of amide I′ band, isotope-labeled amide I′ oscillators, and amino acid side chains, which clearly shows the presence of different processes overlapping in time. Although the amide I′ band only provides averaged information on the backbone secondary structure, circumventing this averaging by isotope labeling in combination with the high time resolution reveals logarithmic oscillations of the transient absorption. This observation highlights the complexity of the transition on a local level. In agreement with previous studies, we find folding of a small α-helix to be considerably thermally activated. However, this finding does not necessarily require the existence of a pronounced rate-limiting barrier. Large thermal activation can also arise from an activated diffusion process on a rugged energy landscape, which is more consistent with our observations of nonexponentiality, kinetic heterogeneity, and strong temperature dependence of β.
The present results call for additional investigation of the cause of nonexponentiality in α-helix formation. A key role of nonnative contacts in secondary structure formation as suggested by recent all-atom molecular dynamics simulations would question simple statistical kinetic models, because folding then strongly depends on nonlocal interactions within the peptide sequence. The present experimental approach is well suited to address these questions further: more detailed isotope-labeling studies can access events at different backbone sites and side chains. The possibility of tailoring the action of the photoswitch should be explored further to drive the system to certain locations on the modified energy landscape: an α-helical peptide in which the photoswitch bridges only four residues has already been reported (19). In this peptide, switching to cis preforms one helical turn. If helix nucleation were a rate-limiting event in folding, switching would mean a jump right onto this free-energy barrier, which should lead to a speed-up of the folding kinetics.
Acknowledgments
We thank Martin Volk for valuable discussions. This work has been supported by Swiss National Science Foundation Grant 2100-067573/1.
Author contributions: J.B., J.H., J.R.K., G.A.W., and P.H. designed research; J.B., J.H., J.R.K., G.A.W., and P.H. performed research; J.B., J.H., J.R.K., G.A.W., and P.H. analyzed data; and J.B. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: T-jump, temperature jump.
References
- 1.Woutersen, S., Mu, Y., Stock, G. & Hamm, P. (2001) Proc. Natl. Acad. Sci. USA 98, 11254-11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daniel, R. M., Dunn, R. V., Finney, J. L. & Smith, J. C. (2003) Annu. Rev. Biophys. Biomol. Struct. 32, 69-92. [DOI] [PubMed] [Google Scholar]
- 3.Karplus, M. (2000) J. Phys. Chem. B 104, 11-27. [Google Scholar]
- 4.Gruebele, M. (2002) Curr. Opin. Struct. Biol. 12, 161-168. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson, N. & Fersht, A. R. (2003) Curr. Opin. Struct. Biol. 13, 75-81. [DOI] [PubMed] [Google Scholar]
- 6.Volk, M. (2001) Eur. J. Org. Chem. 14, 2605-2621. [Google Scholar]
- 7.Werner, J. H., Dyer, R. B., Fesinmeyer, R. M. & Andersen, N. H. (2002) J. Phys. Chem. B 106, 487-494. [Google Scholar]
- 8.Huang, C.-Y, She, H., DeGrado, W. F., McCafferty, D. G. & Gai, F. (2002) J. Am. Chem. Soc. 124, 12674-12675. [DOI] [PubMed] [Google Scholar]
- 9.Volk, M., Kholodenko, Y., Lu, H. S. M., Gooding, E. A., DeGrado, W. F. & Hochstrasser, R. M. (1997) J. Phys. Chem. B 101, 8607-8616. [Google Scholar]
- 10.Williams, S., Causgrove, T. P., Gilmanshin, R., Fang, K. S., Callender, R. H., Woodruff, W. H. & Dyer, R. B. (1996) Biochemistry 35, 691-697. [DOI] [PubMed] [Google Scholar]
- 11.Thompson, P. A., Eaton, W. A. & Hofrichter, J. (1997) Biochemistry 36, 9200-9210. [DOI] [PubMed] [Google Scholar]
- 12.Thompson, P. A., Munoz, V., Jas, G. S., Henry, E. R., Eaton, W. A. & Hofrichter, J. (2000) J. Phys. Chem. B 104, 378-389. [Google Scholar]
- 13.Huang, C.-Y, Getahun, Z., Wang, T., DeGrado, W. F. & Gai, F. (2001) J. Am. Chem. Soc. 123, 12111-12112. [DOI] [PubMed] [Google Scholar]
- 14.Huang, C.-Y, Klemke, J. W., Getahun, Z., DeGrado, W. F. & Gai, F. (2001) J. Am. Chem. Soc. 123, 9235-9238. [DOI] [PubMed] [Google Scholar]
- 15.Huang, C.-Y., Getahun, Z., Zhu, Y., Klemke, J. W., DeGrado, W. F. & Gai, F. (2002) Proc. Natl. Acad. Sci. USA 99, 2788-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu, Y., Alonso, D. O. V., Maki, K., Huang, C.-Y., Lahr, S. J., Daggett, V., Roder, H., DeGrado, W. F. & Gai, F. (2003) Proc. Natl. Acad. Sci. USA 100, 15486-15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petty, S. A. & Volk, M. (2004) Phys. Chem. Chem. Phys. 6, 1022-1030. [Google Scholar]
- 18.Kumita, J. R., Smart, O. S. & Woolley, G. A. (2000) Proc. Natl. Acad. Sci. USA 97, 3803-3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flint, D. G., Kumita, J. R., Smart, O. S. & Woolley, G. A. (2002) Chem. Biol. 9, 391-397. [DOI] [PubMed] [Google Scholar]
- 20.Bredenbeck, J., Helbing, J., Sieg, A., Schrader, T., Zinth, W., Renner, C., Behrendt, R., Moroder, L., Wachtveitl, J. & Hamm, P. (2003) Proc. Natl. Acad. Sci. USA 100, 6452-6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bredenbeck, J., Helbing, J., Behrendt, R., Renner, C., Moroder, L. & Hamm, P. (2003) J. Phys. Chem. B 107, 8654-8660. [Google Scholar]
- 22.Spörlein, S., Carstens, H., Satzger, H., Renner, C., Behrendt, R., Moroder, L., Tavan, P., Zinth, W. & Wachtveitl, J. (2002) Proc. Natl. Acad. Sci. USA 99, 7998-8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wachtveitl, J., Spörlein, S., Satzger, H., Fonrobert, B., Renner, C., Behrendt, R., Moroder, L. & Zinth, W. (2004) Biophys. J. 86, 2350-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bredenbeck, J., Helbing, J. & Hamm, P. (2005) Rev. Sci. Instrum. 75, 4462-4466. [Google Scholar]
- 25.Hagen, S. J. (2003) Proteins 50, 1-4. [DOI] [PubMed] [Google Scholar]
- 26.Marqusee, S. & Baldwin, R. L. (1989) Proc. Natl. Acad. Sci. USA 84, 8898-8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marqusee, S., Robbins, V. H. & Baldwin, R. L. (1989) Proc. Natl. Acad. Sci. USA 86, 5286-5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merutka, G., Shalongo, W. & Stellwagen, E. (1991) Biochemistry 30, 4245-4248. [DOI] [PubMed] [Google Scholar]
- 29.Aurora, R. & Rose, G. D. (1998) Protein Sci. 7, 21-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang, T., Zhu, Y., Getahun, Z., Du, D., Huang, C.-Y., DeGrado, W. F. & Gai, F. (2004) J. Phys. Chem. B 108, 15301-15310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi, J.-H., Ham, S. & Cho, M. (2003) J. Phys. Chem. B 107, 9132-9138. [Google Scholar]
- 32.Chen, E., Kumita, J. R., Woolley, G. A. & Kliger, D. S. (2003) J. Am. Chem. Soc. 125, 12443-12449. [DOI] [PubMed] [Google Scholar]
- 33.Bredenbeck, J. & Hamm, P. (2003) Rev. Sci. Instrum. 74, 3188-3189. [Google Scholar]
- 34.Hamm, P., Kaindl, R. A. & Stenger, J. (2000) Opt. Lett. 25, 1798-1800. [DOI] [PubMed] [Google Scholar]
- 35.Huang, R., Kubelka, J., Barber-Armstrong, W., Silva, R. A. G. D., Decatur, S. M. & Keiderling, T. A. (2004) J. Am. Chem. Soc. 126, 2346-2354. [DOI] [PubMed] [Google Scholar]
- 36.Martinez, G. & Millhauser, G. (1995) J. Struct. Biol. 114, 23-27. [DOI] [PubMed] [Google Scholar]
- 37.Barth, A. (2000) Prog. Biophys. Mol. Biol. 74, 141-173. [DOI] [PubMed] [Google Scholar]
- 38.Hamm, P., Ohline, S. M. & Zinth, W. (1997) J. Chem. Phys. 106, 519-529. [Google Scholar]
- 39.Metzler, R., Klafter, J. & Jortner, J. (1999) Proc. Natl. Acad. Sci. USA 96, 11085-11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satzger, H., Root, C. & Braun, M. (2004) J. Phys. Chem. A 108, 6265-6271. [Google Scholar]
- 41.Burns, D. C., Flint, D. G., Kumita, J. R., Feldman, H. J., Serrano, L., Zhang, Z., Smart, O. S. & Woolley, G. A. (2004) Biochemistry 43, 15329-15338. [DOI] [PubMed] [Google Scholar]
- 42.Šali, A., Shakhnovich, E. & Karplus, M. (1994) Nature 369, 248-251. [DOI] [PubMed] [Google Scholar]
- 43.Onuchic, J. N., Luthey-Schulten, Z. & Wolynes, P. G. (1997) Annu. Rev. Phys. Chem. 48, 545-600. [DOI] [PubMed] [Google Scholar]
- 44.Nymeyer, H., Garcia, A. E. & Onuchic, J. N. (1998) Proc. Natl. Acad. Sci. USA 95, 5921-5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skorobogatiy, M., Guo, H. & Zuckermann, M. (1998) J. Chem. Phys. 109, 2528-2535. [Google Scholar]
- 46.Zhou, Y., Zhang, C., Stell, G. & Wang, J. (2003) J. Am. Chem. Soc. 125, 6300-6305. [DOI] [PubMed] [Google Scholar]
- 47.Gillespie, B. & Plaxco, K. W. (2000) Proc. Natl. Acad. Sci. USA 97, 12014-12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saigo, S. & Shibayama, N. (2003) Biochemistry 42, 9669-9676. [DOI] [PubMed] [Google Scholar]
- 49.Yang, W. Y., Prince, R. B., Sabelko, J., Moore, J. S. & Gruebele, M. (2000) J. Am. Chem. Soc. 122, 3248-3249. [Google Scholar]
- 50.Sabelko, J., Ervin, J. & Gruebele, M. (1999) Proc. Natl. Acad. Sci. USA 96, 6031-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang, W. Y. & Gruebele, M. (2004) Biophys. J. 87, 596-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, W. Y. & Gruebele, M. (2003) Nature 423, 193-197. [DOI] [PubMed] [Google Scholar]
- 53.Wang, T., Du, D. & Gai, F. (2003) Chem. Phys. Lett. 370, 842-848. [Google Scholar]
- 54.Zwanzig, R. (1988) Proc. Natl. Acad. Sci. USA 85, 2029-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bryngelson, J. D. & Wolynes, P. G. (1989) J. Phys. Chem. 93, 6902-6915. [Google Scholar]
- 56.Sheu, S.-Y., Yang, D.-Y., Selzle, H. L. & Schlag, E. W. (2003) Proc. Natl. Acad. Sci. USA 100, 12683-12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cho, J.-H., Sato, S. & Raleigh, D. P. (2004) J. Mol. Biol. 338, 827-837. [DOI] [PubMed] [Google Scholar]
- 58.Baldwin, R. L. (2002) Adv. Protein Chem. 62, 361-367. [DOI] [PubMed] [Google Scholar]
- 59.Shi, Z., Woody, R. W. & Kallenbach, N. R. (2002) Adv. Protein Chem. 62, 163-240. [DOI] [PubMed] [Google Scholar]
- 60.Shortle, D. (1996) FASEB J. 10, 27-34. [DOI] [PubMed] [Google Scholar]
- 61.Daura, X., Glättli, A., Gee, P., Peter, C. & van Gunsteren, W. F. (2002) Adv. Protein Chem. 62, 341-360. [DOI] [PubMed] [Google Scholar]
- 62.van Gunsteren, W. F., Bürgi, R., Peter, C. & Daura, X. (2001) Angew. Chem. Int. Ed. 40, 351-355. [PubMed] [Google Scholar]
- 63.Garcia, A. E. (2004) Proteins 45, 669-676. [Google Scholar]
- 64.Mu, Y., Nguyen, P. & Stock, G. (2005) Proteins 58, 45-52. [DOI] [PubMed] [Google Scholar]
- 65.Hummer, G., Garcia, A. E. & Garde, S. (2001) Proteins 42, 77-84. [PubMed] [Google Scholar]
- 66.Zhang, W., Lei, H., Chowdhury, S. & Duan, Y. (2004) J. Phys. Chem. B 108, 7479-7489. [Google Scholar]
- 67.Bertsch, R. A., Vaidehi, N., Chan, S. I. & Goddard, W. A., III, (1998) Proteins 33, 343-357. [DOI] [PubMed] [Google Scholar]
- 68.Chowdhury, S., Zhang, W., Wu, C., Xiong, G. & Duan, Y. (2003) Biopolymers 68, 63-75. [DOI] [PubMed] [Google Scholar]
- 69.Bicout, D. J. & Szabo, A. (2000) Protein Sci. 9, 452-465. [DOI] [PMC free article] [PubMed] [Google Scholar]