

Abstract
The reconstruction of gene regulatory networks underlying cell differentiation from high-throughput gene expression and chromatin data remains a challenge. Here, we derive dynamic gene regulatory networks for human myeloid differentiation using a 5-day time-series of RNA-seq and ATAC-seq data. We profile HL-60 promyelocytes differentiating into macrophage, neutrophil, monocyte, and monocyte-derived macrophages. We find a rapid response in the expression of key transcription factors and lineage markers that only regulate a subset of their targets at a given time, which is followed by chromatin accessibility changes that occur later along with further gene expression changes. We observe differences between promyelocyte-derived and monocyte-derived macrophages at both the transcriptional and chromatin landscape level, despite using the same differentiation stimulus, which suggest that the path taken by cells in the differentiation landscape defines their end cell state. More generally, our approach of combining neighboring time points and replicates to achieve greater sequencing depth can efficiently infer footprint-based regulatory networks from long series data.
eTOC paragraph
We use a human cell line model of myeloid differentiation time-course to study the dynamics of gene regulation. We integrate neighboring time-points of gene expression and chromatin accessibility data, to generate cell-and time-specific gene regulatory networks that identify changes in transcription factor interactions during myeloid differentiation.
Introduction
Vertebrate developmental commitments are implemented within cells through remodeling of chromatin accessibility that allow transcription factor binding of promoter and enhancer cis-regulatory modules (CRMs) across the genome to allow for transcription factor binding. The identification of CRMs is therefore critical to understanding the complexities of gene regulatory circuits in a variety of organisms (Hardison and Taylor, 2012; Peters and Davidson, 2015). The derivation of transcription factor footprints is a powerful application of open chromatin assays such as ATAC-seq and DNase-seq. DNaseI footprinting has been used to identify transcription factor occupancy (Neph et al., 2012) and to extract transcriptional networks in many biological contexts (Sullivan et al., 2014). Recently, ATAC-seq was also applied to characterizing transcription factor regulation in the mammalian brain (Mo et al., 2015) and identifying variation in primary T cells (Qu et al., 2015). There has been relatively less work in incorporating open chromatin data directly in a dynamic gene regulatory network (GRN). Sullivan et al. characterized light/dark time-specific dynamics in A. thaliana through the generation of chromatin interaction networks (Sullivan et al., 2014). Yet all GRNs are by their very nature dynamic and should ideally capture the many steps of differentiation that have been described in well-defined systems such as T-cell development (Zhang et al., 2012).
The immune system is a complex and interactive network of diverse cell types, with a myriad of functional properties that are crucial to maintaining an immunological-responsive balance within an organism. The coordinated organization of cellular differentiation starting from a hematopoietic stem cell is established early and maintained throughout the development of an organism, resulting in the generation of the interacting innate and adaptive immune systems. Much is known about the vast heterogeneity of surface marker expression throughout hematopoietic cellular differentiation and maturation. Considerable marker and cellular plasticity exists across the adaptive (Zhu and Paul, 2010) and innate immune systems (Ginhoux and Jung, 2014). Due to the difficulty in differentiating primary immune cells ex vivo, recent studies have extracted dynamics of cellular commitment from comparisons of terminally differentiated primary cells (Chen et al., 2014), still limited by sorting of cell populations.
Several studies have looked at genome-wide chromatin accessibility dynamics in myeloid cells, including tissue macrophage populations (Lavin et al., 2014), terminally differentiated immune cells (Lara-Astiaso et al., 2014), neutrophil populations (Wong et al., 1999), chromatin dynamics of macrophages and monocytes (Saeed et al., 2014), immune-responsive late enhancers in macrophages upon stimulation (Ostuni et al., 2013), and immediate responses to lipid A stimulation in mouse macrophages (Tong et al., 2016). Other studies measured chromatin accessibility in the context of immune-responsive late enhancers in macrophages upon stimulation (Ostuni et al., 2013) and immediate responses to lipid A stimulation in mouse macrophages (Tong et al., 2016). While studies have investigated gene networks across immune cell-types (Spooner et al., 2009; Yosef et al., 2013; Zhang et al., 2012), temporal dynamic gene regulatory networks during cell differentiation have not been previously described. Studies have focused on changes in histone modifications and transcription factor binding by comparing terminally differentiated cells, but a comprehensive view of how chromatin elements gain or lose accessibility throughout myeloid differentiation has not been published and would by default not capture any intermediate stages for which appropriate markers have not yet been identified. An unbiased and appropriately controlled model system is needed to probe changes in gene expression and the chromatin landscape for the purpose of mapping the regulatory networks that act during myeloid differentiation.
The HL-60 promyelocytic cell line (Gallagher et al., 1979) has been used extensively to study the properties of granulocyte proliferation, myeloid differentiation, and malignant function over the years (Collins, 1987). One of this cell line’s most interesting properties is its ability to terminally differentiate through the induction of chemical agents into monocyte (Mangelsdorf et al., 1984), macrophage (Murao et al., 1983), and neutrophil (Breitman et al., 1980) lineages. While its aberrant proliferative capacity is not comparable to any normal promyelocytic cell, it shares a required transcriptional program that mediates proper cellular commitment inherent in normal granulocyte differentiation (Collins, 1987). HL-60 cells’ aberrant mutations in the MYC locus and their proliferative capability have been compared to normal cells (Harris and Ralph, 1985). In addition, groups such as the ENCODE Project Consortium have catalogued gene expression, chromatin elements, and transcription factor binding in HL-60 cells (The ENCODE Project Consortium, 2012). Furthermore, a considerable amount of recent single-cell genomic data is available for HL-60 (Buenrostro et al., 2015; Cusanovich et al., 2015).
We use the HL-60 model system to address the transcriptional complexity and the cis-regulatory dynamics in myeloid differentiation using a 5-day time-series of transcriptome and chromatin accessibility by probing from the earliest (3 hours) to the latest (up to 168 hours depending on the cell) stages of cellular differentiation. We observe initially rapid changes in gene expression that are followed by later changes in chromatin accessibility during cellular differentiation. We observe that “direct” macrophages and monocyte-derived macrophages have significantly different gene expression and open chromatin profiles, even though they were differentiated with the same PMA stimulus. We use dynamically expressed regulators and chromatin footprinting in our 192 datasets to build draft gene regulatory networks of human macrophage, monocyte and neutrophil differentiation. Finally, we find that key transcriptional regulators such as PU.1 exhibit definitive roles that both maintain and drive the regulatory circuits specified during human myeloid differentiation.
Results
A transcriptional time course of myeloid differentiation
We induced the differentiation of HL-60 cells into three distinct lineages to measure the transcriptional and open chromatin dynamics that drive cellular commitment into macrophage, monocyte, and neutrophil lineages. We characterized the morphology of each lineage during induction of differentiation using Giemsa staining (Figure 1A) and also measured immune cell function using phagocytosis assays. We observed changes in cellular morphology as early as 3 hours post-differentiation, with all lineages reaching terminal differentiation by 120 hours. Furthermore, to characterize how HL-60 derived macrophages differ epigenetically from monocyte-derived macrophages, we induced the differentiation of monocytes to macrophages (monocyte-derived macrophage) for 48 hours and measured changes in chromatin accessibility, gene expression, and function (Figure 1B). Moreover, we included immune effects induced by LPS-stimulation at various time-points of differentiation (Figure 1B). All measurements were done in triplicate, resulting in 96 RNA-seq and 96 ATAC-seq datasets. This allowed us to reproducibly measure 69,658 accessible chromatin elements across human myeloid differentiation. We used cellular morphology, and the time-series profiling of chromatin and expression patterns to segment each time-point into temporal stages. We observed distinct morphological changes during the intermediate and late stages such as macrophages growing in cell size (15 to 25 microns) and changing neutrophil nuclei morphologies before reaching a terminally differentiated and segmented conformation at 4–5 days of differentiation (Figure 1A). We further observed genome-wide similarities of chromatin accessibility and gene expression for closely timed-samples (3, 6 and 12 hr; 24 and 48 hr; 96 and 120 hr) in monocyte, neutrophil, and macrophage time-series (Figure 1C–D, black boxes). Thus, in order to compare cell stages during differentiation across all myeloid cell-types, we grouped time-points into early (3 to 12 hrs), intermediate (12 to 48 hrs), and late (48 to 120 hrs) temporal stages for both chromatin accessibility and gene expression (Figure 1D). Additionally, because of the high quality of our ATAC-seq data (Figure S6), we used the pooled stages primarily for the purpose of calling transcription factor footprints from the ATAC-seq data in order to build a dynamic gene regulatory network of myeloid differentiation into macrophages, monocytes, and neutrophils.
Figure 1. A dynamic model of human myeloid differentiation using HL-60s.

(A) HL-60 directed differentiation of neutrophil, monocyte, and macrophage show cell intermediates at varying time-points using Giemsa staining. Intermediate progenitors were characterized based on morphology during the course of differentiation and categorized as: (I) immature macrophage, (II) macrophage < 20μm, (III) macrophage > 20μm, (IV) myelocyte, (V) banded neutrophil, (VI) segmented neutrophil, (VII) monoblast, (VIII) promonocyte, (IX) monocyte. Scale indicates 20μm. (B) Schematic outline of study design. Colored cell identifier denotes myeloid cell-types. (C) Temporal staging of time-points for each cell-type was based on the clustering of RNA-seq and ATAC-seq data. (D) Genome-wide clustering reveals inherent structure of the transcriptome and chromatin landscape during myeloid differentiation (Pearson correlation). Black boxes mark samples grouped as temporal stages. Red boxes indicate LPS induced time-points.
Cell-specific trajectories during differentiation reflect the transcriptional differences between macrophage sub-types
We performed a principal component analysis on the RNA-seq and ATAC-seq time-series respectively (Figure 2A–B). We found that the first principal component for the RNA-seq time-series explains the differences between each myeloid cell type (Figure 2A). Conversely, the first component for the ATAC-seq data reflects the temporal attribute of our differentiation system (Figure 2B). Additionally, the principal component analysis of our RNA-seq and ATAC-seq datasets for the different myeloid lineages revealed that while both directly derived macrophage and monocyte-derived macrophages look morphologically identical, they have dramatically different gene expression and open chromatin profiles. This is particularly striking given the gene expression time-points of early directly-derived macrophages (3hr) and late monocytes are quite similar (120hr, right before differentiation into macrophages). In order to identify the candidate regulators that exhibit either shared or specific expression profiles between macrophage and monocyte-derived macrophage cells, we hierarchically clustered 901 expressed transcription factors expressed across our time-series (> 1 FPKM) (Figure 2C). Several regulators were differentially expressed (p-value < 0.05) in monocyte-derived macrophages compared to macrophages, which include the nuclear orphan receptor factors (NR4A1, NR4A2, NR4A3), EGR3, FOSB, HES1, and PRDM8 genes (Figure 2D). A second cluster shows comparable expression in both macrophage sub-types for EGR1, EGR2, MAFB, POU2F2, RELB, NFKB2, and ATF3 regulators (Figure 2E). These regulators are important in the pro-inflammatory function, maturation, and differentiation of macrophages (Aziz et al., 2009; Ginhoux and Jung, 2014). Recently, rapid up-regulation of EGR1, EGR2, and NR4A1 was observed in mouse macrophages within minutes of lipid A stimulation (Tong et al., 2016), which suggests a similar role in function during both stimulation and differentiation of macrophages. Interestingly, we detect differential gene expression (p-value < 0.05) for MAF and TEAD2 only in the latest stages of macrophage specification (Figure 2F), reflecting the specific differences in regulator expression between macrophage and monocyte-derived macrophages in our system.
Figure 2. Distinct cells during myeloid differentiation.

(A and B) Principal component analysis of RNA-seq and ATAC-seq time-series. Time-points were connected serially to illustrate cell-specific trajectories. Cell-types are labeled with distinct colored points. (C) Heatmap of 902 expressed transcription factors during macrophage, monocyte, and monocyte-derived macrophage differentiation. Each column represents the expression for a time-point, whereas each row represents a transcription factor. RNA-seq data is row-mean normalized and row clustered using Euclidean distance. (D) Representative cluster of transcription factors showing the highest expression in monocyte-derived macrophages. (E) Cluster of transcription factors that demonstrate similar expression patterns between macrophage and monocyte-derived macrophage time-points. (F) Representative cluster of transcription factors expressed specifically in macrophage cells.
Measuring the earliest regulatory transitions during cellular commitment
Having identified rapid changes in cellular morphology upon differentiation in our Giemsa staining assays, we focused our attention on characterizing the earliest expression and chromatin dynamics during the first 24 hours of differentiation. We first focused on genes that are up-regulated as early as 3 hours. Transcriptional regulators such as the key myeloid regulator PU.1, the macrophage-specific factor MAFB, and members of the EGR and STAT families show an increase in expression after 3 hours of macrophage induction (Figure S1A). We found a higher number of genes to be up-regulated in macrophages (321 genes p-value < 0.05, Figure S1D–E) when compared to neutrophils (71 genes p-value <0.05, Figure S1F) and monocytes (64 genes p-value <0.05, Figure S1G) at our earliest 3-hour time-point using maSigPro. The number of differentially expressed genes at 6 hours post-differentiation in neutrophils (146 genes p-value <0.05, Figure S1B) and monocytes (306 genes p-value < 0.05, Figure S1C) are more comparable to the number of genes up-regulated after 3 hours post-macrophage differentiation (Figure S1E). Known markers, such as the monocyte markers CD14 and transcription factor CEBPE show increased expression in monocytes while CCR6 and B2M reflect an appropriate neutrophil-specific differentiation response. Thus, the macrophage commitment program changes the expression of a larger number of genes sooner than neutrophil and monocyte terminal differentiation does in our system.
While changes in gene expression can rapidly occur as a result of cell signaling induced by stress or immune stimulus, chromatin accessibility dynamics depend on both transcription factor occupancy and nucleosome remodeling, which may be rate limiting. To this end, we analyzed accessibility dynamics of regulatory chromatin elements over the first 24 hours and identified a total of 893 differentially accessible chromatin regions, 112 of which lose their chromatin accessibility over the first 24 hours. The largest fraction of these differentially accessible regions occurs during macrophage specification (465 chromatin elements p-value < 0.05, Figure S1I), with a majority of chromatin elements becoming accessible by the 24-hour mark in all lineages. A similar enrichment of differential accessible elements 24 hours post-differentiation were also observed for neutrophil (152 chromatin elements p-value < 0.05, Figure S1J) and monocyte (276 chromatin elements p-value < 0.05, Figure S1K) lineages. These numbers represent a fraction of the overall changes in open chromatin detected in our time course, which suggests that changes in chromatin accessibility are more important for controlling the middle and later parts of myeloid differentiation programs.
Time-dependent modules of transcriptional regulation define myeloid differentiation programs
We identified 2,854 genes (e <0.05, FDR 1%) that were differentially expressed across our time-series and partitioned them into 13 clusters using a generalized linear model, which display both temporal and lineage-specific profiles (Figure 3A). We explored co-expression of genes across all clusters and report representative genes that are relevant to cell-specific differentiation programs (Figure 3B). Interestingly, we found that cluster 3 (324 genes, Figure 3B) exhibits maximal expression at 12 hours in macrophages and equivalent 132/144 hour time-point in monocyte-derived macrophage cells, while the expression profiles in neutrophils and monocytes were relatively static. This identifies a set of genes expressed with similar expression patterns, but with different relative magnitudes during the differentiation of our two macrophage sub-types, within the same time-window of differentiation.
Figure 3. Temporal modules of transcriptional regulator expression.

(A) Heatmap of 2,854 differentially expressed genes during myeloid differentiation (α=0.05, FDR < 1%, p-val < 0.01). 13 expression clusters were derived using k-means and denoted by both color and number for all differentially expressed genes. FPKM values log transformed and row-mean normalized for all genes. (B) Gene expression profiles for clusters (3, 6, 7, and 13). Mean expression and standard deviation (error bars) are plotted across all biological replicates and for all genes in each corresponding cluster. Cluster size (n) and representative genes (rg) are denoted. (C) Heatmap of 232 differentially expressed transcriptional regulators (α=0.05, FDR < 1%, p-val < 0.01). Seven regulator clusters were derived using k-means (C1–C7). Representative regulators from each cluster are shown respectively. FPKM values are log transformed and row-mean normalized for all regulators. (D) Schematic of transcriptional regulator classification. Transcriptional classes are denoted as immediate (blue), intermediate (pink), late (Purple) and static (Gray). (E and F) Transcriptional profiles for each cluster based on classification for macrophage and neutrophil cells, respectively. Mean FPKM expression is shown for each cluster. (G) PU.1 gene expression during myeloid differentiation increases across all cell-types. The mean FPKM values for each time-series are shown.
From our previous analysis, we had identified several transcriptional regulators that exhibited clear temporal and lineage dynamics. This prompted us to focus on transcription factor expression in our time-series. We partitioned 232 differentially expressed regulators into 7 clusters (Figure 3C), and identified time-dependent modules of transcriptional regulator expression. Such modules have also been described as ‘waves’ of transcriptional regulation in a previous study of Th17 differentiation (Yosef et al., 2013). We identify immediate, intermediate, and late modules of transcription factor expression during differentiation, and assign transcriptional classes to each cluster and lineage respectively (Figure 3D–F). Regulators that do not exhibit a distinct transcriptional class are considered static. The key myeloid regulator PU.1 shows a unique behavior compared to all transcription factors that set it apart from any class. Interestingly, we observe increased expression of PU.1 across our time-series but with observably distinct kinetics between each cell-type (Figure 3G). Our expression analysis found distinct time-dependent modules of transcriptional regulation that account for the distinct temporal role of key regulators in the differentiation program of our model of myeloid commitment.
Assessing chromatin landscape dynamics during differentiation and enrichment of transcriptional regulators
We addressed the genome-wide dynamics of chromatin accessibility using the same methods applied in deriving our expression results and identify 8,907 differentially accessible chromatin elements (α <0.05, FDR 1%) distributed in 13 clusters using a generalized linear model (Figure 4A). We observe a loss of differential chromatin accessibility in 2,732/8,907 (31%) chromatin elements (clusters; 7 Figure 4B, 9, 11) as cells were induced to differentiate, while 6,175/8,907 (69%) sites show a gain in chromatin accessibility that are shared or lineage-specific during myeloid differentiation. For example, elements from cluster 6 (n=778) show increased chromatin accessibility in both macrophage and monocyte-derived macrophages whereas cluster 5 elements (n=429) are primarily accessible in neutrophils (Figure 4A–B). Interestingly, we observe a cell-specific preference for chromatin element accessibility in macrophage and monocyte-derived macrophage cells (clusters; 2 & 6 Figure 4D). The FC receptor genes (FCRLA, FCRLB), which are enriched for accessibility in macrophages and monocyte-derived macrophages, exhibit shared and cell-specific preferences for chromatin element accessibility. Importantly, the similarity in accessibility between macrophage and monocyte-derived macrophage cells shows a clear demarcation from the chromatin landscape of both differentiated monocytes and neutrophils. Lastly, we detect a subset (cluster 8, n=275) of shared chromatin elements that are accessible in all terminally differentiated cells across the time-series (Figure 4D).
Figure 4. Temporal modules of differential chromatin accessibility.

(A) Heatmap of 8,919 differential accessible chromatin elements during myeloid differentiation (α=0.05, FDR < 1%, p-val < 0.01). 13 expression clusters were derived using k-means and denoted by both color and number for all chromatin elements. 10xRPM (Reads Per Million) values are log transformed and row-mean normalized. (B) Heatmaps showing ATAC-seq read density of chromatin elements from three clusters (5,6, and 7). Mean ATAC-seq read density was derived using temporal stages (e:Early, m: Middle, l: Late). ATAC-seq signal is shown for a window of +/−1.5 kb from the chromatin element center and ranked from strongest to weakest for all comparisons. Cluster size (n) is denoted. (C) Heatmap of de novo motif transcription factor enrichment. Rows indicate cluster of chromatin elements mined for motifs, while columns indicate transcription factor motif of interest. Transcription factor motifs were hierarchically clustered based on significance using a Euclidean distance. Non-significant motifs are represented as white boxes. Motif significance is shown for a q-val < 0.05 and q-val < 5×10−4 denoted by light or dark green boxes respectively. (D) Examples of chromatin element clusters specified during differentiation. Browser tracks of ATAC-seq data for all cell-types are normalized by read density. Chromatin elements from two differing cluster profiles reflect the complex regulatory diversity (left browser panel) during myeloid differentiation. Cell-specific chromatin accessibility is strongly enriched in neutrophils (middle panel), while temporal changes in chromatin element accessibility can be observed across all cell-types (last panel). Colored boxes identify with chromatin cluster.
We performed a de novo motif analysis on each accessible element across all 13 clusters to identify the transcriptional regulators enriched in our differentially accessible chromatin elements. We identified 21 transcription factor motifs de novo (significant; q-value < 0.05, highly significant; q-value < 5.0 × 10−4) enriched in our chromatin clusters (Figure 4C). Motifs for MYC and E2F1 were enriched in chromatin clusters 7 and 11, which exhibit a decrease in accessibility during myeloid differentiation. Since MYC and E2F1 were identified in clusters assigned to the immediate transcriptional class in our expression analysis (Figure 3C), it is likely that a depletion of MYC and E2F1 occupancy occurs at these elements during cellular commitment. Additionally, we observe the PU.1 motif in 12 of 13 chromatin clusters, EGR (11 of 13), STAT (4 of 13), and IRF (8 of 13), among many other transcription factor binding site motif enrichments. Here, an initial analysis of transcription factor motif enrichment in our chromatin accessible clusters, combined with gene expression information provide a series of potential candidates to further understanding immune cell regulation.
Transcriptome and chromatin dynamics in response to LPS stimulation
Lipopolysaccharide (LPS), which is an outer membrane component of Gram negative bacteria, is capable of eliciting a strong immune response in myeloid cells (Fujishima et al., 1993). To understand the responsiveness of all three myeloid lineages under LPS stimulation we measured immediate changes in the transcriptome and chromatin landscape of our differentiated cells after 3 hours in response to LPS treatment for macrophage (48 and 120 hours), neutrophil (120 hours), monocyte (120 hours) and monocyte-derived macrophage (168 hours) cells. We found 25 genes commonly up-regulated in monocyte, macrophage and neutrophil cells during LPS treatment (Figure S2D) including CXCL2, IL23A, CD44, and IL8, which have been shown previously to elicit an LPS-response in the immune system (De Filippo et al., 2013). Gene ontology analysis demonstrates significant term enrichment for immune response (p-value < 1×10−7) and programmed cell death (p-value 5×10−3). We measured differential expression of 48 and 120 hour macrophages under LPS treatment, observing up-regulation of 115 and 210 genes respectively (Figure S2A, S3A–B) such as known LPS-responsive genes IL8, SOD2, TLR2, as well as transcriptional regulators such as MAFB and FOSL1. We then compared the differentiation states of macrophages at 48 and 120 hours treated with LPS. We find that the majority of the up-regulated genes during this comparison are markers of cellular maturation in macrophages (CD68, CSF1, FCGRT), with few up-regulated genes during LPS stimulation (Figure S3B). Based on the assumption that the timing of cellular differentiation for monocyte-derived macrophage cells (168 hours) and macrophages (48 hours) are equivalent, we sought to address the similarity of expression profiles based on the cell-state and LPS response. While LPS-responsive genes (SOD2, PTGS2, IL8) showed no change differential change in expression during stimulation, several up-regulated genes demonstrate changes in expression for markers defining cellular identity (Figure S2C; CSF1: macrophage, CD14: monocyte). We furthermore found 80 genes that are up-regulated in monocytes treated with LPS (Figure S3C), while we detect 687 genes differentially expressed in neutrophils (Figure S2B). Interestingly, our analysis identifies the differential expression of IL6 in neutrophils after LPS treatment, which has been widely debated (Zimmermann et al., 2015), while PU.1 expression is unchanged. Our results demonstrate gene expression dynamics mediated by LPS treatment that are shared and cell-specific during cellular differentiation.
We measured the effects of LPS treatment on the chromatin landscape in each myeloid lineage. We found a total 352 chromatin elements that are differentially accessible in our time-series after 3 hours of LPS treatment. Interestingly, a considerable fraction of differentially accessible chromatin elements are dynamic specifically in neutrophils (266/352, 75%), while only minor changes in the chromatin landscape are significant in macrophages (19/352, 5%), monocytes (6/352, ~2%), and monocyte-derived macrophages (32/352, 9%) (Figure S2F). We also identify a subset of elements that are shared in neutrophils and monocyte-derived macrophages that become accessible upon LPS treatment (31/352, ~9%). Because neutrophils demonstrate a significant change in the chromatin landscape during LPS treatment, we performed an analysis to integrate gene expression and chromatin accessibility profiles (Supplemental methods and Figure S2E). We identified three clusters with dynamic profiles of chromatin accessibility during neutrophil differentiation (Figure S2G). Each differentially accessible chromatin element was associated to the nearest gene TSS (Supplemental methods). We identified several LPS-mediated gene-element associations in neutrophil cells (Figure S2G). We show two examples upon LPS treatment in neutrophils; positively correlated expression and chromatin dynamics (Figure S2H; NFKBIA), dynamic gene expression with a static chromatin landscape (Figure S2H′, SOD2), and unchanged accessibility and gene expression patterns (Figure S3D, S100A9). Our analysis of LPS-treated myeloid cells demonstrates that a measured gene expression response occurs quite rapidly, while the reorganization of the chromatin landscape is both cell-specific and time-sensitive.
Building myeloid gene regulatory networks using chromatin footprinting data
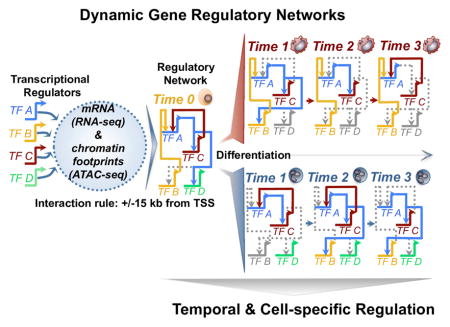
We leveraged our ATAC-seq datasets by merging biological replicates and time-points based on temporal staging (Figure 1C) to achieve ≥ 200 million reads for chromatin footprinting analysis. We identified an average of 85k footprints per time-point, resulting in more than 1.02 million chromatin footprints across our time-series. To infer transcriptional interactions using chromatin footprints, we generated a gene regulatory network of footprints in the promoter and proximal enhancers of key differentially regulated transcription factors (Supplemental methods), allowing us to identify temporal and cell-specific regulatory dynamics during cellular differentiation. Chromatin footprints derived for each transcription factor were associated to the nearest gene TSS. We included regulatory interactions for genomic distances of 15 kb or less between transcriptional regulators. Our strategy captured well-known myeloid regulators such as PU.1, EGR, GFI1, and CEBPα among others. We took into account the changes in expression of each transcription factor during differentiation into our networks. A ‘genome-view’ (Peters and Davidson, 2015) of all identified interactions derived from our networks, consisting of 23 regulators with 158 interactions is illustrated as a circuit diagram (Figure 5A). We found a range in correlation between the number of footprints detected and gene expression for regulator PU.1 during macrophage (R2=0.65) and monocyte (R2=0.6), but not neutrophil (R2=0.18) differentiation. We find that for the remaining 22 transcription factors, we did not observe a strong correlation between the number of active footprints and gene expression. One key difference between our draft GRNs and classical, perturbation-based GRNs is that our footprinting alone do not allow us to assign activating or repressing activity to footprints. We evaluated the precision of our networks in identifying PU.1 interactions using PU.1 HL-60 ChIP-seq data from ENCODE (Figure S4). Approximately ~90% of our PU.1 derived footprints are supported by PU.1 ChIP-seq data in undifferentiated HL-60 cells (Figure S4A, B, C). AUROC analysis with HL-60 PU.1 ChIP-seq and PU.1 ATAC-seq footprints we called support specificity and sensitivity of our analysis (Figure S4C). Furthermore, all five PU.1 edges in our HL-60 gene regulatory network were also supported by PU.1 ChIP-seq data. Several other linkages in our GRNs were previously identified in myeloid cells (Table S1), which suggest that our networks capture known regulatory interactions along with many new candidate interactions.
Figure 5. PU.1–regulated targets change in differentiating human myeloid cell types.

(A) A comprehensive ‘genome-view’ gene regulatory network snapshot of 23 transcriptional regulators and 158 inferred regulatory interactions were generated from ATAC-seq footprinting (Supplemental methods). The ‘genome-view’ network representation shows all inferred interactions during human myeloid differentiation. Each TF regulator is assigned a unique color identifier to track changes in regulatory interactions during differentiation. (B) PU.1 sub-circuit of regulatory interactions in undifferentiated HL-60 cells. (C) Genome-view of the PU.1 sub-circuitry in myeloid cells. We inferred 13 PU.1-mediated core regulatory interactions. (D) PU.1 sub-circuitry of differentiating neutrophils. (E) PU.1 sub-circuitry of differentiating monocytes. (F) PU.1 sub-circuitry of differentiating macrophage. Colored edges indicate that a regulatory interaction was observed for the respective time-point. Dashed grey edges indicate regulatory interactions observed at other time-points or cell-types respectively. Colored grey gene arrows indicate no mRNA detected at a given time-point.
PU.1-targeted regulatory interactions during myeloid differentiation
The PU.1 locus has been extensively characterized for transcription factor regulatory interactions in the context of hematopoietic development (Hoogenkamp et al., 2007; Leddin et al., 2011). To study the PU.1 regulatory interactions recovered in our myeloid time courses, we generated PU.1-specific subnetworks. A total of 21 interactions were observed, of which 8 would regulate the PU.1 locus (Figure 5A) and 13 downstream targets are PU.1-bound (Figure 5C). Among the first set, RUNX1 specifically regulates both the upstream regulatory elements (Huang et al., 2008) and promoter of PU.1 (Koh et al., 2013) in myeloid cells, which we recover for undifferentiated HL-60s as well as for differentiated macrophage, monocyte, monocyte-derived macrophage, and neutrophil subnetworks (Figure 5A). Similarly, CEBPα has also been shown to regulate PU.1 through regulation of its promoter and upstream regulatory elements in hematopoietic cells (Kummalue and Friedman, 2003; Yeamans et al., 2007) and we observe CEBPα regulating PU.1 specifically in differentiated macrophage and neutrophil cells (Figure 5A). Our subnetworks also capture the known PU.1 auto-regulatory feedback loop (Chen et al., 1995; Okuno et al., 2005) (Figure 5B, D–F), maintaining a constant transcriptional burst of expression across differentiation. Interestingly, our networks identify PU.1 occupancy for several additional regulators such as MAFB (Figure 5B), EGR (Figure 5D, F), STAT (Figure 5B, D, E), and VDR (Figure 5E–F). Our gene regulatory network analysis identifies likely candidates regulated by PU.1 during human granulopoiesis.
Myeloid cell specification network analysis
Having observed time-dependent modules of expression for many transcriptional regulators, we investigated the changes in our draft footprinting-based GRNs during early, intermediate, and late time points of differentiation in macrophage, neutrophil, monocyte, and monocyte-derived macrophage differentiation based on the grouping of time-points into temporal stages (Figure 1C). One example out of many dynamics in our footprinting GRN is the behavior of the transcriptional regulator PU.1. Because PU.1 acts as potent regulator of myeloid fate specification, we focused on PU.1-specific regulation of EGR family members in terminally differentiated cells. Neutrophil and macrophage cell fate specification is mediated through concerted regulation of PU.1, EGR (1,2), GFI1, and CEBPα (Laslo et al., 2006). Our PU.1 subnetwork identifies regulation of PU.1 of EGR family members during macrophage and neutrophil differentiation (Figure 5D, F). To this end, we generated both EGR (Figure S5) and GFI1 (Figure S4) subnetworks to illustrate their regulatory interactions in myeloid cells. EGR regulators can directly repress GFI1 promoter activity in NIH3T3 cells in luciferase assays (Laslo et al., 2006), and our EGR subnetwork captures this regulatory interaction using footprinting. We also identify EGR-specific regulation of lineage and temporal mediated regulators such as MAFB, which is specified during macrophage commitment, and JUND, which is specified in both monocyte and macrophage differentiation (Figure S5D, E). Our analysis emphasizes the dynamic expression patterns for each differentially expressed EGR (1,2,3) member during myeloid differentiation (Figure S5G). We observe the most dramatic change (~10–100 fold) in EGR expression in macrophage cells 3 hours post-differentiation. Interestingly, the monocyte-derived macrophage cells also display the same sharp and dramatic increase in EGR expression. We identify auto-regulatory feedback interactions for each EGR members across the different cell-types (Figure S5C–F), a likely explanation for dramatic and sustained EGR expression during differentiation. While the role of EGRs in specifying neutrophil and macrophage fates has been previously described (Laslo et al., 2006, 2008), our analysis provides additional chromatin interactions of EGR regulation during myeloid differentiation.
Similarly, the transcriptional repressor GFI1 has been previously shown to function as an integral regulator in multi-lineage blood cell development through the regulation of gene expression (Van der Meer et al., 2010; Möröy and Khandanpour, 2011). We identify dynamic changes in both the temporal expression (Figure S4C) and GFI1 occupancy for several transcription factors in our gene regulatory networks (Figure SD–H). GFI1 can promote neutrophil differentiation by antagonizing PU.1 and EGR activity via direct protein-protein interactions (Dahl et al., 2007) and through direct transcriptional repression (Kubosaki et al., 2009; Laslo et al., 2006; Spooner et al., 2009). Our analysis identifies this well-studied regulation of PU.1 by GFI1 in our HL-60 and neutrophil networks (Figure S4D, E). We recover the GFI1 auto-regulatory feedback loop in our neutrophil network (Figure S4E), which has been studied in previous work on GFI1 auto-regulation (Yücel et al., 2004), as well as previously unreported GFI1 regulatory interactions for STAT6 and MAFB in neutrophil and monocyte cell-types (Figure S4F, G). Our network analysis recovers many previously identified regulatory interactions fundamental in hematopoietic cells, in addition to many new candidate interactions. It also demonstrates the power of chromatin networks in uncovering the dynamic regulatory circuitry specified during cellular differentiation.
We tested a subset of our predictions by transfecting HL-60 cells with a siRNA targeting PU.1 (PU.1KD) and matching scrambled siRNAs, both in undifferentiated as well as 24-hour differentiated into macrophage, monocyte and neutrophils (Figure 6A). We profiled our PU.1KD 24-hours post-treatment using Smart-seq2 (Picelli et al., 2013) and ATAC-seq in biological replicates. PU.1 gene expression was significantly reduced in PU.1KD cells compared to controls in undifferentiated HL-60 (50%), macrophage (95%), monocyte (65%) and neutrophils (67%) (Figure 6B). Additionally, we observe several PU.1 targets such as STAT1, EGR, MAFB, and IRF2 with significant changes in expression upon PU.1 reduction (Figure 6B). Interestingly, we observe an up-regulation in STAT1, MAFB, and IRF2 transcription factors in both undifferentiated HL-60 and macrophages, while in monocytes we find expression differences for regulators STAT1 and IRF2. We also find cell-specific differences for regulators based on our PU.1 knockdown. E2F8 was significantly down-regulated in PU.1KD macrophage cells, but found that in undifferentiated HL-60 cells E2F8 expression increases (Figure 6B). Similar observations were also detected at the level of chromatin accessibility for PU.1 targets (Figure 6C). Interestingly, we find that chromatin accessibility changes were observed at additional regulatory elements (JUND, GFI1, NFE2) that could explain changes in gene expression not directly specified by PU.1 regulation (Figure 6C). We find that gene expression for STAT1 and MAFB was up-regulated in PU.1KD HL-60, but observe a loss in accessibility for STAT1 and gain in accessibility for MAFB revealing a specific level of regulation by PU.1 in distinct myeloid cells. Our companion ATAC-seq analyses found increased accessibility for 6,642 regions in PU.1KD and increased accessibility for 4,223 regions in control conditions for at least one cell type (Supplementary file 1). Focusing on our core gene regulatory network across myeloid differentiation, we found that our PU.1 knockdown clearly validates that 11 of 13 (85%) regulatory interactions in our core network are distinctly mediated by PU.1, based on differential gene expression and/or chromatin accessibility in one or more myeloid cells (Figure 6D, TFs with at least one asterisk). We observe significant up regulation in MAFB accessibility and gene expression in undifferentiated cells following PU.1 knockdown. Based on these results, PU.1 is thus predicted to act in a repressive manner, where depletion of PU.1 promotes both a change in accessibility and gene expression that leads to MAFB expression in undifferentiated myeloid cells. It has been previously shown that the lineage-specific transcription factor MAFB plays a key role in maintaining cytokine sensitivity thresholds and maintaining a self-renewal state in mouse hematopoietic stem cells through a PU.1 circuit (Sarrazin et al., 2009). Our results reveal that a PU.1 mediated interaction with MAFB is maintained in human undifferentiated HL-60 cells. The reduction of PU.1 levels ultimately results in the up-regulation of MAFB expression. Thus, PU.1 perturbations confirm most of the key inferred interactions of our core network and in addition would allow us to clearly mark edges as repressing or activating.
Figure 6. PU.1–regulated targets change in differentiated human myeloid cell types.

(A) Undifferentiated HL-60 were treated with a PU.1 siRNA or control siRNA. HL-60 cells were then induced to differentiate into macrophages, monocytes, and neutrophils and harvested 24-hours post siRNA treatment. Undifferentiated HL-60 cells were also measured following siRNA treatment for 24 hours. (B) mRNA fold change (Log2 transformed) between PU.1KD and siControl is shown for each PU.1 target from our gene regulatory network. Differential expression significance was calculated using biological replicates (Supplemental Methods). * p-value < 0.05; ** p-value < 0.01. (C) Chromatin accessibility fold change (Log2) between PU.1KD and control conditions is shown for PU.1 associated elements and chromatin elements near regulators. Distance of chromatin element to start of each gene is indicated. Differential accessibility is indicated * p-value < 0.05; ** p-value < 0.01. (D) Genome-view of the PU.1 sub-circuitry in myeloid cells. PU.1 regulatory interactions are specified as red arrows in our circuit diagram. Colored boxes underneath gene names indicate the cell-type origin of the PU.1 mediated interaction. Black asterisk indicates a change in PU.1 target by gene expression only. Blue asterisk indicates a change in PU.1 target by chromatin accessibility only. Orange asterisk indicates a change in PU.1 target detected by both gene expression and chromatin accessibility. Significance is indicated by a p-value < 0.05.
Overall, we found 2,911 genes to be differentially up-regulated in our PU.1KD and 2,870 genes up-regulated in control conditions for at least one cell-type (Supplementary file 2). We found that 13% of differentially expressed genes upon knockdown are direct PU.1 targets based on footprinting for at least one myeloid cell type. We extended our analysis to include regulatory interactions mediated by transcriptional regulators downstream of PU.1 in our gene regulatory network to include indirect targets, which jointly accounted for 32% of differentially expressed genes. Thus, the gene regulatory network nearly triples the number of differentially expressed genes whose change in expression due to our specific PU.1 perturbations can be accounted for.
Discussion
We used transcriptome and open chromatin changes in differentiating HL-60 promyelocytes to map gene regulatory networks of human myeloid cell differentiation into macrophages, monocytes, and neutrophils. Our gene expression time-series allowed us to carefully define temporal profiles of expression in connection with the timing of myeloid cell differentiation. Interestingly, we found greater gene expression changes earlier (~3 hours) during macrophage differentiation than in the monocyte and neutrophil lineages. Immediately up-regulated genes included key transcriptional regulators such as PU.1, MAFB, and EGR that are known to drive macrophage maturation and differentiation. For neutrophil and monocyte genes, we observed greater cell-specific changes in expression 6 hours post-differentiation. While we detected substantial changes in gene expression early (3–6 hours) in differentiation, we found relatively few differential changes in the chromatin landscape until 24 hours post-differentiation in all three lineages. Instead, the majority of changes observed across the accessible chromatin landscape occurred during the middle to late temporal stages of differentiation. This temporal delay in chromatin accessibility of regulatory elements is similar to the temporal delays in histone modifications that ultimately establish T-cell identity (Zhang et al., 2012). Our analysis of the most immediate transcriptome and accessible chromatin dynamics highlights that the earliest changes in gene expression are independent of chromatin remodeling, while intermediate and late changes in gene expression are likely dependent on chromatin organization, enzymatic histone modifications, and the exact levels of transcription factors during cell differentiation. This is likely to be a general principle of cellular specification.
Previously, blood monocytes were believed to predominantly give rise to all resident macrophages in adult tissues (van Furth and Cohn, 1968). Recent evidence now challenges this paradigm and offers insight about macrophage differentiation in the context of both tissue maintenance and development (Ginhoux and Jung, 2014). We leveraged our human myeloid system to study and to understand the derivation of macrophages from two alternative cell states: monocyte-derived and “direct” promyelocyte-derived precursor. Our analysis identified clusters of genes and chromatin elements shared between monocyte-derived macrophages and macrophages (Figure 3, 4, S2), demonstrating similarities in regulatory components that are distinct from both monocyte and neutrophil lineages. However, our PCA analysis demonstrates that the cell trajectories measured during differentiation are quite different for the two macrophage subtypes with respect to the transcriptome and chromatin landscape, even though both differentiation events were triggered by the same stimulus (PMA), resulting in subtype-specific expression for key transcriptional regulators (Figure 2). Furthermore, an analysis comparing LPS-mediated gene expression changes for equivalent time-points of macrophage (48 hours) and monocyte-derived macrophages (168 hours) showed that most of the differentially expressed genes were mediators of macrophage or monocyte specification. We hypothesized that the course of macrophage development and LPS stimulation would result in differing transcriptional profiles, ultimately driving differences in phagocytic function. While the monocyte-derived macrophage cells were able to exhibit phagocytosis functionality and cellular morphology consistent with macrophages (Figure 1), it is still possible that these cells may not have matured fully or reached a terminally differentiated state, as they still show expression for CD14. However, it is remarkable that the cells very near to each other in gene expression and chromatin space (3hr macrophages and 120hr monocytes) follow very different trajectories under the same differentiation stimulus while still maintaining a common cell type. Effectively, monocyte-derived macrophages “remember” that they were derived from monocytes, still able to function as macrophages.
We derived clusters of differentially expressed transcription factors as well as chromatin accessible elements genome-wide, and analyzed the difference in expression of several transcriptional regulators and their corresponding motif enrichment across the chromatin clusters. We built a myeloid differentiation gene regulatory network using open chromatin footprinting using methods previously used to study transcriptional regulatory interactions in several biological contexts (Neph et al., 2012b; Sullivan et al., 2014). In our case, we integrated transcription factor expression and chromatin footprints from ATAC-seq to build gene regulatory networks of myeloid differentiation. By simultaneously inferring regulatory interactions from transcription factor expression and open chromatin footprinting, we were able to reconstruct dynamic gene regulatory networks more robustly.
Our networks include 23 differentially expressed regulators interconnected through 158 footprints. We recovered regulatory interactions that were derived experimentally from previous studies in hematopoietic cells (Table S1), thus demonstrating the accuracy of our analysis framework. For example, our analysis recovered the PU.1 and GFI1 auto-regulatory feedback loops, which have been implicated in the controlled expression of both transcription factors in hematopoietic cells (Chen et al., 1995; Okuno et al., 2005; Yücel et al., 2004). Interestingly, we detect PU.1 footprints in distinctly different target genes in each lineage, despite PU.1 expression increasing to similar end-levels across all cell types in our time-series. These lineage-specific differences could come from a combination of regulation of chromatin opening at these target genes by other TFs, or simply from combinatorial interactions with cofactors. A PU.1 siRNA knockdown in undifferentiated HL-60, macrophage, monocytes, and neutrophils validates most of the PU.1 interactions in our core network and reveals both direct as well as indirect changes in regulator expression and chromatin accessibility for elements associated to regulators in our core network.
In contrast to PU.1, the expression of GFI was highest in terminally differentiated neutrophils, was lower in macrophages, and monocytes, and was not significantly detected in monocyte-derived macrophages. Unlike PU.1, the subnetworks for GFI1 mirror our observations from its expression profile in that the regulatory interactions in neutrophils were unique, showed a similar pattern in macrophages and monocytes, and were absent in monocyte-derived macrophage cells. Our network analysis highlights the contrast between classes of transcription factors represented by GFI1 and PU.1. Some transcription factors such as GFI1 are lineage-constrained, with highest expression and participation in the regulatory network restricted to one lineage. This contrasts with fundamental regulators such as PU.1, which show comparable levels of expression across all lineages, yet exhibit a repertoire of unique regulatory interactions across differentiated myeloid cells. Thus, our network analysis of PU.1 and GFI1 emphasizes the central nature of transcription factors, playing distinct regulatory roles in an otherwise uniform chromatin landscape.
In summary, our study highlights the complex immune regulatory circuitry of human neutrophil, monocyte, and macrophage differentiation. We demonstrate the power of combined time-course open chromatin and gene expression analysis, which allowed us to construct dynamic draft gene regulatory networks that recover both previously established as well as interactions in human myeloid cells that to our knowledge have not been previously described. Our analysis of subnetworks for transcription factors like PU.1 and GFI reflect the intrinsic and dynamic nature of their regulatory function during human myeloid specification. Additionally, these networks of human myeloid differentiation may also serve to further our understanding of transcriptional regulation in cellular differentiation and etiology of human immunopathologies.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Ali Mortazavi (ali.mortazavi@uci.edu). Please also refer to supplemental text for additional STAR methods references.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental design
HL-60 cells (ATCC-CCL240™) were grown in Modified Dulbecco’s Medium in a final concentration of 20% FBS with penicillin antibiotics (1%). Cells were routinely cultured at a density of 1×106 cells/ml in a total 10 ml. Differentiation of HL-60 cells into promyelocyte-derived macrophages was performed using PMA(10μM) (Murao et al., 1983), monocytes using vitamin D3 (100nm) (Mangelsdorf et al., 1984), and neutrophils with 1 μmol/l of retinoic acid and 1–25% (vol/vol) DMSO (Breitman et al., 1980). Differentation was performed for promyelocyte-derived macrophage, monocyte and neutrophils for 5 days. Monocyte-derived macrophages were first differentiated from HL-60 for 5 days into monocytes with vitamin D3 (100nm). Media was changed to only include PMA(10μM) on day 5 and further differentiated from monocytes into macrophages for up to 48 hours. Media was replenished every 48 hours for all differentiation time-courses with each additive included. Cells were monitored daily during the course of differentiation and viability (>95%) was measured during differentiation prior to data collection. LPS (Sigma-Aldrich) stimulation at 48, 120, and 168 hours for specific cell-types was induced at a final concentration of 100ng/ml for ~3h, immediately followed by expression and chromatin analysis. Cells were induced to differentiate from three biological HL-60 cell culture growths over the course of 5–7 days. For each time-point, differentiating cells were collected for both RNA-seq (~2–3 million cells) and ATAC-seq (~50,000 cells) from the same treated cells. All biological replicates were collected for the same time-point simultaneously and libraries were generated from each single biological replicate were processed together for both RNA-seq and ATAC-seq measurements. We assayed a total of 13 cell-specific time-points upon myeloid differentiation. This resulted in the generation of 96 ATAC-seq and 96 RNA-seq datasets.
Morphological and functional characterization of myeloid cells
Cell morphology was profiled during myeloid differentiation using an optimized Giemsa staining procedure. Cells were imaged using the Zeiss Observer at 40X magnification. To test for cell phagocytosis, HL-60 and differentiated cells were tested with the Cayman Phagocytosis Assay kit (IgG FITC) at various time-points of differentiation. Phagocytosis was tracked through interval imaging (5–10 seconds) for a total of 30–60 minutes using the EVOS FL. Videos were compiled from images and assessed for active cell phagocytosis and movement.
METHOD DETAILS
Tandem gene expression and chromatin profiling
Approximately ~2 million HL-60 and differentiated cells were collected for RNA-seq and 50,000 cells were harvested for ATAC-seq for each replicate of each time point. Briefly, cells were assessed for cell viability, counted, and washed with PBS. RNA-seq was performed as previously described (Mortazavi et al., 2008). ATAC-seq was performed as previously described (Buenrostro et al., 2013) with the addition of a DNA size selection step after library generation to enrich for accessible chromatin ranging from 100–400bp. RNA-seq libraries were sequenced as single-end 86bp and ATAC-seq libraries as paired-end 43bp reads on the Nextseq 500 Illumina platform. Approximately 1 billion RNA-seq and 2 billion ATAC-seq reads were generated.
Gene expression analysis of myeloid differentiation
RNA-seq reads were mapped to the hg38 reference genome using STAR (Dobin et al., 2013) aligner and mapped to Gencode version 20 gene annotations using Cufflinks (Trapnell et al., 2010). Batch effects due to the generation of libraries were considered and corrected for using Combat (Johnson et al., 2007). Batch-corrected data was normalized using TMM function in EdgeR strictly(Robinson et al., 2010). maSigPro (Nueda et al., 2014) allows for a two-step regression modeling strategy, which was used in identifying gene expression dynamics across differentiation of all lineages. An alpha of 0.05 for multiple hypothesis testing and a false discovery control of 1% were used, in both gene and transcription factor analysis. A k-cluster of 13 was selected based on previous analysis using hierarchical clustering and k-means clustering on the entire dataset. Gene ontology enrichments were determined for each cluster using DAVID (Huang et al., 2007). Gene expression heatmaps were generated using Tree View 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) and using R software. PCA analysis was performed using R.
ATAC-seq data processing and analysis
ATAC-seq reads were mapped to the hg38 reference genome using bowtie (Langmead et al., 2009). HOMER (Heinz et al., 2010) was used to call open chromatin regions across all replicates. The mean fragment length across all libraries was calculated to be 80 bp. A dual selection strategy was employed for calling open chromatin regions using HOMER. Briefly, HOMER was run on a size setting to enrich for ‘narrow’ regions (120–150bp) then sequentially run on size selecting ‘broad’ regions (500bp) at an FDR 1%. ‘Narrow’ and ‘broad’ regions were then merged into a single bed file for each replicate (Figure S6B). This allowed for an enrichment of diverse accessible chromatin elements. A region identified in all three biological replicates was then considered as a biologically reproducible specific open chromatin region. A master set of open chromatin regions was generated by consolidation of all peaks identified across all time-points. ENCODE ‘blacklist’ regions and open chromatin regions mapping to ChrM were discarded from our analysis. ATAC-seq data quality was measured using a sample efficiency measurement: fraction of uniquely mapped reads in peaks divided by all uniquely mapping reads. The mean efficiency for all ATAC-seq data was 32%, ranging from 25%–65% for all samples (Figure S6A). Read coverage was estimated for each open chromatin region and normalized by sample size and efficiency to detect changes in open chromatin dynamics across all time-points. Data was then corrected for batch effects using Combat and normalized using the TMM function in EdgeR. maSigPro was used to identify open chromatin dynamics across the time-series (alpha=0.05, FDR=1%). Chromatin heatmaps were generated using Deeptools software (Ramírez et al., 2014). PCA analysis was performed with ATAC-seq counts using R functions.
Transcription factor motif enrichment
Chromatin clusters were mined for de novo motifs using EXTREME (Quang and Xie, 2014). Chromatin elements were converted to fasta format using a masked hg38 genome reference. Fasta dinucleotide shuffling was performed on masked data (fasta-dinucleotide-shuffle.py) and k-mer search (GappedKmerSearch.py). PWMs were generated (Consensus2PWM.py) and used to identify motifs de novo (EXTREME.py). Motif similarities were quantified using TOMTOM (Gupta et al., 2007) and recovered based on significant q-values.
Chromatin footprinting and construction of myeloid gene regulatory networks
ATAC-seq data partitioned by early, intermediate, and late stages were merged to achieve ≥ 200 million reads for footprinting analysis. Reads were shifted by +4 bp for the +strand and −5 bp for the –strand (Buenrostro et al., 2013). Chromatin footprints were determined using the Wellington algorithm (Piper et al., 2013) with the following parameters (-fp 6,31,1 -sh 7,36,4 -fdrlimit -2), restricting our analysis to footprints with an FDR 1%. We then scanned chromatin footprints for motifs using FIMO (Grant et al., 2011); Neph et al., 2012b) to identify transcription factor motifs (p-value < 10−6) identified from the most recent JASPAR database (Mathelier et al., 2014). Lastly, we determined the quality (footprints with a posterior of 0.7 and higher) of identified TF-footprints using Centipede (Pique-Regi et al., 2011) and compiled a final set of factor-specific footprints. To build myeloid gene regulatory networks, we focused on a subset of transcriptional regulators that were differentially expressed (2-fold) and enriched in our transcription factor clusters across our time-series (Figure 3C). This allowed us to investigate networks of transcription factors that demonstrated temporal and cell-specific expression dynamics. We used a +/−15kb window around the TSS for identifying regulatory interactions that would include promoter and enhancer interactions. Directed edges were drawn from the first gene node to another gene node when a TF-motif footprint was identified neighboring the first gene within a 15 kb distance of the second gene’s TSS. Networks were generated for undifferentiated, early, intermediate, and late grouped time-points across all cell-types. Edges not identified for a corresponding time-point were drawn and colored as grey dashed lines. HL-60 PU.1 ChIP-seq peaks in biological replicates from the ENCODE consortium (ENCSR000BUW) were retrieved and converted to hg38 genome build using UCSC liftover tool. AUROC was performed using 5,000 PU.1 ChIP-seq peaks in HL-60 cells with an observed PU.1 motif (p-value < 1e-4) and a negative set of 5,000 ATAC-seq peaks in HL-60 cells that did not overlap PU.1 ChIP-seq data and had a PU.1 motif (p-value < 1e-4). The Phypher function from the ROCR package (Sing et al., 2005) was used to calculate the area under the curve. All networks were generated using Biotapestry software (Longabaugh et al., 2005, 2009).
LPS-stimulation analysis
Corresponding time-points that included LPS stimulation were analyzed for both differential chromatin and gene expression comparisons. EdgeR was used to call differentially expressed and accessible chromatin elements with an FDR <1%, p-value < 0.05 and fold change > 1.5. Hierarchical clustering was performed using Euclidean distance on neutrophil chromatin elements. Association of LPS-specific accessible chromatin elements to LPS-specific identified genes was performed using GREAT (McLean et al., 2010). Gene-element associations were then filtered to a distance of 3kb from element to TSS.
PU.1 siRNA knockdown in undifferentiated HL-60, macrophages, monocytes and neutrophils
HL-60 cells were transfected using the Lonza Amaxa nucleofector. Briefly, 1×106 undifferentiated HL-60 cells were washed in 1X PBS and transfected with the Lonza nucleofector cocktail, 1μg of GFP plasmid, and either a human non-targeting siRNA (Dharmacon Inc, D-001206-13-05) or human PU.1 siRNA (Dharmacon Inc, E-010537-00-00). We achieved 50–70% nucleofection efficiency in HL-60 cells using GFP as a marker. Biological replicates were performed for each targeted siRNA and condition. Following transfection with non-targeting or PU.1 siRNA, cells were either differentiated into macrophage, neutrophil, and monocytes, or left without induction. HL-60 and differentiated myeloid cells were harvested 24 hours post-nucleofection. Cell viability was determined using trypan blue staining and counted using a hemocytometer prior to RNA isolation. RNA quality was evaluated for all samples using an Agilent 2100 Bioanalyzer. Samples with a RIN > 9 were used to build RNA-seq libraries from 50,000 cells using the Smart-seq2 protocol (Picelli et al., 2013). ATAC-seq was performed with 50,000 cells for each cell condition after PU.1 siRNA mediated knockdown. Libraries were analyzed on an Agilent 2100 Bioanalyzer for fragment distribution and quantified using Kapa Biosystems universal library quantification kit. Libraries were normalized to 2 nM and sequenced as 40 bp paired-end dual indexed reads using Illumina’s NextSeq 500 at an average depth of ~10 million reads for Smart-seq2 and average of ~15–20 million reads for ATAC-seq. Differential analysis for RNA-seq and ATAC-seq were performed using biological replicates with EdgeR.
QUANTIFICATION AND STATISTICAL ANALYSIS
In each experiment, HL-60 or differentiated cells for each respective condition or time-point were analyzed in biological replicates: n = 3. Graphs show mean ± SEM. Two-step regression analysis performed on gene expression and chromatin accessibility was estimated with significant clusters reporting an alpha < 0.05 for multiple hypothesis testing and an FDR of 1%.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the sequencing data reported in this paper is GEO: GSE79046. This parent directory includes the following data sets: GSE79044 (RNA-seq), GSE79019 (ATAC-seq), GSE87055 (siRNA PU.1 RNA-seq), and GSE87114 (siRNA PU.1 ATAC-seq).
Supplementary Material
Highlight bullet points.
More gene expression changes than open chromatin changes early in differentiation.
Substantial changes in open chromatin later in differentiation.
Integration of ATAC-seq and RNA-seq to generate dynamic gene regulatory networks.
Changes in PU.1 footprint occupancy during myeloid differentiation.
Acknowledgments
The Mortazavi lab at UC Irvine was supported by grants NIH U54HG006998 and DP2 GM111100 as well as EU-FP7 project STATegra (306000) to AM.
Footnotes
AUTHOR CONTRIBUTIONS
AM, RNR, and AC conceived study. RNR, MAM, NE and DW performed experiments. RNR performed all analyses. All authors contributed to writing of the manuscript.
Accession Numbers
All data are accessible through GEO Series accession number GSE79046.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aziz A, Soucie E, Sarrazin S, Sieweke MH. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science (New York, NY) 2009;326:867–871. doi: 10.1126/science.1176056. [DOI] [PubMed] [Google Scholar]
- Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015 doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ray-Gallet D, Zhang P, Hetherington CJ, Gonzalez DA, Zhang DE, Moreau-Gachelin F, Tenen DG. PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene. 1995;11:1549–1560. [PubMed] [Google Scholar]
- Chen L, Kostadima M, Martens JHa, Canu G, Garcia SP, Turro E, Downes K, Macaulay IC, Bielczyk-Maczynska E, Coe S, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014:345. doi: 10.1126/science.1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SJ. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood. 1987;70:1233–1244. [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015:348. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl R, Iyer SR, Owens KS, Cuylear DD, Simon MC. The transcriptional repressor GFI-1 antagonizes PU.1 activity through protein-protein interaction. The Journal of Biological Chemistry. 2007;282:6473–6483. doi: 10.1074/jbc.M607613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121:4930–4937. doi: 10.1182/blood-2013-02-486217. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishima S, Hoffman AR, Vu T, Kim KJ, Zheng H, Daniel D, Kim Y, Wallace EF, Larrick JW, Raffin TA. Regulation of neutrophil interleukin 8 gene expression and protein secretion by LPS, TNF-alpha, and IL-1 beta. Journal of Cellular Physiology. 1993;154:478–485. doi: 10.1002/jcp.1041540305. [DOI] [PubMed] [Google Scholar]
- van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. The Journal of Experimental Medicine. 1968;128:415–435. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, et al. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood. 1979 [PubMed] [Google Scholar]
- Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nature Reviews. Immunology. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics (Oxford, England) 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biology. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nature Reviews Genetics. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P, Ralph P. Human leukemic models of myelomonocytic development: a review of the HL-60 and U937 cell lines. Journal of Leukocyte Biology. 1985;37:407–422. doi: 10.1002/jlb.37.4.407. [DOI] [PubMed] [Google Scholar]
- Hoogenkamp M, Krysinska H, Ingram R, Huang G, Barlow R, Clarke D, Ebralidze A, Zhang P, Tagoh H, Cockerill PN, et al. The Pu.1 locus is differentially regulated at the level of chromatin structure and noncoding transcription by alternate mechanisms at distinct developmental stages of hematopoiesis. Molecular and Cellular Biology. 2007;27:7425–7438. doi: 10.1128/MCB.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Zhang P, Hirai H, Elf S, Yan X, Chen Z, Koschmieder S, Okuno Y, Dayaram T, Growney JD, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nature Genetics. 2008;40:51–60. doi: 10.1038/ng.2007.7. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biology. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics (Oxford, England) 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. Journal of Allergy and Clinical Immunology. 2013;132:264–286. doi: 10.1016/j.jaci.2013.05.044. [DOI] [PubMed] [Google Scholar]
- Koh CP, Wang CQ, Ng CEL, Ito Y, Araki M, Tergaonkar V, Huang G, Osato M. RUNX1 meets MLL: epigenetic regulation of hematopoiesis by two leukemia genes. Leukemia. 2013;27:1793–1802. doi: 10.1038/leu.2013.200. [DOI] [PubMed] [Google Scholar]
- Kubosaki A, Tomaru Y, Tagami M, Arner E, Miura H, Suzuki T, Suzuki M, Suzuki H, Hayashizaki Y. Genome-wide investigation of in vivo EGR-1 binding sites in monocytic differentiation. Genome Biology. 2009;10:R41. doi: 10.1186/gb-2009-10-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummalue T, Friedman AD. Cross-talk between regulators of myeloid development: C/EBPalpha binds and activates the promoter of the PU.1 gene. Journal of Leukocyte Biology. 2003;74:464–470. doi: 10.1189/jlb.1202622. [DOI] [PubMed] [Google Scholar]
- Langmead B, Langmead B, Trapnell C, Trapnell C, Pop M, Pop M, Salzberg SL, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. Chromatin state dynamics during blood formation. Science (New York, NY) 2014:1–10. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage Transcriptional Priming and Determination of Alternate Hematopoietic Cell Fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- Laslo P, Pongubala JMR, Lancki DW, Singh H. Gene regulatory networks directing myeloid and lymphoid cell fates within the immune system. Seminars in Immunology. 2008;20:228–235. doi: 10.1016/j.smim.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-gonen R, David E, Keren-shaul H, Merad M. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leddin M, Perrod C, Hoogenkamp M, Ghani S, Assi S, Heinz S, Wilson NK, Follows G, Schönheit J, Vockentanz L, et al. Two distinct auto-regulatory loops operate at the PU.1 locus in B cells and myeloid cells. Blood. 2011;117:2827–2838. doi: 10.1182/blood-2010-08-302976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longabaugh WJR, Davidson EH, Bolouri H. Computational representation of developmental genetic regulatory networks. Developmental Biology. 2005;283:1–16. doi: 10.1016/j.ydbio.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Longabaugh WJR, Davidson EH, Bolouri H. Visualization, documentation, analysis, and communication of large-scale gene regulatory networks. Biochimica et Biophysica Acta. 2009;1789:363–374. doi: 10.1016/j.bbagrm.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Koeffler HP, Donaldson CA, Pike JW, Haussler MR. 1,25-Dihydroxyvitamin D3-induced differentiation in a human promyelocytic leukemia cell line (HL-60): Receptor-mediated maturation to macrophage-like cells. Journal of Cell Biology. 1984;98:391–398. doi: 10.1083/jcb.98.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen C, Chou A, Ienasescu H, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Research. 2014;42:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnology. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Meer LT, Jansen JH, van der Reijden Ba. Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia. 2010;24:1834–1843. doi: 10.1038/leu.2010.195. [DOI] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron. 2015;86:1369–1384. doi: 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möröy T, Khandanpour C. Growth factor independence 1 (Gfi1) as a regulator of lymphocyte development and activation. Seminars in Immunology. 2011;23:368–378. doi: 10.1016/j.smim.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Murao S, Gemmell MA, Callaham MF, Anderson NL, Huberman E. Control of macrophage cell differentiation in human promyelocytic HL-60 leukemia cells by 1,25-dihydroxyvitamin D3 and phorbol-12-myristate-13-acetate. Cancer Research. 1983;43:4989–4996. [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012a;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012b;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nueda MJ, Tarazona S, Conesa A. Next maSigPro: updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics (Oxford, England) 2014:1–5. doi: 10.1093/bioinformatics/btu333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y, Huang G, Rosenbauer F, Evans EK, Radomska HS, Iwasaki H, Akashi K, Moreau-Gachelin F, Li Y, Zhang P, et al. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol Cell Biol. 2005;25:2832–2845. doi: 10.1128/MCB.25.7.2832-2845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters I, Davidson EH. Genomic Control Process. 2015. [Google Scholar]
- Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Research. 2013;41:e201. doi: 10.1093/nar/gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Degner JF, Pai Aa, Gaffney DJ, Gilad Y, Pritchard JK. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Research. 2011;21:447–455. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K, Zaba LC, Giresi PG, Li R, Longmire M, Kim YH, Greenleaf WJ, Chang HY. Individuality and Variation of Personal Regulomes in Primary Human T Cells. Cell Systems. 2015;1:51–61. doi: 10.1016/j.cels.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D, Xie X. EXTREME: An online em algorithm for motif discovery. Bioinformatics. 2014;30:1667–1673. doi: 10.1093/bioinformatics/btu093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Research. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed S, Quintin J, Kerstens HHD, Rao Na, Aghajanirefah a, Matarese F, Cheng S-C, Ratter J, Berentsen K, van der Ent Ma, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014 doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S, Mossadegh-Keller N, Fukao T, Aziz A, Mourcin F, Vanhille L, Kelly Modis L, Kastner P, Chan S, Duprez E, et al. MafB Restricts M-CSF-Dependent Myeloid Commitment Divisions of Hematopoietic Stem Cells. Cell. 2009;138:300–313. doi: 10.1016/j.cell.2009.04.057. [DOI] [PubMed] [Google Scholar]
- Schwartsmann G, Madan K, Heins Y, Pinedo HM, Leyva A. Double-minute chromatin bodies in HL-60 leukemia cells sensitive and resistant to differentiation inducing agents. Cell Biology International Reports. 1987;11:651–655. doi: 10.1016/0309-1651(87)90099-3. [DOI] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: Visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- Spooner CJ, Cheng JX, Pujadas E, Laslo P, Singh H. A Recurrent Network Involving the Transcription Factors PU.1 and Gfi1 Orchestrates Innate and Adaptive Immune Cell Fates. Immunity. 2009;31:576–586. doi: 10.1016/j.immuni.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan AM, Arsovski AA, Lempe J, Bubb KL, Weirauch MT, Sabo PJ, Sandstrom R, Thurman RE, Neph S, Reynolds AP, et al. Mapping and Dynamics of Regulatory DNA and Transcription Factor Networks in A. thaliana. Cell Reports. 2014;8:2015–2030. doi: 10.1016/j.celrep.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Sung MH, Baek S, Hager GL. Genome-wide footprinting: ready for prime time? Nat Meth. 2016;13:222–228. doi: 10.1038/nmeth.3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, Smale ST. A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell. 2016;165:165–179. doi: 10.1016/j.cell.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Stamatoyannopoulos JA. Genomic footprinting. Nature Methods. 2016:13. doi: 10.1038/nmeth.3768. [DOI] [PubMed] [Google Scholar]
- Wong ET, Jenne DE, Zimmer M, Porter SD, Gilks CB. Changes in chromatin organization at the neutrophil elastase locus associated with myeloid cell differentiation. Blood. 1999;94:3730–3736. [PubMed] [Google Scholar]
- Yeamans C, Wang D, Paz-Priel I, Torbett BE, Tenen DG, Friedman AD. C/EBPα binds and activates the PU.1 distal enhancer to induce monocyte lineage commitment. Blood. 2007;110:3136–3142. doi: 10.1182/blood-2007-03-080291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yücel R, Kosan C, Heyd F, Möröy T. Gfi1: Green fluorescent protein knock-in mutant reveals differential expression and autoregulation of the growth factor independence 1 (Gfi1) gene during lymphocyte development. The Journal of Biological Chemistry. 2004;279:40906–40917. doi: 10.1074/jbc.M400808200. [DOI] [PubMed] [Google Scholar]
- Zhang Ja, Mortazavi A, Williams Ba, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149:467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Research. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Aguilera FB, Castellucci M, Rossato M, Costa S, Lunardi C, Ostuni R, Girolomoni G, Natoli G, Bazzoni F, et al. Chromatin remodelling and autocrine TNFα are required for optimal interleukin-6 expression in activated human neutrophils. Nature Communications. 2015;6:6061. doi: 10.1038/ncomms7061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.