

Abstract
Introduction
Targeting downstream effectors required for oncogenic Ras signaling is a potential alternative or complement to the development of more direct approaches targeting Ras in the treatment of Ras-dependent cancers.
Areas covered
Here we review literature pertaining to the molecular scaffold Kinase Suppressor of Ras (KSR) and its role in promoting signals critical to tumor maintenance. We summarize the phenotypes in knockout models, describe the role of KSR in cancer, and outline the structure and function of the KSR1 and KSR2 proteins. We then focus on the most recent literature that describes the crystal structure of the kinase domain of KSR2 in complex with MEK1, KSR-RAF dimerization particularly in response to RAF inhibition, and novel attempts to target KSR proteins directly.
Expert opinion
KSR is a downstream effector of Ras-mediated tumorigenesis that is dispensable for normal growth and development, making it a desirable target for the development of novel therapeutics with a high therapeutic index. Recent advances have revealed that KSR can be functionally inhibited using a small molecule that stabilizes KSR in an inactive conformation. The efficacy and potential for this novel approach to be used clinically in the treatment of Ras-driven cancers is still being investigated.
Keywords: APS-2-79, cancer therapeutics, ERK, Kinase Suppressor of Ras, KSR1, KSR2, MEK, Raf, Ras-dependent cancers, Ras signaling
1. Introduction
Kinase Suppressor of Ras (KSR) proteins were identified more than 20 years ago in Drosophila (KSR) and Caenorhabditis elegans (KSR1 and KSR2) and shown to modulate Ras-mediated signaling [1–3]. It was immediately recognized that KSR had a more dominant regulatory effect on signaling from mutated Ras than wild type Ras. This result was counterintuitive as the more robust signal of constitutively active Ras was predicted to be more difficult to repress [4]. In Drosophila, heterozygous mutations in KSR revert the phenotype of constitutively active Ras, RasG12V, which speaks to the ability of KSR to regulate mutated Ras signaling [3]. Several groups have shown that KSR acts downstream of Ras as a molecular scaffold for the Raf/MEK/ERK kinase cascade [5–19]. KSR plays a role in regulating several cellular mechanisms to promote cell proliferation and survival including participation in metabolic regulation, cell cycle reinitiation following DNA damage repair, and translational regulation of key mediators of a transformed phenotype such as Myc [15,20–25]. However, the mechanisms behind these effects are not yet fully elucidated.
2. Phenotypic analysis of KSR genetic inactivation
KSR proteins have been studied by genetic inactivation in several models. In Drosophila, there is only one KSR protein and homozygous inactivating or truncating mutations are lethal [3]. In contrast, in a genetic screen to identify modifiers of Ras signaling, it was discovered that heterozygous loss of ksr in Drosophila suppresses RasG12V signaling and prevented the roughening of the eye that is seen with increased Ras signaling [3]. In comparison, two KSR proteins are present (KSR1 and KSR2) in C. elegans as well as mammals and have both unique and overlapping functions [26]. In all cases, KSR proteins contribute positively to ERK phosphorylation and activation [1–3].
Apart from a few minor defects, ksr1-/- knockout mice are fertile and otherwise phenotypically and developmentally normal (Figure 1). Ksr1-/- mice have hair follicle defects similar to the phenotype of egfr-/- mice reinforcing the idea these proteins are within the same pathway [6,27,28]. As a result of reduced ERK signaling, ksr1-/- mice have a marginally impaired immunological response, particularly in regards to T-cell activation [6,16,29,30]. Most importantly, ksr1-/- mice are resistant to Ras-driven tumor formation [6]. This fact is demonstrated by the reduced mammary tumor burden in ksr1-/- mice with transgenic expression of polyomavirus Middle T-Antigen [6]. Induction of skin tumors with v-Ha-Ras was also completely lost in ksr1-/- mice [28]. These observations demonstrate that KSR modulates Ras signaling in vivo, but it is largely dispensable for normal cell survival. Yet the requirement for KSR1 in Ras-driven tumor formation makes it an attractive target for therapeutic intervention.
Figure 1. KSR Knockout Mice.
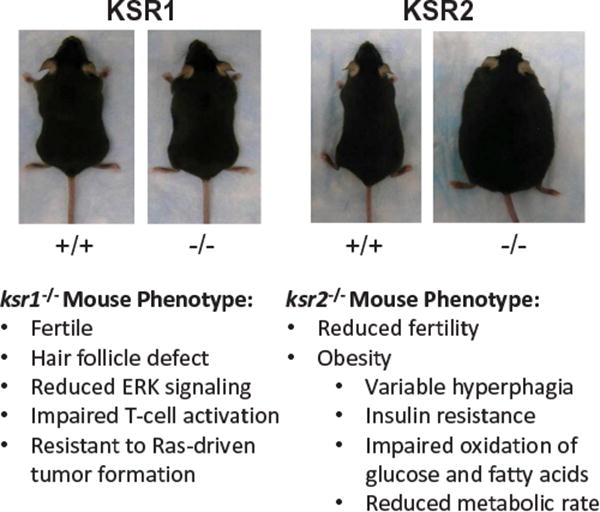
Ksr1-/- mice are largely developmentally and phenotypically normal, yet resistant to Ras-driven transformation. Ksr2- mice have an abnormal metabolic profile and become obese.
In contrast to the mild phenotype of ksr1-/- mice, ksr2-/- mice have reduced fertility and become spontaneously obese [31–34] (Figure 1). Although ksr2-/- mice have not been assessed for their resistance to tumor formation, there is in vitro evidence for a role of KSR2 in promoting tumor formation. In ksr1-/- mouse embryo fibroblasts (MEFs), ectopic expression of KSR2 restored ERK1/2 activation and mutant Ras-dependent anchorage independent growth [35]. KSR2 is expressed in a mouse neuroendocrine cell line (Min-6) and mouse neuroblastoma/rat glioma hybrid cell line (NG108-15) and shRNA-mediated depletion caused reduced proliferation and anchorage-independent growth [35]. Consistent with observations in the knockout mice, RNAi studies of KSR2 in insulinoma cell lines showed defects in both ERK1/2 and AMPK activation leading to reduced metabolic activity. Therefore, while the role of KSR2 in human cancers has not been defined, substantial evidence suggests a potential role for KSR2 in cancer [35,36].
3. The role of KSR in cancer
KSR1 has been extensively implicated in Ras-driven cancers [6,11,14,20,23–25,28,35,37–39]. Consistent with its role as a molecular scaffold for the Raf/MEK/ERK cascade, KSR1 interacts with each kinase in this cascade [13,14,40], and increasing levels of KSR1 enhance Ras signaling to a maximum point [14]. As predicted of a scaffold, exceeding the optimal cellular KSR1 concentration disrupts ERK signaling and inhibits Ras transformation by separating and sequestering the members of the kinase cascade from one another [14,39]. Comparing KSR1 expression in a panel of colon cancer cell lines to nontransformed human colon epithelial cell lines suggests that enhanced ERK signaling may be accomplished, at least in part, by upregulation of KSR1 in colorectal cancer [25]. Ksr1-/- mouse embryo fibroblasts are resistant to Ras-transformation, maintain contact inhibition, and fail to form colonies in soft agar [14]. In preclinical mechanistic studies, stable depletion of KSR1 using multiple shRNA sequences reduces the ability of colon cancer cells expressing mutant K-Ras to grow in an in vitro soft agar assay as well as in an in vivo xenograft mouse model [25]. Finally, RNAi-mediated depletion of KSR1 robustly induces cell death in the Ras-mutated colon cancer cell line HCT116 cells, but not in nontransformed human colon epithelial cells [25]. Taken together, these results support the conclusion that KSR1 is required for the complete oncogenic potential of Ras-driven cancer cells. These observations demonstrate that specific targeting of KSR1 could be selectively toxic to Ras-driven cancers with relatively little toxicity to the patient [28].
4. Structural analysis of KSR proteins
KSR is highly conserved from invertebrates to mammals. However, Drosophila express only one KSR protein, while C. elegans and mammals encode two members, KSR1 and KSR2 [3,13]. KSR proteins are structurally related to Raf proteins; however, they have diverged to obtain significant structural and functional differences (Figure 2). KSR proteins are highly homologous, containing five conserved areas (CA1-CA5) [3]. The first conserved region on the N-terminus end is CA1. B-Raf binding to KSR1 depends on a 40 amino acid sequence within CA1 as well as MEK binding to the CA5 area of KSR1 [10,41,42]. Within the CA1 domain, amino acids 25–170, termed CA1α, contain a coiled coil and sterile-α-motif (SAM), which promotes KSR1 membrane association that is essential for its positive effects on MAPK signaling [43]. The CA2 is a proline-rich region with an unknown function. CA3 is a cysteine-rich atypical C1 motif that mediates the membrane localization of KSR by recruiting phospholipids and is largely homologous with the CR1 cysteine-rich region in Raf [44,45]. Studies on the cysteine-rich region of KSR, in contrast to C1 regions in Raf and PKCγ, have demonstrated that the KSR1 C1 domain is structurally unique, particularly within the ligand binding region, does not react to phorbol esters or ceramide, and does not directly interact with Ras [45]. CA4 is a serine/threonine rich region containing a FXFP motif that mediates interaction with ERK [10,46,47] and is similar to the CR2 region in Raf proteins. Interaction of KSR1 with ERK is not constitutive and requires Ras activation [48,49]. The CA5 domain in KSR proteins encodes a kinase domain highly homologous to Raf family CR3 kinase domains [2,3]. CA5 contains a putative kinase domain; however, there are multiple mutations within this domain including an important lysine to arginine exchange in a lysine residue that is generally required for kinase activity (Table 1) [3,10]. Substantial effort has been exerted to clarify if KSR can or does phosphorylate any substrates within cells, and if so whether this activity contributes to the downstream effects of KSR (c.f., Section 8).
Figure 2. KSR Structure.

KSR proteins are structurally similar to Raf proteins with the CA3, CA4, and CA5 regions in KSR sharing significant homology with the CR1, CR2, and CR3 regions in Raf respectively. CR1–3: Conserved Regions 1–3; RBD: Ras-binding domain; CRD: Cysteine-rich domain; Ser/Thr-rich: Serine/Threonine-rich domain; CA1–5: Conserved Areas 1-5; CC-SAM: Coiled coil-sterile alpha motif domain; Pro-rich: Proline-rich domain
Table 1.
KSR mutations and associated characteristics.
| Mutation | Effects | Location | Species | References |
|---|---|---|---|---|
| W255X, R277 H | Decrease constitutively active Ras-mediated signaling | CA3 | C. elegans | (Kornfeld et.al. 1995) |
| G549E, P696 L, Q733X | Decrease constitutively active Ras-mediated signaling | CA5 | C. elegans | (Kornfeld et al. 1995) |
| G484E | Decrease constitutively active Ras-mediated signaling | CA5, ATP-binding region | C. elegans | (Sundaram and Han 1995) |
| R531 H | Decrease constitutively active Ras-mediated signaling | CA5 | C. elegans | (Sundaram and Han 1995) |
| G494E | Decrease constitutively active Ras-mediated signaling | CA5 | C. elegans | (Sundaram and Han 1995) |
| P630S, P630 L | Decrease constitutively active Ras-mediated signaling | CA5 | C. elegans | (Kornfeld et al. 1995, Sundaram and Han 1995) |
| Intron 12 Change | Decrease constitutively active Ras-mediated signaling | CA5 | C. elegans | (Sundaram and Han 1995) |
| G→A generating a stop codon following G678 | ||||
| C727Y | Decrease constitutively active Ras-mediated signaling | CA5 | C. elegans | (Sundaram and Han 1995) |
| A696 V | Decrease constitutively active Ras-mediated signaling | CA5 | Drosophila | (Therrien et al. 1995) |
| A703 T | Decrease constitutively active Ras-mediated signaling | CA5 | Drosophila | (Therrien et al. 1995) |
| S721 + 10bp in N727 → frameshift | Decrease constitutively active Ras-mediated signaling | CA5 | Drosophila | (Therrien et al. 1995) |
| S548 + 4bp→ L50G, R51S | Weak disruption of constitutively active Ras-mediated signaling | N-terminus | Drosophila | (Therrien et al. 1995) |
| C359S and C362S (CRM-KSR1) | Prevented the enhanced RasV12-mediated signaling seen with the expression of exogenous WT KSR1, but did not disrupt RasV12-mediated maturation Loss of KSR1 membrane localization |
CA3 | Xenopus oocyte meiotic maturation assays using exogenous mouse KSR1 | (Michaud et al. 1997) |
| CA3 domain (amino acids 319-390) | Augments Ras signaling | CA3-Only | Mouse KSR1 | (Michaud et al. 1997) |
| CRM CA3 domain | Abolished augmented Ras signaling | CA3 | Mouse KSR1 | (Michaud et al. 1997) |
| Myristylated N-Terminus KSR1 (Myr-KSR) | Constitutively localized to the plasma membraneAccelerates RasV12-induced maturation Expression of Myr-KSR alone was unable to promote oocyte maturation |
N-terminus | Xenopus oocyte meiotic maturation assays using exogenous mouse KSR1 | (Michaud et al. 1997) |
| CRM Myr-KSR | Abolished the positive effect of Myr-KSR on Ras signaling | CA3, N-terminus | Mouse KSR1 | (Michaud et al. 1997) |
| R589 M, R589 L | Inactivates kinase domain and blocks MEK binding | CA5, ATP-binding site | Mouse KSR1 | Yu et al. 1998, Voile et al. 1999) |
| G580 V and A587 T | Decreased MEK:KSR association Decreased ERK activation |
CA5 | Mouse | (Yu et al. 1998) |
| S190, T256, T274, S297, S320, T411, S429, S434, S518 | KSR phosphorylation sites confirmed with mutagenesis | N-Terminus to the CA5 domain | Mouse | (Voile et al. 1999) |
| C540 | Suppressed ERK and MEK activation Interacts with MEK1Decreased Ras signaling (suppressed Xenopus oocyte maturation, cellular transformation, and Drosophila eye development) | Truncated C-terminus, kinase domain preserved | Mouse | (Therrien et al. 1996, Joneson et al. 1998, Yu et al. 1998, Voile et al. 1999) |
| N539 | No effect on the activation of ERK Fails to interact with MEK1 Unable to bind B-Raf Interacts with ERK2 |
Truncated N-terminus | Mouse | (Therrien et al. 1996, Joneson et al. 1998, Yu et al. 1998, Voile et al. 1999, McKay et al. 2009) |
| C809Y | Lacks KSR:MEK interaction Unable to bind B-Raf Increased Ras signaling | CA5 MEK docking site | Mouse | (Yu et al. 1998, Stewart et al. 1999, Muller et al. 2000, Kortum et al. 2006, McKay et al. 2009) |
| FSFP/AAAP DEF docking motif for activated ERK (FxFP) (AxAP-KSR1) |
Lacks KSR:ERK interaction Decreased Ras signaling (decreased RasG12V-induced senescence associated β-galactosidase activity) Required for Ras-induced senescence Decreased proliferative rate with activated Ras Increased binding to endogenous B-Raf |
FXFP Motif ERK docking site CA4 | Mouse | (Jacobs et al. 1999, Fantz et al. 2001, Kortum et al. 2006, McKay et al. 2009) |
| S392A, S392A/S297A | Defective 14-3-3 binding Enhanced growth-factor mediated binding to B-Raf Increased plasma membrane localization even without growth factor stimulation Accelerates Ras-induced oocyte maturation Unable to promote oocyte maturation without activated Ras Promotes ERK activation and cell cycle progression following growth factor treatment |
CA3 | Xenopus oocyte meiotic maturation assays using exogenous mouse KSR1 | (Cacace et al. 1999, Muller et al. 2001, Razidlo et al. 2004, McKay et al. 2011) |
| L360A/R363A (KR/AA) L360Q/R363G (KR/QG) | Abolished CK2 bindingNo decrease in MEK, ERK, or 14-3-3 binding Decreased Ras-mediated MEK and ERK activation |
CA3 | Xenopus oocyte meiotic maturation assay | (Ritt et al. 2007) |
| S518A (CK2 phosphorylation site) | No apparent effects on Ras signaling Biological effect is unknown | CA5 | Mouse | (Ritt et al. 2007) |
| L56G and R57S | Disrupted binding of mammalian KSR1 to endogenous B-Raf. | CA1 | Mouse | (McKay et al. 2009) |
| T260A/T274A/S320A/S443A (FBm-KSR1) | Increased/Prolonged plasma membrane localization | CA5 | Mouse | (Cacace et al. 1999, McKay et al. 2009) |
| ERK-dependent S/TP sites mutated to alanine | Increased association with B-Raf | |||
| Increased Ras signaling | ||||
| Loss of CA3 | Disrupt or reduce AMPK binding to KSR2 | CA3 | Mouse | (Costanzo-Garvey et al. 2009, Dougherty et al. 2009, Pearce et al. 2013) |
| Frameshift mutations and nonsense mutation that disrupt the kinase domain | Region between CA2 and CA3 | Human | ||
| E667 V, A373 T | ||||
| Asp-529A | Inhibition of caspase cleavage | CA5 | Mouse | (McKay and Morrison 2007) |
| DEVA mutant | Reduced apoptotic signaling in response to tumor necrosis factor and cycloheximide treatment due to decreased caspase cleaved C-terminal KSR fragments | C-Terminus | ||
| DEVD site for caspase-mediated cleavage | ||||
| C-terminal KSR1 fragment (CTF-KSR1) | Reduced ERK activation and enhanced apoptotic signaling | C-Terminus | Mouse | (McKay and Morrison 2007) |
| Result of caspase cleavage during apoptosis |
MEK1/2 bind to the CA5 region of KSR proteins and the interaction is constitutive in both quiescent and cells activated with growth factors [40,48,49]. Mutations within the CA5 region that abrogate binding of KSR to MEK also reduce ERK signaling (Table 1) [1–3,40,49]. However, due to the location of these mutations either within or near the ATP binding domain, they potentially also interfere with ATP binding or other KSR functions that may be independent of interaction with MEK (Table 1). Mutation C809Y within the C terminal tail of KSR1, and distal to the ATP binding domain, also disrupts MEK binding to KSR, yet allows for increased Ras-mediated ERK signaling (Table 1) [39]. These data suggest that the interaction between KSR and MEK is dispensable for Ras-induced ERK signaling and raises the possibility that this interaction reflects a negative regulatory role for KSR1 to control the timing and spatial location of MEK activation. The CA5 domain is also required for KSR to bind to Raf, but the mechanism is incompletely understood [41].
Another region has been identified in KSR2 between CA2 and CA3 that is required for interaction of KSR proteins with AMPK. Mutations in this and nearby regions reduced the binding of AMPK to KSR (Table 1) [33,36,42,50]. Tissue-specific splice variants have also been identified. B-KSR1 is a splice variant of murine KSR1 that is preferentially expressed in neural tissues that largely acts like KSR1 in regards to Raf/MEK/ERK interactions and signaling regulation, but specifically plays a role in cells within the central nervous system [49]. A truncated version of KSR2 (T-KSR2) was found in mouse testes, which may play a role in male fertility [51].
5. KSR proteins are molecular scaffolds of the Raf/MEK/ERK kinase cascade
Substantial evidence has demonstrated that KSR proteins act as molecular scaffolds for the Raf/MEK/ERK kinase cascade [5–19]. KSR promotes Raf phosphorylation of MEK [8,44,46] and is required for maximal Ras-mediated ERK phosphorylation and activation by MEK [3,6,13,14,20,46]. Prior to experiments that controlled the level of KSR1 expression, publications reported a conflicting role for KSR overexpression, suggesting that KSR1 could both promote and inhibit Ras signaling [1–3,14,38,44,46,48,52–54]. These data were consistent with the idea that KSR1 acts as a scaffold for the Raf/MEK/ERK kinase cascade as increasing this scaffold to an optimum level increases signaling; however, once the optimal level is exceeded, models predict that the scaffold will dilute and sequester the individual signaling components and disrupt signaling. This dose-dependent action of KSR1 was demonstrated in ksr1-/- mouse embryo fibroblasts expressing various levels of a transgene KSR1 [14]. ERK signaling and proliferation, as well as KSR1 interaction with Raf, MEK, and ERK, all increased with increasing KSR1 expression until KSR1 was approximately 14-fold higher than endogenous levels in wild type mouse embryo fibroblast cells. ERK signaling and cell proliferation then dramatically decreased, while the interaction of Raf, MEK and ERK with KSR1 plateaued when KSR1 was further increased to a level 20-fold higher than endogenous levels [14]. When excessively high levels of KSR inhibit signaling of the MAPK cascade, these inhibitory effects can be abrogated by over-expressing additional components of the MAPK pathway. This was elegantly demonstrated in Drosophila S2 cells where the over-expression of Raf and MEK in conjunction with KSR overexpression still demonstrates robust MEK phosphorylation by Raf even with levels of KSR that would normally interfere with MEK phosphorylation [13]. These characteristics are consistent with the defining features of scaffolding proteins [55,56]. Furthermore, the scaffolding activity is both temporally and spatial regulated and is dependent upon Ras activation status and KSR subcellular localization to the plasma membrane, allowing for additional levels of regulation of the Ras pathway.
In resting conditions, KSR1 is bound to MEK and an autocatalytic ubiquitin ligase, IMP, and is sequestered in the cytoplasm as a result of C-TAK1 phosphorylating KSR1 at S392, which promotes 14–3-3 binding [40,41,48,52,54,57,58] (Figure 3(a)). The crystal structure of KSR2 kinase domain bound to MEK demonstrated that these proteins interact at two primary locations: the activation segments within their kinase domains and the alpha G helixes on the C-terminal lobe of each protein [8], and mutations within the alpha G helix of MEK [41], or mutations that either disrupt the secondary structure of or are within the alpha G helix of KSR inhibits the binding of KSR to MEK (Table 1) [8]. Of note, when KSR is bound to MEK, the activation segments of both proteins are constrained. MEK cannot be phosphorylated and activated, and KSR is in an inactive conformation [8]. This inactive state may reflect KSR1:MEK heterodimers in the cytoplasm of quiescent cells. Upon Ras activation, Raf is phosphorylated, and PP2A dephosphorylates KSR1 at S392, such that 14–3-3 no longer binds [59] (Figure 3(b)). IMP dissociates simultaneously from KSR1, interacts with GTP-bound Ras, autoubiquitinates, and is targeted to the proteasome for degradation [57] (Figure 3(b)). KSR1 is then free to move to the plasma membrane with MEK in tow [5,13,14] (Figure 3(c)). The localization of KSR1 to the plasma membrane is dependent upon its interaction with Caveolin-1 and is required for KSR1-mediated ERK activation and Ras-driven transformation [18]. Once KSR1 localizes to the plasma membrane, MEK is phosphorylated by activated Raf [5,13,14,44,63]. Based on the observation that KSR1 bearing C809Y mutations fails to interact with MEK and promotes ERK activation better than WT KSR1, MEK is predicted to phosphorylate ERK when it is dissociated from KSR1 (Table 1). Once phosphorylated by MEK, ERK interacts with KSR1, and this interaction is required for normal ERK signaling [39,41] (Figure 3(d)). The sustained and coordinated activation of ERK ultimately promotes the transformation, survival, and proliferation of Ras-driven cancer cells [6,14,21,28]. The activation of ERK also controls a negative feedback loop, in which activated ERK when bound to KSR1 phosphorylates and inhibits both KSR1 and B-Raf [41,64]. This phosphorylation by ERK causes KSR1 and B-Raf to dissociate from the plasma membrane and halts additional ERK activation.
Figure 3. KSR1 as a scaffold for the Raf/MEK/ERK kinase cascade.

(a) When Ras is bound to GDP and inactive, KSR1 is constitutively bound to MEK1/2 and IMP and is phosphorylated (yellow circle) at S392 by C-TAK1 allowing for 14–3-3 binding and cytoplasmic sequestration. (b) Upon Ras activation, PP2A dephosphorylates KSR1 at S392 causing 14–3-3 and IMP to dissociate and IMP to autoubiquitinate and be degraded. (c) Through interaction with caveolin-1, KSR1 and MEK1/2 then move to the plasma membrane where Raf activates MEK1/2 by phosphorylation at S217/S221. (d) MEK1/2 then dissociates from KSR1 and activates ERK1/2 through phosphorylation at T202/Y204 and T185/Y187. Activated ERK1/2 then associates with KSR1, which allows ERK1/2 to phosphorylate KSR1 and Raf and initiate a negative feedback loop.
6. KSR proteins form heterodimers with Raf proteins to regulate MEK and ERK activation
Dimerization of Raf proteins is thought to be crucial for wild type Raf activation. This dimerization is not unique to Raf proteins within the MAPK pathway as both Ras and ERK have also been shown to form dimers [65–69]. The similarity between KSR and Raf proteins is specifically conserved within the region required for Raf dimerization and KSR has been shown to form heterodimers with Raf, particularly B-Raf [8,37,63]. This dimerization regulates an allosteric conformational change in KSR that allows for the phosphorylation of MEK [8]. Specifically, when KSR forms a dimer with a Raf protein (cis interaction), the conformational change in KSR facilitates the exposure of the activation site on MEK and allows for its phosphorylation. However, the dimerization of KSR and Raf orients the Raf protein such that the catalytic site of Raf is not in proximity to its phosphorylation target site on MEK [8]. Therefore, this phosphorylation must be completed by another Raf protein (trans interaction) [8]. More recently, it has been shown that KSR2 is also able to homodimerize through a side-to-side interface that is specifically dependent upon Arg718 [8]. In a genetic screen, mutations at this site were previously shown to suppress Ras signaling, suggesting that dimerization of KSR proteins is required to promote Ras signaling [1-3]. This is consistent with results that mutations that inhibit the KSR-Raf heterodimerization decrease Raf activity (Table 1) [63]; however, the functional role of KSR homodimers is still incompletely understood.
7. The effects of Raf kinase inhibitors on KSR and Raf dimerization
Raf kinase inhibitors were developed in the hopes they would be able to suppress Ras signaling even in the presence of oncogenic, activating Ras or Raf mutations. Unfortunately, while these inhibitors were shown to strongly antagonize Raf activity in RafV600E mutant cells, paradoxical increases in Raf activity and downstream Ras signaling were often seen in cells with Ras or Raf mutations following treatment with Raf kinase inhibitors [70–72]. Several follow-up studies demonstrated that this effect was due to increased Raf dimerization and subsequent activation [70,72–74]. This led to several studies that revealed there are multiple classes of Raf mutations including highly activating mutations (e.g. V600E) that resulted in monomeric activated Raf as well as additional mutations that only weakly improved the catalytic activity of Raf or even decreased it, yet promoted dimer formation and the subsequent Raf activation [70,72]. These Raf mutants would then increase the activation of its binding partner, which could be a wild type or mutant Raf protein, to ultimately increase overall Raf activation and ERK signaling. Therefore, the Raf kinase inhibitors are only functional against highly activating Raf mutations, and can lead to an increase in Raf activity by increasing Raf dimer formation [72]. It is important to note, that Raf inhibitor binding also demonstrates inverse cooperativity as binding of the inhibitor to one Raf protein in a Raf protein dimer, decreases the affinity of the other protein to inhibitor binding [75,76]. The inhibitor, therefore, promotes increased dimer formation and subsequent Raf activation where one Raf protein is still catalytically active, which can increase overall Raf activity even in cells that have Raf mutations.
Raf inhibitors also promote KSR1-B-Raf heterodimer formation [9]. In this manner, KSR1 can compete with C-Raf for inhibitor-induced dimerization to B-Raf. The dimerization between C-Raf and B-Raf promotes ERK signaling; however, complex formation of KSR and B-Raf actually limits ERK activation [9], which suggests that KSR in cells being treated with Raf kinase inhibitors may actually limit paradoxical, rebound ERK activation in response to inhibitor-induced Raf dimerization and activation. While dimerization of B-Raf with KSR2 allosterically alters the orientation of KSR2 into a more catalytically active conformation, the overall effects of KSR expression reducing Raf inhibitor-induced ERK signaling suggests that the phenotypic effects of KSR in this scenario is not likely to be a result of its kinase activity [9].
8. Non-canonical functions of KSR proteins
Recent work has demonstrated additional functions for KSR that are either independent or downstream of its role as a scaffold for the Raf/MEK/ERK kinase cascade. Both KSR1 and KSR2 have been shown to promote tumorigenesis through positive regulation of AMPK [16,25,33,35]. While the role of AMPK in cancer is still controversial, multiple groups have shown that after transformation, AMPK can promote tumor cell survival by mediating an increase in the overall metabolic capacity allowing the cells to survive in stressful conditions. One mechanism by which this occurs is through the upregulation of PGC1β and its transcriptional partner ERRα [23,25]. An additional KSR-regulated mechanism promoting the expression of PGC1β has recently been described where KSR1 promotes ERK activation in colon cancer cells, which is required for increased MYC translation. MYC then acts as a transcription factor and increases PGC1β transcript levels [24]. KSR proteins also plays a role as an overall metabolic regulator in cells by regulating glucose metabolism [22] and adipogenesis [15]. The scaffold function of KSR1 promotes adipogenesis by coordinating the timing and intensity of ERK-dependent p90 RSK activation with the expression of its key adipogenic substrate C/EBPβ. Defective adipogenesis in vitro caused by loss of KSR1 can be rescued by adding back low levels of KSR1. Increasing KSR1 levels above the optimal level inhibits adipogenesis through sustained ERK signaling while inducing the phosphorylation and inhibition of the key adipogenic transcription factor PPARγ [15].
The presence of a kinase domain within the CA5 domain of KSR has led numerous groups to examine the potential of KSR to act as a kinase. Initial reports suggested KSR was a ceramide-activated kinase [74] even though amino acids critical for phosphotransferase activity, including the lysine involved in exchange of the gamma phosphate of ATP with a substrate are not conserved within the kinase domain of KSR proteins [3,26,49]. More recently, a crystal structure of the kinase domain of KSR2 and MEK1 with ATP bound within the catalytic site combined with in vitro assays and chemical genetics data suggest KSR2 has the potential to phosphorylate MEK [8]. However, the evidence supporting a role for KSR as a kinase has shown very low levels of substrate phosphorylation in vitro (low stoichiometry), has demonstrated phosphorylation on sites different than those required to activate MEK, has largely been based on experiments performed outside of cellular systems, and may be a consequence of co-precipitating kinases as many subsequent experiments have demonstrated KSR1 lacks catalytic activity [44,78]. This raises the question as to whether any residual intrinsic kinase activity within KSR proteins has biological relevance. Expression of the isolated kinase domain of KSR inhibited ERK signaling and suppressed Ras-dependent Xenopus oocyte maturation, cellular transformation, and Drosophila eye development [46]. This supports the role of KSR proteins as molecular scaffolds, where the isolated expression of the CA5 domain, which could only bind and sequester MEK, but not regulate its cellular localization or coordinate interactions with Raf, would be expected to antagonize MAPK signaling [40,46,54]. In contrast, the conservation of the binding site residues for ATP within KSR and the prevalence of mutations within the ATP binding pocket in loss-of-function KSR mutants do suggest that ATP binding itself may play an important role in KSR activity (Table 1) [2,3,46]. Thus, KSR proteins have been considered pseudokinases.
KSR1 has been shown to travel through the nucleus when in complex with MEK [79]. The functional significance of this subcellular localization is unknown, but may facilitate activity of ERK toward nuclear substrates. Further investigation may provide additional understanding of the complex role KSR plays in modulating ERK signaling as well as other processes. These and additional undiscovered pathways could reveal novel approaches for targeting of KSR-dependent actions specific to tumor cell maintenance.
9. KSR as a target for therapy
Based on the role KSR1 plays in modulating signaling through the Raf/MEK/ERK kinase cascade and the fact that ksr1-/- mice are largely phenotypically normal, targeting KSR1 in Ras-driven cancers appears to be a reasonable approach to selectively target cancer cells without subjecting patients to the side effects that normally accompany chemotherapeutics. Supporting this strategy, RNAi approaches depleting cancer cells of KSR both within in vitro and in vivo models demonstrated a decrease in tumor growth [25]. Further, a continuous infusion of phosphorothioate antisense oligonucleotides that inhibited KSR1 expression caused regression of established tumors and inhibited metastases without overt toxicity in Ras-driven PANC-1 pancreatic and A549 non-small cell lunch cancer xenografts in nude mice [80].
Recent studies have attempted to target KSR proteins directly for therapy. The small molecule APS-2–79 is able to bind and stabilize KSR in an inactive state, interfere with KSR:Raf heterodimerization, and inhibit oncogenic Ras signaling [37]. Based on the finding that mutations in KSR that suppress oncogenic Ras signaling largely mapped to a region adjacent to the ATP-binding pocket, it was hypothesized that a small molecule that bound KSR within the ATP-binding pocket could interfere with Ras signaling. APS-2–79 blocks heterodimerization with Raf and conformationally biases KSR towards an inactive state [37] similar to the conformation of the KSR2 kinase domain bound to MEK1 and ATP [8]. In this conformational state, MEK cannot be phosphorylated because the active segments of both MEK and KSR2 interact directly with additional stabilization provided by interactions between the alpha G helices on the C-terminal lobe of each protein [8]. The efficacy of APS-2–79 has been demonstrated with a simplified cell-based reconstitution system that monitored KSR-dependent MEK and ERK signaling. KSR enhanced MEK phosphorylation at Ser218/Ser222 by Raf in a dose-dependent manner. This increased phosphorylation was inhibited by APS-2– 79, but not a similar small molecule that due to small modifications was no longer able to bind to KSR2. The effect of APS-2–79 was also lost when KSR was mutated within the active site (A690F) such that KSR can promote MEK phosphorylation even in the absence of ATP binding. APS-2–79 had no ability to affect MEK phosphorylation in the absence of KSR, suggesting that the effect of APS-2–79 on ERK signaling is dependent on KSR, and APS-2–79 does not inhibit MEK phosphorylation by interacting directly with Raf proteins even though Raf shares a high degree of homology with KSR1 and KSR2 [37]. Unfortunately, APS-2–79 was only modestly able to decrease cell viability in two Ras-mutated cancer cell lines (HCT116 and A549) and did not affect Raf-mutated cancer cells (A375 and SK-MEL-293) [37]. In contrast to APS-2–79, in HCT116 cells, transient siRNA-mediated depletion of KSR1 causes a reduced viability in vitro and stable shRNA-mediated depletion of KSR1 reduces tumor growth in vivo [25]. There are several reasons that may explain the discrepancy between APS-2–79 treatment and RNAi-induced protein loss. The ability of APS-2–79 to bind and directly inhibit KSR1 has not yet been demonstrated, and therefore it may not bind and inhibit KSR1 as well as it does KSR2, limiting its efficacy. The limited efficacy of APS-2–79 could also result from compensation by alternative MAPK pathway scaffolds. Several scaffolds have been shown to allow for increased Ras-mediated signaling such as IQ motif-containing GTPase activating protein 1 (IQGAP1), Sef, dystroglycan, β-arrestin, MEK partner 1 (MP1), and Paxillin [11,55,56,65,66,81]. However, this possibility is less likely as the RNAi-mediated depletion of KSR1 robustly disrupts Ras signaling even with potential compensation by other scaffolds. It is also possible that APS-2–79 only inhibits a subset of KSR-dependent signaling events and therefore is less effective than RNAi-mediated depletion of KSR1. However, APS-2–79 treatment shows substantial synergy in Ras-mutated cancer cells with MEK inhibitors, particularly trametinib. This observation suggests that robustly inhibiting Ras-mediated ERK signaling in conjunction with inhibition of non-canonical components of KSR1-dependent signaling is efficacious though the exact mechanism behind this synergy is not known [37]. Demonstrating the binding and inhibition of APS-2–79 on KSR1 and evaluating its ability to inhibit both canonical and non-canonical KSR1-dependent effects will be important moving forward.
10. Conclusions
Despite being identified more than 20 years ago, the functions of KSR proteins and the mechanisms by which they modulate Ras signaling and promote cancer cell survival are not fully understood. It is well established that KSR acts as a scaffold for the Raf/MEK/ERK kinase cascade and promotes phosphorylation and activation of Ras downstream effectors through protein:protein interactions and subcellular trafficking. However, understanding the dynamics of these interactions, the control of the subcellular localization of KSR, and the regulation of KSR expression in cells is vital because KSR has the potential to both promote or inhibit ERK signaling. This behavior is consistent with KSR acting as a scaffold protein and suggests that since KSR modulates Ras signaling in normal cells, KSR itself must be tightly regulated. These regulatory mechanisms, while likely still at play in cancer cells, are at least somewhat disrupted as evidenced by increased KSR1 protein expression. This increased expression in cancer and selective requirement for KSR1 for cancer cell survival, but not normal cell survival, suggests that KSR1 would be a selective, efficacious therapeutic target in cancers of the pancreas, colon, and lung where Ras mutations are commonly required for tumor growth and survival.
Recent work has further characterized KSR proteins, and several new functional and physical interactions have been identified. The introduction of Raf inhibitors has expanded our understanding of KSR:B-Raf heterodimerization and the mechanisms behind Raf activation. The study completed by Brennan et al. presented the crystal structure of the kinase domain of KSR2 complexed with MEK1, which provides novel insights into this physical interaction. The recent publication by Dhawan et al. demonstrated the possibility of targeting KSR proteins with a small molecular inhibitor that stabilizes KSR in an inactive state to effectively limit Ras signaling. The limited efficacy of APS-2–79 as monotherapy against Ras-and Raf-mutated cancer cells is disappointing; however, more selective targeting of KSR1 may substantially improve its effectiveness and is still a viable therapeutic approach based on the promising signaling studies. Further elucidating the roles of KSR in both cancer and normal cells will allow for additional approaches to be employed for the development of additional KSR inhibitors that have the potential to transform the clinical treatment of Ras-driven cancers.
11. Expert opinion
The recognition that KSR modulates Ras signaling and acts as a scaffold for Raf/MEK/ERK kinase cascade has led to KSR being evaluated as a therapeutic target in Ras-driven cancers. Ksr1-/- mice are grossly normal, but resistant to Ras-driven tumori-genesis, and in vitro studies have demonstrated a selective requirement for KSR1 in Ras-mutated cancer cells making KSR1 an attractive target for therapeutic intervention in cancer. KSR is constitutively bound to MEK in an inactive conformation until Ras is activated. Upon Ras activation, KSR localizes to the plasma membrane and KSR and B-Raf form heterodimers that causes a conformational change and makes the active site on MEK available for phosphorylation. Recently, a small molecule inhibitor, APS-2–79, was identified that binds to KSR2 when it is in complex with MEK and locks it in an inactive conformation. This prevents KSR2:Raf dimerization and does not allow for the conformational change that must occur for MEK to be phosphorylated and activated when it is bound to KSR2.
The next big step for the current KSR inhibitor will be to prove its selectivity and efficacy. Since the APS-2–79 inhibitor fits within the ATP-binding region of the pseudokinase domain of KSR2, it is reasonable to assume there could be some nonspecific interactions with other kinases and ATP-binding proteins. As KSR2 has been primarily studied in obesity models, it will be important to evaluate the inhibitory effects of APS-2–79 specifically on KSR1. Perhaps most importantly, this small molecule inhibitor needs to prove its anticancer efficacy in both cellular and animal models, since the current work has largely focused specifically on MEK and ERK activation.
An insufficient understanding of the differences between KSR1 and KSR2 is a limitation of the current research. These proteins have substantially different expression profiles. KSR2 is largely expressed in the brain including within the pituitary. While KSR1 is also highly expressed in the brain, it is also expressed at relatively low levels in most other tissues. The profound differences in the phenotype of ksr1-/- and ksr2-/- mice demonstrates that while there is likely significant overlap in their functions, KSR1 and KSR2 have unique and distinct physiological roles.
Developing an inhibitor that preferentially targets KSR1 more than KSR2 and other related proteins will be vitally important as selectivity will impact both its efficacy as a cancer therapeutic and its potential side effects. To identify and develop a specific, efficacious KSR1 inhibitor that lacks major side effects, additional work evaluating the shared and specific roles of KSR1 and KSR2 is necessary. This will include understanding the regulation behind the tissue specific expression of KSR2 and upregulation of KSR1 in cancer, as well as the profound effects on energy balance observed with disruption of KSR2, but not with loss of KSR1. It will also be important to examine the effects of KSR inhibition in a global context, particularly in light of the evidence that KSR expression antagonizes Raf kinase inhibitor-induced Raf dimerization and paradoxical activation. Future work will further clarify the relationship between these structurally similar proteins.
Targeting scaffolding proteins and interfering selectively with protein:protein interactions has proven significantly more difficult than targeting kinases and is one of the reasons that drugs directly targeting mutated Ras proteins are not yet clinically available. Many kinase inhibitors fit within a pocket in the protein. This allows for tight, specific binding of the small molecule that often competes with ATP, enzyme cofactors, substrates, or other proteins that interact with the kinase. Additional approaches could be employed to target KSR. Potential allosteric effects between the extended amino terminal regions and the carboxyl terminal kinase domain have not been exploited. The extensive regulation of KSR expression, protein:protein interactions, and subcellular localization also create additional opportunities for the inhibition of Ras-dependent and KSR-regulated signals. For example, impeding KSR1 localization to the plasma membrane could disrupt Ras-driven ERK activation. This might be achieved by stabilizing 14-3-3 binding or preventing the interaction between KSR1 and Caveolin-1, which has been previously shown to be required for KSR localization to the plasma membrane.
Specific targeting of KSR1 is likely to hold significant promise as a selective therapeutic in Ras-driven cancers. Approximately 30% of all human cancers possess activating Ras mutations. If KSR1 is critical for the transduction of signals from oncogenic Ras even in a fraction of these cancers, selective disruption of this molecular scaffold would represent a significant advancement in the treatment of cancer in many patients. A significant advantage of this approach is the possibility of treating the tumors without subjecting patients to the harsh side effects that are experienced with current therapies. This promising approach is likely to be explored extensively in the coming years following the sentinel paper by Dhawan et al. that demonstrates targeting and stabilizing KSR in an inactive state is a viable approach.
highlights.
Though ksr1-/- mice are largely phenotypically normal, they have reduced ERK signaling and are resistant to Ras-driven tumorigenesis.
Ksr2-/- mice serve as an obesity model due to their increased adiposity, associated insulin resistance, and the detection of ksr2 mutations in some obese humans.
KSR proteins are highly conserved from invertebrates to mammals and are structurally related to Raf proteins.
The canonical function of KSR is as a scaffold for the Raf/MEK/ERK kinase cascade.
KSR requires Ras activation and plasma membrane localization for activity where it forms a heterodimer with Raf to promote MEK and ERK phosphorylation and activation.
The small molecule APS-2-79 binds to the ATP-binding domain of KSR proteins stabilizing them in an inactive state complexed with MEK, interfering with KSR:Raf heterodimerization, and inhibiting downstream Ras signaling.
Acknowledgments
Funding: This manuscript was supported by funding from the National Institutes of Health, National Cancer Institute (F30 CA203397, P30 CA036727, RO1 CA157774, T32 CA009476).
Footnotes
Declaration of interest The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Kornfeld K, Hom DB, Horvitz HR. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell. 1995 Dec 15;83:903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- 2.Sundaram M, Han M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell. 1995 Dec 15;83:889–901. doi: 10.1016/0092-8674(95)90205-8. [DOI] [PubMed] [Google Scholar]
- 3••.Therrien M, Chang HC, Solomon NM, et al. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995 Dec 15;83:879–888. doi: 10.1016/0092-8674(95)90204-x. This report was the first to identify KSR in Drosophila, demonstrate it is required for normal Ras signaling, and describe the five conserved areas within the protein sequence. [DOI] [PubMed] [Google Scholar]
- 4.Downward J. KSR: a novel player in the RAS pathway. Cell. 1995 Dec 15;83:831–834. doi: 10.1016/0092-8674(95)90198-1. [DOI] [PubMed] [Google Scholar]
- 5.Morrison DK. KSR: a MAPK scaffold of the Ras pathway? J Cell Sci. 2001 May;114:1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- 6••.Nguyen A, Burack WR, Stock JL, et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002 May;22:3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. This study describes the phenotype of ksr-/-mice that are characterized by decreased ERK signaling, attenuated T-cell activation, and inhibited tumor development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritt DA, Daar IO, Morrison DK. KSR regulation of the Raf-MEK-ERK cascade. Methods Enzymol. 2006;407:224–237. doi: 10.1016/S0076-6879(05)07019-9. [DOI] [PubMed] [Google Scholar]
- 8••.Brennan DF, Dar AC, Hertz NT, et al. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011 Apr 21;472:366–369. doi: 10.1038/nature09860. This report published the crystal structure of the kinase domain of KSR2 in complex with MEK1, suggests KSR forms both homodimers and heterodimers with B-Raf, and highlights B-Raf:KSR2 heterodimerization triggers a conformational change in the KSR2:MEK complex and promotes the phosphorylation and activation of MEK by an additional B-Raf protein. [DOI] [PubMed] [Google Scholar]
- 9•.McKay MM, Freeman AK, Morrison DK. Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small Gtpases. 2011 Sep;2:276–281. doi: 10.4161/sgtp.2.5.17740. This report shows Raf kinase inhibitors promote KSR1:B-Raf heterodimerization, which restricts B-Raf from dimerizing with C-Raf reducing the paradoxical MAPK signaling following Raf kinase inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nature reviews. Mol Cell Bio. 2005 Nov;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 11.Claperon A, Therrien M. KSR and CNK: two scaffolds regulating RAS-mediated RAF activation. Oncogene. 2007 May 14;26:3143–3158. doi: 10.1038/sj.onc.1210408. [DOI] [PubMed] [Google Scholar]
- 12.Raabe T, Rapp UR. KSR-a regulator and scaffold protein of the MAPK pathway. Sci STKE Signal Trans Knowledge Environ. 2002 Jun 11;2002:pe28. doi: 10.1126/stke.2002.136.pe28. [DOI] [PubMed] [Google Scholar]
- 13.Roy F, Laberge G, Douziech M, et al. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002 Feb 15;16:427–438. doi: 10.1101/gad.962902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Kortum RL, Lewis RE. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol. 2004 May;24:4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. This study describes the role of KSR1 as a scaffold for Raf/MEK/ERK signaling by demonstrating a concentration-dependent increase in ERK signaling by adding up to 14 times the wild type level of KSR1 to ksr1-/- mouse embryo fibroblasts. Increasing KSR1 more than 14-fold causing inhibition of ERK signaling, which is consistent with scaffold protein behaviors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kortum RL, Costanzo DL, Haferbier J, et al. The molecular scaffold kinase suppressor of Ras 1 (KSR1) regulates adipogenesis. Mol Cell Biol. 2005;25:7592–7604. doi: 10.1128/MCB.25.17.7592-7604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Channavajhala PL, Rao VR, et al. Proteomic characterization of the dynamic KSR-2 interactome, a signaling scaffold complex in MAPK pathway. Biochim Biophys Acta. 2009;1794:1485–1495. doi: 10.1016/j.bbapap.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 17.Huang L, Pan CQ, Li B, et al. Simulating EGFR-ERK signaling control by scaffold proteins KSR and MP1 reveals differential ligand-sensi-tivity co-regulated by Cbl-CIN85 and endophilin. Plos One. 2011;6:e22933. doi: 10.1371/journal.pone.0022933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortum RL, Fernandez MR, Costanzo-Garvey DL, et al. Caveolin-1 is required for kinase suppressor of Ras 1 (KSR1)-mediated extracellular signal-regulated kinase 1/2 activation, H-RasV12-induced senescence, and transformation. Mol Cell Biol. 2014 Sep 15;34:3461–3472. doi: 10.1128/MCB.01633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karthik D, Majumder P, Palanisamy S, et al. Targeting cysteine rich C1 domain of Scaffold protein Kinase Suppressor of Ras (KSR) with anthocyanidins and flavonoids - a binding affinity characterization study. Bioinformation. 2014;10:580–585. doi: 10.6026/97320630010580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Razidlo GL, Kortum RL, Haferbier JL, et al. Phosphorylation regulates KSR1 stability, ERK activation, and cell proliferation. J Biol Chem. 2004 Nov 12;279:47808–47814. doi: 10.1074/jbc.M406395200. [DOI] [PubMed] [Google Scholar]
- 21.Razidlo GL, Johnson HJ, Stoeger SM, et al. KSR1 is required for cell cycle reinitiation following DNA damage. J Biol Chem. 2009 Mar 13;284:6705–6715. doi: 10.1074/jbc.M806457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klutho PJ, Costanzo-Garvey DL, Lewis RE. Regulation of glucose homeostasis by KSR1 and MARK2. Plos One. 2011;6:e29304. doi: 10.1371/journal.pone.0029304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher KW, Das B, Kortum RL, et al. Kinase suppressor of ras 1 (KSR1) regulates PGC1alpha and estrogen-related receptor alpha to promote oncogenic Ras-dependent anchorage-independent growth. Mol Cell Biol. 2011;31:2453–2461. doi: 10.1128/MCB.05255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCall JL, Gehring D, Clymer BK, et al. KSR1 and EPHB4 regulate Myc and PGC1beta to promote survival of human colon tumors. Mol Cell Biol. 2016 Sep 1;36:2246–2261. doi: 10.1128/MCB.00087-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Fisher KW, Das B, Kim HS, et al. AMPK promotes aberrant PGC1beta expression to support human colon tumor cell survival. Mol Cell Biol. 2015 Nov;35:3866–3879. doi: 10.1128/MCB.00528-15. This manuscript demonstrates the level of KSR1 expression and selective requirement for KSR1 in colon cancer cells and a xenograft mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohmachi M, Rocheleau CE, Church D, et al. C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr Bio: CB. 2002 Mar 5;12:427–433. doi: 10.1016/s0960-9822(02)00690-5. [DOI] [PubMed] [Google Scholar]
- 27.Hansen LA, Alexander N, Hogan ME, et al. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am J Pathol. 1997;150:1959–1975. [PMC free article] [PubMed] [Google Scholar]
- 28••.Lozano J, Xing R, Cai Z, et al. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 2003 Jul 15;63:4232–4238. This manuscript describes ksr1-/- mice describing their minor hair follicle defect and resistance to Ras-driven, but not MT-driven, tumorigenesis. [PubMed] [Google Scholar]
- 29.Fusello AM, Mandik-Nayak L, Shih F, et al. The MAPK scaffold kinase suppressor of Ras is involved in ERK activation by stress and proinflammatory cytokines and induction of arthritis. J Immunology. 2006 Nov 01;177:6152–6158. doi: 10.4049/jimmunol.177.9.6152. [DOI] [PubMed] [Google Scholar]
- 30.Le Borgne M, Filbert EL, Shaw AS. Kinase suppressor of Ras 1 is not required for the generation of regulatory and memory T cells. Plos One. 2013;8:e57137. doi: 10.1371/journal.pone.0057137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Revelli JP, Smith D, Allen J, et al. Profound obesity secondary to hyperphagia in mice lacking kinase suppressor of ras 2. Obesity. 2011;19:1010–1018. doi: 10.1038/oby.2010.282. [DOI] [PubMed] [Google Scholar]
- 32.Henry MD, Costanzo-Garvey DL, Klutho PJ, et al. Obesity-dependent dysregulation of glucose homeostasis in kinase suppressor of ras 2-/- mice. Physiol Reports. 2014 Jul 1;2:e12053. doi: 10.14814/phy2.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costanzo-Garvey DL, Pfluger PT, Dougherty MK, et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo L, Costanzo-Garvey DL, Smith DR, et al. Cell non-autonomous regulation of hepatic IGF-1 and neonatal growth by Kinase Suppressor of Ras 2 (KSR2) Sci Rep. 2016 Aug 26;6:32093. doi: 10.1038/srep32093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez MR, Henry MD, Lewis RE. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol Cell Biol. 2012 Sep;32:3718–3731. doi: 10.1128/MCB.06754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dougherty MK, Ritt DA, Zhou M, et al. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell. 2009 Jun 26;34:652–662. doi: 10.1016/j.molcel.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Dhawan NS, Scopton AP, Dar AC. Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling. Nature. 2016 Aug 24;537:112–116. doi: 10.1038/nature19327. This is the first report demonstrating that a small molecule inhibitor can stabilize KSR2 in an inactive state and reduce oncogenic Ras signaling by reducing Raf:KSR heterodimerization and prevent conformational changes required for phosphorylation and activation of KSR-bound MEK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joneson T, Fulton JA, Volle DJ, et al. Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J Biol Chem. 1998 Mar 27;273:7743–7748. doi: 10.1074/jbc.273.13.7743. [DOI] [PubMed] [Google Scholar]
- 39.Kortum RL, Johnson HJ, Costanzo DL, et al. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol Cell Biol. 2006;26:2202–2214. doi: 10.1128/MCB.26.6.2202-2214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Stewart S, Sundaram M, Zhang Y, et al. Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell Biol. 1999 Aug;19:5523–5534. doi: 10.1128/mcb.19.8.5523. This report demonstrates KSR binds with MEK and may promote the translocation of MEK to the plasma membrane highlighting the ability of KSR to act as a molecular scaffold for MAPK signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci USA. 2009 Jul 7;106:11022–11027. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Udell CM, Rajakulendran T, Sicheri F, et al. Mechanistic principles of RAF kinase signaling. Cell Mol Life Sci: CMLS. 2011;68:553–565. doi: 10.1007/s00018-010-0520-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koveal D, Schuh-Nuhfer N, Ritt D, et al. A CC-SAM, for coiled coil-sterile alpha motif, domain targets the scaffold KSR-1 to specific sites in the plasma membrane. Sci Signal. 2012 Dec 18;5:ra94. doi: 10.1126/scisignal.2003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michaud NR, Therrien M, Cacace A, et al. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc Natl Acad Sci U S A. 1997 Nov 25;94:12792–12796. doi: 10.1073/pnas.94.24.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou M, Horita DA, Waugh DS, et al. Solution structure and functional analysis of the cysteine-rich C1 domain of kinase suppressor of Ras (KSR) J Mol Biol. 2002 Jan 18;315:435–446. doi: 10.1006/jmbi.2001.5263. [DOI] [PubMed] [Google Scholar]
- 46.Therrien M, Michaud NR, Rubin GM, et al. KSR modulates signal propagation within the MAPK cascade. Genes Dev. 1996 Nov 1;10:2684–2695. doi: 10.1101/gad.10.21.2684. [DOI] [PubMed] [Google Scholar]
- 47.Jacobs D, Glossip D, Xing H, et al. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999 Jan 15;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- 48.Cacace AM, Michaud NR, Therrien M, et al. Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller J, Cacace AM, Lyons WE, et al. Identification of B-KSR1, a novel brain-specific isoform of KSR1 that functions in neuronal signaling. Mol Cell Biol. 2000;20:5529–5539. doi: 10.1128/mcb.20.15.5529-5539.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearce LR, Atanassova N, Banton MC, et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell. 2013 Nov 7;155:765–777. doi: 10.1016/j.cell.2013.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo L, Volle DJ, Lewis RE. Identification of a truncated kinase suppressor of Ras 2 mRNA in sperm. FEBS Open Bio. 2014;4:420–425. doi: 10.1016/j.fob.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denouel-Galy A, Douville EM, Warne PH, et al. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr Bio: CB. 1998 Jan 1;8:46–55. doi: 10.1016/s0960-9822(98)70019-3. [DOI] [PubMed] [Google Scholar]
- 53.Sugimoto T, Stewart S, Han M, et al. The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. Embo J. 1998 Mar 16;17:1717–1727. doi: 10.1093/emboj/17.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu W, Fantl WJ, Harrowe G, et al. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr Bio: CB. 1998 Jan 1;8:56–64. doi: 10.1016/s0960-9822(98)70020-x. [DOI] [PubMed] [Google Scholar]
- 55.Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may bipha-sically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci U S A. 2000 May 23;97:5818–5823. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 57.Matheny SA, Chen C, Kortum RL, et al. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature. 2004 Jan 15;427:256–260. doi: 10.1038/nature02237. [DOI] [PubMed] [Google Scholar]
- 58.Muller J, Ory S, Copeland T, et al. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell. 2001;8:983–993. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 59.Ory S, Zhou M, Conrads TP, et al. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Bio: CB. 2003 Aug 19;13:1356–1364. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- 60.Fantz DA, Jacobs D, Glossip D, et al. Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphory-late specific residues. J Biol Chem. 2001 Jul 20;276(29):27256–65. doi: 10.1074/jbc.M102512200. [DOI] [PubMed] [Google Scholar]
- 61.Ritt DA, Zhou M, Conrads TP, et al. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007 Jan 23;17(2):179–84. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 62.McKay MM, Morrison DK. Caspase-dependent cleavage disrupts the ERK cascade scaffolding function of KSR1. J Biol Chem. 2007 Sep 7;282(36):26225–34. doi: 10.1074/jbc.M702692200. [DOI] [PubMed] [Google Scholar]
- 63.Rajakulendran T, Sahmi M, Lefrancois M, et al. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009 Sep 24;461:542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- 64.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nature reviews. Mol Cell Bio. 2015 May;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 65.Casar B, Pinto A, Crespo P. ERK dimers and scaffold proteins: unexpected partners for a forgotten (cytoplasmic) task. Cell Cycle. 2009 Apr 01;8:1007–1013. doi: 10.4161/cc.8.7.8078. [DOI] [PubMed] [Google Scholar]
- 66.Casar B, Pinto A, Crespo P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaf-fold complexes. Mol Cell. 2008 Sep 05;31:708–721. doi: 10.1016/j.molcel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 67.Lin WC, Iversen L, Tu HL, et al. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci U S A. 2014 Feb 25;111:2996–3001. doi: 10.1073/pnas.1321155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nan X, Tamguney TM, Collisson EA, et al. Ras-GTP dimers activate the mitogen-activated protein kinase (MAPK) pathway. Proc Natl Acad Sci U S A. 2015 Jun 30;112:7996–8001. doi: 10.1073/pnas.1509123112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santos E. Dimerization opens new avenues into Ras signaling research. Sci Signal. 2014 May 06;7:pe12. doi: 10.1126/scisignal.2005318. [DOI] [PubMed] [Google Scholar]
- 70.Kortum RL, Morrison DK. Path forward for RAF therapies: inhibition of monomers and dimers. Cancer Cell. 2015 Sep 14;28:279–281. doi: 10.1016/j.ccell.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gibney GT, Messina JL, Fedorenko IV, et al. Paradoxical oncogen-esis–the long-term effects of BRAF inhibition in melanoma. Nature reviews. Clin Oncol. 2013;10:390–399. doi: 10.1038/nrclinonc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013 Feb 21;49:751–758. doi: 10.1016/j.molcel.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freeman AK, Ritt DA, Morrison DK. The importance of Raf dimerization in cell signaling. Small Gtpases. 2013;4:180–5. doi: 10.4161/sgtp.26117. (Jul-Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lavoie H, Thevakumaran N, Gavory G, et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol. 2013;9:428–436. doi: 10.1038/nchembio.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015 Sep 14;28:370–383. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kholodenko BN. Drug resistance resulting from kinase dimerization is rationalized by thermodynamic factors describing allosteric inhibitor effects. Cell Rep. 2015 Sep 22;12:1939–1949. doi: 10.1016/j.celrep.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Y, Yao B, Delikat S, et al. Kinase suppressor of Ras is cer-amide-activated protein kinase. Cell. 1997 Apr 04;89:63–72. doi: 10.1016/s0092-8674(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 78.Volle DJ, Fulton JA, Chaika OV, et al. Phosphorylation of the kinase suppressor of ras by associated kinases. Biochemistry. 1999 Apr 20;38:5130–5137. doi: 10.1021/bi983050d. [DOI] [PubMed] [Google Scholar]
- 79.Brennan JA, Volle DJ, Chaika OV, et al. Phosphorylation regulates the nucleocytoplasmic distribution of kinase suppressor of Ras. J Biol Chem. 2002 Feb 15;277:5369–5377. doi: 10.1074/jbc.M109875200. [DOI] [PubMed] [Google Scholar]
- 80.Zhang J, Zafrullah M, Yang X, et al. Downregulation of KSR1 in pancreatic cancer xenografts by antisense oligonucleotide correlates with tumor drug uptake. Cancer Biol Ther. 2008;7:1490–1495. doi: 10.4161/cbt.7.9.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009 Apr;21:462–469. doi: 10.1016/j.cellsig.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]