

Abstract
Metastasis causes more than 90% of cancer-related deaths and most prostate cancer (PCa) patients also die from metastasis. The ‘metastatic cascade’ is a complex biological process that encompasses tumor cell dissociation (from the primary tumor), local invasion, intravasation, transport in circulation, extravasation, colonization, and overt growth in end organs. It has become clear that successful metastasis not only involves many tumor cell-intrinsic properties but also depends on productive interactions between cancer cells and the tumor microenvironment. In this Review, we begin with a general summary on cancer metastasis and a specific discussion on PCa metastasis. We then discuss recent advances in our knowledge of the cellular determinants of PCa metastasis and the importance of tumor microenvironment, especially an immunosuppressive tumor microenvironment, in shaping metastatic propensities. We conclude with a presentation of current and future therapeutic options for patients with PCa metastasis, emphasizing the development of novel, mechanism-based combinatorial strategies for treating metastatic and castration-resistant PCa.
Keywords: metastasis, prostate cancer, cancer stem cells, microenvironment
Introduction
Despite the overwhelming prevalence of metastases-associated deaths in cancer patients, many biological programs underlying this complex process remain unknown. Research has been hindered in part by the complexities surrounding the metastatic process and the complex nature and heterogeneity of metastatic tumors. With the advent and continuous augmentation of the field of ‘cancer genomics’, along with other advancements, recent progress has been made in the field of metastasis in elucidating cellular programs that drive cancer metastasis. Several fundamental concepts of dissemination and metastatic outgrowth of cancer have been outlined. Metastasis is a complex process that occurs through a multi-step process, wherein the fate of a metastatic cell is influenced by and depends significantly on its interaction with components of the primary tumor, circulatory/lymphatic, and distant organ environment. The complexity increases due to the cell-intrinsic properties within that cancer cell as well as adaptive programs that impel that cancer cell to survive during metastatic colonization, both of which vary between various cancer types. These mechanisms, guiding cell-intrinsic and adaptive programs, have proven more difficult to elucidate. This Review aims to summarize the recent progress in elucidating unique cellular mechanisms and adaptive programs that drive prostate cancer (PCa) metastasis. This progress can be partially attributed to improvements in experimental models of metastasis, and we will provide experimental data confirming the value of these models. As these novel mechanisms inevitably offer potential therapeutic targets and strategies for the management of metastasis, the Review will end with a discussion on current therapeutic efficacies and future proposals.
Metastatic cascade: A primer
Metastasis occurs when cancer cells leave the primary tumor mass, travel, and survive in other locations in the body. This process is complex comprising many stochastic events that are dependent on both intrinsic properties of tumor cells as well as reciprocal responses from and interactions with numerous other cell types in the microenvironments of both primary tumors and end organs [1–4]. Metastatic cells can be generated via clonal evolution or clonal selection, wherein time-dependent acquisition of mutated genetic drivers within tumor cells, confer proliferative and invasive properties. Alternatively, cancer stem cells (CSCs), which pre-exist in primary tumors, may be endowed with intrinsic metastatic propensity. Both clonal selection and CSCs likely cooperate to generate cells that have metastatic capabilities. The ultimate success of metastasis also depends on cross-talk between metastatic cancer cells (the ‘seeds’) and specific end-organ microenvironments (the ‘soil’) [1]. Thus, through evolutionary time, genetic diversity, epigenetics, and the tumor microenvironment together contribute to metastatic success. Metastasis consists of many interrelated events including dissociation of prospective metastasizing cells from primary tumors, local invasion, intravasation, transport in the circulation, arrest in microvessels of various organs, extravasation, seeding and latency, formation of micrometastasis, and colonization and subsequent formation of macrometastasis [3]. With numerous variables at each step, metastasis is a highly inefficient process and tumor cells must constantly adapt in order to successfully colonize distant organs and form clinically overt lesions.
In the first stage of metastasis, the pre-colonization phase including local invasion and intravasation into the tumor vasculature or lymphatic system [3], tumor cells can be activated by the local microenvironment and inflammatory signaling to invade and migrate through the stroma via cytoskeletal rearrangements and the secretion of proteases that degrade extracellular matrix (ECM) proteins. Once shed into the circulation, tumor cells are called circulating tumor cells (CTCs), which must survive a variety of stresses especially the hemodynamic shear stress. CTCs can transport as CTC clusters containing tumor cells or mix of tumor cells and host cells such as macrophages, platelets, and leukocytes, which can provide several benefits [5–7]. For example, platelets may physically protect CTC clusters from shear forces, can induce reversible metabolic changes in tumor cells that increase their ability to withstand oxidative stress in the bloodstream, and can increase invasiveness via releasing signaling molecules [5, 7].
The next phase, organ colonization, includes arrest of CTCs in capillaries at distant sites, extravasation, seeding and latency, and overt colonization [3]. The first capillary bed that a CTC encounters is largely determined by patterns of blood circulation, which influences the final destination of metastasis. Cancer cells then exit capillaries into the tissue parenchyma by penetrating the endothelial cell and pericyte layers, a process known as extravasation. The differing structures of the capillary walls in each organ, and the capacity of CTCs to pass through endothelial walls both influence the organ tropism of metastasis. Additionally, cancer cells may possess the ability to specifically target niches, such as the bone marrow hematopoietic stem cell (HSC) niche, as their final metastatic destination. Thus, a combination of priming signals from the tumor stroma, the composition of CTC clusters, blood-circulation patterns, the structure of target-organ capillary walls, cancer-cell-autonomous functions, and metastatic niches, together, determine metastatic infiltration of specific organs.
If CTCs infiltrate distant organs and survive, they are called disseminated tumor cells (DTCs). The foreign microenvironment, which encompasses stromal cells, ECM constituents, growth factors and cytokines, and even the microarchitecture of the tissue itself, are all factors that influence the survival and tumor-initiating activity of DTCs. After extravasation, DTCs must develop resistance to immunity (i.e., immune surveillance) and other host-tissue defenses. DTCs must also remain in supportive specialized niches, in which pro-metastatic stromal mediators would ultimately activate stem-cell support pathways and pathways that integrate cell metabolism and survival. DTCs can also enhance their own survival by expressing autocrine factors or by recruiting stromal cells as a source of soluble activators and amplifiers. DTCs then enter a latent state, during which they must achieve long-term survival [8]. In the final stages, cells break out of latency, reinitiate overt outgrowth, overtake the local tissue microenvironment and expand into large macroscopic metastases. The initiation of overt colonization differs in each organ and involves the selection of organ-specific metastatic traits, which gives rise to organ-specific populations of metastatic cells. When macroscopic metastases are detected, the patient is treated with combinations of conventional chemotherapy, targeted therapy and immunotherapy, which can reduce metastatic burden. Nevertheless, a population of residual cancer cells will withstand treatment via alteration of intracellular pathways for survival and via survival signals from non-neoplastic stromal cells until drug-resistant clones emerge. As a result, the cure rates of patients with metastasis remain disappointingly low.
These sequential steps outlining the metastatic cascade are the basis for all cancer types. However, the effect of specific environmental interactions with cancer cells harboring inherent attributes, lead to novel mechanistic differences between different cancer types. In the following section, we highlight specific examples of the adaptive programs found in PCa cells that lead to metastatic PCa.
Prostate cancer metastasis: Recent advances and experimental assays
PCa remains the most prevalent non-cutaneous cancer in men in North America and the second most common cause of cancer death worldwide. Age is the greatest risk factor for PCa, as the majority (64%) of PCa patients are over 70 years and <1% are under age 50. The growth of normal and malignant prostate tissue is regulated by androgens through action of the androgen receptor (AR) in both epithelial and stromal cells. Thus, the primary treatment for metastatic PCa (mPCa) is androgen-deprivation therapy (ADT), and in the majority of patients, this provides a temporary control of the disease. However, cancer cells eventually become castration resistant resulting in disease progression to metastatic castration-resistant prostate cancer (mCRPC). The survival rate for both patients with mPCa at diagnosis and patients with mCRPC upon ADT failure is poor. Interestingly, overall survival (OS) time in men with mCRPC is associated with sites of metastasis, with a shorter OS observed for lung and liver metastases as compared with bone and non-visceral involvement [9].
The development of an efficacious cancer therapy critically relies on the existing paradigm of cancer pathogenesis. The oligometastatic state, first proposed in 1995, was defined as an intermediate stage of cancer spread between locally confined disease and widely metastatic disease [10] At the time, the cell-of-origin, the specific cellular and molecular mechanisms as well as the importance of the microenvironment leading to the development of cancer were unknown or excluded, and tumor size was the principle basis for tumor staging. The clinical implication was that ablation of these limited and treatable cancer metastases, along with primary tumor resection, could potentially result in a cure. Today, the emergence of high-resolution genome technologies has revolutionized the field of cancer genomics. Within the PCa field, this technology has led to data generally supporting a monoclonal origin of multifocal PCa [11]. These studies suggest that primary tumors are composed of many different subclones, each one comprised of genetically identical cells, distinguishable from other subclones by their specific acquired mutations. Subclones with advantageous survival attributes such as intrinsic drug resistance, become dominant and survive. Interestingly, recent studies also provide evidence that PCa, in the context of ADT-associated metastasis, displays dynamic patterns of evolution [12]. Metastasis-to-metastasis spread was found to be common via two mechanisms. First, subclones within a metastasis can originate from another metastatic site rather than the primary tumor, a process called ‘cross-metastatic seeding’ [13]. This phenomenon was also demonstrated in response to therapy in a patient with lethal PCa [14]. Second, the same sets of subclones can seed multiple sites of metastasis, a process called ‘polyclonal seeding’ [13]. Multiple subclones may be shared between such polyclonal seeds for two or more metastases, suggesting that these subclones might functionally cooperate with one another to promote metastatic progression. Distant metastases could also reseed the surgical bed suggesting that PCa cells may take advantage of pre-existing supportive niches [14]. This process of ‘tumor self-seeding’ has previously been observed with PCa CTCs, which could lead to additional recruitment of CTCs and confer enhanced tumor growth [15]. Despite these breakthroughs in analysis of the PCa genome, clinical diagnoses for oligometastatic PCa are still based on the number of extrapelvic lesions, as a gap still remains between genomic data and specific properties of an individual patient’s disease.
Lack of an accurate definition of oligometastatic PCa emphasizes the critical need for more efficacious biomarkers to monitor mPCa disease behavior, optimize duration of treatment and assess benefit from therapy. In this regard, CTCs could be used to track the biological evolution of mPCa. Despite technical challenges due to the rarity and molecular heterogeneity of CTCs [6, 16], significant technological innovation has created several reliable methods for their isolation and detection. CTCs have recently been shown to have diagnostic and predictive values in patients with mCRPC [17]. Whole genome sequencing (WGS) at the single cell level demonstrated the shared genomic alterations between CTCs and serial tissue biopsies obtained from a patient with mCRPC [18]. As bone is the most frequent site of mPCa, data obtained from bone marrow aspirates can also be relevant. Interestingly, tumor cell clusters have been found to be more prevalent and bigger in bone marrow aspirates than in blood, express higher levels of the AR protein per tumor cell, and are prognostic in mCRPC. This suggests that such DTC clusters are key mediators in PCa progression, and that methods that allow for serial monitoring of the disease composition in the bone marrow can provide insight into relevant determinants of PCa progression [19]. Several predictive biomarkers have also been proposed for the identification of PCa patients at high risk for metastatic progression. A recent report describes the first study implicating that autoantibodies specific for fetuin-A, a liver secretory protein that accumulates in bone, may be effective prognostic indicators for PCa patients prone to metastasis [20]. Another recent study validates eight differentially methylated CpGs including CpGs in five genes (ALKBH5, ATP11A, FHAD1, KLHL8, and PI15) and three intergenic regions, which could be used as prognostic biomarkers to distinguish PCa patients with metastatic-lethal from non-recurrent tumors [21].
The functional validation of WGS data has been partly inhibited by the difficulty in developing patient derived models that capture the biological heterogeneity and mutation landscape in advanced PCa. Animal models offer an advantage of more accurately mimicking results in a living organism, and include genetically engineered mouse models (GEMMs), xenograft models, and patient-derived xenografts (PDXs). The major limitation of most PCa cell lines and xenografts is that they are derived from clinically aggressive lesions. In an effort to increase the clinical translatability of animal models of PCa metastasis, several recent reports provide evidence for novel models that overcome this limitation. RapidCap, a novel GEMM for mPCa analysis that displays highly penetrant metastatic disease progression and emergence of castration-resistant metastasis, was used to successfully show that Myc is a driver of Pten-mutant metastasis [22]. Spontaneous metastasis in the popular human xenograft model of primary PCa, CWR22, was recently reported and characterized to represent the full spectrum of disease from primary PCa, to CRPC, to metastatic disease [23]. Recently, PDX lines called the Living Tumor Laboratory (LTL), based on the subrenal capsule grafting technique, have been reported [24]. This collection of more than 200 transplantable PDX models of various low to high-grade human cancers, including 45 PCa lines, could be a powerful platform for translational cancer research. PDXs derived from sequential biopsies from prostate metastases from a single patient can also provide individual metastatic tumor expression signatures that may be helpful in predicting efficacy of drugs, and data confirms that the patient’s genomic and transcriptomic alterations are preserved in these sequential PDXs [25].
How, in reality, can human metastasis be accurately experimentally assayed? Two major metastasis assays have been employed (Figure 1A). In experimental metastasis assays, human cancer cells are directly injected into the circulation, either intra-cardiacally (IC) or through tail vein (TV) of recipient immunodeficient mice. Such assays bypass key early steps of metastasis including local invasion and intravasation but do assess the survival of tumor cells in the circulation and all subsequent steps of the metastatic cascade. The IC-injected human cancer cells would disseminate into many organs whereas TV-injected cancer cells become ‘trapped’ mainly in the lung (Figure 1A). In contrast, spontaneous metastasis assays, in which human cancer cells are implanted in immune-compromised mice either ectopically (e.g., subcutaneous or SC) or orthotopically (e.g., in the dorsal prostate or DP), allow the analysis of most steps in the metastatic cascade (Figure 1A). In both types of metastasis assays, the sites and extent of metastasis can be qualitatively, semi-quantitatively, or quantitatively assessed depending on whether human cancer cells are labeled or not or labeled with GFP/FRP vs. luciferase (Figure 1B). For example, GFP-labeled PC3 (PC3-GFP) human PCa cells injected into the heart ‘metastasized’ to most of the 9 organs analyzed whereas the same cells injected through the TV were only found in the lung, as semi-quantitatively assessed by estimating the abundance of GFP+ cells (Table 1). Similarly, we employed luciferase-tagged (Figure 1C–E), GFP-labeled (Figure 2; Table 2, Table S1), or dual-labeled (Figure S1) human PCa cells to perform spontaneous metastasis assays. Of interest, we observed that PC3-luc/GFP cells implanted in the DP, i.e., the orthotopic site, metastasized to the lung, kidney, lymph node (LN), and some other organs whereas the same cells implanted SC, despite developing similar-sized primary tumors, rarely metastasized (Figure 1C–E; data not shown). Similar differential metastasis between DP vs. SC tumors was observed using GFP-labeled (Table 2) or Luc/GFP-tagged (Figure S1) cells. Human PCa cells implanted into the mouse anterior prostate (AP) lobes also manifested greater metastatic potential compared to SC implanted isogenic cells (Figure 2; Table S1). Time course studies of DP metastasis revealed that metastasis to the lung and pancreas was observed first in the second week after implantation, after which time increasing metastasis to many other organs was noted (Table S2). By 7 weeks, the lung, LN, and kidney showed highest levels of metastasis. Importantly, human PCa cells recovered from end organs in both IC injection (Figure 3) and AP-implantation (Figure S2) models could re-establish clonogenic growth. These studies support the well-established concept that the orthotopically implanted human tumor cells produce more widespread metastasis than ectopically implanted isogenic tumor cells [26] and highlight the importance of primary tumor microenvironment in dictating metastatic spread. In addition to assessing end organs for labeled metastatic cells, metastatic route tracking allows visualization of the specific routes cancer cells take when migrating from the primary tumor to distant sites. Recent data utilizing this technique on orthotopically transplanted human PCa-GFP cells in mice was shown to accurately mimic metastasis in hormone-independent advanced-stage PCa seen in the clinic [27].
Figure 1.
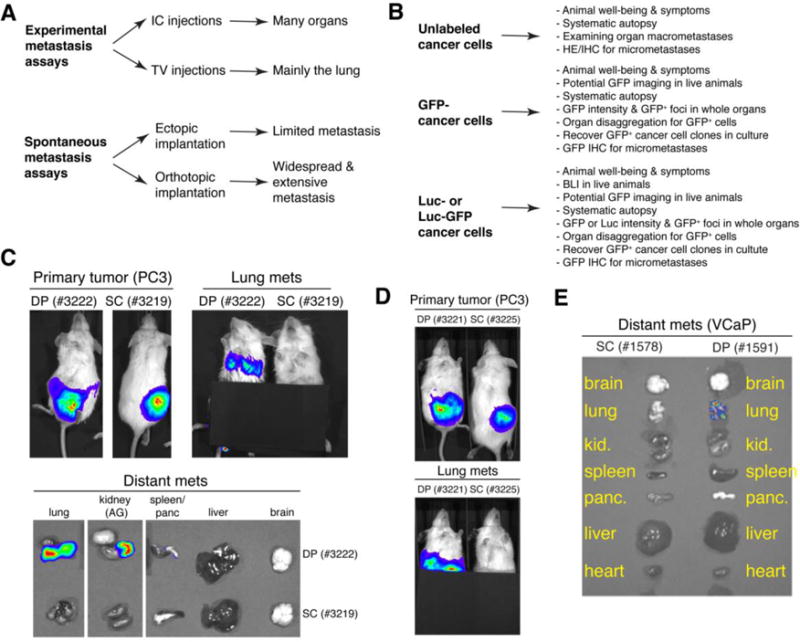
Experimental assays assessing metastasis of human cancer cells. (A) Two major metastasis assays. Experimental metastasis assays can be carried out via IC or TV injection leading to metastasis in many organs or mainly lung metastasis, respectively. Spontaneous metastasis assays can be carried out via ectopic or orthotopic implantation of human cancer cells leading to limited vs. widespread metastasis in many organs, respectively. (B) The level of metastasis can be qualitatively, semi-quantitatively, or quantitatively assessed depending on whether the starting cells are labeled or not. (C) DP-implanted (dorsal prostate) human PCa cells show more widespread metastasis than SC-implanted (subcutaneum) PCa cells. 500,000 PC3-Luc/GFP cells were implanted into the DP or SC in separate NOD/SCID mice. Shown are IVIS images taken at week 7 after injection (top panels). Primary tumors are on the left and corresponding lung metastasis is on the right for both DP and SC implanted mice. Note that DP-implanted PC3-Luc/GFP cells metastasized to the lungs and kidney, whereas SC-implanted PC3-Luc/GFP cells did not metastasize. Distant metastases to the lung, kidney, spleen/pancreas, liver, and brain in these mice are shown (bottom panels). Note that DP-implanted PC3-Luc/GFP cells metastasized to the lung and kidney (AG; adrenal gland) whereas SC-implanted cells did not metastasize. The fluorescence emitted from the lung was much less than from the kidney and thus the exposure time had to be increased to see positivity; however, all other organs remained negative for Luc/GFP expression even after this increase. (D) The experiment was repeated in two additional mice in order to confirm that DP-implanted human PCa cells show more widespread metastasis than SC-implanted PCa cells. (E) 500,000 VCaP-Luc/GFP cells were implanted into the DP or SC in NOD/SCID mice. Shown are representative IVIS images of brain, lung, kidney, spleen, pancreas, liver, and heart from one mouse 11 weeks post injection. Note that DP-implanted VCaP-Luc/GFP cells metastasized to the lungs whereas SC implanted VCaP-Luc/GFP cells did not metastasize.
Table 1.
Metastasis profiles of IC or TV injected PC3-GFP cells
| Cell typea (Route & number) | Incidenceb (Inj. & term. dates) | Metastasisc
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Femur | Tibia | Lung | Kidney | Liver | Pancreas | Brain | Spleen | LNd | ||
| PC3-GFP (IC) | ||||||||||
| 10,000 cells | 1/1 (Inj. 07/28/05) | |||||||||
| 0001 | term: 03/07/06 | − | − | + | − | − | + | − | − | − |
| 100,000 cells | 1/1 (Inj. 07/28/05) | |||||||||
| 0002 | term: 11/19/05 | + | + | +++ | ++ | + | +++ | + | + | +++ |
| 1,000,000 cells | 2/2 (inj. 08/04/05) | |||||||||
| 0003 | term: 10/03/05 | N.D | N.D | +++ | ++ | + | +++ | ++ | ++ | +++ |
| 0004 | term: 10/06/05 | N.D | N.D | +++ | ++ | + | +++ | +++ | ++ | +++ |
| PC3-GFP (TV) | ||||||||||
| 5,000,000 cells | 2/2 (Inj. 08/19/05) | |||||||||
| 0005 | term: 10/24/05 | − | − | ++ | − | − | − | − | − | − |
| 0006 | term: 10/26/05 | − | − | ++ | − | − | − | − | − | − |
For intracardiac (IC) injection, cells were injected directly into to left heart ventricle. A total of 4 animals were injected with 3 cell doses indicated. Note increased levels of metastasis at 100,000 and 1,000,000 cells compared to 10,000 cells. N.D, not determined. For tail vein (TV) injection, a total of 2 animals were injected with 5,000,000 cells each. Numbers (e.g., 0001) beneath refer to animal tags.
Animals were terminated when they became moribund. The injection (inj.) and termination (term.) dates were indicated. Ratios represent the number of animals bearing tumors/the total number of animals injected.
Metastasis to different organs was semi-quantified, at necropsy, as “−, +, ++, +++” upon harvesting and disaggregating the end organs into single cells followed by examining the GFP+ PCa cells of the whole organ under a fluorescence microscope. −, no GFP+ cells; ±, 1–5 GFP+ cells; +, dozens of GFP+ cells; ++, hundreds of GFP+ cells; +++, thousands or tens of thousands of GFP+ cells (see images below for examples).
Lymph nodes (LN) examined including the renal, sciatic, mesenteric, inguinal, and/or pyloric LN.
![]()
Figure 2.
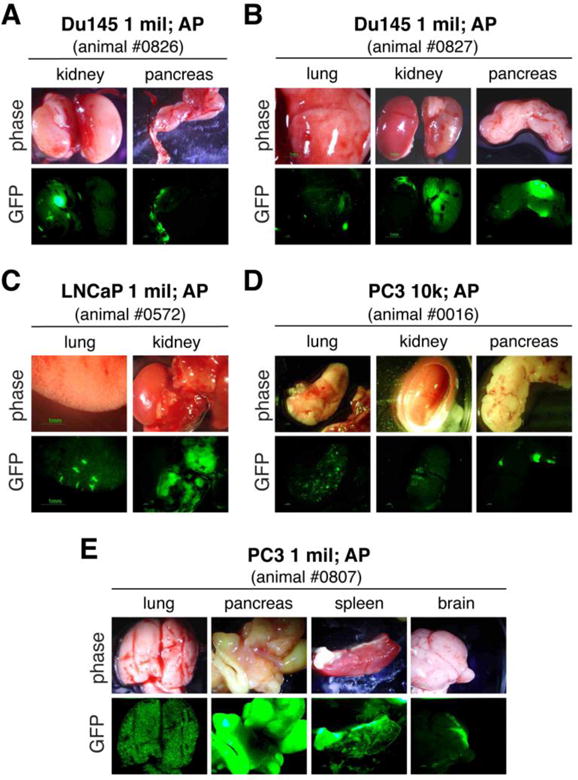
AP-implanted PCa cells metastasize to multiple organs. Shown are representative organ images (upper panels, phase; lower panels, GFP) of 1 million Du145-GFP cells (A and B), 1 million LNCaP-GFP cells (C) and 10,000 (D) or 1 million (E) of PC3-GFP cells implanted in the AP of NOD/SCID mice and harvested ~60 days post implantation. Animal tag numbers are indicated.
Table 2.
DP-implanted PCa cells show more metastasis than SC-injected PCa cells.
| Cell type (Route & number)a | Terminationb (Inj. & term. dates; tumor weights) | Metastasisc
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Femur | Tibia | Lung | Kidney | Liver | Pancreas | Brain | Spleen | LNd | ||
| PC3-GFP (SC) | ||||||||||
| 1,000,000 cells | 4/4 (Inj. 01/27/06) | |||||||||
| 0417 | term: 02/28/06 (0.2g, 0.3g) | − | − | + | − | − | − | − | − | − |
| 0419 | term: 03/06/06 (0.7g, 1.4g) | − | − | +++ | + | − | ± | − | − | + |
| 0414 | term: 03/21/06 (3.2g) | − | − | ++ | + | − | − | − | − | ++ |
| 0418 | term: 03/21/06 (1.3g) | − | − | ++ | − | ± | − | − | − | − |
| PC3-GFP (DP) | ||||||||||
| 1,000,000 cells | 4/4 (Inj. 12/27/06) | |||||||||
| 0431 | term: 01/31/07 (0.5g) | + | + | +++ | + | ± | + | − | + | ++ |
| 0433 | term: 01/31/07 (0.3g) | ± | ± | +++ | + | + | +++ | ± | + | + |
| 0428 | term: 02/09/07 (1.1g) | − | − | +++ | +++ | ++ | +++ | ± | +++ | +++ |
| 0429 | term: 02/09/07 (0.9g) | − | − | +++ | ++ | + | +++ | ± | ++ | +++ |
| Du145-GFP (SC) | ||||||||||
| 2,000,000 cells | 1/1 (Inj: 12/30/05) | |||||||||
| 0007 | term: 04/18/06 (0.7g) | − | − | ± | − | − | − | − | − | − |
| 4,000,000 cells | 1/1 (Inj: 12/30/05) | |||||||||
| 0008 | term: 03/29/06 (4.2g) | − | − | − | − | − | ± | − | − | − |
| Du145-GFP (DP) | ||||||||||
| 2,000,000 cells | 3/3 (Inj: 08/11/05) | |||||||||
| 0009 | term: 09/29/05 | N.D | N.D | +++ | ++ | + | +++ | − | ++ | − |
| 0010 | term: 10/20/05 | − | + | +++ | ++ | + | +++ | − | + | − |
| 0011 | term: 10/29/05 | + | − | ++ | ++ | + | ||||
The indicated numbers of PCa cells were implanted in 50% Matrigel either SC or into the DP of NOD/SCID mice. Numbers (e.g., 0417) beneath refer to animal tags.
Animals were terminated when they became moribund or tumor burden became overwhelming. Both injection (Inj.) and termination (term.) dates are indicated. Note that for SC injections each animal received either one or two implantations (tumor weights in grams indicated in parentheses). The ratio indicates the number of animals bearing tumors/the total number of animals injected.
Metastasis to different organs was evaluated independently by two investigators via observing and enumerating GFP+ cells of the freshly disaggregated whole organs under a fluorescence microscope. Relative levels of metastasis were semi-quantitatively indicated as “−” to “+++”.−, no GFP+ cells; ±, <10 GFP+ cells; +, dozens of GFP+ cells; ++, hundreds of GFP+ cells; +++, thousands or tens of thousands of GFP+ cells. N.D, not determined.
Lymph nodes examined, including renal, caudal, sciatic, mesenteric, inguinal, and pyloric LN.
Figure 3.
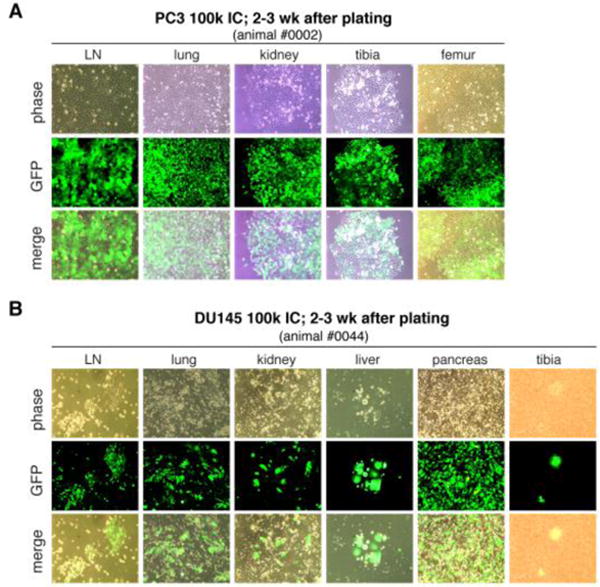
IC-injected PCa cells recovered from end organs formed colonies. Shown are representative images of PC3-GFP (A) and DU145-GFP (B) cells cultured (2–3 weeks after plating) from the organs indicated. Animals were injected with 100,000 GFP-tagged PCa cells and terminated ~4 months post injection. Organs were disaggregated into single cells, which were plated and cultured in RPMI1640 containing 7% FBS. The GFP− cells on the background in some images are the host cells. Original magnifications, ×200.
One caveat with mouse xenograft studies is the lack of a functional immune system in host animals. The future use of humanized mice, immunodeficient mouse strains with engrafted human immune systems, can allow researchers to examine xenograft growth in the context of a human immune system and resultant tumor microenvironment, and recent studies have highlighted the increased similarities in attendant tumor structure, metastasis, and signaling to those features in cancer patients [28]. Further improvements in PDX models and GEMMs of metastasis, enhanced imaging technologies and advanced genomic sequencing, including the ability to analyze single cells [29], will produce a deeper understanding of the cellular basis of PCa metastasis. Below we discuss the nature of metastatic cells, i.e., cellular determinants of PCa metastasis (Table 3).
Table 3.
Cellular determinants of PCa metastasis.
| Cell | Role in mPCa | Mechanism | Ref | |
|---|---|---|---|---|
| PCa Cells | PCSC | re-initiation of tumor growth after dissemination in distant organs | CD44+ (xenograft models): enhanced clonogenicity, enriched in tumor- and metastasis-initiating cells | [31] |
| ALDHhigh (human PCa): enhanced clonogenicity, enriched in tumor- and metastasis-initiating cells | [32] | |||
| TOP2Ahigh | metastasis-initiating cells | defined by high expression of TOP2A, phenotype of recurrent/mPCa and a marker of rapid proliferation, distinct from CSCs (a subpopulation of TOP2Aneg cells), display a higher frequency of abnormal cell divisions due to increased aneuploidy formation, ability to acquire stemness features and generate TOP2Aneg cells | [35] | |
| Epithelial-EMT phenotype | a capacity to transition through programs endowing properties for dissemination and initiating metastatic colonies | defined by a transitory state between epithelial and mesenchymal programs and not a fixed state, enhanced stemness characteristics and tumor-initiating capacity compared to non-EMT epithelial cells, only EMT tumor cells have the capacity to revert to an epithelial state and proliferate to form macro-metastases (non-EMT epithelial and mesenchymal-PCa cells do not) | [42] | |
| EMT-CTC | dissemination via circulatory system | exist in phenotypic states intermediate to epithelial and mesenchymal states (i.e. display both epithelial and mesenchymal markers): epithelial in nature allowing an epithelial cell–cell adhesion and a mesenchymal motility trait | [43] | |
| DTC | maintain dormancy and stemness in distant organs | target the BM-HSC niche to remain dormant and chemoresistant; bind osteoblasts to induce the expression of TBK1 and inhibit mTOR signaling leading to dormancy, maintain PCSC attributes | [47,48] | |
| Mitotic quiescence | bone metastasis-initiating cells | CD133−/CD44+/α2/β1+ phenotype: ability to home to bone via high expression of CXCR4 | [49] | |
| Microenvironment | CAF | increase oncogenic potential of PCa cells | create a niche for PCa cells via secreting inflammatory cytokines, angiogenic factors, ECM-proteins and their proteases, and GFs; promote tumorigenesis, EMT, cancer stemness, bone metastasis; secrete TGF-β to accelerate FGFR1-driven adenocarcinoma; secrete CXCL12, which binds to CXCR4 on PCa cells and induces EMT thus promoting metastasis to secondary tumor sites; novel source of GDF15, a TGFβ/BMP family pro-metastatic GF, which increases PCa cell migration, invasion, tumor growth, and systemic effects on the outgrowth of distant and indolent PCa cells; release TGF-β to induce FOXP3 expression in CD8+ T cells, converting them into immunosuppressive T-regs; can induce monocyte recruitment via SDF-1 and promote monocyte trans-differentiation toward the M2 immunosuppressive macrophage phenotype | [51–56,62,63] |
| CAM | increase oncogenic potential of PCa cells | interact with PCa cells and CAFs to promote cell motility, escape from the primary tumor, enhanced malignancy, metastatic spread, and activation of endothelial cells and BM-derived precursors to drive angiogenesis; induce prostate tumorigenesis via AR-inflammatory cytokine CCL4–STAT3 activation, downregulation of p53/PTEN tumor suppressors, and promotion of EMT; found in CTC clusters to aide in invasion and migration via modification of the adhesion function of integrins interacting with laminin 511, and docetaxel resistance | [63–65] | |
| MDSC | immunosuppression | inhibit proliferation of CD8+ T cells, display high levels of activated STAT3 which induce expression and enzymatic activity of Arginase-1, a potent T-cell inhibitor | [69] | |
| MC | increase oncogenic potential of PCa cells | intratumoral MCs negatively regulate angiogenesis and tumor growth, peritumoral MCs express high levels of FGF-2 which may contribute to the expansion of PCa; enhance PCa invasion and increase PCSC populations in ADT-treated cells via alteration of LncRNA-HOTAIR/PRC2-AR-MMP9 signals | [72,73] | |
| B-cell | increase oncogenic potential of PCa cells | release lymphotoxin to promote survival and proliferation via activation of IKKα and STAT3 in androgen-deprived PCa cells; activate the IKKα-E2F1-BMI1 cascade in PCSCs to induce proliferation and tumor recurrence in AR-deprived conditions; mediate chemotherapy resistance via IL-10 and PD-L1 | [75–77] | |
| BM-MSC | increase oncogenic potential of PCa cells | produce TGF-β1, leading to MMP-2 and −3 upregulation in PCa cells; produce CCL5 which alters HIF2α signaling within PCa cells and increases PCSCs and metastasis via AR signaling; ability to transdifferentiate into CAFs via TGF-β1 in the environment to increase tumor invasiveness, perform vascular mimicry, and recruit monocytes | [79,80] | |
| Osteoclast in BM-HSC niche | contribute to mPCa growth after DTCs establish in the bone | play a role in HSC mobilization and osteoclast activation by PCa cells releases GFs and cytokines sequestered in bone matrix; activation is not necessary for the earliest seeding of PCa into the HSC niche | [47,81] | |
| Osteoblast in BM-HSC niche | regulate CSC activities of DTCs | activate conversion of DTCs to PCSCs in the HSC niche via cell-to-cell contact, expression of GAS6 and activation of the Mer/mTOR pathway | [82] | |
| Adipocyte | increase oncogenic potential of PCa cells | pre-adipocytes recruited by PCa cells to the TME increase PCa cell invasion and metastasis via modulating miR-301a/AR/TGF-β1/Smad/MMP9 signals | [83] | |
| adipocytes in periprostatic adipose tissue direct PCa cell migration via secreting CCL7 which stimulates CCR3-expressing PCa cells | [84] | |||
| BM adipocytes drive osteoclastogenesis via expression of CXCL1 and CXCL2 and tumor-driven osteolysis of the bone; induce metabolic reprogramming via paracrine signaling with PCa cells to induce increased expression of glycolytic enzymes via HIF-1α activation | [85,86] | |||
| Autonomic nerve fibers | increase oncogenic potential of PCa cells | SNS adrenergic fibers promote survival via stromal β2- and β3-adrenergic receptors, PNS cholinergic fibers promote invasion, migration, distant metastases via nerve ending–derived acetylcholine–activated CHRM1 | [87] |
Abbreviations: ADT, androgen deprivation therapy; ALDH, aldehyde dehydrogenase; AR, androgen receptor; BM, bone marrow; BM-MSC, bone marrow derived mesenchymal stem cell; BMP, bone morphogenetic protein; CAF, cancer associated fibroblast; CAM, cancer-associated macrophage; CCL, chemokine (C-C motif) ligand; CCR, chemokine (C-C motif) receptor type; CD, cluster of differentiation; CHRM3, cholinergic muscarinic receptor 3; CTC, circulating tumor cell; CXCL4, C-X-C motif chemokine ligand type 4; CXCR4, C-X-C chemokine receptor type 4; DTC, disseminated tumor cell; EMT, epithelial-mesenchymal transition; FGF-2, fibroblast growth factor 2; FGFR1, fibroblast growth factor receptor 1; FOXP3, forkhead box protein P3; GAS6, growth arrest specific 6; GDF15, growth/differentiation factor 15; GF, growth factor; HIF2α, hypoxia inducible factor alpha; IFNγ, interferon gamma; IKKα, inhibitor of nuclear factor kappa-B kinase subunit alpha; MC, mast cell; MDSC, myeloid-derived suppressor cell; MMP, matrix metallopeptidase; mTOR, mechanistic target of rapamycin; MSC, mesenchymal stem cell; PCSC, prostate cancer stem cell; PD-L1, programmed death ligand 1; PNS, parasympathetic nervous system; PRC2, polycomb repressive complex 2; PTEN, phosphatase and tensin homolog; SDF-1, stromal cell-derived factor-1 or CXCL12; SNS, sympathetic nervous system; STAT3, signal transducer and activator of transcription 3; T-reg, T regulatory cell; TBK1,TANK binding kinase 1; TGF-β, transforming growth factor-beta; TME, tumor microenvironment; TOP2A, DNA topoisomerase 2-alpha.
Cellular determinants of PCa metastasis
What is the inter-relationship between metastatic PCa cells versus prostate cancer stem cells (PCSCs), cells undergoing EMT (epithelial-mesenchymal transition), and the above-discussed CTCs and DTCs? In general, PCSCs seem to possess enhanced metastatic propensities (reviewed in [30]). For example, CD44+ PCa cells purified from several xenograft models are enriched not only in tumor-initiating cells but also in metastasis-initiating cells [31]. Similarly, high aldehyde dehydrogenase (i.e., ALDH) activity has been associated with both tumor-initiating and metastasis-initiating cells in human PCa [32]. A subpopulation of PSA−/lo cells characterized by a lack of prostate specific antigen (PSA) expression, a differentiation marker, is enriched in CSCs that can undergo asymmetric cell division [33]; and cells within the PSA−/lo subpopulation bearing the ALDHhiCD44+α2β1+ phenotype (or TM+ cells) have been shown to both initiate and propagate CRPC [34]. Whether PSA−/lo or TM+ PCa cells also possess enhanced metastatic capacities has not been prospectively addressed. It should be noted that stemness represents a “state” rather than a fixed phenotype defined by biomarkers. Thus, stemness can be lost or acquired during metastasis. Indeed, not all tumor-initiating cells (i.e., CSCs) posses the ability to metastasize. For example, PCa cells expressing high levels of DNA topoisomerase 2-alpha (TOP2A) are the cells that can initiate metastasis but the TOP2Aneg cell population is enriched in CSCs [35]. Interestingly, a population of PCa cells with enhanced metastatic potential, i.e., metastases-initiating cells (also called MICs), appears to have the special ability to recruit and reprogram non-tumorigenic cancer as well as non-cancerous epithelial and stromal cells to participate in the metastatic process [36, 37].
The relationship between metastatic PCa cells and EMT is more complicated. Despite overwhelming preclinical evidence, the true role of EMT in cancer progression and metastasis remains hotly debated as recent genetic studies suggest that EMT is not required for lung or pancreatic cancer metastasis but instead is associated with chemoresistance [38, 39]. PCa cells with an EMT phenotype were shown to display stem cell features characterized by increased expression of Sox2, Nanog, Oct4, Lin28B and/or Notch1 [40]. However, cancer cells in metastatic outgrowths are epithelial-like suggesting that mesenchymal cells must revert to the epithelial phenotype via the mesenchymal-to-epithelial transition (MET), in order to initiate colonization. Indeed, constitutive Snail1 activation, an EMT inducer, actually suppresses stemness in PCa cell lines [41]. These discussions highlight EMT and MET as a dynamic process in which cancer cells may reside at different stages of mesenchymal - epithelial continuum. A recent study in an autochthonous genetic murine model of PCa reported the isolation and characterization of mesenchymal-like and EMT tumor cells, the latter of which harbor both epithelial and mesenchymal characteristics [42]. Both populations displayed enhanced stemness and enriched tumor-initiating capacity. Nevertheless, only epithelial EMT tumor cells had the ability to form macrometastases [42], suggesting that mesenchymal and epithelial states in cancer cells contribute differentially to their capacities for tumor initiation versus metastatic seeding.
The relationship between CTCs, EMT-PCa cells, and PCSCs is also intricate. Cohesive-clusters of CTCs from primary prostate tumors are reported to employ a partial EMT, adopting some mesenchymal characteristics but maintaining an “intermediate phenotype” that is epithelial in nature, thereby allowing an epithelial cell–cell adhesion and a mesenchymal motility trait [43]. CTCs from patients with advanced PCa display both epithelial and mesenchymal markers [44]. The distinction between CTCs and PCSCs is blurry as well. In one study, 60% of the CTCs profiled from PCa samples express putative stem-like markers such as ALDH7A1 and KLF4 but whether such CSC marker-expressing CTCs truly possess enhanced CSC and metastasis properties remains unknown [45].
DTCs are known to be largely dormant and this dormancy may be induced by an unfavorable tumor microenvironment, epigenetic changes caused by the end organ microenvironment, inadequate nutrient supplies, or by the inherent nature of the DTCs [46]. PCa DTCs and PCSCs share many common biological traits such as dormancy, and the tumor microenvironment may play a major role in regulating stem cell functions in DTCs. For example, PCa DTCs may target the normal HSC niche for survival and compete with resident HSCs to establish their own niche [47]. DTCs expressing a PCSC phenotype were shown to represent a small fraction of total DTCs, but were able to better block HSC binding to osteoblasts, an interaction HSCs use to localize to the niche, thus displacing HSCs from their niche. Importantly, there was a time-dependent enrichment in the frequency of this PCSC population in bone marrow, further enhancing the ability to displace HSCs. Additionally, PCa cell binding to components of the HSC niche increases the dormant population of PCa and their ability to withstand multiple chemotherapeutic insults. ‘Dormancy enriched’ populations of human PCa cells bind osteoblasts in the HSC niche in the bone marrow, which induced the expression of TBK1 (TANK binding kinase 1), subsequently inhibiting mTOR signaling leading to dormancy, maintenance of PCSC attributes (CD133+/CD44+), and drug resistance [48]. Dormancy in DTCs and PCSCs partially explains why not all patients with DTCs develop overt clinical metastasis. It should be noted that metastatic ability may not necessarily be solely attributed to PCSC phenotypes, as a recent study demonstrates that a subpopulation of bone metastasis-initiating PCa cells is defined by mitotic quiescence, but not by ‘stemness’ [49]. This mitotic quiescence was associated with high levels of CXCR4 production, a molecule that plays a role in the homing of quiescent tumor cells to bone. Thus it appears that both DTCs and PCSCs can be cellular determinants of PCa metastasis, due to their common nature of dormancy.
Microenvironmental regulation of PCa metastasis
The microenvironment can influence cells in both primary tumors and in the end organs during the successful establishment of metastasis. Both types of tumor microenvironment consist of a network of stromal fibroblasts, fibroblast-like cells, smooth muscle cells, immune cells, blood vessels, mesenchymal stem cells (MSCs), fat cells, neural cells, and tumor cells that all communicate with each other via both direct cell-cell contact and soluble factors including chemokines, cytokines, and the ECM components. These reciprocal interactions create a pro-tumor and inflammatory-sustained microenvironment, which leads to altered tumor cell survival, proliferation and angiogenesis, resistance to therapy, evasion of immune surveillance and increased metastatic spread largely by virtue of epigenetic mechanisms. Data from a recent study clearly displayed evidence of epigenetic reprogramming wherein highly metastatic variants of human PCa cells were generated via serial cycles of LN metastasis in mice, ultimately producing a selected cell line that developed 100% metastasis in several metastatic sites [50]. Intriguingly, this metastatic selection shifted an initial heterogeneous population of PCa cells to be highly enriched in giant cells resistant to chemotherapy drugs and specific for bone metastasis.
In primary tumor microenvironment, the growth and invasive potential of carcinoma cells are strongly influenced by the local accumulation of connective tissue cells and extracellular material, a phenomenon called the ‘reactive tumor stroma’. The reactive stroma in PCa is composed of fibroblasts, myofibroblasts, endothelial cells and immune cells, but consists largely of activated cancer-associated fibroblasts (CAFs). CAFs support the survival of PCa cells by secreting soluble factors including inflammatory cytokines, angiogenic factors and ECM-proteins and their proteases, and growth factors, creating a niche that modulates the oncogenic potential of adjacent epithelia in PCa. Reciprocal activation of PCa cells and CAFs promotes tumorigenesis, EMT, cancer stemness, and bone metastasis of human PCa [51–53]. Human reactive stroma exhibits increased transforming growth factor-beta (TGF-β) signaling adjacent to pathologic lesions, and is enhanced by WNT signaling, which accelerates FGFR1-driven (fibroblast growth factor receptor 1) adenocarcinoma [54]. Additionally, MSCs can be recruited into PCa tumors, converted into CAFs via CXCR6 signaling, and secrete CXCL12, which binds to CXCR4 on tumor cells and induces EMT, promoting metastasis to secondary tumor sites [55]. Finally, CAFs can be a novel source of GDF15, a TGFβ/BMP family pro-metastatic growth factor, which produces prominent paracrine effects on PCa cell migration, invasion, and tumor growth in human PCa, including systemic in vivo effects on the outgrowth of distant and otherwise indolent PCa cells [56].
Despite their metastasis-abetting roles, the stromal cells in primary PCa surprisingly do not seem to sustain prevalent clonal somatic copy-number alterations [57]. The genomes of stromal cells associated with PCa were found to be generally stable and indistinguishable from the benign stroma and benign epithelial cells from the same patient [57], suggesting that molecular mechanisms other than chromosomal aberrations, most likely epigenetic, cause reactive stroma initiation and PCa progression. For example, IL-6 can contribute to the epigenetic silencing of TGF-β type II receptor (TGFBR2) in adjacent fibroblastic cells, and this loss of prostatic stromal TGF-β signaling was found to mediate the further epigenetic silencing of DNA damage repair and reactive oxygen-metabolism genes [58]. It is important to note that the exact function of CAFs in PCa progression may depend upon the stage, grade, hormonal and differentiation status of the primary tumor. This complexity is further augmented by the fact that the interaction between CAFs and PCa cells is mediated in part by androgen receptor (AR), which is heterogeneously expressed and whose role in tumor progression may be context-dependent [59].
The overall immune-microenvironment (i.e., the composition of immune cell types) in primary tumors may also play a significant part in PCa metastasis. Mutations in PCa epithelial cells can generate patient-specific neoantigens that can trigger immune recognition [60]. Additionally, remodeling of the tissue ECMs releases a range of soluble and insoluble factors, some being ‘danger signals’ alerting the immune system leading to the recruitment of infiltrating inflammatory cells [61]. Despite this, anti-tumor immune activation is inefficient in PCa, suggesting that regulatory networks within the reactive stroma generally inhibit immune cell functions locally. For example, TGF-β released by CAFs or PCa cells, can induce FOXP3 (forkhead box protein P3) expression in CD8+ T cells, which may convert CD8+ effector T cells into T regulatory cells (T-regs), thus creating an immunosuppressive tumor microenvironment [62]. Furthermore, CAFs in PCa can induce monocyte recruitment via SDF-1 (stromal cell-derived factor-1 or CXCL12) delivery, and promote monocyte trans-differentiation toward the M2 immunosuppressive macrophage phenotype, cells causally responsible for establishing an immunosuppressive pro-tumor microenvironment [63]. The reciprocal interplay between PCa cells, CAFs and these cancer-associated macrophages (CAMs), promotes cell motility, escape from the primary tumor, enhanced malignancy and metastatic spread. Infiltrating macrophages alone have been shown to induce prostate tumorigenesis via AR-inflammatory cytokine CCL4–STAT3 (C-C motif chemokine ligand 4)–(signal transducer and activator of transcription 3) activation, downregulation of p53/PTEN (phosphatase and tensin homolog) tumor suppressors, and promotion of EMT signaling pathways [64]. CAMs within CTC clusters also aide in PCa-cell invasion and migration through modification of the adhesion function of laminin binding integrins that interact with laminin 511, and recent data suggests a significant role for CAMs in the development of docetaxel resistance in CRPC [65].
Precise quantification and characterization of the immune infiltrates in PCa is crucial to improve treatment efficacy; however, the composition of this immune infiltrate in PCa varies widely with disease stage and patient age. Additionally, infiltrating lymphocytes are poorly characterized due in part to the immunosuppressive microenvironment. It has been documented that clusters of CD25+ and FOXP3+ T-regs, and quiescent CD3+ lymphocytes, surround PCa islets [66]. Cells expressing programmed cell death 1 (PD-1) and its ligand (PD-L1), whose binding results in reduced T-cell activation, were also detected in these clusters. PCa patients with higher numbers of CD4+ T-regs in the PCa environment have an increased risk of mortality, supporting the role of this immunosuppressed microenvironment in PCa [67]. High fractions of T-regs and blood monocytic myeloid-derived suppressor cells (MDSCs) in peripheral blood have been related to poor prognosis [68]. In a recent study, mCRPC-associated MDSCs potently inhibited autologous CD8+ T cell proliferation and CD8+ T cell production of IFNγ and granzyme-B [69]. Circulating MDSCs also displayed high levels of activated STAT3, which induced expression and enzymatic activity of Arginase-1, a potent T-cell inhibitor, conferring further MDSC-mediated immunosuppression.
High expression levels of natural killer (NK) cell activating receptors (NKp46, NKG2D, DNAM-1 and NKp30) and high cytotoxicity, are associated with good prognosis in patients with mPCa, suggesting that NK cells play an important role in reducing PCa progression and improving response to therapy [70]. However, recent detection of tissue tolerance mechanisms against NK cells further implicates the presence of a strong immunosuppressive environment in PCa. For instance, NK cells infiltrating the prostate gland display an immature phenotype with low or no cytotoxic potential, and PCa cells may also induce the expression of NK inhibitory receptors and downregulate the expression of NK activating receptors [71]. The function of mast cells (MCs) in mPCa is clearly determined by the microenvironment. For example, intratumoral MCs negatively regulate angiogenesis and tumor growth, while peritumoral MCs express high levels of the angiogenic factor FGF-2, which may contribute to the expansion of prostate tumors [72]. In ADT treated cells, infiltrating MCs have been shown to enhance PCa invasion via altering LncRNA-HOTAIR/PRC2-AR-MMP9 signals and increasing PCSC populations [73]. Higher B-cell infiltration is present PCa tissue, which correlates with higher grades and higher risk of recurrence [74]. CXCL13 induced B-cell recruitment, and Lymphotoxin released from B-cells was shown to promote CRPC via activation of IKKα and STAT3 in androgen-deprived PCa cells, thereby promoting survival and proliferation [75]. Also, activation of the IKKα-E2F1-BMI1 cascade by infiltrating B-cells occurs in PCSCs and controls tumor recurrence [76]. Finally, B-cells can mediate chemotherapy resistance through the production of cytokine IL-10 and expression of the inhibitory ligand PD-L1 [77]. Again, it is important to note that the precise function of each immune cell type is context dependent. Overall, CAMs, neutrophils and MCs mostly exhibit pro-tumor qualities, whereas NKs exhibit anti-tumor effects.
Once prospective metastatic PCa cells leave the primary tumor and extravasate into the end organs, most of the cells would remain dormant, which can last for years to decades. This dormancy obviously entails PCa cells in a foreign microenvironment to first develop the ability to escape the immune surveillance, a process that we generally know little of at this moment. As PCa cells have a proclivity to metastasize to the bone, we shall focus our subsequent discussions mainly on how the bone marrow (BM) microenvironment may be conducive in promoting PCa bony metastasis. The BM microenvironment contains a highly heterogeneous population of cells, including HSCs, MSCs, vascular endothelial cells, fibroblasts, adipocytes and nerve fibers, all of which have the potential to influence prostate tumor growth in the marrow. In PCa bone metastases, expression of TGFB2R is lost in CAFs, causing loss of TGF-β responsiveness and an alteration in chemokine levels, thus facilitating development of mixed osteoblastic/osteolytic PCa bone lesions [78]. MSCs can play a pro-oncogenic role in bone metastasis by producing TGF-β1, which subsequently leads to MMP-2 and MMP-3 upregulation in PCa cells. CCL5 produced by infiltrating MSC cells altered HIF2α signaling within PCa cells, which might play a key role in increasing PCSCs and PCa metastasis via altering AR signals [79]. MSC tropism can also be regulated by TGF-β1 [80]. TGF-β1 secreted by both PCa cells and tumor stroma cells, attracts MSCs and can induce their transdifferentiation into CAFs, which can increase tumor invasiveness, perform vascular mimicry, and recruit monocytes.
Bone metastases from PCa are associated with increased osteoblast and osteoclast activity. Osteoclasts are thought to play a role in HSC mobilization, and the activation of osteoclastic activity by tumor cells can release growth factors and cytokines sequestered in bone matrix, which contributes to the growth of PCa in the bone, a process termed “the vicious cycle” [47]. Osteoclasts play a significant role in driving tumor growth once DTCs become established in bone, but are not critical for the earliest seeding of DTCs into the HSC niche [81]. Osteoblastic niche-derived GAS6 and the Mer/mTOR pathway can convert PCa DTCs to PCSCs in the HSC niche, highlighting the significant role of these cells and the HSC niche in promoting PCa metastasis [82]. It is becoming increasing clear that adipocytes also have a definite and significant role in PCa progression. For example, infiltrating pre-adipocytes in the prostate tumor microenvironment can be recruited to PCa and increase PCa metastasis via modulation of the miR-301a/AR/TGF-β1/Smad/MMP9 signals [83]. Adipocytes located in the periprostatic adipose tissue were also recently shown to direct the migration of PCa cells, a process dependent on the stimulation of CCR3 expressing cells by adipocyte-secreted-CCL7 [84]. In human PCa, expression of the CCR3 receptor was found to be associated with the occurrence of aggressive disease, implicating adipocytes in PCa aggressiveness. Additionally, increased marrow adiposity due to high fat diet was found to accelerate growth and progression of prostate tumors in bone. Bone marrow adipocytes were found to be a significant source of CXCL1 and CXCL2, which drives osteoclastogenesis and prostate tumor-driven osteolysis of the bone in mPCa [85]. Bone marrow adipocytes also play a role in metabolic reprogramming by interacting with mPCa cells in a paracrine signaling fashion to induce increased expression of glycolytic enzymes, via the oxygen-independent mechanism of HIF-1α activation [86].
Finally, formation of autonomic nerve fibers in the tumor microenvironment may also regulate PCa development and dissemination [87]. In the initial phases of cancer development, adrenergic fibers from the SNS (sympathetic nervous system), acting through stromal β2- and β3-adrenergic receptors, promote tumor cell survival. Cholinergic fibers of the PNS (peripheral nervous system) subsequently play predominant roles in tumor cell invasion, migration, and distant metastases via the involvement of nerve ending–derived acetylcholine–activated cholinergic muscarinic receptor 1 (CHRM1) in mesenchymal cells in the tumor microenvironment. Intriguingly, expression of CHRM3 in tumor cells themselves may increase PCa cell aggressiveness and invasion via autocrine response to acetylcholine and perhaps other neurotransmitters [88].
Potential therapeutic intervention of PCa metastasis
Death by PCa in most patients is accompanied by metastasis. To date, there is no effective therapy available for the treatment of CRPC and patients generally die within 2 to 4 years. The substantial degree of PCa heterogeneity and the existence of divergent cancer clones within a primary tumor may explain development of rapid treatment resistance and hinder therapeutic targeting [89, 90]. Heterogeneity in CRPC is observed clinically, ranging from patients who are asymptomatic and do not have metastases to those with substantial symptoms and both bony and visceral metastases. While several molecular tests have been introduced commercially, currently none are used to assist clinicians in establishing efficacious treatment options. In the future, molecular stratification, which will rely on the identification of key molecular drivers and mechanisms underlying PCa metastasis, will guide therapeutic options for maximum efficacy. Based on our current knowledge, the most efficacious therapy should target cellular, molecular and epigenetic determinants, as well as microenvironmental regulators, with a preferential focus on those linked to ‘stemness’.
Currently, patients diagnosed with organ-confined PCa of low Gleason grade follow an indolent clinical course and can be managed by ‘active surveillance’, which involves simply monitoring the tumor over time, preventing unnecessary therapy and their serious side effects [91]. For metastatic disease, current treatment options consist of AR-directed therapies (abiraterone acetate, enzalutamide), taxane tubulin-binding chemotherapies (docetaxel, cabazitaxel), immunotherapies (sipuleucel-T), and bone-targeting radiopharmaceuticals (radium-223). PCa is still one of the few solid tumors treated with a mono-treatment approach. With the ease of prescribing oral agents with less toxicity, over administering agents intravenously with serious side effects, chemotherapy has remained secondary to second-generation androgen receptor signaling inhibitors [92]. But at best, the ‘improvement in survival’ for any one of these treatment options only adds a few months to patients’ median survival time.
With recent advances in characterizing PCa cellular and molecular heterogeneity, and in the understanding of the mechanisms of PCa adaptation to ADT, it is becoming clear that therapeutic efforts should focus on combination regimens, ultimately leading to precision medicine. As PCa heterogeneity increases throughout PCa progression, these combinatorial approaches may be more efficacious if used in the early stages of PCa [93]. A greater understanding of PCa heterogeneity provided the rationale for two large randomized clinical trials assessing the addition of chemotherapy to ADT. The CHAARTED randomized phase III trial revealed the remarkable efficacy of docetaxel combined with ADT, versus ADT alone, in patients with newly diagnosed mPCa [94]. Additionally, early administration of docetaxel to patients with mPCa was shown to significantly benefit the outcomes of those patients. The STAMPEDE study also demonstrated an unprecedented survival benefit in favor of the combination [95]. In another recent trial, abiraterone was shown to have direct bone antiresorptive and anabolic activity [96], and a randomized phase 3 trial is currently examining the survival benefit of abiraterone and ADT versus ADT plus a placebo in men with newly diagnosed mCRPC (NCT01715285). In comparison with bicalutamide, enzalutamide has proven to be a more potent AR blocker with no AR agonist activity in mPCa [97, 98]. Based on these results, a current randomized phase 3 trial in patients with mCRPC seeks to compare ADT plus enzalutamide versus ADT plus a conventional anti-androgen (NCT02446405). The need for improving AR-directing tumor growth inhibition has also led to current trials investigating combinations of hormonal agents. The rationale for these trials is based on preclinical observations that abiraterone treatment can activate AR in CRPC via intratumoral de novo steroid synthesis [99]. Finally, a new generation of AR pathway-targeted agents, including ODM-201, a novel AR inhibitor that displays high-affinity binding and inhibition of AR nuclear translocation, and galeterone, a multi-targeted agent that combines CYP19 inhibition and AR antagonism with an ability to increase AR protein degradation, are being tested in clinical trials in patients with mPCa [100, 101].
The lack of a survival benefit from ADT as a monotherapy could be due to androgen-independent PCSCs already present at early stages of the disease. While ADT destroys the bulk of AR+ cells, continued survival of PCSCs promotes CRPC. Thus, it is essential to develop combinatorial therapeutics, which include agents specifically targeting these cells, a challenging feat as many PCSCs characteristics preclude current targeting methods. Small-cell neuroendocrine differentiation in prostatic carcinoma is an increasingly common resistance mechanism to potent ADT as AR−/lo cells demonstrate stem cell like characteristics. A recent study showed that these cells and high-grade/NE prostate tumors are characterized by elevated FOXC2 (TF Forkhead Box Protein C2) [102]. Targeting FOXC2 using a p38 inhibitor restored epithelial attributes and ADT-sensitivity, and reduced the shedding of CTCs with significant shrinkage in the tumor mass, thus specifying a mechanism to target PCSCs.
The recent progress in next-generation sequencing has provided a more accurate genomic landscape of PCa suggesting that future mPCa treatments will likely be molecularly driven. For example, one study demonstrated that the majority of mCRPCs analyzed contained potentially clinically actionable mutations that could be targeted by available drugs [103]. Importantly, approximately 20% of patients had somatic mutations in DNA repair genes (DRGs), including BRCA1, BRCA2, ATM, FANCA, RAD51B, and RAD51C [103]. The rate of DRG germline aberrations in mCRPC was recently shown to be 11.8% [104]. Thus, inherited and acquired defects in DNA damage repair are key mechanisms in the genesis of malignant tumors, and detection of germline mutations in DRGs in men with PCa has important clinical implications. Patients with mPCa and DRG mutations have been shown to respond to poly(ADP-ribose) polymerase (PARP) inhibitors (PARP facilitates DNA damage repair) [105]. These tumors also appear to be responsive to platinum-based chemotherapy [106]. A recent phase II study shows that the PARP inhibitor olaparib has significant anticancer activity in patients with mCRPC harboring aberrations in DRG [107]. Also, a DNA damage and repair pathway signature could be used as a prognostic signature biomarker of long-term outcomes of metastasis-free and OS in high-risk PCa, further supporting the important role of this altered pathway in mPCa [108].
Deaths from PCa are often due to bone disease and its complications, and development of therapeutics for mCRPC reflects a growing appreciation for the crucial role of cross-talk between tumor cells and bone microenvironment in mPCa (reviewed in [109]). Targeting the bone microenvironment in metastatic disease, has generally aimed at abnormal osteoclast activity. The first agent approved for the management of bone metastases in CRPC patients was zoledronic acid (ZA), which reduced skeletal-related events in PCa patients with bone metastases, but was ineffective for the prevention of bone metastases in high-risk nonmetastatic PCa patients. Denosumab, a fully human monoclonal antibody against the receptor activator nuclear kappa B ligand (RANKL) pathway which inhibits osteoclast formation, function and survival, was better than ZA for prevention of skeletal-related events in men with bone metastases from CRPC. Unfortunately, the administration of these potent osteolysis inhibitors reduce the burden of bone metastatic disease and relieve pain, but this benefit does not translate in an improvement in survival. Radium-223, an α-emitter that leads to irradiation of osteoblastic metastases in a highly localized effect, not only showed efficacy in preventing symptomatic skeletal events, but was the first bone-targeted therapy associated with a significant OS improvement when used in men with CRPC and bone metastases. Trials testing the concomitant administration of radium-223 with ADT (NCT02582749), abiraterone (NCT02043678), enzalutamide (NCT02194842) and docetaxel (NCT01106352) are ongoing.
Studies suggest that stromal targeting agents are only modestly effective when used as monotherapy in patients with mCRPC. Likewise, blocking angiogenesis with non-hormonal agents such as tyrosine kinase inhibitors to inhibit PCa bone metastasis has produced dismal results. As the BM microenvironment contains multiple proangiogenic factors and soluble cytokines, it may be necessary to block multiple pathways simultaneously and in combination with chemotherapy in patients with mCRPC. Cabozantinib, an oral inhibitor of multiple receptor tyrosine kinases, failed to improve survival in men with mCRPC [110], but the combination of cabozantinib and ADT is currently being tested in a phase 2 study (NCT01630590) in men with mCRPC. In a pre-clinical study, one group successfully created a gene therapy approach that co-targets cancer and its stromal cells in CRPCs [111]. The authors designed a novel human osteonectin promoter (hON-522E) based on the observation that human osteonectin is overexpressed in PCa epithelium and in tumor stroma. The hON-522E promoter was highly active in AR− mPCa cells and bone stromal cells, but not AR+ PCa cells, and thus co-targeted PCa epithelium and bone stroma, effectively inhibiting androgen-independent PCa growth [111]. Recent development and use of 3D in vitro models of PDXs have been shown to recapitulate important pathological properties of PCa bone metastasis and can be used for systematic interrogation of complex in vivo tumor-stromal interactions [112]. Advances such as these will facilitate the development of efficacious bone-targeting therapeutics.
Development of therapeutic resistance is inevitable in mPCa. Approximately half of the men diagnosed with mCRPC benefit from treatment with docetaxel but all develop docetaxel-resistant disease. The prevailing approach to addressing secondary drug resistance in cancer focuses on treating the resistance mechanisms at relapse. Acquired docetaxel resistance in PCa was linked to a subpopulation of cells in PCa tissues that lacked differentiation markers and HLA class I antigens, which were correlated to tumor aggressiveness, poor patient prognosis, and displayed potent tumor-initiating capacity [113]. Importantly, suppression of the Notch and Hedgehog signaling pathways in these tumor-initiating cells, suppressed acquired docetaxel resistance in PCa [113]. Expression of AR splice variant 7 (AR-V7) in CTCs from men with mCRPC was associated with primary resistance to enzalutamide and abiraterone therapy [114]. An option for men with AR-V7 is the drug galeterone (TOK-001), which degrades the AR itself [115]. Interestingly, AR blockade via enzalutamide paradoxically enhanced metastasis, mediated by the chemoattractant CCL2 [116], and a TNFα-CCL2 paracrine loop was induced in response to ADT leading to a metastatic phenotype, and might account for some forms of PCa therapy resistance [117]. AR gene aberrations in circulating cell-free DNA in patients’ blood are associated with resistance to enzalutamide and abiraterone in mCRPC, illustrating the possibility for interrogating additional mechanisms [118]. Recent studies have shown that small-molecular inhibitors of the PI3K, Akt, and mTOR pathway currently in the clinic, produce reactivation of the pathway they are intended to suppress. PI3K antagonists have been shown to drive mitochondrial metabolic reprogramming resulting in tumor adaptation, drug resistance, and acquisition of metastatic competency in PCa [119].
The observation of EMT plasticity in cancer presents a challenge for anti-EMT therapeutics. Deciphering the full spectrum of intermediate EMT states and how PCa cells make these transitions will help define an EMT therapeutic window. A more advantageous option would be impeding EMT plasticity by targeting master EMT regulators. EMT plasticity is regulated epigenetically through the activity of the chromatin remodeling protein HMGA2, which is highly upregulated in tumors from men with mCRPC, suggesting the use of HDAC inhibitors [120]. This is important because EMT also confers resistance to mPCa therapeutics. EMT mediates docetaxel resistance and high risk of relapse in PCa [121]. There is also evidence that androgen deprivation induces EMT in PCa via a ZEB1-AR feedback loop, revealing a potentially important consequence of a standard-of-care for PCa [122]. Hypoxic stress caused by ADT can also promote EMT and drive selection of more malignant PCa tumors [123]. AR splice variants can contribute to PCa aggressiveness through induction of EMT and expression of stem cell marker genes [124]. Therefore, targeting regulators of these transitions may increase the therapeutic efficacy of ADT. Twist1/AR signaling is also augmented in castration resistant as well as mesenchymal-type PCa, indicating the molecular mechanism of mutual and functional crosstalk between EMT and castration resistance [125]. The EMT master regulator, Snail, promotes enzalutamide resistance and mPCa, as a consequence of increased full-length AR and AR-V7 expression and nuclear localization, in preclinical models of PCa and in patients [126]. On the other hand, ligand-activated AR regulates expression of several key EMT factors whereas antiandrogens counteract AR activity only on selected EMT genes [127].
Resistance to targeted therapy in patients with bone metastasis invariably develops likely due to the unique tumor microenvironment consisting of new bone formation [128]. A recent study indicates that PCa-induced bone provides a ‘resistance niche’ that allows PCa cells to survive under cabozantinib treatment and perhaps other targeted therapies [129]. Following cabozantinib treatment, only tumor cells positioned adjacent to the newly formed woven bone remained viable and expressed high levels of pFAK-Y397 and pTalin-S425, mediators of integrin signaling. By combining cabozantinib with FAK inhibitors that target integrins, authors observed delayed tumor recurrence in contrast to cabozantinib treatment alone [129]. This data suggests that identifying paracrine de novo resistance mechanisms may significantly contribute to the generation of a broader set of potent therapeutic tools that act combinatorially to inhibit mPCa.
Although very little is known about the immune cell profile in PCa metastases, there are noticeable correlations between PCa prognosis and immune infiltrate, pointing to the rationale for using immunotherapy for treatment of PCa. Unfortunately, the lack of more powerful clinical benefits of therapeutic vaccination may be accredited to the immune suppressive tumor microenvironment in mPCa. Although preclinical studies supported the antitumor activity of ipilimumab, a humanized monoclonal antibody that binds to cytotoxic T-lymphocyte antigen 4 (CTLA4), the subsequent phase 1/2 studies of ipilimumab in mCRPC yielded quite disappointing results [130, 131]. However, ipilimumab is currently being investigated in combination with a luteinizing hormone-release hormone agonist before radical prostatectomy for men with high-risk PCa in a phase 2 trial [93]. Despite these trials, immunotherapy for mCRPC remains a ‘premature’ approach. The lack of clinical benefits of immunotherapy may also be due to resistance mechanisms. A recent study shows that the PD-1/PD-L1 axis, an effective checkpoint blockade in cancer, increases PCa cell resistance to doxorubicin and docetaxel [132]. These findings indicate that combinations of chemotherapy and immune checkpoint blockade may limit chemoresistance and progression to metastatic PCa disease.
Despite the highly dynamic process of metastasis and the emergence of resistant populations, the prevailing approach to addressing drug resistance in cancer focuses on treating resistance mechanisms at relapse. The application of mathematical analysis of the evolutionary dynamics of tumor populations including tumor microenvironmental influences and adaptations to therapeutics in PCa cells, may provide an more optimal treatment strategy for mPCa. For example, ‘adaptive therapy’ is an alternative approach in PCa to combat tumor cells that have evolutionarily adapted to changing tumor environmental conditions and treated by therapeutics. This strategy consists of modifying a continuous treatment in order to maintain a fixed population of chemosensitive cells that suppress the growth of chemoresistant cells [133]. Indeed, recent work highlights an approach for combining extensive drug resistance selection experiments with pharmacological profiles to identify novel vulnerabilities during the course of tumor clonal evolution [134].
Conclusions & perspectives
Despite advances in detection and therapy, many facets of mPCa remain unknown. The inability to discern between indolent cancer and aggressive PCa tumors leads to many inefficient therapeutic regimens, and unnecessary, significant side effects in men. Advanced PCa is currently considered incurable. Chemical castration can prolong life by several years but almost all tumors become resistant. Drugs designed to treat castration-resistant tumors are also facing a resistance problem, and their efficacy is eventually lost in all men. This acquired resistance to anti-cancer drugs constitutes important barriers to the successful management of cancer. The importance of the tumor microenvironment in promoting PCa metastasis has been validated. However, the heterogeneity of both stroma cells and tumor cells at different stages of progression vary widely, complicating therapeutics directly targeting them. The ability of PCa cells to escape the immune system is also a major hindrance. In an ideal situation, the key to an effective treatment for mPCa would target a particular mutation or variation in a tumor. In reality, the enormous cellular and molecular mechanisms currently defined in mPCa make this scenario unrealistic. These observations suggest that preventing metastasis might be easier than attempting to treat it.
The establishment of whole-exome sequencing precision medicine trials of metastatic tumors requiring prospective follow-up of patients can aide in understanding molecular mechanisms of response and resistance to anticancer therapies, improving decision making, and identifying novel biomarkers [135]. Additionally, continuous updates based on emerging treatment and disease phenotype data from CRPC clinical trails will move drug development closer to unmet needs in clinical practice [136]. Our continued efforts in the development of more advanced models, the discovery of novel cellular and molecular determinants of metastasis and drug-resistance, paying close attention to ‘stemness’, and a focus on combinatorial therapeutics, all within a collaborative scientific environment, are fundamental to resolving these issues in the future.
Supplementary Material
Acknowledgments
Work in the authors’ lab was supported, in part, by grants from the NIH (NCI R01-CA155693), DOD (W81XWH-13-1-0352, W81XWH-14-1-0575, and W81XWH-16-1-0575), the Roswell Park Cancer Institute, and the Chinese Ministry of Science and Technology (MOST) Grant (2016YFA0101203).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None.
References
- 1.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 2.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rycaj K., TD . Metastasis and metastatic cells: a historical perspective and current analysis. In: Huiping L LJ, editor. Cancer Stem Cells: Targeting the Roots of Cancer, Seeds of Metastasis, and Sources of Therapy Resistance. Academic Press/Elsevier; 2016. pp. 317–40. [Google Scholar]
- 5.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–90. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351–6. doi: 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–91. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–22. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halabi S, Kelly WK, Ma H, Zhou H, Solomon NC, Fizazi K, et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men With Castration-Resistant Prostate Cancer. J Clin Oncol. 2016;34:1652–9. doi: 10.1200/JCO.2015.65.7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyes DK, Pienta KJ. The biology and treatment of oligometastatic cancer. Oncotarget. 2015;6:8491–524. doi: 10.18632/oncotarget.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyd LK, Mao X, Xue L, Lin D, Chaplin T, Kudahetti SC, et al. High-resolution genome-wide copy-number analysis suggests a monoclonal origin of multifocal prostate cancer. Genes Chromosomes Cancer. 2012;51:579–89. doi: 10.1002/gcc.21944. [DOI] [PubMed] [Google Scholar]
- 12.Shen MM. Cancer: The complex seeds of metastasis. Nature. 2015;520:298–9. doi: 10.1038/nature14377. [DOI] [PubMed] [Google Scholar]
- 13.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–26. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, A’Hern R, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–8. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- 17.Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2016.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang R, Lu YT, Ho H, Li B, Chen JF, Lin M, et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015;6:44781–93. doi: 10.18632/oncotarget.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuhn P, Carlsson A, Luttgen MS, Dizon KK, Troncoso P, Corn P, et al. Paired high-content analysis of prostate cancer cells in bone marrow and blood characterizes increased androgen receptor expression in tumor cell clusters. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mintz PJ, Rietz AC, Cardo-Vila M, Ozawa MG, Dondossola E, Do KA, et al. Discovery and horizontal follow-up of an autoantibody signature in human prostate cancer. Proc Natl Acad Sci U S A. 2015;112:2515–20. doi: 10.1073/pnas.1500097112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao S, Geybels MS, Leonardson A, Rubicz R, Kolb S, Yan Q, et al. Epigenome-wide Tumor DNA Methylation Profiling Identifies Novel Prognostic Biomarkers of Metastatic-lethal Progression in Men with Clinically Localized Prostate Cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho H, Herzka T, Zheng W, Qi J, Wilkinson JE, Bradner JE, et al. RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov. 2014;4:318–33. doi: 10.1158/2159-8290.CD-13-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seedhouse SJ, Affronti HC, Karasik E, Gillard BM, Azabdaftari G, Smiraglia DJ, et al. Metastatic phenotype in CWR22 prostate cancer xenograft following castration. Prostate. 2016;76:359–68. doi: 10.1002/pros.23127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Wang JX, Xue H, Lin D, Dong X, Gout PW, et al. Subrenal capsule grafting technology in human cancer modeling and translational cancer research. Differentiation. 2016;91:15–9. doi: 10.1016/j.diff.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Kohli M, Wang L, Xie F, Sicotte H, Yin P, Dehm SM, et al. Mutational Landscapes of Sequential Prostate Metastases and Matched Patient Derived Xenografts during Enzalutamide Therapy. PLoS One. 2015;10:e0145176. doi: 10.1371/journal.pone.0145176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–7. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Wang X, Hoffman RM, Seki N. Real Time Metastatic Route Tracking of Orthotopic PC-3-GFP Human Prostate Cancer Using Intravital Imaging. J Cell Biochem. 2016;117:1027–32. doi: 10.1002/jcb.25391. [DOI] [PubMed] [Google Scholar]
- 28.Morton JJ, Bird G, Refaeli Y, Jimeno A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Res. 2016;76:6153–8. doi: 10.1158/0008-5472.CAN-16-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanay A, Regev A. Scaling single-cell genomics from phenomenology to mechanism. Nature. 2017;541:331–8. doi: 10.1038/nature21350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Tang DG. Prostate cancer stem cells and their potential roles in metastasis. J Surg Oncol. 2011;103:558–62. doi: 10.1002/jso.21806. [DOI] [PubMed] [Google Scholar]
- 31.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 32.van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010;70:5163–73. doi: 10.1158/0008-5472.CAN-09-3806. [DOI] [PubMed] [Google Scholar]
- 33.Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, et al. The PSA(−/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–69. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K, et al. Defining a Population of Stem-like Human Prostate Cancer Cells That Can Generate and Propagate Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016;22:4505–16. doi: 10.1158/1078-0432.CCR-15-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Liu Y, Chen W, Fang Y, Xu H, Zhu HH, et al. TOP2Ahigh is the phenotype of recurrence and metastasis whereas TOP2Aneg cells represent cancer stem cells in prostate cancer. Oncotarget. 2014;5:9498–513. doi: 10.18632/oncotarget.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu GC, Chung LW. RANK-mediated signaling network and cancer metastasis. Cancer Metastasis Rev. 2014;33:497–509. doi: 10.1007/s10555-013-9488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chu GC, Zhau HE, Wang R, Rogatko A, Feng X, Zayzafoon M, et al. RANK- and c-Met-mediated signal network promotes prostate cancer metastatic colonization. Endocr Relat Cancer. 2014;21:311–26. doi: 10.1530/ERC-13-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–6. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–30. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Celia-Terrassa T, Meca-Cortes O, Mateo F, Martinez de Paz A, Rubio N, Arnal-Estape A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122:1849–68. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015;75:2749–59. doi: 10.1158/0008-5472.CAN-14-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alix-Panabieres C, Pantel K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016;6:479–91. doi: 10.1158/2159-8290.CD-15-1483. [DOI] [PubMed] [Google Scholar]
- 44.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyamoto DT, Sequist LV, Lee RJ. Circulating tumour cells-monitoring treatment response in prostate cancer. Nat Rev Clin Oncol. 2014;11:401–12. doi: 10.1038/nrclinonc.2014.82. [DOI] [PubMed] [Google Scholar]
- 46.Ruppender NS, Morrissey C, Lange PH, Vessella RL. Dormancy in solid tumors: implications for prostate cancer. Cancer Metastasis Rev. 2013;32:501–9. doi: 10.1007/s10555-013-9422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121:1298–312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013;15:1064–74. doi: 10.1593/neo.13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang N, Docherty F, Brown HK, Reeves K, Fowles A, Lawson M, et al. Mitotic quiescence, but not unique “stemness,” marks the phenotype of bone metastasis-initiating cells in prostate cancer. FASEB J. 2015;29:3141–50. doi: 10.1096/fj.14-266379. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Wu C, Hoffman RM. Prostate Cancer Heterogeneous High-Metastatic Multi-Organ-Colonizing Chemo-Resistant Variants Selected by Serial Metastatic Passage in Nude Mice Are Highly Enriched for Multinucleate Giant Cells. PLoS One. 2015;10:e0140721. doi: 10.1371/journal.pone.0140721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 52.Gururajan M, Josson S, Chu GC, Lu CL, Lu YT, Haga CL, et al. miR-154* and miR-379 in the DLK1-DIO3 microRNA mega-cluster regulate epithelial to mesenchymal transition and bone metastasis of prostate cancer. Clin Cancer Res. 2014;20:6559–69. doi: 10.1158/1078-0432.CCR-14-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Josson S, Gururajan M, Sung SY, Hu P, Shao C, Zhau HE, et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene. 2015;34:2690–9. doi: 10.1038/onc.2014.212. [DOI] [PubMed] [Google Scholar]
- 54.Carstens JL, Shahi P, Van Tsang S, Smith B, Creighton CJ, Zhang Y, et al. FGFR1-WNT-TGF-beta signaling in prostate cancer mouse models recapitulates human reactive stroma. Cancer Res. 2014;74:609–20. doi: 10.1158/0008-5472.CAN-13-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruzzese F, Hagglof C, Leone A, Sjoberg E, Roca MS, Kiflemariam S, et al. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res. 2014;74:3408–17. doi: 10.1158/0008-5472.CAN-13-2259. [DOI] [PubMed] [Google Scholar]
- 57.Bianchi-Frias D, Basom R, Delrow JJ, Coleman IM, Dakhova O, Qu X, et al. Cells Comprising the Prostate Cancer Microenvironment Lack Recurrent Clonal Somatic Genomic Aberrations. Mol Cancer Res. 2016;14:374–84. doi: 10.1158/1541-7786.MCR-15-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banerjee J, Mishra R, Li X, Jackson RS, 2nd, Sharma A, Bhowmick NA. A reciprocal role of prostate cancer on stromal DNA damage. Oncogene. 2014;33:4924–31. doi: 10.1038/onc.2013.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol. 2014;11:365–76. doi: 10.1038/nrclinonc.2014.72. [DOI] [PubMed] [Google Scholar]
- 60.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 61.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 62.Shafer-Weaver KA, Anderson MJ, Stagliano K, Malyguine A, Greenberg NM, Hurwitz AA. Cutting Edge: Tumor-specific CD8+ T cells infiltrating prostatic tumors are induced to become suppressor cells. J Immunol. 2009;183:4848–52. doi: 10.4049/jimmunol.0900848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423–31. doi: 10.1038/onc.2013.191. [DOI] [PubMed] [Google Scholar]
- 64.Fang LY, Izumi K, Lai KP, Liang L, Li L, Miyamoto H, et al. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013;73:5633–46. doi: 10.1158/0008-5472.CAN-12-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahon KL, Lin HM, Castillo L, Lee BY, Lee-Ng M, Chatfield MD, et al. Cytokine profiling of docetaxel-resistant castration-resistant prostate cancer. Br J Cancer. 2015;112:1340–8. doi: 10.1038/bjc.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ebelt K, Babaryka G, Frankenberger B, Stief CG, Eisenmenger W, Kirchner T, et al. Prostate cancer lesions are surrounded by FOXP3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur J Cancer. 2009;45:1664–72. doi: 10.1016/j.ejca.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 67.Davidsson S, Ohlson AL, Andersson SO, Fall K, Meisner A, Fiorentino M, et al. CD4 helper T cells, CD8 cytotoxic T cells, and FOXP3(+) regulatory T cells with respect to lethal prostate cancer. Mod Pathol. 2013;26:448–55. doi: 10.1038/modpathol.2012.164. [DOI] [PubMed] [Google Scholar]
- 68.Idorn M, Kollgaard T, Kongsted P, Sengelov L, Thor Straten P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol Immunother. 2014;63:1177–87. doi: 10.1007/s00262-014-1591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hossain DM, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin Cancer Res. 2015;21:3771–82. doi: 10.1158/1078-0432.CCR-14-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pasero C, Gravis G, Granjeaud S, Guerin M, Thomassin-Piana J, Rocchi P, et al. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget. 2015;6:14360–73. doi: 10.18632/oncotarget.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P, et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016;76:2153–65. doi: 10.1158/0008-5472.CAN-15-1965. [DOI] [PubMed] [Google Scholar]
- 72.Johansson A, Rudolfsson S, Hammarsten P, Halin S, Pietras K, Jones J, et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am J Pathol. 2010;177:1031–41. doi: 10.2353/ajpath.2010.100070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li L, Dang Q, Xie H, Yang Z, He D, Liang L, et al. Infiltrating mast cells enhance prostate cancer invasion via altering LncRNA-HOTAIR/PRC2-androgen receptor (AR)-MMP9 signals and increased stem/progenitor cell population. Oncotarget. 2015;6:14179–90. doi: 10.18632/oncotarget.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woo JR, Liss MA, Muldong MT, Palazzi K, Strasner A, Ammirante M, et al. Tumor infiltrating B-cells are increased in prostate cancer tissue. J Transl Med. 2014;12:30. doi: 10.1186/1479-5876-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–5. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ammirante M, Kuraishy AI, Shalapour S, Strasner A, Ramirez-Sanchez C, Zhang W, et al. An IKKalpha-E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes Dev. 2013;27:1435–40. doi: 10.1101/gad.220202.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature. 2015;521:94–8. doi: 10.1038/nature14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li X, Sterling JA, Fan KH, Vessella RL, Shyr Y, Hayward SW, et al. Loss of TGF-beta responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol Cancer Res. 2012;10:494–503. doi: 10.1158/1541-7786.MCR-11-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luo J, Lee SO, Cui Y, Yang R, Li L, Chang C. Infiltrating bone marrow mesenchymal stem cells (BM-MSCs) increase prostate cancer cell invasion via altering the CCL5/HIF2alpha/androgen receptor signals. Oncotarget. 2015;6:27555–65. doi: 10.18632/oncotarget.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V, et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-beta1. Stem Cells. 2016;34:2536–47. doi: 10.1002/stem.2412. [DOI] [PubMed] [Google Scholar]
- 81.Zalucha JL, Jung Y, Joseph J, Wang J, Berry JE, Shiozawa Y, et al. The Role of Osteoclasts in Early Dissemination of Prostate Cancer Tumor Cells. J Cancer Stem Cell Res. 2015;3 doi: 10.14343/jcscr.2015.3e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shiozawa Y, Berry JE, Eber MR, Jung Y, Yumoto K, Cackowski FC, et al. The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.9251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xie H, Li L, Zhu G, Dang Q, Ma Z, He D, et al. Infiltrated pre-adipocytes increase prostate cancer metastasis via modulation of the miR-301a/androgen receptor (AR)/TGF-beta1/Smad/MMP9 signals. Oncotarget. 2015;6:12326–39. doi: 10.18632/oncotarget.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Laurent V, Guerard A, Mazerolles C, Le Gonidec S, Toulet A, Nieto L, et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat Commun. 2016;7:10230. doi: 10.1038/ncomms10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis. 2015;32:353–68. doi: 10.1007/s10585-015-9714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Huttemann M, Podgorski I. Bone marrow adipocytes promote the warburg phenotype in metastatic prostate tumors via HIF-1alpha activation. Oncotarget. 2016 doi: 10.18632/oncotarget.11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 88.Wang N, Yao M, Xu J, Quan Y, Zhang K, Yang R, et al. Autocrine Activation of CHRM3 Promotes Prostate Cancer Growth and Castration Resistance via CaM/CaMKK-Mediated Phosphorylation of Akt. Clin Cancer Res. 2015;21:4676–85. doi: 10.1158/1078-0432.CCR-14-3163. [DOI] [PubMed] [Google Scholar]
- 89.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. 2015;47:367–72. doi: 10.1038/ng.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tosoian JJ, Trock BJ, Landis P, Feng Z, Epstein JI, Partin AW, et al. Active surveillance program for prostate cancer: an update of the Johns Hopkins experience. J Clin Oncol. 2011;29:2185–90. doi: 10.1200/JCO.2010.32.8112. [DOI] [PubMed] [Google Scholar]
- 92.Graff JN, Beer TM. Metastatic prostate cancer in 2015: The new and the old that is new again. Nat Rev Clin Oncol. 2016;13:73–4. doi: 10.1038/nrclinonc.2015.226. [DOI] [PubMed] [Google Scholar]
- 93.Francini E, Taplin ME. Prostate cancer: Developing novel approaches to castration-sensitive disease. Cancer. 2017;123:29–42. doi: 10.1002/cncr.30329. [DOI] [PubMed] [Google Scholar]
- 94.Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N Engl J Med. 2015;373:737–46. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387:1163–77. doi: 10.1016/S0140-6736(15)01037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iuliani M, Pantano F, Buttigliero C, Fioramonti M, Bertaglia V, Vincenzi B, et al. Biological and clinical effects of abiraterone on anti-resorptive and anabolic activity in bone microenvironment. Oncotarget. 2015;6:12520–8. doi: 10.18632/oncotarget.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Penson DF, Armstrong AJ, Concepcion R, Agarwal N, Olsson C, Karsh L, et al. Enzalutamide Versus Bicalutamide in Castration-Resistant Prostate Cancer: The STRIVE Trial. J Clin Oncol. 2016;34:2098–106. doi: 10.1200/JCO.2015.64.9285. [DOI] [PubMed] [Google Scholar]
- 98.Shore ND, Chowdhury S, Villers A, Klotz L, Siemens DR, Phung D, et al. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): a randomised, double-blind, phase 2 study. Lancet Oncol. 2016;17:153–63. doi: 10.1016/S1470-2045(15)00518-5. [DOI] [PubMed] [Google Scholar]
- 99.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014;15:975–85. doi: 10.1016/S1470-2045(14)70240-2. [DOI] [PubMed] [Google Scholar]
- 101.Montgomery B, Eisenberger MA, Rettig MB, Chu F, Pili R, Stephenson JJ, et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016;22:1356–63. doi: 10.1158/1078-0432.CCR-15-1432. [DOI] [PubMed] [Google Scholar]
- 102.Paranjape AN, Soundararajan R, Werden SJ, Joseph R, Taube JH, Liu H, et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene. 2016 doi: 10.1038/onc.2015.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med. 2016;375:443–53. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373:1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cheng HH, Pritchard CC, Boyd T, Nelson PS, Montgomery B. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2016;69:992–5. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–50. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Evans JR, Zhao SG, Chang SL, Tomlins SA, Erho N, Sboner A, et al. Patient-Level DNA Damage and Repair Pathway Profiles and Prognosis After Prostatectomy for High-Risk Prostate Cancer. JAMA Oncol. 2016;2:471–80. doi: 10.1001/jamaoncol.2015.4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vignani F, Bertaglia V, Buttigliero C, Tucci M, Scagliotti GV, Di Maio M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat Rev. 2016;44:61–73. doi: 10.1016/j.ctrv.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 110.Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J Clin Oncol. 2016;34:3005–13. doi: 10.1200/JCO.2015.65.5597. [DOI] [PubMed] [Google Scholar]
- 111.Sung SY, Chang JL, Chen KC, Yeh SD, Liu YR, Su YH, et al. Co-Targeting Prostate Cancer Epithelium and Bone Stroma by Human Osteonectin-Promoter-Mediated Suicide Gene Therapy Effectively Inhibits Androgen-Independent Prostate Cancer Growth. PLoS One. 2016;11:e0153350. doi: 10.1371/journal.pone.0153350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fong EL, Wan X, Yang J, Morgado M, Mikos AG, Harrington DA, et al. A 3D in vitro model of patient-derived prostate cancer xenograft for controlled interrogation of in vivo tumor-stromal interactions. Biomaterials. 2016;77:164–72. doi: 10.1016/j.biomaterials.2015.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373–88. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bambury RM, Rathkopf DE. Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urol Oncol. 2016;34:348–55. doi: 10.1016/j.urolonc.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 116.Lin TH, Lee SO, Niu Y, Xu D, Liang L, Li L, et al. Differential androgen deprivation therapies with anti-androgens casodex/bicalutamide or MDV3100/Enzalutamide versus anti-androgen receptor ASC-J9(R) Lead to promotion versus suppression of prostate cancer metastasis. J Biol Chem. 2013;288:19359–69. doi: 10.1074/jbc.M113.477216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sha K, Yeh S, Chang C, Nastiuk KL, Krolewski JJ. TNF signaling mediates an enzalutamide-induced metastatic phenotype of prostate cancer and microenvironment cell co-cultures. Oncotarget. 2015;6:25726–40. doi: 10.18632/oncotarget.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2015;21:2315–24. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 119.Caino MC, Altieri DC. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin Cancer Res. 2016;22:540–5. doi: 10.1158/1078-0432.CCR-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ruscetti M, Dadashian EL, Guo W, Quach B, Mulholland DJ, Park JW, et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene. 2016;35:3781–95. doi: 10.1038/onc.2015.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marin-Aguilera M, Codony-Servat J, Reig O, Lozano JJ, Fernandez PL, Pereira MV, et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol Cancer Ther. 2014;13:1270–84. doi: 10.1158/1535-7163.MCT-13-0775. [DOI] [PubMed] [Google Scholar]
- 122.Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72:527–36. doi: 10.1158/0008-5472.CAN-11-3004. [DOI] [PubMed] [Google Scholar]
- 123.Byrne NM, Nesbitt H, Ming L, McKeown SR, Worthington J, McKenna DJ. Androgen deprivation in LNCaP prostate tumour xenografts induces vascular changes and hypoxic stress, resulting in promotion of epithelial-to-mesenchymal transition. Br J Cancer. 2016;114:659–68. doi: 10.1038/bjc.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E, et al. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate. 2015;75:161–74. doi: 10.1002/pros.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shiota M, Itsumi M, Takeuchi A, Imada K, Yokomizo A, Kuruma H, et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr Relat Cancer. 2015;22:889–900. doi: 10.1530/ERC-15-0225. [DOI] [PubMed] [Google Scholar]
- 126.Ware KE, Somarelli JA, Schaeffer D, Li J, Zhang T, Park S, et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Colditz J, Rupf B, Maiwald C, Baniahmad A. Androgens induce a distinct response of epithelial-mesenchymal transition factors in human prostate cancer cells. Mol Cell Biochem. 2016;421:139–47. doi: 10.1007/s11010-016-2794-y. [DOI] [PubMed] [Google Scholar]
- 128.Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2015;34:3617–26. doi: 10.1038/onc.2014.314. [DOI] [PubMed] [Google Scholar]
- 129.Lee YC, Lin SC, Yu G, Cheng CJ, Liu B, Liu HC, et al. Identification of Bone-Derived Factors Conferring De Novo Therapeutic Resistance in Metastatic Prostate Cancer. Cancer Res. 2015;75:4949–59. doi: 10.1158/0008-5472.CAN-15-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fong L, Kwek SS, O’Brien S, Kavanagh B, McNeel DG, Weinberg V, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69:609–15. doi: 10.1158/0008-5472.CAN-08-3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol. 2013;24:1813–21. doi: 10.1093/annonc/mdt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, et al. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget. 2016;7:10557–67. doi: 10.18632/oncotarget.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res. 2009;69:4894–903. doi: 10.1158/0008-5472.CAN-08-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao B, Sedlak JC, Srinivas R, Creixell P, Pritchard JR, Tidor B, et al. Exploiting Temporal Collateral Sensitivity in Tumor Clonal Evolution. Cell. 2016;165:234–46. doi: 10.1016/j.cell.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Beltran H, Eng K, Mosquera JM, Sigaras A, Romanel A, Rennert H, et al. Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response. JAMA Oncol. 2015;1:466–74. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34:1402–18. doi: 10.1200/JCO.2015.64.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.