

Abstract
CARMILs are large multidomain proteins that regulate the actin-binding activity of capping protein (CP), a major capper of actin filament barbed ends in cells. CARMILs bind directly to CP and induce a conformational change that allosterically decreases but does not abolish its actin-capping activity. The CP-binding domain of CARMIL consists of the CP-interaction (CPI) and CARMIL-specific interaction (CSI) motifs, which are arranged in tandem. Many cellular functions of CARMILs require the interaction with CP; however, a more surprising result is that the cellular function of CP in cells appears to require binding to a CARMIL or another protein with a CPI motif, suggesting that CPI-motif proteins target CP and modulate its actin-capping activity. Vertebrates have three highly conserved genes and expressed isoforms of CARMIL with distinct and overlapping localizations and functions in cells. Various domains of these CARMIL isoforms interact with plasma membranes, vimentin intermediate filaments, SH3-containing class I myosins, the dual-GEF Trio, and other adaptors and signaling molecules. These biochemical properties suggest that CARMILs play a variety of membrane-associated functions related to actin assembly and signaling. CARMIL mutations and variants have been implicated in several human diseases. We focus on roles for CARMILs in signaling in addition to their function as regulators of CP and actin.
INTRODUCTION
The dynamics of actin filament assembly and disassembly play important roles in many biological processes, both normal and pathological (Pollard and Cooper, 2009). Actin filaments grow and shrink by addition and loss, respectively, of actin subunits at the ends of filaments. The barbed (plus) end of the filament is favored over the pointed (minus) end for assembly, both thermodynamically and kinetically (Pollard, 2016), and cells control their shape and migration by regulating barbed-end filament assembly spatially and temporally (Shekhar et al., 2016). Examples of such regulation are numerous and affect a wide range of processes, including development and differentiation (Harris et al., 2009), immunity and inflammation (Marcos-Ramiro et al., 2014), and cancer cell invasion and metastasis (Kumar and Weaver, 2009; Mierke, 2013).
The growth of actin filament barbed ends by the addition of subunits is a major mechanism by which actin filaments form and assemble to do work in cells. A free actin subunit is able to add to a free barbed end on its own, and a number of factors that promote this process have been discovered and play important roles (Pollard, 2016). In cells, the creation of free barbed ends correlates with and appears to be sufficient to induce actin filament polymerization (Bailly et al., 1999; Huang et al., 1999; Ghosh et al., 2004). Free barbed ends can be generated in several ways: de novo assembly of a new actin filament from free actin subunits, severing of an existing actin filament, and uncapping of the barbed end of a capped filament. Free barbed ends will stop growing if they are functionally capped, and capping is a feature of both termination and promotion of actin assembly, depending on the context of polymerases and other regulators (Pollard, 2016).
One major mediator for capping the barbed end of filaments is capping protein (CP), an obligate α/β heterodimer found in essentially all eukaryotic cells. A small number of direct-binding inhibitors of CP have been discovered (reviewed in Cooper and Sept, 2008; Edwards et al., 2014), and in vitro, purified CP can be inhibited by phosphatidylinositol (4,5)-bisphosphate and other anionic phospholipids (Heiss and Cooper, 1991; Kuhn and Pollard, 2007; Li et al., 2012), by the protein V-1 (Bhattacharya et al., 2006; Fujiwara et al., 2014), and by a diverse set of proteins that contain a conserved capping protein interaction (CPI) motif (Hernandez-Valladares et al., 2010; Edwards et al., 2014). CPI-motif proteins can remove CP from the barbed end (Edwards et al., 2014) and appear to be required for normal CP function (Edwards et al., 2015). This review focuses on the CPI-containing CP, Arp2/3, myosin-I linker (CARMIL) family of proteins and their roles as regulators of CP activity and scaffolding molecules for signaling pathways.
CARMIL-FAMILY PROTEINS
CARMIL-family proteins are large, highly conserved, multidomain homodimers. CARMILs were discovered in Acanthamoeba (Acan125) and Dictyostelium (p116) based on direct binding of their proline-rich domain (PRD) to the Src homology 3 (SH3) domain of a subset of class I myosins (Xu et al., 1995, 1997; Zot et al., 2000; Jung et al., 2001). A myosin I SH3 domain was used as an affinity ligand in the purification of amoeba CARMILs from cells in a tight complex with CP and Arp2/3 complex (Jung et al., 2001). Amoeba CARMILs possess verprolin-like and acidic regions that are capable of activating Arp2/3 complex for nucleation of actin polymerization (Jung et al., 2001); however, these two regions do not appear to be present in vertebrate CARMILs, and CARMIL1 does not appear to bind or activate Arp2/3 complex (Yang et al., 2005; Liang et al., 2009).
Amoebozoa and invertebrates have one gene encoding CARMIL, whereas vertebrates have three (Liang et al., 2009; Edwards et al., 2014; Stark and Cooper, 2015), and the genomes of fungi and plants appear to lack CARMIL homologues altogether. The three vertebrate isoforms, CARMIL1, 2, and 3, can be defined and distinguished from each other by conserved differences in their amino acid sequences (Figure 1).
FIGURE 1:

Conservation and domain architecture of CARMIL proteins. (A) Domain architecture of CARMIL proteins, illustrating the arrangement of the PH domain, linker (L), N-cap (N), LRR domain, C-cap (C), HD, CBR consisting of a CPI motif and a CSI motif, MBD, and a PRD. Sequence alignment of the CBR for selected CARMILs, including the three vertebrate isoforms in zebrafish, mouse, and human (encoded by three separate genes). The alignment includes sequences for invertebrates, which lack the CSI motif region or lack residues known to be required for the CSI-CP interaction (Zwolak et al., 2010b). Shaded residues are identical to the consensus sequences. (B) Unrooted phylogenetic tree showing the relationships among CARMIL proteins, revealing five groups of CARMIL genes. Vertebrate genomes have three genes that encode three conserved isoforms—CARMIL1, CARMIL2, and CARMIL3—and invertebrates have a single CARMIL gene and isoform. Invertebrate CARMILs can be further classified into those that contain a CSI motif and those that do not.
CARMIL-protein domain architecture
Vertebrate CARMILs share a common domain architecture (Zwolak et al., 2013), illustrated in Figure 1. The N-terminus has a noncanonical pleckstrin-homology (PH) domain, followed by a leucine-rich repeat (LRR) domain. The PH and LRR domains are connected by an apparently rigid linker; this linker and the N-cap region of the LRR domain contain conserved amino acid sequences that are highly distinctive for CARMILs, but they serve a function as yet unknown. These sequences have been used to aid in the identification of CARMILs from various organisms (Liang et al., 2009; Zwolak et al., 2013; Stark and Cooper, 2015). The LRR domain is followed by a helical dimerization (HD) domain, and the C-terminal half of the protein consists of an extended intrinsically disordered region. The disordered region contains a CP-binding region (CBR) made up of a CPI motif and a CARMIL-specific interaction (CSI) motif arranged in tandem, followed by a short basic and hydrophobic membrane-binding domain (MBD) and a proline-rich domain (PRD) that binds SH3 domains of class I myosins and other proteins (Liang et al., 2009; Edwards et al., 2013; Zwolak et al., 2013; Roncagalli et al., 2016). We discuss these domains in detail, with consideration for the differences between isoforms.
Pleckstrin-homology domain
The noncanonical PH domain was identified by biochemical and structural studies of mouse CARMIL1, including x-ray crystallography (Zwolak et al., 2013). The PH domain binds monophosphorylated phosphatidylinositides—phosphatidylinositol 3-phosphate, phosphatidylinositol 4-phosphate, and phosphatidylinositol 5-phosphate—with high specificity. In contrast to canonical PH domains, the residues important for phospholipid binding are not in the traditionally recognized lipid-binding pocket (Ferguson et al., 2000; Lemmon, 2007); instead, the key residues lie near the interface of the PH domain and the LRR domain (Zwolak et al., 2013). Removal of the PH domain from the CARMIL1 N-terminus decreases the degree of plasma membrane localization in cells, confirming the physiological importance of phospholipid binding (Zwolak et al., 2013).
In contrast, the PH domain of CARMIL2 lacks the conserved residues needed for phospholipid binding, based on sequence alignments (Zwolak et al., 2013). As predicted, the CARMIL2 PH domain is not sufficient to target green fluorescent protein (GFP) to the plasma membrane (Lanier et al., 2015). For CARMIL3, the PH domain has not been studied experimentally, but its sequence is similar to that of CARMIL1, including the conservation of the phospholipid-binding residues.
Leucine-rich repeat domain
LRRs, defined by the consensus sequence LxxLxLxx(N/C)xxL, are generally involved in protein–protein interactions (Kobe and Kajava, 2001; Enkhbayar et al., 2004; Bella et al., 2008). In the crystal structure of mouse CARMIL1, the 16-repeat LRR assumes a horseshoe shape as found for other LRR domains (Kobe and Deisenhofer, 1996; Zwolak et al., 2013). Within this horseshoe, the LRR adopts a typical structure, with α-helices on the convex surface and β-strands on the concave surface (Zwolak et al., 2013). Although the secondary structure features of the repeats are characteristic of LRR domains in general, the amino acid sequences of LRR domains in CARMILs are more similar to one another than to those in unrelated proteins (Kobe and Kajava, 2001), suggesting the possibility of functional differences rather than simple structural conservation.
The notion of distinct functions for CARMIL LRR domains is supported by studies in human cultured cells, in which CARMIL2 localizes to vimentin filaments via its LRR domain (Lanier et al., 2015). This conclusion is based on the finding that a chimeric protein in which the LRR comes from CARMIL2 and the remainder of the polypeptide is taken from CARMIL1 colocalizes with vimentin in cells and rescues the loss of CARMIL2 function. In contrast, the converse chimera, composed of the LRR domain of CARMIL1 fused with all other domains from CARMIL2, fails to localize with vimentin in cells and is unable to rescue the loss of CARMIL2 function (Lanier et al., 2015). In this regard, the cellular functions of all the domains of CARMIL 1 and 2 other than the LRR domain can be considered as overlapping. This conclusion is necessarily limited to the setting of the cell culture assays and cell types examined.
LRR domains are bounded by ends with distinct flanking sequences, referred to as N-cap and C-cap, respectively (Bella et al., 2008; Zwolak et al., 2013; Dao et al., 2014). N-caps are believed to either stabilize the hydrophobic core or promote correct folding of the LRR domain (Bella et al., 2008; Dao et al., 2014). Among CARMILs, the N-cap sequences are very similar to one another (Liang et al., 2009; Zwolak et al., 2013; Stark and Cooper, 2015). For this reason, the N-cap region has been useful in BLAST searches for CARMIL family members in the genomes of various organisms (Liang et al., 2009; Stark and Cooper, 2015), and it has been referred to as a CARMIL homology domain (CHD; Liang et al., 2009). Unlike the N-cap, the sequences of the C-caps are not conserved among CARMILs, nor are they similar to C-cap sequences in other LRR domains; nevertheless, their secondary structure resembles that of the C-cap in the LRR domain of tropomodulin (Krieger et al., 2002; Zwolak et al., 2013).
Helical dimerization domain
CARMILs exist as homodimers in cells, and biochemical studies reveal that homodimerization is mediated by the HD domain of CARMIL (Zwolak et al., 2013). Isolated HD domains dimerize (Zwolak et al., 2013), and full-length CARMILs, purified or isolated from cells, are found as homodimers (Roncagalli et al., 2016; Wang et al., 2016). The antiparallel nature of the dimer, revealed by small-angle x-ray scattering (SAXS) of mouse CARMIL1, indicates that dimerization places the N-terminal PH domains in an orientation allowing both of them to interact with a planar structure, such as the plasma membrane (Liang et al., 2009). Indeed, the PH domain of CARMIL1 is important for membrane localization in cells (Zwolak et al., 2013).
Homodimerization of CARMILs has been documented for Acanthamoeba CARMIL (Remmert et al., 2004), mammalian CARMIL1 (Liang et al., 2009; Zwolak et al., 2013), and mammalian CARMIL2 (Roncagalli et al., 2016; Wang et al., 2016). Heterodimers of CARMIL1 and CARMIL2 were not observed for human cells expressing the two isoforms at endogenous levels (Liang et al., 2009); however, heterodimers can be induced to form in small amounts when CARMILs are expressed at artificially high levels (Edwards, Lanier, and Cooper, unpublished data). The amino acid sequences of the HD domains of CARMIL1 and CARMIL3 are more similar to one another than to the sequence of the CARMIL2 HD domain (Zwolak et al., 2013), raising the possibility that CARMIL1-CARMIL3 heterodimers may form under physiological conditions. However, structural models of HD homodimers based on SAXS lack the resolution to address this question, and heterodimer formation has not been properly investigated, either with purified proteins or in cells.
Capping protein–binding region
The CARMIL CBR is part of the extended intrinsically disordered C-terminal portion of the polypeptide that follows the HD domain. The CBR includes two conserved sequences in tandem termed the capping protein interaction (CPI) and CARMIL-specific interaction (CSI) motifs (Hernandez-Valladares et al., 2010; Figure 1). This region has also been called CAH3 (for CARMIL homology domain 3), and the portions corresponding to the CPI and CSI motifs have been called CAH3a and CAH3b, respectively (Zwolak et al., 2010b).
The CPI motif, with the consensus sequence LxHxTxxRPK(x)6P, is found in a diverse set of otherwise unrelated proteins, all of which bind directly to CP (Bruck et al., 2006). For CARMIL1 and CARMIL2, a number of cellular functions have been demonstrated to depend on the ability of the motif to bind CP (Edwards et al., 2013, 2014; Lanier et al., 2015). Two cocrystal structures of CP, one in complex with the full CBR of CARMIL1 and the other with the CPI motif of CD2AP, revealed a conserved set of close contacts between the CPI motifs and CP (Hernandez-Valladares et al., 2010; Takeda et al., 2010).
In contrast to the more common CPI motif, the CSI motif (consensus sequence RxDEGxEEFFxKR; Hernandez-Valladares et al., 2010; Zwolak et al., 2010b) is present only in proteins of the CARMIL family. Moreover, the CSI is limited to CARMILs from vertebrates and higher invertebrates; CARMILs from lower eukaryotes, including Acanthamoeba, Dictyostelium, and Caenorhabditis, lack the consensus sequence (Figure 1; Zwolak et al., 2010b; Edwards et al., 2014). Whereas the CPI motif on its own is sufficient to bind to CP and decrease its actin-binding activity, the CSI motif binds CP weakly and has relatively little ability to inhibit its activity. Nevertheless, as part of the CBR, the CSI makes an important contribution to the binding and inhibition of CP (Hernandez-Valladares et al., 2010; Zwolak et al., 2010b).
Actin and the CBR bind to distinct sites on the mushroom-shaped CP α/β heterodimer. The barbed end of the actin filament binds to the top surface of the mushroom (Narita et al., 2006; Kim et al., 2010), whereas the CBR binds to the mushroom stalk via its CPI and CSI motifs (Hernandez-Valladares et al., 2010; Takeda et al., 2010). The CPI motif binds along the surface of about half of the perimeter of the mushroom stalk, covering parts of both the CP α- and β-subunits. Contact residues between CPI and CP, identified in the cocrystal structure (Hernandez-Valladares et al., 2010; Takeda et al., 2010), have been documented to be important for this interaction in biochemical and cell biological assays (Edwards et al., 2013, 2015; Lanier et al., 2015).
The short linker sequence that connects the CPI and CSI motifs was not resolved in cocrystal structures, suggesting that it may not bind residues on the surface of CP (Hernandez-Valladares et al., 2010). Replacing the residues of the linker sequence with a set of alternating Ala and Gly residues did not affect the ability of the CBR to bind and inhibit CP activity in actin-capping assays (Kim et al., 2012), confirming the absence of functionally important close contacts in this region. However, it remains possible that the physical properties of the linker affect the kinetics and thermodynamics of CBR binding through effects on the physical properties of the intrinsically disordered domain, including the adjacent CPI and CSI motifs.
The binding of CARMIL to CP has an allosteric effect on the conformation of CP’s actin-binding surface. CARMIL binding promotes occupancy of a CP state that is more similar to unbound CP than to actin-bound CP (Figure 2). This state has structural and dynamic properties that decrease the binding affinity of CP for the barbed end of the actin filaments (Hernandez-Valladares et al., 2010; Kim et al., 2012; Edwards et al., 2013). Evidence for this conclusion includes the observation that the CBR domain of CARMIL decreases the actin capping activity of CP by 250-fold (Kim et al., 2012). In addition, CBR binding leads to an increase in the dissociation rate of CP from the barbed end, suggesting that a molecule of CBR can bind to a molecule of CP bound to a capped actin filament, forming a trimolecular complex (Fujiwara et al., 2010; Hernandez-Valladares et al., 2010; Kim et al., 2012; Lanier et al., 2015). This is an important point, relevant to cellular function—CARMIL binding to CP decreases but does not abolish the capping activity of CP, and this trimolecular complex can exist in vitro and, presumably, in vivo (Yang et al., 2005).
FIGURE 2:
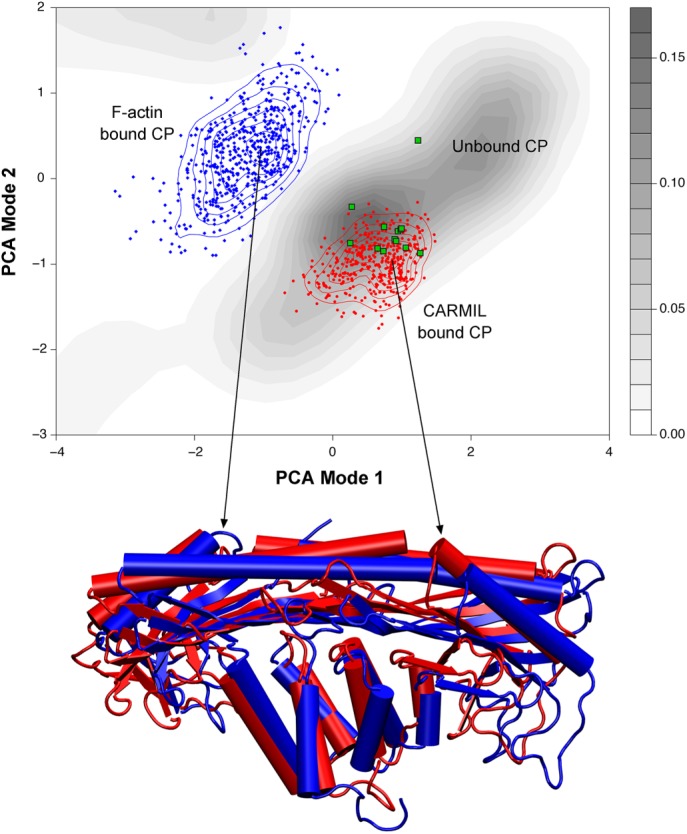
Principal component analysis (PCA) of CP. Top, PCA of molecular dynamics simulations of the structural conformations of CP. The simulations were begun with CP that was either free (gray contour shading), bound to F-actin (blue dots and contour lines), or bound to CARMIL (red dots and contour lines). The results were projected onto the same conformational space; existing CP structures were overlaid as green dots. Based on this analysis, CARMIL-bound CP exists in a conformational space more like that of unbound CP (gray) than to that of F-actin–bound CP. Bottom, overlay of protein structures, depicting the conformations that correspond to the peaks of CARMIL-bound and F-actin bound CP. This analysis supports the findings that CARMIL allosterically inhibits CP by eliciting changes to the actin-binding surface, holding CP in a conformation similar to the free state. Used with permission from Kim et al. (2012).
These biochemical observations have two potential implications for the function of CARMIL (and other CPI proteins) in cells. First, CARMIL may recruit or target the barbed-end capping activity of CP to a site of actin assembly. In the case of Arp2/3-nucleated actin assembly, capping barbed ends is necessary for the reconstitution of actin polymerization and actin-based motility from purified components (Loisel et al., 1999). In the case of formin-nucleated actin assembly, CP and formins antagonize each other functionally (Breitsprecher and Goode, 2013), and they can simultaneously bind to the barbed end of an actin filament, forming a so-called decision complex. The decision complex resolves with either capping of the filament by CP and dissociation of formin from the tip, or removal of CP from the barbed end, followed by formin-mediated actin polymerization (Bombardier et al., 2015; Shekhar et al., 2015). Both Arp2/3- and formin-mediated actin polymerization presumably rely on CP to limit the length of the actin filaments and help to maintain a pool of free actin monomers.
The second implication of the foregoing biochemical observations is that CARMILs may modulate the capping activity of CP, “tuning it down” to levels that are physiologically relevant for the cytoplasmic concentrations of CP and barbed ends (Yang et al., 2005; Uruno et al., 2006; Hernandez-Valladares et al., 2010; Kim et al., 2012; Lanier et al., 2015). Consistent with this model, when the CBR-CP complex binds to actin barbed ends in vitro, CP dissociates from the barbed end at a faster rate, similar to the time frame for interactions of CP with the actin cytoskeleton in cells (Miyoshi et al., 2006; Fujiwara et al., 2010). This action of CARMIL may contribute to the disassembly and turnover of actin filaments in cells by making the capping of barbed ends more dynamic.
The potential models of “targeting” CP and “tuning” its capping activity are not exclusive and may both exist in cells. The relevance of these models is supported by the cellular phenotypes of a CP point mutant that is unable to bind to CPI motifs (Edwards et al., 2015). Although this mutant has normal actin-capping activity in biochemical assays, it provides little or no actin-related function in cells. Dominant-negative and expression-rescue approaches showed that the CP mutant phenocopies the simple loss of CP. These findings led to the conclusion that CP function in cells depends on an interaction with a protein that contains a CPI motif.
CP can be inhibited by the protein V-1 in vitro, and V-1 binds to the actin-binding surface of CP, acting as a direct competitor for the capping of barbed ends (Bhattacharya et al., 2006; Takeda et al., 2010; Zwolak et al., 2010a; Fujiwara et al., 2014). If CP is inhibited by the binding of V-1 in cells, as revealed by a study with Dictyostelium (Jung et al., 2016), then one attractive model is that CARMILs, along with other proteins containing CPI motifs, activate rather than inhibit CP, as first suggested by Fujiwara et al. (2014). In this model, activation of CP is a consequence of a CPI-motif protein binding to the CP/V-1 complex, increasing the rate of V-1 dissociation by the same allosteric mechanism that decreases actin capping and promotes uncapping (Takeda et al., 2010; Fujiwara et al., 2014; Edwards et al., 2015).
Membrane-binding domain
CARMILs have membrane-binding domains that consist of unstructured protein regions with basic and hydrophobic residues. This type of membrane-binding domain binds to lipid bilayers through a combination of electrostatic and hydrophobic interactions (Heo et al., 2006). In this context, binding requires a high density of basic and hydrophobic residues, but it is largely independent of the exact sequence (Papayannopoulos et al., 2005; Heo et al., 2006; Brzeska et al., 2010). A search algorithm designed to detect amino acid sequences with these features identified one in mouse CARMIL1, and the predicted region was found to bind to acidic phospholipids with a binding affinity in the micromolar range (Brzeska et al., 2010). This initial study used in vitro lipid-binding assays and a relatively large fragment of mouse CARMIL1. Later, smaller fragments of human CARMIL1, CARMIL2, and CARMIL3 containing the MBDs were found to be sufficient to target GFP to the plasma membrane in cells, including to the leading edge of migrating cells, which is an active site of actin polymerization (Lanier et al., 2016).
These studies showed that the CARMIL MBD is sufficient for membrane and lipid binding. In addition, the MBD has been shown to be necessary for membrane localization of full-length CARMILs in cells and is also required for the function of CARMIL in cells. Specifically, for CARMIL2, a series of point mutations in the MBD revealed that the MBD is essential for membrane localization and for function (Lanier et al., 2016). Both full-length CARMIL2 and the C-terminal half alone required the MBD for their membrane localization in cells. For full-length CARMIL2, the MBD was required to rescue the phenotypes caused by the loss of endogenous CARMIL2, revealing its requirement for CARMIL2 function within cells (Lanier et al., 2016).
For CARMIL1 and CARMIL3, the necessity of the MBD for function in cells has not been investigated directly. However, for CARMIL1, observations from the literature strongly suggest that the MBD is important. Specifically, two studies, using different approaches to examine the function of CARMIL1 mutants deficient for CP binding, provide evidence for MBD function as well. In the first study, a CARMIL1 mutant with a 123-amino acid internal deletion that encompasses both the CBR and the MBD failed to rescue cell migration phenotypes of CARMIL1-depleted cells in wound-healing assays (Yang et al., 2005). In the second study, a CARMIL1 CP-binding mutant with substitutions for two highly conserved residues of the CPI consensus sequence, leaving both the CSI motif and the MBD intact, was largely able to rescue the wound-healing cell migration phenotype in CARMIL1-depleted cells (Edwards et al., 2013). One key difference between the CARMIL1 mutant constructs in the two studies is the presence of the MBD in the expression construct for the latter but not the former. Taken together, these studies support the hypothesis that the MBD is necessary for the cellular function of CARMIL1, consistent with the results obtained for CARMIL2 described earlier.
The MBDs of different CARMIL isoforms have functions that are sufficiently similar as to be interchangeable in cells in some settings. As discussed earlier, the MBD of CARMIL1 was able to substitute for that of CARMIL2 in a chimera consisting of the PH domain of CARMIL1, the LRR domain of CARMIL2, and the C-terminal half of CARMIL1 (including the CBR and MBD), rescuing cellular phenotypes caused by depletion of endogenous CARMIL2 (Lanier et al., 2016).
The presence of the MBD and the CSI motif is a distinctive feature of CARMILs among the diverse and otherwise unrelated set of CPI motif–containing proteins (Lanier et al., 2016). Thus the MBD and the CSI motif may provide functions that distinguish the cellular roles of CARMILs from those of other CPI-motif proteins. The close proximity of the MBD to the CBR, which includes both CPI and CSI motifs, raises the possibility that the function of the MBD is to target regulation of CP and actin assembly to the vicinity of the membrane (Lanier et al., 2016).
Proline-rich domain
The C-terminal domains of CARMILs are rich in proline residues. Indeed, the initial discoveries of CARMILs in Acanthamoeba and Dictyostelium were based on direct physical interactions between the PRDs of CARMILs with the SH3 domains of the tails of certain class I myosins (Xu et al., 1995, 1997; Zot et al., 2000; Jung et al., 2001). Acanthamoeba and Dictyostelium CARMILs have their CBR at the extreme end of the C-terminus, and they lack the extended C-terminal PRD seen in vertebrates (Figure 1); however, PxxP motifs responsible for SH3 binding are present just upstream of the CBR (Xu et al., 1995, 1997; Zot et al., 2000; Jung et al., 2001).
In vertebrates, the CARMIL1 PRD binds to myosin-IE, a class I myosin with an SH3 domain in its tail (Liang et al., 2009). Whereas CARMIL1 PRD has many PxxP motifs, the one(s) critical for SH3 binding have not been identified. The physiological importance of this interaction has not been assessed; however, the tail domain of myosin-IE appears to stabilize focal adhesions at the leading edge of migrating cells (Gupta et al., 2013).
CARMIL2 does not bind to myosin-IE, based on a direct comparison with CARMIL1 (Liang et al., 2009). However, the CARMIL2 PRD includes 10 PxxP motifs, and it interacts with the signaling adaptor GRB2, which contains two SH3 domains. This interaction with GRB2 appears to couple CARMIL2 to CD28 signaling (Roncagalli et al., 2016), as discussed later. One can imagine that future studies will uncover other binding partners for the PRDs of CARMILs.
ROLES OF CARMILS AT THE CELLULAR AND ORGANISMAL LEVELS
CARMIL expression during development and differentiation
The patterns and timing of expression of CARMIL isoforms in vertebrate cells and tissues have been addressed, but only to a limited extent (Table 1). As noted earlier, vertebrates have three genes from which three highly conserved isoforms of CARMIL are expressed (Figure 1).
TABLE 1:
Summary of CARMIL functions in cells and organisms.
| Cellular functions | ||||
|---|---|---|---|---|
| Isoform | Expression | Actin | Signaling | Human disease |
| Invertebrate | NA | NA | Decreased NF-κB signaling | NA |
| CARMIL1 | Skin, testis, spleen | CP regulation, macropinocytosis | Wound healing, increased Trio signaling | Gout, ARDS |
| CARML2 | Brain, spinal cord, kidney, thymus, spleen, bone marrow, eyes | CP regulation, cell polarity | Wound healing, CD28-mediated T-cell activation, increased NF-κB signaling | Immune function, T-cell development |
| CARMIL3 | Brain, eyes, spinal cord, heart, testis | NA | NA | Oncofetal gene |
NA, not available.
In zebrafish, the three isoforms show distinct spatial and temporal expression patterns through development and differentiation (Stark and Cooper, 2015). Of the three, CARMIL1 is most widely expressed, being present in the skin, fins, branchial arches, gut, and cloaca (Stark and Cooper, 2015). An RNA-sequencing study of zebrafish skin found only the CARMIL1 isoform ( Wang, Cokus, and Sagasti, personal communication). CARMIL2 is expressed at high levels in brain and spinal cord and at lower levels in eye and developing kidney (Trinh et al., 2011; Stark and Cooper, 2015). CARMIL3 is expressed at high levels in brain and eye, with lower levels in the spinal cord. It is also expressed in the developing heart and is the only isoform observed in this organ (Stark and Cooper, 2015).
Studies of the expression of CARMIL isoforms in other vertebrates have been more limited. An analysis of five isolated mouse tissues revealed that CARMIL1 was highly expressed in testis and weakly in spleen and brain; CARMIL2 was highly expressed in thymus and spleen and weakly in bone marrow; and CARMIL3 was expressed in brain and testis (Liang et al., 2013). In a study of human tissues, CARMIL2 expression, assayed by reverse transcription PCR (RT-PCR), was detected in 30 different tissues, including skin (Matsuzaka et al., 2004). A study of CARMIL3 expression in human cells identified it as an oncofetal gene; it was found in brain, colon, heart, kidney, liver, lung, skeletal muscle, and spinal cord of both adult and fetal tissue samples, based on RT-PCR assays (Hsu et al., 2011).
The patterns of expression of CARMIL isoforms in vertebrates can also be obtained from the RNA-sequencing and microarray data curated in the Expression Atlas at the European Bioinformatics Institute (Kapushesky et al., 2012; Petryszak et al., 2014). In spite of certain inconsistencies across data sets, even for the same organism and tissue, several trends are clear. First, CARMIL1 is the most widely expressed isoform, detectable within a variety of tissues across multiple species. Analyses of skin revealed CARMIL1 expression in sheep (Bakhtiarizadeh et al., 2016), mice (Huntley et al., 2016), and humans (FANTOM Consortium and the RIKEN PMI and CLST (DGT) et al., 2014; Uhlen et al., 2015; Human Protein Atlas [www.proteinatlas.org]). Second, CARMIL2 and CARMIL3 are the isoforms most commonly expressed in brain tissues across a variety of organisms (Kapushesky et al., 2012; Petryszak et al., 2014).
Collectively the expression studies reveal that the three CARMIL isoforms have isoform-specific expression patterns, but that, in some cases, two or three isoforms are found within the same cells and tissues. For example, several human cultured cell lines express all three CARMIL isoforms (Liang et al., 2009). Further studies of the expression of CARMIL proteins within organisms would have value, in part to address apparent inconsistencies among previous studies.
Cellular functions of CARMILs
Because CARMILs are large, multidomain proteins, one might expect them to play multiple roles within cells. Indeed, although the CBR domain and actin-based functions have received the most attention, recent studies reveal roles in signaling networks and in human disease that may involve additional CARMIL domains (Table 1).
Regulation of actin assembly via CP.
CARMILs contain CPI motifs; therefore their actin-related cellular functions need to be considered in the context of the otherwise-unrelated proteins with CPI motifs, as previously reviewed (Edwards et al., 2014). All CPI-motif proteins localize to membranes and may serve as scaffolds for the recruitment and activation of CP at sites of actin assembly. The physiological importance of the CPI motif–CP interaction in cells has been demonstrated for several of these proteins. For CD2AP, a rescue construct bearing mutations that affect CPI residues fails to rescue CP localization in CD2AP-null cells (Zhao et al., 2013). For CKIP-1, interactions between the CPI motif and CP are required for increases in levels of cellular actin and F-actin caused by overexpression (Canton et al., 2006). For both CARMIL1 (Edwards et al., 2013) and CARMIL2 (Lanier et al., 2015), loss of CPI-CP interactions results in phenotypes reminiscent of the loss of CP function or localization.
In addition to the aforementioned studies investigating the role of the CPI residues in the interaction with CP, a complementary study probed CP residues for their relevance by alanine substitutions for two CP β-subunit residues in close contact with CPI in cocrystal structures (Hernandez-Valladares et al., 2010; Edwards et al., 2015). This mutant CP retained its full actin-binding capability; however, when expressed in cells, it mimicked the phenotype of loss of function of CP (Edwards et al., 2015). This study concluded that CP requires an interaction with a CPI-motif protein to function.
On the other hand, the CPI motifs of CARMIL1 or CARMIL2 do not appear to be necessary for all cellular functions. In particular, cell migration during wound-healing assays with cultured fibroblasts showed little dependence on the CPI motif (Edwards et al., 2013; Lanier et al., 2015). Similarly, in T-cells, the CARMIL2 CPI motif was not required for the CARMIL2 functions in CD28 costimulation and downstream signaling during T-cell activation (Roncagalli et al., 2016).
CARMIL proteins as signaling scaffolds
NF-κB signaling pathway.
In insects, treatment with a nicotinic acetylcholine receptor agonist, clothianidin, leads to increased expression of the single CARMIL gene (in flies, Dmel/LRR; in bees, Amel/LRR), and this leads to a decrease in NF-κB activation (Di Prisco et al., 2013). This deficiency in NF-κB activation was found to account for an increase in the effects of pathogens on insects previously exposed to neonicotinoids such as clothianidin. In this setting, CARMIL functions as a negative regulator of NF-κB signaling downstream of receptor-based signaling.
Signaling promoting T-cell development.
In higher organisms, CARMIL2 (also known as RLTPR) contributes to the immune system by controlling the development of certain T-cell subsets: Tregs and effector memory CD4+ T-cells (Liang et al., 2013; Roncagalli et al., 2016; Sorte et al., 2016; Wang et al., 2016). Within murine T-cells, CARMIL2 is required for proper localization of PKCθ and CARMA1 after CD28 stimulation (Liang et al., 2013). This study identified a point mutation that affects the mouse CARMIL2 LRR domain (RltprBas) and abrogates the effects of CD28 stimulation while retaining the CP-binding activity of CARMIL2. Ligand-induced CD28 internalization was accelerated in cells expressing the mutant CARMIL2, presumably due to decreases in “drag” resulting from loss of interactions with PKCθ, CARMA1, and downstream molecules (Liang et al., 2013; Roncagalli et al., 2016). CARMIL2 was required for the production of interleukin-2 downstream of the CD28 signaling pathway; this required its PH, LRR, and PRD domains, but the CP-binding activity of the CPI motif was dispensable (Roncagalli et al., 2016). Of interest, another CPI-motif protein, CapZIP, was found to be required, suggesting that the presence of the CPI motif of CapZIP allows for the loss of the CARMIL2 CPI motif (Tian et al., 2015; Roncagalli et al., 2016).
Rho-family GTPase signaling pathway.
Several studies have implicated CARMIL in Rac signaling via the dual–guanine nucleotide exchange factor (GEF) protein Trio. First, the single CARMIL gene in Caenorhabditis elegans (CRML-1) was detected in a genetic screen for inhibitors of the migration of neurons and axon growth cones (Vanderzalm et al., 2009). Genetic and biochemical interactions revealed that the inhibitory effect of CARMIL was mediated by Trio (UNC-73; Vanderzalm et al., 2009). A subsequent genetic study, investigating epithelial intercalation in the epidermis of this organism, also revealed that CARMIL inhibits Trio (Walck-Shannon et al., 2015). In this case, loss of CARMIL, and thus loss of Trio inhibition, led to loss of the polarized actin-rich protrusions that are required for intercalation among epithelial cells. In both studies, CARMIL functioned as a net negative regulator of Rac and RhoG, proteins that are activated by Trio’s GEF domains and promote Arp2/3-mediated actin polymerization (Walck-Shannon et al., 2015).
In cultured human cells, loss of CARMIL1, but not CARMIL2, led to the loss of Rac1 activation that occurs when cells spread on a fibronectin-coated substrate (Liang et al., 2009). In that study, Trio also interacted biochemically with CARMIL1 but not with CARMIL2. Later, the ability of CARMIL1 to enhance Rac1 activation was found to be independent of the ability of CARMIL1 to bind CP (Edwards et al., 2013). These results for CARMILs in humans and worms differ in one paradoxical respect. Whereas in human cells, the effect of CARMIL1 on Rac1- and Arp2/3-mediated actin assembly was positive, in worms, the net effect of CARMIL was negative. This discrepancy in outcome may reflect differences in the organisms, including the fact that humans have three CARMIL isoforms, which have both distinct and overlapping functions (Walck-Shannon et al., 2015).
CARMIL proteins in human disease.
CARMIL genes and proteins have been implicated in several human diseases. A variant of the gene encoding CARMIL1 (LRRC16A) has been associated with increased susceptibility to gout (Kolz et al., 2009; Sakiyama et al., 2014). This led to the proposal that CARMIL1-based regulation of CP, and thus of actin, influences the function of the urate-transporting macromolecular complex through an unknown mechanism (Sakiyama et al., 2014).
CARMIL1 variants have also been implicated in acute respiratory distress syndrome (ARDS; Wei et al., 2015, 2017); single-nucleotide polymorphisms (SNPs) have been associated with blood platelet count, an important parameter of clinical outcome. One SNP, studied in detail, causes a single–amino acid change in the LRR domain, decreased expression of mRNA, increased patient survival, and a decreased rate of platelet loss during the course of the illness (Wei et al., 2017). One potential scenario is that decreased CARMIL1 function impairs the actin cytoskeleton of the platelet, which lessens platelet activation and therefore mitigates the loss of circulating blood platelets. Paradoxically, poor platelet function has a beneficial effect for the ARDS patient because loss of platelets is a critical negative determinant.
In the case of the CARMIL2 gene (RLTPR), three studies identified mutations in families with primary immunodeficiencies (Sorte et al., 2016; Wang et al., 2016; Schober et al., 2017). In one of these studies, a missense mutation affecting the LRR domain (L603H) was found in four patients from three Norwegian families. The patients presented with warts, molluscum contagiosum, dermatitis, and evidence of immune dysfunction and susceptibility to viral infection in other systems (Sorte et al., 2016). In these individuals, levels of regulatory and CD4+ T-cells were low, as was the synthesis of interferon-γ in CD4+ T-cells and NK cells.
In the second study of CARMIL2, three point mutations (L372R, L489Q, and Q817X) were found in six individuals from three families (Wang et al., 2016). Homozygous individuals experienced mucocutaneous infections associated with decreased levels of regulatory and memory CD4+ T-cells as well as memory B-cells. The latter were unable to activate NF-κB after stimulation of B-cell receptors, indicating that B-cell dysfunction was independent of activation by CD4+ T-cells (Wang et al., 2016). Two of the mutations (L372R and L489Q) affect the LRR domain of CARMIL2, and the third (Q817X) introduces a stop codon in the HD domain. The missense alleles are associated with low levels of CARMIL2 protein, and the nonsense allele is expected to produce a truncated protein that lacks important functional domains in the intrinsically disordered C-terminal half of the protein.
In a third study of immunodeficiencies caused by CARMIL2 mutations, nonsense mutations in the linker/N-cap region or the N-terminal half of the LRR domain affected CD28-based activation, development, and function of T-cells (Schober et al., 2017). Patients lacked regulatory T-cells and suffered from immunodeficiency syndromes similar to those in patients in the studies discussed earlier (Sorte et al., 2016; Wang et al., 2016; Schober et al., 2017). In T-cells isolated from patients, levels of F-actin at the leading edge were decreased and the microtubule network was disorganized. When migrating, the T-cells were often multipolar, with decreased directed motion (Schober et al., 2017), reminiscent of the effects of CARMIL2 knockdown in a cultured cell line (Liang et al., 2009). These mutations also resulted in decreased activation of NF-κB signaling (Schober et al., 2017).
Overall these three studies of human patients suggest that the effects of CARMIL2 mutations in are likely related to the role of CARMIL2 in CD28 costimulation and T-cell development, as discussed earlier.
Molecular models of CARMIL function in cells
Here we consider working models for CARMIL function in cells as a framework for current knowledge and a basis for future studies. The biochemical, structural, and cellular properties of CARMIL proteins suggest that they interact with signaling networks to regulate the dynamics of the actin cytoskeleton at membranes. CARMILs move to the membrane either by passive diffusion or active transport. Once near the membrane, CARMILs may bind directly to membrane lipids and then recruit and/or inhibit CP via their CBR domains, as well as interact with signaling molecules.
One open question is whether the CBR domains of full-length CARMILs are found in an autoinhibited state in cells. In the case of Acanthamoeba CARMIL, a set of biochemical experiments with purified proteins provided compelling evidence that autoinhibition does occur (Uruno et al., 2006). In that study, a C-terminal fragment of CARMIL, which included the CBR, bound to CP with ∼20-fold greater affinity than did full-length CARMIL. Moreover, mild proteolytic cleavage of full-length CARMIL generated the more active C-terminal fragment. On the other hand, in a separate study with mouse CARMIL1, full-length CARMIL1 and its CBR fragment were similarly effective in actin polymerization-based assays measuring CP inhibition (Yang et al., 2005). The difference in the results from the two studies may be attributable to a notable difference between the proteins—the CBR of Acanthamoeba CARMIL is at the C-terminus and lacks the CSI-motif, whereas the CBR of mouse CARMIL1 is separated from the C-terminus by ∼300 amino acid residues and contains the CSI motif. Of course, differences in solution and other conditions may also affect the accessibility of the CBR in the full-length protein because the C-terminal regions of both proteins are intrinsically disordered.
We next consider and propose models with additional details for vertebrate CARMIL1 and CARMIL2, based on findings from published studies. For CARMIL3, the paucity of published data prevents a detailed discussion of models. The domain structure of CARMIL3 is similar to those of CARMIL1 and CARMIL2; however, the isoforms display conserved sequence differences that suggest the presence of distinct functions.
Model for CARMIL1.
This model proposes that CARMIL1 homodimers are transported to the plasma membrane along actin filaments. Class I myosins, namely myosin-1E and myosin-1F, bind PxxP motifs of CARMIL1’s PRD via their SH3 domains, and they carry CARMIL1 toward the membrane-associated barbed ends of actin filaments (Figure 3). The PH domain and MBD of CARMIL1 then bind directly to membrane lipids. Arp2/3 complex is activated at or near the membrane by signals from receptors transduced by small GTPases. Arp2/3 nucleates actin polymerization, and its branched network of actin filaments requires CP for proper assembly and force production.
FIGURE 3:

Models for CARMIL1 and CARMIL2 function within cells. (A) CARMIL1 is transported to the membrane via myosin-IE. CARMIL1 interacts with the membrane via PH domain and MBD. At the membrane, CARMIL1 recruits CP and relieves it from inhibition by V-1. Released V-1 then activates NF-κB signaling. CARMIL1 also activates Trio, which leads to an increase in Rac1 activation and Arp2/3-mediated actin assembly. (B) CARMIL2 is transported to the membrane via an interaction with vimentin filaments, where it then associates with the membrane via its MBD. In migrating cells, the CARMIL2 model is similar to CARMIL1 in that the CBR interacts with CP bound by V-1. This activates CP, allowing for barbed-end capping. (C) During CD28 costimulation to activate T-cells, membrane-localized CARMIL2 interacts with the adaptor protein GRB2, leading to downstream activation of CARMA1, PKCθ, and NF-κB activation.
In this model, CP is recruited to the membrane by CARMIL1. Binding of CARMIL1 to CP promotes dissociation of the CP inhibitor V-1, which activates CP for barbed-end capping. In addition, the fact that CP is bound to CARMIL1 provides for capping with kinetic rate constants and binding affinities that are relevant to the time scale of actin-based motility and the physiological concentrations of the reacting. Experimental evidence supports the existence of a pool of CP/V-1 complex in cells; most of the cellular population of CP is bound to V-1 (Fujiwara et al., 2014), and modeling studies suggest that a pool of CP diffuses slowly (McMillen and Vavylonis, 2016).
While at the membrane, CARMIL1 also interacts with the dual-GEF Trio, promoting signaling by RhoG, Rac1, and/or RhoA. The resulting increase in Rac1 activity promotes actin assembly by positively regulating the WAVE complex, creating a positive feedback loop for Arp2/3-mediated actin nucleation (Figure 3).
Model for CARMIL2.
Certain aspects of the model proposed for CARMIL2 function resemble those of CARMIL1, including the ability to recruit and regulate CP to promote cell migration, lamellipodial assembly, membrane ruffling, and macropinocytosis, based on the loss-of-function phenotypes for CARMIL1 and CARMIL2. Overlapping functions for CARMIL1 and CARMIL2 were also suggested by the observation that a chimera consisting of the PH domain of CARMIL1, the LRR domain of CARMIL2, and the C-Terminal half of CARMIL1 (including the CBR and MBD), rescues cellular phenotypes caused by the depletion of endogenous CARMIL2 (Lanier et al., 2015)
Transport of CARMILs 1 and 2 to the membrane are likely to differ. Only CARMIL1 associates with myosin 1E (Liang et al., 2009), and only CARMIL2 associates with dynamic vimentin filaments (Liang et al., 2009; Lanier et al., 2015). Thus vimentin filaments may carry CARMIL2 toward the membrane by interacting with its LRR domain (Figure 3), for which the biochemical mechanism is not yet understood.
At the membrane of migrating cells, CARMIL2 is proposed to contribute to actin assembly by regulating CP in the manner described for CARMIL1 (Figure 3). However, both CARMIL1 and CARMIL2 are necessary for lamellipodial dynamics and ruffling, and neither protein is able to rescue the loss of the other, revealing that certain aspects of their function must be distinct (Liang et al., 2009). CARMIL2 is known to differ from CARMIL1 in ways that are potentially important for regulation of actin assembly. Unlike CARMIL1, CARMIL2 does not contribute to Rac1 activation, but it does affect the level of expression of myosin-IIB (Liang et al., 2009).
CARMIL2 plays a scaffolding role in T-cell development, which does not require the function of the CPI motif and is thus independent of CP-based regulation of actin. Instead, this role requires the PH, LRR, and PRD domains of CARMIL2 (Roncagalli et al., 2016; Wang et al., 2016). This role may involve CARMIL2-interacting proteins such as transmembrane receptors (CD28 and CD8B), GEFs (VAV1 and DOCK8), phosphatases (PRPRF, PRPRC, and PTPN6), adaptor proteins (FYB, SIT1, GRAP2, GRB2, and Carma1), and a GAP (RASL3; Roncagalli et al., 2016). During CD28 costimulation of T-cells, CARMIL2 is recruited to the activated receptor and binds an adaptor protein (GRB2 or GRAP2). Here CARMIL2 serves as a binding partner for CARMA1, which activates protein kinase Cθ (PKCθ) and thus leads to NF-ΚB activation, thereby promoting the activation and proliferation of T-cells (Figure 3).
These models suggest that CARMIL1 and CARMIL2 activate NF-κB signaling; however, as described earlier, work in insects shows that CARMIL negatively regulates NF-κB (Di Prisco et al., 2013). In an alternative model that accounts for negative regulation of NF-κB, CARMIL removes CP from the barbed ends of filaments, allowing it to bind to V-1, thereby preventing V-1 from activating NF-κB (Bhattacharya et al., 2006).
SUMMARY, CONCLUSIONS, AND FUTURE DIRECTIONS
CARMILs are a family of large, multidomain proteins with important cellular functions involved in development, differentiation, and disease. All CARMILs bind to CP and regulate actin assembly in biochemical experiments with purified proteins; in cells, this interaction is important for the regulation of actin assembly and actin-based motility. CARMILs are often localized to membranes, where they regulate membrane-associated actin assembly. The roles of CARMILs as CP regulators have been documented relatively well; however, regulation of CP by CARMIL is likely to be complementary to and distinct from CP regulation by other CPI-motif proteins (Edwards et al., 2014). Exciting newer work has begun to uncover novel CP-independent roles for CARMILs.
CARMIL-family proteins are present in many eukaryotes, with the notable exceptions of plants and fungi. Vertebrates have three highly conserved genes that encode CARMILs, and these isoforms have cellular and biochemical functions that are overlap partially but also have distinct functions. For example, only CARMIL2 interacts with vimentin filaments within the cell, and it may mediate the effects of vimentin filaments on actin assembly at membranes.
It is likely that many functions of CARMILs remain to be identified. Indeed, the effects of mutations in model organisms and human patients have only recently come to light. In terms of human disease, mutations of CARMIL1 and CARMIL2 have been implicated in several different disorders. Much also remains to be learned about the functional implications of the interactions of CARMILs with known binding partners. Finally, the functions of many of the CARMILs domains are not yet known. This new information will be important for understanding the molecular mechanisms of a diverse set of biological and pathological processes, many of which depend on the function of the actin cytoskeleton.
Acknowledgments
We are grateful to David Sept (University of Michigan) for the modified version of the plot in Figure 2 and Alvaro Sagasti (UCLA) for communicating unpublished results. The writing of this article was supported by National Institutes of Health Grants GM R35118171 and GM R0195509.
Abbreviations used:
- Arp2/3
myosin-I linker protein
- CAH3
CARMIL homology domain 3
- CARMIL
capping protein, Arp2/3, myosin-I linker
- CBR
capping protein–binding region
- CP
capping protein
- CPI
capping protein interaction motif
- CSI
CARMIL-specific interaction motif
- GEF
guanine nucleotide exchange factor
- LRR
leucine-rich region
- MBD
membrane-binding domain
- PRD
proline-rich domain
- SH3
Src homology 3.
Footnotes
REFERENCES
- Bailly M, Macaluso F, Cammer M, Chan A, Segall JE, Condeelis JS. Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation. J Cell Biol. 1999;145:331–345. doi: 10.1083/jcb.145.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhtiarizadeh MR, Hosseinpour B, Arefnezhad B, Shamabadi N, Salami SA. In silico prediction of long intergenic non-coding RNAs in sheep. Genome. 2016;59:263–275. doi: 10.1139/gen-2015-0141. [DOI] [PubMed] [Google Scholar]
- Bella J, Hindle KL, McEwan PA, Lovell SC. The leucine-rich repeat structure. Cell Mol Life Sci. 2008;65:2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya N, Ghosh S, Sept D, Cooper JA. Binding of myotrophin/V-1 to actin-capping protein: implications for how capping protein binds to the filament barbed end. J Biol Chem. 2006;281:31021–31030. doi: 10.1074/jbc.M606278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombardier JP, Eskin JA, Jaiswal R, Corrêa IR, Xu M-Q, Goode BL, Gelles J. Single-molecule visualization of a formin-capping protein “decision complex” at the actin filament barbed end. Nat Commun. 2015;6:8707. doi: 10.1038/ncomms9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Goode BL. Formins at a glance. J Cell Sci. 2013;126:1–7. doi: 10.1242/jcs.107250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck S, Huber TB, Ingham RJ, Kim K, Niederstrasser H, Allen PM, Pawson T, Cooper JA, Shaw AS. Identification of a novel inhibitory actin-capping protein binding motif in CD2-associated protein. J Biol Chem. 2006;281:19196–19203. doi: 10.1074/jbc.M600166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzeska H, Guag J, Remmert K, Chacko S, Korn ED. An experimentally based computer search identifies unstructured membrane-binding sites in proteins: application to class I myosins, PAKS, and CARMIL. J Biol Chem. 2010;285:5738–5747. doi: 10.1074/jbc.M109.066910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton DA, Olsten MEK, Niederstrasser H, Cooper JA, Litchfield DW. The role of CKIP-1 in cell morphology depends on its interaction with actin-capping protein. J Biol Chem. 2006;281:36347–36359. doi: 10.1074/jbc.M607595200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Sept D. New insights into mechanism and regulation of actin capping protein. Int Rev Cell Mol Biol. 2008;267:183–206. doi: 10.1016/S1937-6448(08)00604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao TP, Majumdar A, Barrick D. Capping motifs stabilize the leucine-rich repeat protein PP32 and rigidify adjacent repeats. Protein Sci. 2014;23:801–811. doi: 10.1002/pro.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prisco G, Cavaliere V, Annoscia D, Varricchio P, Caprio E, Nazzi F, Gargiulo G, Pennacchio F. Neonicotinoid clothianidin adversely affects insect immunity and promotes replication of a viral pathogen in honey bees. Proc Natl Acad Sci USA. 2013;110:18466–18471. doi: 10.1073/pnas.1314923110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Liang Y, Kim T, Cooper JA. Physiological role of the interaction between CARMIL1 and capping protein. Mol Biol Cell. 2013;24:3047–3055. doi: 10.1091/mbc.E13-05-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, McConnell P, Schafer DA, Cooper JA. CPI motif interaction is necessary for capping protein function in cells. Nat Commun. 2015;6:8415. doi: 10.1038/ncomms9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, Cooper JA. Capping protein regulators fine-tune actin assembly dynamics. Nat Rev Mol Cell Biol. 2014;15:677–689. doi: 10.1038/nrm3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkhbayar P, Kamiya M, Osaki M, Matsumoto T, Matsushima N. Structural principles of leucine-rich repeat (LRR) proteins. Proteins. 2004;54:394–403. doi: 10.1002/prot.10605. [DOI] [PubMed] [Google Scholar]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT) Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, Lemmon MA. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell. 2000;6:373–384. doi: 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, Hammer JA. Direct observation of the uncapping of capping protein-capped actin filaments by CARMIL homology domain 3. J Biol Chem. 2010;285:2707–2720. doi: 10.1074/jbc.M109.031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, Piszczek G, Hammer JA. Capping protein regulatory cycle driven by CARMIL and V-1 may promote actin network assembly at protruding edges. Proc Natl Acad Sci USA. 2014;111:E1970–E1979. doi: 10.1073/pnas.1313738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- Gupta P, Gauthier NC, Cheng-Han Y, Zuanning Y, Pontes B, Ohmstede M, Martin R, Knölker H-J, Döbereiner H-G, Krendel M, et al. Myosin 1E localizes to actin polymerization sites in lamellipodia, affecting actin dynamics and adhesion formation. Biol Open. 2013;2:1288–1299. doi: 10.1242/bio.20135827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TJ, Sawyer JK, Peifer M. How the cytoskeleton helps build the embryonic body plan: models of morphogenesis from Drosophila. Curr Top Dev Biol. 2009;89:55–85. doi: 10.1016/S0070-2153(09)89003-0. [DOI] [PubMed] [Google Scholar]
- Heiss SG, Cooper JA. Regulation of CapZ, an actin capping protein of chicken muscle, by anionic phospholipids. Biochemistry. 1991;30:8753–8758. doi: 10.1021/bi00100a006. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Valladares M, Kim T, Kannan B, Tung A, Aguda AH, Larsson M, Cooper JA, Robinson RC. Structural characterization of a capping protein interaction motif defines a family of actin filament regulators. Nat Struct Mol Biol. 2010;17:497–503. doi: 10.1038/nsmb.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C-C, Chiang C-W, Cheng H-C, Chang W-T, Chou C-Y, Tsai H-W, Lee C-T, Wu Z-H, Lee T-Y, Chao A, et al. Identifying LRRC16B as an oncofetal gene with transforming enhancing capability using a combined bioinformatics and experimental approach. Oncogene. 2011;30:654–667. doi: 10.1038/onc.2010.451. [DOI] [PubMed] [Google Scholar]
- Huang M, Yang C, Schafer DA, Cooper JA, Higgs HN, Zigmond SH. Cdc42-induced actin filaments are protected from capping protein. Curr Biol. 1999;9:979–982. doi: 10.1016/s0960-9822(99)80428-x. [DOI] [PubMed] [Google Scholar]
- Huntley MA, Lou M, Goldstein LD, Lawrence M, Dijkgraaf GJP, Kaminker JS, Gentleman R. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics. 2016;17:61. doi: 10.1186/s12864-015-2291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Alexander CJ, Wu XS, Piszczek G, Chen B-C, Betzig E, Hammer JA. V-1 regulates capping protein activity in vivo. Proc Natl Acad Sci USA. 2016;113:E6610–E6619. doi: 10.1073/pnas.1605350113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Remmert K, Wu X, Volosky JM, Hammer JA. The Dictyostelium CARMIL protein links capping protein and the Arp2/3 complex to type I myosins through their SH3 domains. J Cell Biol. 2001;153:1479–1498. doi: 10.1083/jcb.153.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapushesky M, Adamusiak T, Burdett T, Culhane A, Farne A, Filippov A, Holloway E, Klebanov A, Kryvych N, Kurbatova N, et al. Gene Expression Atlas update–a value-added database of microarray and sequencing-based functional genomics experiments. Nucleic Acids Res. 2012;40:D1077–D1081. doi: 10.1093/nar/gkr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Cooper JA, Sept D. The interaction of capping protein with the barbed end of the actin filament. J Mol Biol. 2010;404:794–802. doi: 10.1016/j.jmb.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Ravilious GE, Sept D, Cooper JA. Mechanism for CARMIL protein inhibition of heterodimeric actin-capping protein. J Biol Chem. 2012;287:15251–15262. doi: 10.1074/jbc.M112.345447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J. Mechanism of ribonuclease inhibition by ribonuclease inhibitor protein based on the crystal structure of its complex with ribonuclease A. J Mol Biol. 1996;264:1028–1043. doi: 10.1006/jmbi.1996.0694. [DOI] [PubMed] [Google Scholar]
- Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M, Mangino M, Albrecht E, Wallace C, Farrall M, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5:e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger I, Kostyukova A, Yamashita A, Nitanai Y, Maéda Y. Crystal structure of the C-terminal half of tropomodulin and structural basis of actin filament pointed-end capping. Biophys J. 2002;83:2716–2725. doi: 10.1016/S0006-3495(02)75281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn JR, Pollard TD. Single molecule kinetic analysis of actin filament capping. Polyphosphoinositides do not dissociate capping proteins. J Biol Chem. 2007;282:28014–28024. doi: 10.1074/jbc.M705287200. [DOI] [PubMed] [Google Scholar]
- Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier MH, Kim T, Cooper JA. CARMIL2 is a novel molecular connection between vimentin and actin essential for cell migration and invadopodia formation. Mol Biol Cell. 2015;26:4577–4588. doi: 10.1091/mbc.E15-08-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier MH, McConnell P, Cooper JA. Cell migration and invadopodia formation require a membrane-binding domain of CARMIL2. J Biol Chem. 2016;291:1076–1091. doi: 10.1074/jbc.M115.676882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007;74:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pleskot R, Henty-Ridilla JL, Blanchoin L, Potocký M, Staiger CJ. Arabidopsis capping protein senses cellular phosphatidic acid levels and transduces these into changes in actin cytoskeleton dynamics. Plant Signal Behav. 2012;7:1727–1730. doi: 10.4161/psb.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Cucchetti M, Roncagalli R, Yokosuka T, Malzac A, Bertosio E, Imbert J, Nijman IJ, Suchanek M, Saito T, et al. The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nat Immunol. 2013;14:858–866. doi: 10.1038/ni.2634. [DOI] [PubMed] [Google Scholar]
- Liang Y, Niederstrasser H, Edwards M, Jackson CE, Cooper JA. Distinct roles for CARMIL isoforms in cell migration. Mol Biol Cell. 2009;20:5290–5305. doi: 10.1091/mbc.E08-10-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Marcos-Ramiro B, García-Weber D, Millán J. TNF-induced endothelial barrier disruption: beyond actin and Rho. Thromb Haemost. 2014;112:1088–1102. doi: 10.1160/TH14-04-0299. [DOI] [PubMed] [Google Scholar]
- Matsuzaka Y, Okamoto K, Mabuchi T, Iizuka M, Ozawa A, Oka A, Tamiya G, Kulski JK, Inoko H. Identification, expression analysis and polymorphism of a novel RLTPR gene encoding a RGD motif, tropomodulin domain and proline/leucine-rich regions. Gene. 2004;343:291–304. doi: 10.1016/j.gene.2004.09.004. [DOI] [PubMed] [Google Scholar]
- McMillen LM, Vavylonis D. Model of turnover kinetics in the lamellipodium: implications of slow- and fast-diffusing capping protein and Arp2/3 complex. Phys Biol. 2016;13:066009. doi: 10.1088/1478-3975/13/6/066009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierke CT. Physical break-down of the classical view on cancer cell invasion and metastasis. Eur J Cell Biol. 2013;92:89–104. doi: 10.1016/j.ejcb.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Miyoshi T, Tsuji T, Higashida C, Hertzog M, Fujita A, Narumiya S, Scita G, Watanabe N. Actin turnover-dependent fast dissociation of capping protein in the dendritic nucleation actin network: evidence of frequent filament severing. J Cell Biol. 2006;175:947–955. doi: 10.1083/jcb.200604176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita A, Takeda S, Yamashita A, Maéda Y. Structural basis of actin filament capping at the barbed-end: a cryo-electron microscopy study. EMBO J. 2006;25:5626–5633. doi: 10.1038/sj.emboj.7601395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos V, Co C, Prehoda KE, Snapper S, Taunton J, Lim WA. A polybasic motif allows N-WASP to act as a sensor of PIP(2) density. Mol Cell. 2005;17:181–191. doi: 10.1016/j.molcel.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E, Huber W, Jupp S, Keays M, Kryvych N, et al. Expression Atlas update—a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42:D926–D932. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Actin and actin-binding proteins. Cold Spring Harb Perspect Biol. 2016;8:a018226. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert K, Olszewski TE, Bowers MB, Dimitrova M, Ginsburg A, Hammer JA. CARMIL is a bona fide capping protein interactant. J Biol Chem. 2004;279:3068–3077. doi: 10.1074/jbc.M308829200. [DOI] [PubMed] [Google Scholar]
- Roncagalli R, Cucchetti M, Jarmuzynski N, Grégoire C, Bergot E, Audebert S, Baudelet E, Menoita MG, Joachim A, Durand S, et al. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J Exp Med. 2016;213:2437–2457. doi: 10.1084/jem.20160579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakiyama M, Matsuo H, Shimizu S, Chiba T, Nakayama A, Takada Y, Nakamura T, Takada T, Morita E, Naito M, et al. Common variant of leucine-rich repeat-containing 16A (LRRC16A) gene is associated with gout susceptibility. Hum Cell. 2014;27:1–4. doi: 10.1007/s13577-013-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, Weisser T, Fehlner K, Mautner J, Walz C, et al. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat Commun. 2017;8:14209. doi: 10.1038/ncomms14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar S, Kerleau M, Kühn S, Pernier J, Romet-Lemonne G, Jégou A, Carlier M-F. Formin and capping protein together embrace the actin filament in a ménage à trois. Nat Commun. 2015;6:8730. doi: 10.1038/ncomms9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar S, Pernier J, Carlier M-F. Regulators of actin filament barbed ends at a glance. J Cell Sci. 2016;129:1085–1091. doi: 10.1242/jcs.179994. [DOI] [PubMed] [Google Scholar]
- Sorte HS, Osnes LT, Fevang B, Aukrust P, Erichsen HC, Backe PH, Abrahamsen TG, Kittang OB, Øverland T, Jhangiani SN, et al. A potential founder variant in CARMIL2/RLTPR in three Norwegian families with warts, molluscum contagiosum, and T-cell dysfunction. Mol Genet Genomic Med. 2016;4:604–616. doi: 10.1002/mgg3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark BC, Cooper JA. Differential expression of CARMIL-family genes during zebrafish development. Cytoskeleton. 2015;72:534–541. doi: 10.1002/cm.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Minakata S, Koike R, Kawahata I, Narita A, Kitazawa M, Ota M, Yamakuni T, Maéda Y, Nitanai Y. Two distinct mechanisms for actin capping protein regulation–steric and allosteric inhibition. PLoS Biol. 2010;8:e1000416. doi: 10.1371/journal.pbio.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Wang H, Gish GD, Petsalaki E, Pasculescu A, Shi Y, Mollenauer M, Bagshaw RD, Yosef N, Hunter T, et al. Combinatorial proteomic analysis of intercellular signaling applied to the CD28 T-cell costimulatory receptor. Proc Natl Acad Sci USA. 2015;112:E1594–E1603. doi: 10.1073/pnas.1503286112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh LA, Hochgreb T, Graham M, Wu D, Ruf-Zamojski F, Jayasena CS, Saxena A, Hawk R, Gonzales-Serricchio A, Dixson A, et al. A versatile gene trap to visualize and interrogate the function of the vertebrate proteome. Genes Dev. 2011;25:2306–2320. doi: 10.1101/gad.174037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Uruno T, Remmert K, Hammer JA. CARMIL is a potent capping protein antagonist: identification of a conserved CARMIL domain that inhibits the activity of capping protein and uncaps capped actin filaments. J Biol Chem. 2006;281:10635–10650. doi: 10.1074/jbc.M513186200. [DOI] [PubMed] [Google Scholar]
- Vanderzalm PJ, Pandey A, Hurwitz ME, Bloom L, Horvitz HR, Garriga G. C. elegans CARMIL negatively regulates UNC-73/Trio function during neuronal development. Development. 2009;136:1201–1210. doi: 10.1242/dev.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walck-Shannon E, Reiner D, Hardin J. Polarized Rac-dependent protrusions drive epithelial intercalation in the embryonic epidermis of C. elegans. Development. 2015;142:3549–3560. doi: 10.1242/dev.127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ma CS, Ling Y, Bousfiha A, Camgioglu Y, Jacquot S, Payne K, Crestani E, Roncagalli R, Belkadi A, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 2016;213:2413–2435. doi: 10.1084/jem.20160576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Tejera P, Wang Z, Zhang R, Chen F, Su L, Lin X, Bajwa EK, Thompson BT, Christiani DC. A missense genetic variant in LRRC16A/CARMIL1 improves acute respiratory distress syndrome survival by attenuating platelet count decline. Am J Respir Crit Care Med. 2017;195:1353–1361. doi: 10.1164/rccm.201605-0946OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Wang Z, Su L, Chen F, Tejera P, Bajwa EK, Wurfel MM, Lin X, Christiani DC. Platelet count mediates the contribution of a genetic variant in LRRC 16A to ARDS risk. Chest. 2015;147:607–617. doi: 10.1378/chest.14-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Mitchelhill KI, Kobe B, Kemp BE, Zot HG. The myosin-I-binding protein Acan125 binds the SH3 domain and belongs to the superfamily of leucine-rich repeat proteins. Proc Natl Acad Sci USA. 1997;94:3685–3690. doi: 10.1073/pnas.94.8.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Zot AS, Zot HG. Identification of Acan125 as a myosin-I-binding protein present with myosin-I on cellular organelles of Acanthamoeba. J Biol Chem. 1995;270:25316–25319. doi: 10.1074/jbc.270.43.25316. [DOI] [PubMed] [Google Scholar]
- Yang C, Pring M, Wear MA, Huang M, Cooper JA, Svitkina TM, Zigmond SH. Mammalian CARMIL inhibits actin filament capping by capping protein. Dev Cell. 2005;9:209–221. doi: 10.1016/j.devcel.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bruck S, Cemerski S, Zhang L, Butler B, Dani A, Cooper JA, Shaw AS. CD2AP links cortactin and capping protein at the cell periphery to facilitate formation of lamellipodia. Mol Cell Biol. 2013;33:38–47. doi: 10.1128/MCB.00734-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zot HG, Bhaskara V, Liu L. Acan125 binding to the SH3 domain of acanthamoeba myosin-IC. Arch Biochem Biophys. 2000;375:161–164. doi: 10.1006/abbi.1999.1648. [DOI] [PubMed] [Google Scholar]
- Zwolak A, Fujiwara I, Hammer JA, Tjandra N. Structural basis for capping protein sequestration by myotrophin (V-1) J Biol Chem. 2010a;285:25767–25781. doi: 10.1074/jbc.M110.135848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Uruno T, Piszczek G, Hammer JA, Tjandra N. Molecular basis for barbed end uncapping by CARMIL homology domain 3 of mouse CARMIL-1. J Biol Chem. 2010b;285:29014–29026. doi: 10.1074/jbc.M110.134221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Yang C, Feeser EA, Ostap EM, Svitkina T, Dominguez R. CARMIL leading edge localization depends on a non-canonical PH domain and dimerization. Nat Commun. 2013;4:2523. doi: 10.1038/ncomms3523. [DOI] [PMC free article] [PubMed] [Google Scholar]