

Abstract
Background
Horizontal gene transfer is central to evolution in most bacterial species. The detection of exchanged regions is often based upon analysis of compositional characteristics and their comparison to the organism as a whole. In this study we describe a new methodology combining aspects of established signature analysis with textual analysis approaches. This approach has been used to analyze the two available genome sequences of H. pylori.
Results
This gene-by-gene analysis reveals a wide range of genes related to both virulence behaviour and the strain differences that have been relatively recently acquired from other sequence backgrounds. These frequently involve single genes or small numbers of genes that are not associated with transposases or bacteriophage genes, nor with inverted repeats typically used as markers for horizontal transfer. In addition, clear examples of horizontal exchange in genes associated with 'core' metabolic functions were identified, supported by differences between the sequenced strains, including: ftsK, xerD and polA. In some cases it was possible to determine which strain represented the 'parent' and 'altered' states for insertion-deletion events. Different signature component lengths showed different sensitivities for the detection of some horizontally transferred genes, which may reflect different amelioration rates of sequence components.
Conclusion
New implementations of signature analysis that can be applied on a gene-by-gene basis for the identification of horizontally acquired sequences are described. These findings highlight the central role of the availability of homologous substrates in evolution mediated by horizontal exchange, and suggest that some components of the supposedly stable 'core genome' may actually be favoured targets for integration of foreign sequences because of their degree of conservation.
Background
Helicobacter pylori is a bacterial pathogen associated with gastritis, peptic ulcers, gastric adenocarcinoma, and rare lymphomas [1]. It has a highly panmictic population structure in which homologous recombination makes the predominant contribution to sequence differences within a highly diverse population structure [2]. The acquisition of genes from other strains and species is by far the most rapid evolutionary process. This occurs frequently without loss of existing functions, is central to the evolution of niche-adaptive and pathogenic characteristics of bacteria, and greatly influences inter-strain differences in gene complement [3-5]. In this context, it is notable that none of the traits typically used to differentiate E. coli from Salmonella can be attributed to point mutation genes but are broadly attributable to horizontal exchange [6]. H. pylori is relatively unusual in that it is a naturally transformable Gram-negative species that does not appear to have a species-specific DNA uptake sequence and appears to rely upon its niche separation as a transformation barrier [7]. Disease associated H. pylori strains have been divided into two types, type I being those that carry the cag pathogenicity island [8] (cag PAI), which has a foreign species origin, and are associated with more severe disease.
Dinucleotide composition is highly stable within a genome and can distinguish between sequences from different species. Based upon its constancy the species composition is referred to as a 'genome signature' [9,10]. This characteristic has been applied to assessments of DNA metabolic processes such as methylation and base conversion, DNA structure, and evolutionary relationships. It has also become established as a method for the identification of sequences that have been acquired by inter-species horizontal transfer. For example, lateral transfer has recently been shown using these methods for a tryptophan pathway operon [11], the gain of additional metabolic functions in Pseudomonas putida [12], a determination that many gain of function genes have been acquired by E. coli rather than lost from S. typhi [13], and more recently developed Bayesian methods based upon similar premises have been used to assess global signatures and determine the origins of some lateral transfer events [14,15]. However there are problems associated with this and other methods that use progressive 'walking windows', and the larger the window the greater the problems. These result from the inclusion of intergenic sequence, the inability to distinguish divergences due to a single highly divergent gene from that from a cluster of less divergent ones, and an inability to identify the limits of the abnormal regions. In practice additional features are necessary to determine the ends of such regions, such as the location of repeats typical of pathogenicity islands in H. pylori [16], or comparisons with other sequences as in N. meningitidis strain MC58 [17]. In addition, divergence scores are influenced by the size of the sampling window used such that sampling effects limit analysis of sequences shorter than about 800 bp (data not presented), and the need to use fixed window sizes prevents gene by gene studies.
We describe the use of a linear implementation of signature analysis that can efficiently address a range of walking window sizes using dinucleotide signatures (DNS) and longer signatures. In addition, use of a new approach based upon classical text analysis that allows analysis of genomes gene-by-gene is described. Analysis of H. pylori sequences, combined with comparisons of the identified genes between genomes, reveals complex changes that influence both niche-adaptive and core functions illustrating a previously unpredicted range of functions which are continuously undergoing variation and selection.
Results and discussion
Genes were ranked on the basis of their divergence from the mean genome composition. The degree of divergence that is indicative of acquisition from other species is not an absolute. The frequency with which genes are acquired, the untypicality of the donated material, and the rate at which they are ameliorated to the host sequence composition influence it. Strains J99 and 26695 had 53 (Table 1) and 60 (Table 4) genes respectively with DNS that were >2 SD from the mean. Those with annotated functions included genes from the cag pathogenicity island (6 and 5), vac and related toxins (3 and 4), and restriction-modification genes (2 and 4). On the basis of the similarities determined in the H. pylori strain J99 sequence annotation, 7 of the most divergent genes as determined by DNS are not present in strain 26695. Likewise, 2 of the 50 most divergent genes in strain 26695 are not present in strain J99. This is consistent with the identification of genes acquired from other species that have not extended to both sequenced strains. It also suggests that a significant proportion of the 6 to 7% of genes unique to one or other strain [18] are inherent to the Helicobacter gene pool, but are variably present in different strains rather than reflecting recent foreign origins. Comparisons of a selection of identified orthologous genes in the two strains are shown in Figure 1.
Table 1.
The 53 most divergent (>2 SD) genes in H. pylori strain J99 by DNS showing their ranking in strain 26695 and in TNS and HNS analysis
| DNS order | JHP # | annotation | 26695 # | 26695 DNS order | TNS order | HNS order |
| 1 | JHP0952 | hypothetical protein | HP0427 | 14 | 3 | 1355 |
| 2 | JHP0476 | cag pathogenicity island protein (cag7) | HP0527 | 1 | 2 | 2 |
| 3 | JHP0556 | vacuolating cytotoxin (vacA) paralog | HP0609/10 | 4/13 | 5 | 4 |
| 4 | JHP0274 | vacuolating cytotoxin (vacA) paralog | HP0289 | 2 | 6 | 5 |
| 5 | JHP0305 | hypothetical protein | HP0322 | 3 | 8 | 10 |
| 6 | JHP0942 | hypothetical protein | HP0996 | 5 | 13 | 27 |
| 7 | JHP0856 | vacuolating cytotoxin (vacA) paralog | HP0922 | 6 | 9 | 6 |
| 8 | JHP0050 | hypothetical protein | HP0058 | 88 | 7 | 84 |
| 9 | JHP1300 | hypothetical protein | HP1408 | 15 | 1 | 1 |
| 10 | JHP1044 | hypothetical protein | HP1116 | 8 | 14 | 8 |
| 11 | JHP0928 | hypothetical protein | NAH | - | 12 | 9 |
| 12 | JHP0074 | hypothetical protein | HP0080 | 9 | 32 | 125 |
| 13 | JHP0440 | hypothetical protein | HP0488 | 7 | 16 | 17 |
| 14 | JHP1042 | hypothetical protein | HP1115 | 20 | 25 | 694 |
| 15 | JHP1321 | histidine and glutamine-rich metal-binding protein | HP1432 | 46 | 4 | 49 |
| 16 | JHP0934 | hypothetical protein | NAH | - | 15 | 95 |
| 17 | JHP0495 | cag island protein (cagA) | HP0547 | 31 | 20 | 12 |
| 18 | JHP0931 | topoisomerase I (topA 3) | NAH | - | 18 | 20 |
| 19 | JHP0693 | hypothetical protein | HP0756 | 24 | 59 | 1490 |
| 20 | JHP0632 | N-methylhydantoinase | HP0696 | 19 | 44 | 36 |
| 21 | JHP0471 | cag pathogenicity island protein (cag3) | HP0522 | 11 | 35 | 62 |
| 22 | JHP0438 | outer membrane protein | HP0486 | 26 | 67 | 145 |
| 23 | JHP0026 | hypothetical protein | HP0030 | 45 | 36 | 64 |
| 24 | JHP1084 | outer membrane protein (omp26) | HP1157 | 34 | 17 | 24 |
| 25 | JHP0481 | cag island protein (cagT) | HP0532 | 23 | 70 | 558 |
| 26 | JHP0052 | hypothetical protein | HP0059 | 43 | 24 | 120 |
| 27 | JHP0336 | hypothetical protein | HP1089 | 12 | 51 | 54 |
| 28 | JHP1426 | iron(III) dicitrate transport protein (fecA) | HP1400 | 32 | 78 | 111 |
| 29 | JHP0174 | hypothetical protein | HP0187 / 8 / 6 | 47&1127&596 | 88 | 90 |
| 30 | JHP1297 | type III restriction enzyme (res) | NAH | - | 63 | 28 |
| 31 | JHP0953 | hypothetical protein | NAH | - | 26 | 1463 |
| 32 | JHP0067 | urease beta subunit (urea amidohydrolase) (ureB) | HP0072 | 21 | 37 | 70 |
| 33 | JHP0941 | integrase/recombinase (xerD) | HP0995 | 25 | 100 | 541 |
| 34 | JHP0548 | flagellin A (flaA) | HP0601 | 33 | 40 | 154 |
| 35 | JHP0299 | hypothetical protein | HP061/2 | 230&765 | 11 | 275 |
| 36 | JHP1033 | hypothetical protein | HP1106 | 59 | 262 | 342 |
| 37 | JHP1409 | type II restriction enzyme (methyltransferase) | NAH | - | 55 | 15 |
| 38 | JHP0626 | iron(III) dicitrate transport protein (fecA) | HP0686 | 62 | 89 | 47 |
| 39 | JHP0940 | hypothetical protein | NAH | - | 53 | 393 |
| 40 | JHP1253 | hypothetical protein | HP1333 | 40 | 75 | 384 |
| 41 | JHP0132 | cytochrome oxidase (cbb3 type) (fixN) | HP0144 | 27 | 206 | 209 |
| 42 | JHP0842 | hypothetical protein | HP0906 | 42 | 29 | 21 |
| 43 | JHP0925 | hypothetical protein | NAH | - | 130 | 990 |
| 44 | JHP0613 | hypothetical protein | HP0669 | 69 | 42 | 33 |
| 45 | JHP0565 | DNA mismatch repair protein (mutS) | HP0621 | 22 | 227 | 82 |
| 46 | JHP1363 | DNA polymerase I (polA) | HP1470 | 30 | 81 | 46 |
| 47 | JHP0489 | cag island protein (cagH) | HP0541 | 71 | 137 | 398 |
| 48 | JHP1260 | siderophore-mediated iron transport protein (tonB) | HP1341 | 85 | 1260 | 402 |
| 49 | JHP0492 | DNA transfer protein (cagE) | HP0544 | 104 | 95 | 50 |
| 50 | JHP1121 | DNA-directed RNA polymerase, beta subunit (rpoB) | HP1198 | 84 | 23 | 16 |
| 51 | JHP1434 | DNA repair protein (recN) | HP1393 | 35 | 177 | 160 |
| 52 | JHP0491 | cag island protein (cagF) | HP0543 | 82 | 170 | 828 |
| 53 | JHP0191 | hypothetical protein | HP0205 | 57 | 33 | 7 |
Genes with > 2 SD divergence indicated in bold
NAH indicates No Annotated Homologue in the other sequence
Table 4.
Top 60 most divergent (>2 SD) genes by DNS in H. pylori strain 26695 plus those additional genes in the top 50 genes from TNS and HNS
| DNS order | annotation | HP# | J99 # | J99 DNS order | TNS order | HNS order |
| 1 | cag pathogenicity island protein (cag7) | HP0527 | JHP0476 | 2 | 1 | 1 |
| 2 | vacuolating cytotoxin (vacA) paralog | HP0289 | JHP0274 | 4 | 2 | 4 |
| 3 | poly E-rich hypothetical protein | HP0322 | JHP0305 | 5 | 8 | 5 |
| 4 | hypothetical protein | HP0609 | JHP0556* | 3 | 6 | 9 |
| 5 | hypothetical protein | HP0996 | JHP0942 | 6 | 14 | 46 |
| 6 | vacuolating cytotoxin (vacA) paralog | HP0922 | JHP0856 | 7 | 5 | 3 |
| 7 | hypothetical protein | HP0488 | JHP0440 | 13 | 10 | 12 |
| 8 | hypothetical protein | HP1116 | JHP1044 | 10 | 11 | 13 |
| 9 | hypothetical protein | HP0080 | JHP0074 | 12 | 18 | 122 |
| 10 | hypothetical protein | HP0489 | JHP0441 | 115 | 36 | 582 |
| 11 | cag pathogenicity island protein (cag3) | HP0522 | JHP0471 | 21 | 48 | 100 |
| 12 | hypothetical protein | HP1089 | JHP0336 | 27 | 67 | 59 |
| 13 | vacuolating cytotoxin (vacA) paralog | HP0610 | JHP0556* | 3 | 12 | 17 |
| 14 | hypothetical protein | HP0427 | JHP0952 | 1 | 3 | 737 |
| 15 | hypothetical protein | HP1408 | JHP1300 | 9 | 4 | 738 |
| 16 | type III restriction enzyme R protein (res) | HP0592 | NAH | - | 30 | 35 |
| 17 | hypothetical protein | HP0119 | NAH | - | 7 | 2 |
| 18 | vacuolating cytotoxin (vacA) | HP0887 | JHP0819 | 59 | 25 | 34 |
| 19 | N-methylhydantoinase | HP0696 | JHP0632 | 20 | 35 | 43 |
| 20 | hypothetical protein | HP1115 | JHP1042 | 14 | 33 | 866 |
| 21 | urease beta subunit (urea amidohydrolase) (ureB) | HP0072 | JHP0067 | 32 | 38 | 87 |
| 22 | DNA mismatch repair protein (MutS) | HP0621 | JHP0565 | 45 | 137 | 64 |
| 23 | cag island protein (cagT) | HP0532 | JHP0481 | 25 | 87 | 693 |
| 24 | hypothetical protein | HP0756 | JHP0693 | 19 | 71 | 1548 |
| 25 | integrase/recombinase (xerD) | HP0995 | JHP0941 | 33 | 39 | 448 |
| 26 | outer membrane protein | HP0486 | JHP0438 | 22 | 147 | 142 |
| 27 | cytochrome oxidase (cbb3 type) (fixN) | HP0144 | JHP0132 | 41 | 102 | 168 |
| 28 | type IIS restriction enzyme R and M protein (ECO57IR) | HP1517 | NAH | - | 42 | 14 |
| 29 | DNA transfer protein (cagE) | HP0441 | JHP0492 | 49 | 51 | 22 |
| 30 | DNA polymerase I (polA) | HP1470 | JHP1363 | 46 | 77 | 54 |
| 31 | cag island protein (cagA) | HP0547 | JHP0495 | 17 | 15 | 7 |
| 32 | iron(III) dicitrate transport protein (fecA) | HP1400 | JHP1426 | 28 | 99 | 129 |
| 33 | flagellin A (flaA) | HP0601 | JHP0548 | 34 | 40 | 180 |
| 34 | outer membrane protein (omp26) | HP1157 | JHP1084 | 24 | 17 | 25 |
| 35 | DNA repair protein (recN) | HP1393 | JHP1434 | 51 | 154 | 207 |
| 36 | type I restriction enzyme R protein (hsdR) | HP0464 | NAH | - | 90 | 26 |
| 37 | cell division protein (ftsK) | HP1090 | JHP0335 | 67 | 181 | 90 |
| 38 | hypothetical protein | HP1003 | NAH | - | 61 | 170 |
| 39 | histidine-rich, metal binding polypeptide (hpn) | HP1427 | NAH | - | 26 | 1449 |
| 40 | hypothetical protein | HP1333 | JHP1253 | 40 | 53 | 296 |
| 41 | hypothetical protein | HP0788 | JHP0725 | 68 | 72 | 256 |
| 42 | hypothetical protein | HP0906 | JHP0842 | 42 | 22 | 16 |
| 43 | hypothetical protein | HP0059 | JHP0052 | 26 | 21 | 320 |
| 44 | GMP reductase (guaC) | HP0854 | JHP0790 | 107 | 169 | 451 |
| 45 | hypothetical protein | HP0030 | JHP0026 | 23 | 24 | 39 |
| 46 | histidine and glutamine-rich metal-binding protein | HP1432 | JHP1321 | 15 | 9 | 1432 |
| 47 | hypothetical protein | HP0186 | JHP0174 | 29 | 130 | 276 |
| 48 | fucosyltransferase | HP0651 | JHP0596 | 105 | 43 | 75 |
| 49 | translation elongation factor EF-Tu (tufB) | HP1205 | JHP1128 | 81 | 64 | 166 |
| 50 | virulence associated protein homolog (vacB) | HP1248 | JHP1169 | 79 | 164 | 160 |
| 51 | hypothetical protein | HP0449 | NAH | - | 81 | 449 |
| 52 | type III restriction enzyme R protein | HP1371 | JHP1285 | 55 | 119 | 23 |
| 53 | virB4 homolog (virB4) | HP0459 | NAH | - | 49 | 28 |
| 54 | 2',3'-cyclic-nucleotide 2'-phosphodiesterase (cpdB) | HP0104 | JHP0096 | 56 | 73 | 68 |
| 55 | hypothetical protein | HP1479 | JHP1372 | 135 | 153 | 127 |
| 56 | RNA polymerase sigma-70 factor (rpoD) | HP0088 | JHP0081 | 62 | 55 | 31 |
| 57 | hypothetical protein | HP0205 | JHP0191 | 53 | 78 | 8 |
| 58 | hypothetical protein | HP1143 | JHP1071 | 78 | 29 | 41 |
| 59 | hypothetical protein | HP1106 | JHP1033 | 36 | 272 | 277 |
| 60 | cag pathogenicity island protein (cag13) | HP0534 | JHP0482 | 71 | 225 | 1021 |
| 63 | DNA topoisomerase I (topA) | HP0440 | NAH | - | 149 | 24 |
| 68 | outer membrane protein (omp3) | HP0079 | JHP0073 | 796 | 45 | 99 |
| 69 | hypothetical protein | HP0669 | JHP0613 | 44 | 60 | 42 |
| 74 | cag pathogenicity island protein (cag8) | HP0528 | JHP0477 | 72 | 50 | 27 |
| 75 | hypothetical protein | HP0453 | NAH | - | 58 | 10 |
| 84 | DNA-directed RNA polymerase, beta subunit (rpoB) | JHP1121 | 50 | 23 | 19 | |
| 91 | hypothetical protein | HP1142 | JHP1070 | 60 | 19 | 6 |
| 97 | multidrug resistance protein (spaB) | HP0600 | JHP0547 | 75 | 41 | 30 |
| 103 | type I restriction enzyme R protein (hsdR) | HP1402 | JHP1424 | 195 | 86 | 21 |
| 109 | adenine/cytosine DNA methyltransferase | HP0054 | NAH | - | 120 | 20 |
| 119 | preprotein translocase subunit (secA) | HP0786 | JHP0723 | 159 | 176 | 49 |
| 121 | hypothetical protein | HP0058 | JHP0051 | 394 | 16 | 53 |
| 122 | hypothetical protein | HP0513 | JHP0462 | 104 | 28 | 15 |
| 125 | type I restriction enzyme M protein (hsdM) | HP1403 | JHP1423 | 299 | 340 | 44 |
| 132 | hypothetical protein | HP0731 | JHP0668 | 110 | 80 | 32 |
| 139 | hypothetical protein | HP0508 | JHP0458 | 84 | 32 | 77 |
| 142 | hypothetical protein | HP1187 | JHP1113 | 274 | 31 | 38 |
| 167 | hypothetical protein | HP1520 | NAH | - | 20 | 33 |
| 179 | hypothetical protein | HP0118 | JHP0110 | 64 | 27 | 36 |
| 195 | type III restriction enzyme R protein (res) | HP1521 | JHP1410 | 161 | 210 | 18 |
| 209 | outer membrane protein (omp17) | HP0725 | JHP0662 | 257 | 47 | 101 |
| 224 | hypothetical protein | HP0733 | JHP0670 | 769 | 222 | 48 |
| 230 | hypothetical protein | HP0611 | JHP0299 | 35 | 37 | 1129 |
| 249 | hypothetical protein | HP0345 | NAH | - | 46 | 1338 |
| 283 | hypothetical protein | HP0120 | NAH | - | 44 | 50 |
| 291 | translation initiation factor IF-2 (infB) | HP1048 | JHP0377 | 330 | 332 | 45 |
| 297 | DNA polymerase III alpha-subunit (dnaE) | HP1460 | JHP1353 | 509 | 219 | 47 |
| 342 | type I restriction enzyme R protein (hsdR) | HP0846 | JHP0784 | 244 | 101 | 37 |
| 363 | adenine specific DNA methyltransferase (mod) | HP1522 | JHP1411 | 857 | 207 | 11 |
| 410 | secreted protein involved in flagellar motility | HP1192 | JHP1117 | 614 | 13 | 1256 |
| 593 | hypothetical protein | HP1516 | NAH | - | 34 | 1090 |
| 631 | hypothetical protein | HP0586 | JHP0534 | 577 | 163 | 29 |
| 1080 | type II restriction enzyme (methyltransferase) | HP0478 | JHP0430 | 953 | 220 | 40 |
* probably frame shifted components of the same vacA related gene
Genes with > 2 SD divergence in each analysis are indicated in bold
NAH indicates No Annotated Homologue in the other sequence
Figure 1.

Comparisons using LAlign between a representative selection of orthologous genes with divergent DNA present in both H. pylori strains J99 and 26695 (presented in descending order of divergence as determined in strain J99).
It cannot be assumed that all genes identified in this manner have been recently acquired. It is necessary to assess the nature of the sequence to determine if its divergence might be accounted for on the basis of features of the encoded protein. For example, JHP0476/HP0527, JHP1300/HP1408 and JHP0074/HP0080 include repetitive sequences likely to account for their DNS divergence. This type of analysis cannot be used to determine the possible foreign origin of such genes. Notably, the most divergent cag PAI gene (the 1st and 2nd most divergent gene in the whole genomes of strain 26695 and J99 respectively, JHP0476/HP0527) has a highly complex repetitive structure and the size of the large divergent peak associated with this island using previous methods is largely due to the presence of this gene.
While a significant proportion of the genes identified in this analysis are associated with regions including several such genes and which share characteristics of islands of horizontal transfer or pathogenicity islands, this is far from universally true. There are many instances of single genes or small numbers of genes that are present that are not associated with any features that might otherwise have been used as indicators of horizontal acquisition such as transposases and flanking repeats.
Our initial goal was to identify recently acquired and exchanged genes as candidates likely to be important in niche-adaptation, host interactions, and alterations in bacterial fitness. It has been argued that essential genes are unlikely to be transferred successfully since recipient taxa would already bear functional orthologues, which would have experienced long-term co-evolution with the rest of the cellular machinery. In contrast, it is proposed that those under weak or transient selection – like those associated with nonessential catabolic processes, new operons, and those providing new niche-adaptive changes are likely to be successfully transferred and retained [19]. This leads to a model in which a stable 'core genome' comprised of essential metabolic, regulatory, and cell division genes provides a stable context for the more labile non-essential and niche adaptive genes. On this basis such genes are used for phylogenetic studies and are thought to provide a relatively constant background in which species evolution occurs. Many of the genes identified for which functions are known affect virulence or niche adaptive genes, including: the vacuolating cytotoxin and related toxins (2 and 3), urease and flagellar components, and genes involved in iron acquisition. However, we also find clear evidence, confirmed by differences between the two genome sequences, that recent, and therefore relatively frequent, horizontal transfer is not limited to genes associated with niche adaptation and virulence. Amongst the core function genes identified were mutS, ftsK, xerD, and polA. The comparisons of the latter three between the sequence strains are shown in Figure 1f,g &1j. These comparisons support the results suggesting that these genes have been the substrates for horizontal exchange between species.
Tetranucleotide composition has been used for the consideration of the presence of palindromic sequences that might be substrates for restriction systems and Chi sites and the presence of unstable repeats mediating phase variation [10], but the use of longer component signatures has not been used to identify horizontally acquired regions in bacterial genomes. Following analysis of eukaryotic sequences it was concluded that DNS captures most of the departure from randomness in DNA sequences and that longer component lengths correlate highly with the DNS results [20]. Also, analysis of dinucleotides separated by no, one, or two other nucleotides showed that separated pairs are more nearly random than adjacent pairs and were concluded to be relatively uninformative [9]. However, in preliminary analyses, while results using the typically long walking windows gave concordant results as previously reported, we found that the use of smaller walking windows generated progressively more different patterns of divergence with other length components. Using tetranucleotide (TNS) and hexanucleotide (HNS) signature analysis we find that, while in some instances there is significant overlap between the genes identified using the different component lengths, there are substantial differences that indicate additional horizontally transferred genes not identified by DNS alone (Tables 2 to 6).
Table 2.
Top 50 most divergent genes by TNS in H. pylori strain J99 plus those additional genes > 2 SD greater than the mean by DNS and the 50 most divergent by HNS
| TNS order | Annotation | JHP # | 26695 # | DNS order | HNS order |
| 1 | hypothetical protein | JHP1300 | HP1408 | 9 | 1 |
| 2 | cag pathogenicity island protein (cag7) | JHP0476 | HP0527 | 2 | 2 |
| 3 | hypothetical protein | JHP0952 | HP0427 | 1 | 1355 |
| 4 | histidine and glutamine-rich metal-binding protein | JHP1321 | HP1432 | 15 | 49 |
| 5 | vacuolating cytotoxin (vacA) paralog | JHP0556 | HP0609/10 | 3 | 4 |
| 6 | vacuolating cytotoxin (vacA) paralog | JHP0274 | HP0289 | 4 | 5 |
| 7 | hypothetical protein | JHP0050 | HP0058 | 8 | 84 |
| 8 | hypothetical protein | JHP0305 | HP0322 | 5 | 10 |
| 9 | vacuolating cytotoxin (vacA) paralog | JHP0856 | HP0922 | 7 | 6 |
| 10 | type I restriction enzyme (hsdS) | JHP1422 | NAH | 319 | 3 |
| 11 | hypothetical protein | JHP0299 | HP061/2 | 35 | 275 |
| 12 | hypothetical protein | JHP0928 | NAH | 11 | 9 |
| 13 | hypothetical protein | JHP0942 | HP0996 | 6 | 27 |
| 14 | hypothetical protein | JHP1044 | HP1116 | 10 | 8 |
| 15 | hypothetical protein | JHP0934 | NAH | 16 | 95 |
| 16 | hypothetical protein | JHP0440 | HP0488 | 13 | 17 |
| 17 | outer membrane protein (omp26) | JHP1084 | HP1157 | 24 | 24 |
| 18 | topoisomerase I (topA 3) | JHP0931 | NAH | 18 | 20 |
| 19 | hypothetical protein | JHP0318 | NAH | 286 | 293 |
| 20 | cag island protein (cagA) | JHP0495 | HP0547 | 17 | 12 |
| 21 | hypothetical protein | JHP0110 | HP0118 | 64 | 19 |
| 22 | hypothetical protein | JHP1208 | HP1288 | 91 | 830 |
| 23 | DNA-directed RNA polymerase, beta subunit (rpoB) | JHP1121 | HP1198 | 50 | 16 |
| 24 | hypothetical protein | JHP0052 | HP0059 | 26 | 120 |
| 25 | hypothetical protein | JHP1042 | HP1115 | 14 | 694 |
| 26 | hypothetical protein | JHP0953 | NAH | 31 | 1463 |
| 27 | hypothetical protein | JHP1070 | HP1142 | 60 | 14 |
| 28 | hypothetical protein | JHP1113 | HP1187 | 274 | 39 |
| 29 | hypothetical protein | JHP0842 | HP0906 | 42 | 21 |
| 30 | type II restriction enzyme | JHP0630 | NAH | 173 | 588 |
| 31 | histidine-rich, metal binding polypeptide (hpn) | JHP1320 | HP1427 | 70 | 1404 |
| 32 | hypothetical protein | JHP0074 | HP0080 | 12 | 125 |
| 33 | hypothetical protein | JHP0191 | HP0205 | 53 | 7 |
| 34 | hypothetical protein | JHP0376 | HP1049 | 235 | 1128 |
| 35 | cag pathogenicity island protein (cag3) | JHP0471 | HP0522 | 21 | 62 |
| 36 | hypothetical protein | JHP0026 | HP0030 | 23 | 64 |
| 37 | urease beta subunit (urea amidohydrolase) (ureB) | JHP0067 | HP0072 | 32 | 70 |
| 38 | hypothetical protein | JHP0939 | HP0991 | 116 | 156 |
| 39 | multidrug resistance protein (spaB) | JHP0547 | HP0600 | 75 | 18 |
| 40 | flagellin A (flaA) | JHP0548 | HP0601 | 34 | 154 |
| 41 | hypothetical protein | JHP1071 | HP1143 | 78 | 61 |
| 42 | hypothetical protein | JHP0613 | HP0669 | 44 | 33 |
| 43 | hypothetical protein | JHP0623 | HP0682 | 231 | 1186 |
| 44 | N-methylhydantoinase | JHP0632 | HP0696 | 20 | 36 |
| 45 | hypothetical protein | JHP1049 | NAH | 278 | 470 |
| 46 | vacuolating cytotoxin (vacA) | JHP0819 | HP0887 | 59 | 38 |
| 47 | putative restriction enzyme | JHP0164 | NAH | 88 | 43 |
| 48 | type I restriction enzyme R protein (hsdR) | JHP0784 | HP0846 | 244 | 35 |
| 49 | hook assembly protein, flagella (flgD) | JHP0843 | HP0907 | 103 | 175 |
| 50 | hypothetical protein | JHP0458 | HP0508 | 84 | 44 |
| 51 | hypothetical protein | JHP0336 | HP1089 | 27 | 54 |
| 53 | hypothetical protein | JHP0940 | NAH | 39 | 393 |
| 54 | hypothetical protein | JHP0462 | HP0513 | 104 | 11 |
| 55 | type II restriction enzyme (methyltransferase) | JHP1409 | NAH | 37 | 15 |
| 58 | hypothetical protein | JHP1285 | HP1371 | 55 | 25 |
| 59 | hypothetical protein | JHP0693 | HP0756 | 19 | 1490 |
| 62 | cag pathogenicity island protein (cag8) | JHP0477 | HP0528 | 72 | 31 |
| 63 | type III restriction enzyme (res) | JHP1297 | NAH | 30 | 28 |
| 64 | hypothetical protein | JHP0668 | HP0731 | 110 | 32 |
| 67 | outer membrane protein | JHP0438 | HP0486 | 22 | 145 |
| 70 | cag island protein (cagT) | JHP0481 | HP0532 | 25 | 558 |
| 71 | RNA polymerase sigma-70 factor (rpoD) | JHP0081 | HP0088 | 62 | 37 |
| 75 | hypothetical protein | JHP1253 | HP1333 | 40 | 384 |
| 78 | iron(III) dicitrate transport protein (fecA) | JHP1426 | HP1400 | 28 | 111 |
| 81 | DNA polymerase I (polA) | JHP1363 | HP1470 | 46 | 46 |
| 85 | type I restriction enzyme (hsdS) | JHP0414 | NAH | 275 | 30 |
| 88 | hypothetical protein | JHP0174 | HP0187/8/6 | 29 | 90 |
| 89 | iron(III) dicitrate transport protein (fecA) | JHP0626 | HP0686 | 38 | 47 |
| 95 | DNA transfer protein (cagE) | JHP0492 | HP0544 | 49 | 50 |
| 100 | integrase/recombinase (xerD) | JHP0941 | HP0995 | 33 | 541 |
| 104 | type III restriciton enzyme (mod) | JHP1411 | HP1522 | 857 | 13 |
| 105 | type I restriction enzyme R protein (hsdR) | JHP0416 | HP0464 | 63 | 29 |
| 122 | adenine specific DNA methyltransferase (mod) | JHP0244 | HP0260 | 236 | 48 |
| 130 | hypothetical protein | JHP0925 | NAH | 43 | 990 |
| 137 | cag island protein (cagH) | JHP0489 | HP0541 | 47 | 398 |
| 138 | type I restriction enzyme (hsdR) | JHP1424 | HP1402 | 195 | 22 |
| 158 | hypothetical protein | JHP0540 | NAH | 674 | 26 |
| 170 | cag island protein (cagF) | JHP0491 | HP0543 | 52 | 828 |
| 177 | DNA repair protein (recN) | JHP1434 | HP1393 | 51 | 160 |
| 190 | type III restriction enzyme (mod) | JHP1296 | NAH | 121 | 34 |
| 196 | role in outermembrane permeability (imp) | JHP1138 | HP1215/6 | 208 | 45 |
| 206 | cytochrome oxidase (cbb3 type) (fixN) | JHP0132 | HP0144 | 41 | 209 |
| 227 | DNA mismatch repair protein (mutS) | JHP0565 | HP0621 | 45 | 82 |
| 230 | hypothetical protein | JHP0534 | HP0586 | 577 | 40 |
| 258 | type III restriction enzyme (res) | JHP1410 | HP1521 | 161 | 23 |
| 262 | hypothetical protein | JHP1033 | HP1106 | 36 | 342 |
| 281 | translation initiation factor IF-2 (infB) | JHP0377 | HP1048 | 330 | 42 |
| 290 | type II restriction enzyme (methyltrasferase) | JHP1284 | NAH | 750 | 41 |
| 1260 | siderophore-mediated iron transport protein (tonB) | JHP1260 | HP1341 | 48 | 402 |
Genes with > 2 SD divergence in each analysis are indicated in bold
NAH indicates No Annotated Homologue in the other sequence
Table 6.
Top 50 most divergent genes by HNS in H. pylori strain 26695 plus those additional genes > 2 SD greater than the mean by DNS and the 50 most divergent by HNS
| HNS order | annotation | HP# | J99 # | DNS order | TNS order |
| 1 | cag pathogenicity island protein (cag7) | HP0527 | JHP0476 | 1 | 1 |
| 2 | hypothetical protein | HP0119 | NAH | 17 | 7 |
| 3 | vacuolating cytotoxin (vacA) paralog | HP0922 | JHP0856 | 6 | 5 |
| 4 | vacuolating cytotoxin (vacA) paralog | HP0289 | JHP0274 | 2 | 2 |
| 5 | poly E-rich hypothetical protein | HP0322 | JHP0305 | 3 | 8 |
| 6 | hypothetical protein | HP1142 | JHP1070 | 91 | 19 |
| 7 | cag island protein (cagA) | HP0547 | JHP0495 | 31 | 15 |
| 8 | hypothetical protein | HP0205 | JHP0191 | 57 | 78 |
| 9 | hypothetical protein | HP0609 | JHP0556* | 4 | 6 |
| 10 | hypothetical protein | HP0453 | NAH | 75 | 58 |
| 11 | adenine specific DNA methyltransferase (mod) | HP1522 | JHP1411 | 363 | 207 |
| 12 | hypothetical protein | HP0488 | JHP0440 | 7 | 10 |
| 13 | hypothetical protein | HP1116 | JHP1044 | 8 | 11 |
| 14 | type IIS restriction enzyme R and M protein (ECO57IR) | HP1517 | NAH | 28 | 42 |
| 15 | hypothetical protein | HP0513 | JHP0462 | 122 | 28 |
| 16 | hypothetical protein | HP0906 | JHP0842 | 42 | 22 |
| 17 | vacuolating cytotoxin (vacA) paralog | HP0610 | JHP0556* | 13 | 12 |
| 18 | type III restriction enzyme R protein (res) | HP1521 | JHP1410 | 195 | 210 |
| 19 | DNA-directed RNA polymerase, beta subunit (rpoB) | HP1198 | JHP1121 | 84 | 23 |
| 20 | adenine/cytosine DNA methyltransferase | HP0054 | NAH | 109 | 120 |
| 21 | type I restriction enzyme R protein (hsdR) | HP1402 | JHP1424 | 103 | 86 |
| 22 | DNA transfer protein (cagE) | HP0441 | JHP0492 | 29 | 51 |
| 23 | type III restriction enzyme R protein | HP1371 | JHP1285 | 52 | 119 |
| 24 | DNA topoisomerase I (topA) | HP0440 | NAH | 63 | 149 |
| 25 | outer membrane protein (omp26) | HP1157 | JHP1084 | 34 | 27 |
| 26 | type I restriction enzyme R protein (hsdR) | HP0464 | NAH | 36 | 90 |
| 27 | cag pathogenicity island protein (cag8) | HP0528 | JHP0477 | 74 | 50 |
| 28 | virB4 homolog (virB4) | HP0459 | NAH | 53 | 49 |
| 29 | hypothetical protein | HP0586 | JHP0534 | 631 | 163 |
| 30 | multidrug resistance protein (spaB) | HP0600 | JHP0547 | 97 | 41 |
| 31 | RNA polymerase sigma-70 factor (rpoD) | HP0088 | JHP0081 | 56 | 55 |
| 32 | hypothetical protein | HP0731 | JHP0668 | 132 | 80 |
| 33 | hypothetical protein | HP1520 | NAH | 167 | 20 |
| 34 | vacuolating cytotoxin | HP0887 | JHP0819 | 18 | 25 |
| 35 | type III restriction enzyme R protein (res) | HP0592 | NAH | 16 | 30 |
| 36 | hypothetical protein | HP0118 | JHP0110 | 179 | 27 |
| 37 | type I restriction enzyme R protein (hsdR) | HP0846 | JHP0784 | 342 | 101 |
| 38 | hypothetical protein | HP1187 | JHP1113 | 142 | 31 |
| 39 | hypothetical protein | HP0030 | JHP0026 | 45 | 24 |
| 40 | HP0478 | JHP0430 | 1080 | 220 | |
| 41 | hypothetical protein | HP1143 | JHP1071 | 58 | 29 |
| 42 | hypothetical protein | HP0669 | JHP0613 | 69 | 60 |
| 43 | N-methylhydantoinase | HP0696 | JHP0632 | 19 | 35 |
| 44 | type I restriction enzyme M protein (hsdM) | HP1403 | JHP1423 | 125 | 340 |
| 45 | translation initiation factor IF-2 (infB) | HP1048 | JHP0377 | 291 | 332 |
| 46 | hypothetical protein | HP0996 | JHP0942 | 5 | 14 |
| 47 | DNA polymerase III alpha-subunit (dnaE) | HP1460 | JHP1353 | 297 | 219 |
| 48 | hypothetical protein | HP0733 | JHP0670 | 224 | 222 |
| 49 | preprotein translocase subunit (secA) | HP0786 | JHP0723 | 119 | 176 |
| 50 | hypothetical protein | HP0120 | NAH | 283 | 44 |
| 53 | hypothetical protein | HP0058 | JHP0051 | 121 | 16 |
| 54 | DNA polymerase I (polA) | HP1470 | JHP1363 | 30 | 77 |
| 59 | hypothetical protein | HP1089 | JHP0336 | 12 | 67 |
| 64 | DNA mismatch repair protein (MutS) | HP0621 | JHP0565 | 22 | 137 |
| 68 | 2',3'-cyclic-nucleotide 2'-phosphodiesterase (cpdB) | HP0104 | JHP0096 | 54 | 73 |
| 75 | fucosyltransferase | HP0651 | JHP0596 | 48 | 43 |
| 77 | hypothetical protein | HP0508 | JHP0458 | 139 | 32 |
| 87 | urease beta subunit (urea amidohydrolase) (ureB) | HP0072 | JHP0067 | 21 | 38 |
| 90 | cell division protein (ftsK) | HP1090 | JHP0335 | 37 | 181 |
| 99 | outer membrane protein (omp3) | HP0079 | JHP0073 | 68 | 45 |
| 100 | cag pathogenicity island protein (cag3) | HP0522 | JHP0471 | 11 | 48 |
| 101 | outer membrane protein (omp17) | HP0725 | JHP0662 | 209 | 47 |
| 122 | hypothetical protein | HP0080 | JHP0074 | 9 | 18 |
| 127 | hypothetical protein | HP1479 | JHP1372 | 55 | 153 |
| 129 | iron(III) dicitrate transport protein (fecA) | HP1400 | JHP1426 | 32 | 99 |
| 142 | outer membrane protein | HP0486 | JHP0438 | 26 | 147 |
| 160 | virulence associated protein homolog (vacB) | HP1248 | JHP1169 | 50 | 164 |
| 166 | translation elongation factor EF-Tu (tufB) | HP1205 | JHP1128 | 49 | 64 |
| 168 | cytochrome oxidase (cbb3 type) (fixN) | HP0144 | JHP0132 | 27 | 102 |
| 170 | hypothetical protein | HP1003 | NAH | 38 | 61 |
| 180 | flagellin A (flaA) | HP0601 | JHP0548 | 33 | 40 |
| 207 | DNA repair protein (recN) | HP1393 | JHP1434 | 35 | 154 |
| 256 | hypothetical protein | HP0788 | JHP0725 | 41 | 72 |
| 276 | hypothetical protein | HP0186 | JHP0174 | 47 | 130 |
| 277 | hypothetical protein | HP1106 | JHP1033 | 59 | 272 |
| 296 | hypothetical protein | HP1333 | JHP1253 | 40 | 53 |
| 320 | hypothetical protein | HP0059 | JHP0052 | 43 | 21 |
| 448 | integrase/recombinase (xerD) | HP0995 | JHP0941 | 25 | 39 |
| 449 | hypothetical protein | HP0449 | NAH | 51 | 81 |
| 451 | GMP reductase (guaC) | HP0854 | JHP0790 | 44 | 169 |
| 582 | hypothetical protein | HP0489 | JHP0441 | 10 | 36 |
| 693 | cag island protein (cagT) | HP0532 | JHP0481 | 23 | 87 |
| 737 | hypothetical protein | HP0427 | JHP0952 | 14 | 3 |
| 738 | hypothetical protein | HP1408 | JHP1300 | 15 | 4 |
| 866 | hypothetical protein | HP1115 | JHP1042 | 20 | 33 |
| 1021 | cag pathogenicity island protein (cag13) | HP0534 | JHP0482 | 60 | 225 |
| 1090 | hypothetical protein | HP1516 | NAH | 593 | 34 |
| 1129 | hypothetical protein | HP0611 | JHP0299 | 230 | 37 |
| 1256 | secreted protein involved in flagellar motility | HP1192 | JHP1117 | 410 | 13 |
| 1338 | hypothetical protein | HP0345 | NAH | 249 | 46 |
| 1432 | histidine and glutamine-rich metal-binding protein | HP1432 | JHP1321 | 46 | 9 |
| 1449 | histidine-rich, metal binding polypeptide (hpn) | HP1427 | NAH | 39 | 26 |
| 1548 | hypothetical protein | HP0756 | JHP0693 | 24 | 71 |
* probably frame shifted components of the same vacA related gene
Genes with > 2 SD divergence in each analysis are indicated in bold
NAH indicates No Annotated Homologue in the other sequence
Table 3.
Top 50 most divergent genes by HNS in H. pylori strain J99 plus those additional genes >2 SD greater than the mean by DNS and top 50 by TNS
| HNS order | J99 annotation | JHP # | 26695 # | DNS order | TNS order |
| 1 | hypothetical protein | JHP1300 | HP1408 | 9 | 1 |
| 2 | cag pathogenicity island protein (cag7) | JHP0476 | HP0527 | 2 | 2 |
| 3 | type I restriction enzyme (hsdS) | JHP1422 | NAH | 319 | 10 |
| 4 | vacuolating cytotoxin (vacA) paralog | JHP0556 | HP0609/10 | 3 | 5 |
| 5 | vacuolating cytotoxin (vacA) paralog | JHP0274 | HP0289 | 4 | 6 |
| 6 | vacuolating cytotoxin (vacA) paralog | JHP0856 | HP0922 | 7 | 9 |
| 7 | hypothetical protein | JHP0191 | HP0205 | 53 | 33 |
| 8 | hypothetical protein | JHP1044 | HP1116 | 10 | 14 |
| 9 | hypothetical protein | JHP0928 | NAH | 11 | 12 |
| 10 | hypothetical protein | JHP0305 | HP0322 | 5 | 8 |
| 11 | hypothetical protein | JHP0462 | HP0513 | 104 | 54 |
| 12 | cag island protein (cagA) | JHP0495 | HP0547 | 17 | 20 |
| 13 | type III restriciton enzyme (mod) | JHP1411 | HP1522 | 857 | 104 |
| 14 | hypothetical protein | JHP1070 | HP1142 | 60 | 27 |
| 15 | type II restriction enzyme (methyltransferase) | JHP1409 | NAH | 37 | 55 |
| 16 | DNA-directed RNA polymerase, beta subunit (rpoB) | JHP1121 | HP1198 | 50 | 23 |
| 17 | hypothetical protein | JHP0440 | HP0488 | 13 | 16 |
| 18 | multidrug resistance protein (spaB) | JHP0547 | HP0600 | 75 | 39 |
| 19 | hypothetical protein | JHP0110 | HP0118 | 64 | 21 |
| 20 | topoisomerase I (topA 3) | JHP0931 | NAH – check | 18 | 18 |
| 21 | hypothetical protein | JHP0842 | HP0906 | 42 | 29 |
| 22 | type I restriction enzyme (hsdR) | JHP1424 | HP1402 | 195 | 138 |
| 23 | type III restriction enzyme (res) | JHP1410 | HP1521 | 161 | 258 |
| 24 | outer membrane protein (omp26) | JHP1084 | HP1157 | 24 | 17 |
| 25 | hypothetical protein | JHP1285 | HP1371 | 55 | 58 |
| 26 | hypothetical protein | JHP0540 | NAH | 674 | 158 |
| 27 | hypothetical protein | JHP0942 | HP0996 | 6 | 13 |
| 28 | type III restriction enzyme (res) | JHP1297 | NAH | 30 | 63 |
| 29 | type I restriction enzyme R protein (hsdR) | JHP0416 | HP0464 | 63 | 105 |
| 30 | type I restriction enzyme (hsdS) | JHP0414 | NAH | 275 | 85 |
| 31 | cag pathogenicity island protein (cag8) | JHP0477 | HP0528 | 72 | 62 |
| 32 | hypothetical protein | JHP0668 | HP0731 | 110 | 64 |
| 33 | hypothetical protein | JHP0613 | HP0669 | 44 | 42 |
| 34 | type III restriction enzyme (mod) | JHP1296 | NAH | 121 | 190 |
| 35 | type I restriction enzyme R protein (hsdR) | JHP0784 | HP0846 | 244 | 48 |
| 36 | N-methylhydantoinase | JHP0632 | HP0696 | 20 | 44 |
| 37 | RNA polymerase sigma-70 factor (rpoD) | JHP0081 | HP0088 | 62 | 71 |
| 38 | vacuolating cytotoxin (vacA) | JHP0819 | HP0887 | 59 | 46 |
| 39 | hypothetical protein | JHP1113 | HP1187 | 274 | 28 |
| 40 | hypothetical protein | JHP0534 | HP0586 | 577 | 230 |
| 41 | type II restriction enzyme (methyltrasferase) | JHP1284 | NAH | 750 | 290 |
| 42 | translation initiation factor IF-2 (infB) | JHP0377 | HP1048 | 330 | 281 |
| 43 | restriction enzyme | JHP0164 | NAH | 88 | 47 |
| 44 | hypothetical protein | JHP0458 | HP0508 | 84 | 50 |
| 45 | role in outermembrane permeability (imp) | JHP1138 | HP1215/6 | 208 | 196 |
| 46 | DNA polymerase I (polA) | JHP1363 | HP1470 | 46 | 81 |
| 47 | iron(III) dicitrate transport protein (fecA) | JHP0626 | HP0686 | 38 | 89 |
| 48 | adenine specific DNA methyltransferase (mod) | JHP0244 | HP0260 | 236 | 122 |
| 49 | histidine and glutamine-rich metal-binding protein | JHP1321 | HP1432 | 15 | 4 |
| 50 | DNA transfer protein (cagE) | JHP0492 | HP0544 | 49 | 95 |
| 54 | hypothetical protein | JHP0336 | HP1089 | 27 | 51 |
| 62 | cag pathogenicity island protein (cag3) | JHP0471 | HP0522 | 21 | 35 |
| 64 | hypothetical protein | JHP0026 | HP0030 | 23 | 36 |
| 70 | urease beta subunit (urea amidohydrolase) (ureB) | JHP0067 | HP0072 | 32 | 37 |
| 82 | DNA mismatch repair protein (mutS) | JHP0565 | HP0621 | 45 | 227 |
| 84 | hypothetical protein | JHP0050 | HP0058 | 8 | 7 |
| 90 | hypothetical protein | JHP0174 | HP0187/8/6 | 29 | 88 |
| 95 | hypothetical protein | JHP0934 | NAH | 16 | 15 |
| 111 | iron(III) dicitrate transport protein (fecA) | JHP1426 | HP1400 | 28 | 78 |
| 120 | hypothetical protein | JHP0052 | HP0059 | 26 | 24 |
| 125 | hypothetical protein | JHP0074 | HP0080 | 12 | 32 |
| 145 | Outer membrane protein | JHP0438 | HP0486 | 22 | 67 |
| 154 | flagellin A (flaA) | JHP0548 | HP0601 | 34 | 40 |
| 160 | DNA repair protein (recN) | JHP1434 | HP1393 | 51 | 177 |
| 209 | cytochrome oxidase (cbb3 type) (fixN) | JHP0132 | HP0144 | 41 | 206 |
| 275 | hypothetical protein | JHP0299 | HP061/2 | 35 | 11 |
| 342 | hypothetical protein | JHP1033 | HP1106 | 36 | 262 |
| 384 | hypothetical protein | JHP1253 | HP1333 | 40 | 75 |
| 393 | hypothetical protein | JHP0940 | NAH | 39 | 53 |
| 398 | cag island protein (cagH) | JHP0489 | HP0541 | 47 | 137 |
| 402 | siderophore-mediated iron transport protein (tonB) | JHP1260 | HP1341 | 48 | 1260 |
| 541 | integrase/recombinase (xerD) | JHP0941 | HP0995 | 33 | 100 |
| 558 | cag island protein (cagT) | JHP0481 | HP0532 | 25 | 70 |
| 694 | hypothetical protein | JHP1042 | HP1115 | 14 | 25 |
| 828 | cag island protein (cagF) | JHP0491 | HP0543 | 52 | 170 |
| 990 | hypothetical protein | JHP0925 | NAH | 43 | 130 |
| 1355 | hypothetical protein | JHP0952 | HP0427 | 1 | 3 |
| 1463 | hypothetical protein | JHP0953 | NAH | 31 | 26 |
| 1490 | hypothetical protein | JHP0693 | HP0756 | 19 | 59 |
Genes with > 2 SD divergence in each analysis are indicated in bold
NAH indicates No Annotated Homologue in the other sequence
Table 5.
Top 50 most divergent genes by TNS in H. pylori strain 26695 plus those additional genes > 2 SD greater than the mean by DNS and the 50 most divergent by HNS
| TNS order | annotation | HP# | J99 # | DNS order | HNS order |
| 1 | cag pathogenicity island protein (cag7) | HP0527 | JHP0476 | 1 | 1 |
| 2 | vacuolating cytotoxin (vacA) paralog | HP0289 | JHP0274 | 2 | 4 |
| 3 | hypothetical protein | HP0427 | JHP0952 | 14 | 737 |
| 4 | hypothetical protein | HP1408 | JHP1300 | 15 | 738 |
| 5 | vacuolating cytotoxin (vacA) paralog | HP0922 | JHP0856 | 6 | 3 |
| 6 | hypothetical protein | HP0609 | JHP0556* | 4 | 9 |
| 7 | hypothetical protein | HP0119 | NAH | 17 | 2 |
| 8 | poly E-rich hypothetical protein | HP0322 | JHP0305 | 3 | 5 |
| 9 | histidine and glutamine-rich metal-binding protein | HP1432 | JHP1321 | 46 | 1432 |
| 10 | hypothetical protein | HP0488 | JHP0440 | 7 | 12 |
| 11 | hypothetical protein | HP1116 | JHP1044 | 8 | 13 |
| 12 | vacuolating cytotoxin (vacA) paralog | HP0610 | JHP0556* | 13 | 17 |
| 13 | secreted protein involved in flagellar motility | HP1192 | JHP1117 | 410 | 1256 |
| 14 | hypothetical protein | HP0996 | JHP0942 | 5 | 46 |
| 15 | cag island protein (cagA) | HP0547 | JHP0495 | 31 | 7 |
| 16 | hypothetical protein | HP0058 | JHP0051 | 121 | 53 |
| 17 | outer membrane protein (omp26) | HP1157 | JHP1084 | 34 | 25 |
| 18 | hypothetical protein | HP0080 | JHP0074 | 9 | 122 |
| 19 | hypothetical protein | HP1142 | JHP1070 | 91 | 6 |
| 20 | hypothetical protein | HP1520 | NAH | 167 | 33 |
| 21 | hypothetical protein | HP0059 | JHP0052 | 43 | 320 |
| 22 | hypothetical protein | HP0906 | JHP0842 | 42 | 16 |
| 23 | DNA-directed RNA polymerase, beta subunit (rpoB) | HP1198 | JHP1121 | 84 | 19 |
| 24 | hypothetical protein | HP0030 | JHP0026 | 45 | 39 |
| 25 | vacuolating cytotoxin (vacA) | HP0887 | JHP0819 | 18 | 34 |
| 26 | histidine-rich, metal binding polypeptide (hpn) | HP1427 | NAH | 39 | 1449 |
| 27 | hypothetical protein | HP0118 | JHP0110 | 179 | 36 |
| 28 | hypothetical protein | HP0513 | JHP0462 | 122 | 15 |
| 29 | hypothetical protein | HP1143 | JHP1071 | 58 | 41 |
| 30 | type III restriction enzyme R protein (res) | HP0592 | NAH | 16 | 35 |
| 31 | hypothetical protein | HP1187 | JHP1113 | 142 | 38 |
| 32 | hypothetical protein | HP0508 | JHP0458 | 139 | 77 |
| 33 | hypothetical protein | HP1115 | JHP1042 | 20 | 866 |
| 34 | hypothetical protein | HP1516 | NAH | 593 | 1090 |
| 35 | N-methylhydantoinase | HP0696 | JHP0632 | 19 | 43 |
| 36 | hypothetical protein | HP0489 | JHP0441 | 10 | 582 |
| 37 | hypothetical protein | HP0611 | JHP0299 | 230 | 1129 |
| 38 | urease beta subunit (urea amidohydrolase) (ureB) | HP0072 | JHP0067 | 21 | 87 |
| 39 | integrase/recombinase (xerD) | HP0995 | JHP0941 | 25 | 448 |
| 40 | flagellin A (flaA) | HP0601 | JHP0548 | 33 | 180 |
| 41 | multidrug resistance protein (spaB) | HP0600 | JHP0547 | 97 | 30 |
| 42 | type IIS restriction enzyme R and M protein (ECO57IR) | HP1517 | NAH | 28 | 14 |
| 43 | fucosyltransferase | HP0651 | JHP0596 | 48 | 75 |
| 44 | hypothetical protein | HP0120 | NAH | 283 | 50 |
| 45 | outer membrane protein (omp3) | HP0079 | JHP0073 | 68 | 99 |
| 46 | hypothetical protein | HP0345 | NAH | 249 | 1338 |
| 47 | outer membrane protein (omp17) | HP0725 | JHP0662 | 209 | 101 |
| 48 | cag pathogenicity island protein (cag3) | HP0522 | JHP0471 | 11 | 100 |
| 49 | virB4 homolog (virB4) | HP0459 | NAH | 53 | 28 |
| 50 | cag pathogenicity island protein (cag8) | HP0528 | JHP0477 | 74 | 27 |
| 51 | DNA transfer protein (cagE) | HP0441 | JHP0492 | 29 | 22 |
| 53 | hypothetical protein | HP1333 | JHP1253 | 40 | 296 |
| 55 | RNA polymerase sigma-70 factor (rpoD) | HP0088 | JHP0081 | 56 | 31 |
| 58 | hypothetical protein | HP0453 | NAH | 75 | 10 |
| 60 | hypothetical protein | HP0669 | JHP0613 | 69 | 42 |
| 61 | hypothetical protein | HP1003 | NAH | 38 | 170 |
| 64 | translation elongation factor EF-Tu (tufB) | HP1205 | JHP1128 | 49 | 166 |
| 67 | hypothetical protein | HP1089 | JHP0336 | 12 | 59 |
| 71 | hypothetical protein | HP0756 | JHP0693 | 24 | 1548 |
| 72 | hypothetical protein | HP0788 | JHP0725 | 41 | 256 |
| 73 | 2',3'-cyclic-nucleotide 2'-phosphodiesterase (cpdB) | HP0104 | JHP0096 | 54 | 68 |
| 77 | DNA polymerase I (polA) | HP1470 | JHP1363 | 30 | 54 |
| 78 | hypothetical protein | HP0205 | JHP0191 | 57 | 8 |
| 80 | hypothetical protein | HP0731 | JHP0668 | 132 | 32 |
| 81 | hypothetical protein | HP0449 | NAH | 51 | 449 |
| 86 | type I restriction enzyme R protein (hsdR) | HP1402 | JHP1424 | 103 | 21 |
| 87 | cag pathogenicity island protein (cag12) | HP0532 | JHP0481 | 23 | 693 |
| 90 | type I restriction enzyme R protein (hsdR) | HP0464 | NAH | 36 | 26 |
| 99 | iron(III) dicitrate transport protein (fecA) | HP1400 | JHP1426 | 32 | 129 |
| 101 | type I restriction enzyme R protein (hsdR) | HP0846 | JHP0784 | 342 | 37 |
| 102 | cytochrome oxidase (cbb3 type) (fixN) | HP0144 | JHP0132 | 27 | 168 |
| 119 | type III restriction enzyme R protein | HP1371 | JHP1285 | 52 | 23 |
| 120 | adenine/cytosine DNA methyltransferase | HP0054 | NAH | 109 | 20 |
| 130 | hypothetical protein | HP0186 | JHP0174 | 47 | 276 |
| 137 | DNA mismatch repair protein (MutS) | HP0621 | JHP0565 | 22 | 64 |
| 147 | outer membrane protein | HP0486 | JHP0438 | 26 | 142 |
| 149 | DNA topoisomerase I (topA) | HP0440 | NAH | 63 | 24 |
| 153 | hypothetical protein | HP1479 | JHP1372 | 55 | 127 |
| 154 | DNA repair protein (recN) | HP1393 | JHP1434 | 35 | 207 |
| 163 | hypothetical protein | HP0586 | JHP0534 | 631 | 29 |
| 164 | virulence associated protein homolog (vacB) | HP1248 | JHP1169 | 50 | 160 |
| 169 | GMP reductase (guaC) | HP0854 | JHP0790 | 44 | 451 |
| 176 | preprotein translocase subunit (secA) | HP0786 | JHP0723 | 119 | 49 |
| 181 | cell division protein (ftsK) | HP1090 | JHP0335 | 37 | 90 |
| 207 | adenine specific DNA methyltransferase (mod) | HP1522 | JHP1411 | 363 | 11 |
| 210 | type III restriction enzyme R protein (res) | HP1521 | JHP1410 | 195 | 18 |
| 219 | DNA polymerase III alpha-subunit (dnaE) | HP1460 | JHP1353 | 297 | 47 |
| 220 | type II restriction enzyme (methyltransferase) | HP0478 | JHP0430 | 1080 | 40 |
| 222 | hypothetical protein | HP0733 | JHP0670 | 224 | 48 |
| 225 | cag pathogenicity island protein (cag13) | HP0534 | JHP0482 | 60 | 1021 |
| 272 | hypothetical protein | HP1106 | JHP1033 | 59 | 277 |
| 332 | translation initiation factor IF-2 (infB) | HP1048 | JHP0377 | 291 | 45 |
| 340 | type I restriction enzyme M protein (hsdM) | HP1403 | JHP1423 | 125 | 44 |
* probably frame shifted components of the same vacA related gene
Genes with > 2 SD divergence in each analysis are indicated in bold
NAH indicates No Annotated Homologue in the other sequence
The 50 most divergent J99 ORFs by HNS included 26 (52%) that were not in the 53 (>2 SD) most divergent by DNS, these included 11 restriction-modification system genes and 6 others that were not annotated within the strain 26695 genome sequence. The identification of genes of a type known to be horizontally exchanged, and different between the gene complements of the strains, is strong corroboration for the foreign origin of the additional genes identified by HNS. In several instances (Tables 2 to 6) the DNS did not detect these genes at all e.g. restriction enzymes that were the 3rd, 13th and 41st most divergent genes by HNS, were 319th, 857th and 750th most divergent by DNS, respectively. In some instances the TNS gave intermediate results and in others identified other genes as more divergent than the other methods. The TNS was most sensitive for the detection of rpoB (HP1198 / JHP1121) which is associated with a significantly different gene length in the two strains (Figure 1h). One explanation for this observation is that while the DNS may initially be the most sensitive indicator of horizontal exchange it may become ameliorated to the new sequence characteristics more rapidly that the longer component features, which are probably detecting qualitatively different sequence characteristics.
The differences in the analyses using different length components, and a comparison of the results from the two sequenced strains, suggest a complex evolutionary history for the cag pathogenicity island. These suggest that it probably has mosaic structure including sequences from more than one species background, in addition to sequence that is entirely typical of H. pylori.
It is normally impossible to determine the chronology of events to distinguish insertions and deletions when comparing strains. In strain 26695 there are two open reading frames that are both good candidate coding sequences. There is only one gene in this location in strain J99 composed of the 5' gene from strain 26695 and the 3' end of the subsequent gene. This could have arisen from either a deletion or an insertion event. However, the normal DNS of the J99 gene (JHP0073, 799th in divergence) and the 5' 26695 gene (HP0079, 751st in divergence), and the high divergence of the 3' 26695 gene (HP0078, 68th in divergence), indicate that the most likely event is an insertion into strain 26695 (Figure 1l). Likewise HP0119 is likely to contain an insertion and JHP1113 probably reflects the original sequences (Figure 1k).
The inclusion of two DNA metabolism genes associated with recombination and repair is notable. Both mutS and recN were identified in both strains (22nd and 35th, and 45th and 51st most divergent genes by DNS in strains 26695 and J99 respectively). When the homologous genes were compared between the strains, extensive divergences were evident between more than one region of each protein. That these genes have divergent signatures in both strains suggests that neither has a wholly native composition. This observation is consistent with the models of rapid evolution which suggest that transient competitive advantages are enjoyed by organisms that are hypermutators under conditions of environmental stress and transitions, and that these states which can be produced by mutations in DNA repair genes [21-26]. However, such states have to be reversed so that an unsustainable mutational burden is not attained, and it has been proposed that this reversal is mediated by repair following horizontal transfer and homologous recombination, and that such strains are hyper-recombinogenic [27-29]. The untypicality of mutS and recN suggest that H. pylori is another species that can make use of this strategy for diversification under stressful conditions.
The identification of RNA polymerase genes, with associated differences between the strains, is striking. The divergence of phylogenetic trees based upon different sequences has been highlighted, and particularly the differences between the trees associated with RNA polymerase genes and rRNA [30,31]. It has been argued that RNA polymerase is as essential to cell function as is rRNA and that there is no compelling reason to chose rRNA as the more reliable marker [32]. While the DNS analysis does not address the stability of rRNA (and specifically excludes the rRNA sequences because their differing coding requirements and evolutionary pressures generate a divergent signature for other reasons), it does indicate that RNA polymerase can be a substrate for horizontal transfer, and that trees based upon this gene, or other essential genes, need not necessarily be considered a challenge to rRNA based phylogenies.
Conclusions
The spectrum of recently horizontally acquired sequences identified emphasizes the two driving forces of horizontal exchange: the transfer of a phenotype which alters or enhances bacterial fitness resulting in increased competitive fitness or altered niche adaptation, and the presence of a substrate for homologous recombination. Because of the focus upon, and relative ease of identifying, large islands associated with readily identifiable features and phenotypes, the importance of the latter component has perhaps been underestimated. The genes that have been considered to code for 'core metabolic' 'house-keeping' functions are amongst those most likely to be changed by horizontal transfer events because of the presence of homologous substrates, and changes are likely to persist even when the change is phenotypically neutral. Equally, changes in the genes involved in core functions such as gene expression and DNA metabolism may have pleotropic effects and there may be significant differences in strain behaviour, that are not simply the consequence of differences in their respective gene complements. The selection of genes for phylogenetic analysis on the basis of their coding for conserved core functions is also problematic because these are also frequently the genes most likely to share the high homology that facilitates recombination and horizontal exchange.
Methods
A traditional nucleotide signature is generated by segmenting a sequence of DNA into k equal-sized subsequences (or 'windows'). The mathematical basis for the signature is an odds ratio – pi – calculated by dividing the frequency of a length-L oligonucleotide by its expected frequency. The odds ratios for each of the 4L oligonucleotides in each window (w) are compared with the odds ratios for the overall sequence (s) [9,10,33]. The normalized difference δ is plotted and thus a nucleotide signature consists of a k-length sequence of δ values: δ(w,s) = (1/4L)Σ(4L,i:x)|pi(w) - pi(s)|, where x is the set of all permutations of length L and i is one such permutation.
There are interesting parallels between signature-style genome analysis and stylometric techniques previously used to determine the authorship of controversial literary texts. This is analogous with the biological problem and it is from this that our method is derived. Rather than using a fixed-window signature, signature scores are calculated for each coding open reading frame (ORF) and weighted with variance estimates so that the scores for shorter ORFs confer with their longer counterparts. Bissell's weighted cusum (cumulative sum) [34], 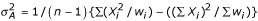 , is modified so that n denotes the number of ORFs in the genome, Xi the number of oligonucleotides in ORF i, and wi the number of nucleotides in ORF i. The results are scaled according to ORF size using the standard error σ = √(
, is modified so that n denotes the number of ORFs in the genome, Xi the number of oligonucleotides in ORF i, and wi the number of nucleotides in ORF i. The results are scaled according to ORF size using the standard error σ = √(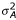 *#ORF). In this way false positives are abrogated by normalizing for over-representation of lower order peptides.
*#ORF). In this way false positives are abrogated by normalizing for over-representation of lower order peptides.
The method is implemented in Java and efficiency is maintained through an O(N) (N = sequence length) refinement: probabilities for the complete sequence are calculated in O(N) steps for any length-L oligonucleotide, and maintain O(N) when 4L>N through a hashing function; the second part of the program calculates σ for each ORF using a loop flattening technique, thereby avoiding the program having to recalculate overlapping sub-expressions. The program is available from ftp://ftp.dcs.warwick.ac.uk/people/Stephen.Jarvis/ and http://www.molbiol.ox.ac.uk/~saunders/.
Sequence alignments, as shown in Figure 1, were performed and displayed using the programs: Lalign and viewed using Lalignview [35].
Abbreviations
ORF, Open Reading Frame; DNS, Dinucleotide Signature; TNS, tetranucleotide signature; HNS, hexanucleotide signature.
Authors' contributions
NJS initiated the project, performed the genome sequence analyses, compared the two strains, interpreted the results, and prepared the biological aspects of the manuscript. PB was a DPhil student who worked on the coding aspects of the new methodology. JFP contributed to the bioinformatics discussions and planning stage of this project. SAJ directed and primarily developed the analysis strategy and the implementation of the new computational basis of the methodology, and prepared the computational aspects of the manuscript.
Acknowledgments
Acknowledgements
At the time most of this study was performed NJS was supported by a Wellcome Trust Advanced Research Fellowship.
Contributor Information
Nigel J Saunders, Email: Nigel.Saunders@path.ox.ac.uk.
Prawit Boonmee, Email: Prawit.Boonmee@warwick.ac.uk.
John F Peden, Email: John.Peden@well.ox.ac.uk.
Stephen A Jarvis, Email: Stephen.Jarvis@warwick.ac.uk.
References
- Cremonini F, Gasbarrini A, Armuzzi A, Gasbarrini G. Helicobacter pylori-related diseases. Eur J Clin Invest. 2001;31:431–437. doi: 10.1046/j.1365-2362.2001.00835.x. [DOI] [PubMed] [Google Scholar]
- Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG. Gene transfer, speciation, and the evolution of bacterial genomes. Curr Opin Microbiol. 1999;2:519–523. doi: 10.1016/S1369-5274(99)00010-7. [DOI] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Lan R, Reeves PR. Gene transfer is a major factor in bacterial evolution. Mol Biol Evol. 1996;13:47–55. doi: 10.1093/oxfordjournals.molbev.a025569. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NJ, Peden JF, Moxon ER. Absence in Helicobacter pylori of an uptake sequence for enhancing uptake of homospecific DNA during transformation. Microbiology. 1999;145:3523–3528. doi: 10.1099/00221287-145-12-3523. [DOI] [PubMed] [Google Scholar]
- Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Burge C. Dinucleotide relative abundance extremes: a genomic signature. Trends in Genetics. 1995;11:283–290. doi: 10.1016/S0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- Karlin S, Mrazek J, Campbell AM. Compositional biases of bacterial genomes and evolutionary implications. J Bacteriol. 1997;179:3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Grettin T, Gottardo R, Keyhani NO, Jensen RA. Lateral gene transfer and ancient paralogy of operons containing redundant copies of tryptophan-pathway genes in Xylela species and in heterocystous cyanobacteria. Genome Biol. 2003;4:R14. doi: 10.1186/gb-2003-4-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinel C, Nelson KE, Tümmler B. Global features of the Pseudomonas putida KT2440 genome sequence. Environmental Microbiology. 2002;4:809–818. doi: 10.1046/j.1462-2920.2002.00331.x. [DOI] [PubMed] [Google Scholar]
- Hooper SD, Berg OG. Detection of genes with atypical nucleotide sequence in microbial genomes. J Mol Evol. 2002;54:365–375. doi: 10.1007/s00239-001-0051-8. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Winberg G, Bräden C-I, Kaske A, Ernberg I, Cöster J. Capturing whole-genome characteristics in short sequences using a naïve Bayesian classifier. Genome Research. 2001;11:1404–1409. doi: 10.1101/gr.186401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Bräden CI, Erngerg I, Cöster J. Quantifying the species-specificity in genomic signatures, synonymous codon choice, amino acid usage and G + C content. Gene. 2003;311:35–42. doi: 10.1016/S0378-1119(03)00581-X. [DOI] [PubMed] [Google Scholar]
- Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, Bukanov NO, Drazek ES, Roe BA, Berg DE. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol. 1998;28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, Ketchum KA, Hood DW, Peden JF, Dodson RJ, Nelson WC, Gwinn ML, DeBoy R, Peterson JD, Hickey EK, Haft DH, Salzberg SL, White O, Fleischmann RD, Dougherty BA, Mason T, Ciecko A, Parksey DS, Blair E, Cittone H, Clark EB, Cotton MD, Utterback TR, Khouri H, Qin H, Vamathevan J, Gill J, Scarlato V, Masignani V, Pizza M, Grandi G, Sun L, Smith HO, Fraser CM, Moxon ER, Rappuoli R, Venter JC. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science. 2000;287:1809–1815. doi: 10.1126/science.287.5459.1809. [DOI] [PubMed] [Google Scholar]
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- Lawrence JG. Gene transfer, speciation, and evolution of bacterial genomes. Curr Opin Microbiol. 1999;2:519–523. doi: 10.1016/S1369-5274(99)00010-7. [DOI] [PubMed] [Google Scholar]
- Karlin S, Ladunga I. Comparisons of eukaryotic genomic sequences. Proc Natl Acad Sci USA. 1994;91:12832–12836. doi: 10.1073/pnas.91.26.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L, Cox EC. Competition between high and low mutation strains of Escherichia coli. Evolution. 1983;37:125–134. doi: 10.1111/j.1558-5646.1983.tb05521.x. [DOI] [PubMed] [Google Scholar]
- Giraud A, Matic I, Tenaillon O, Clara A, Radman M, Fons M, Taddei F. Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science. 2001;291:2606–2608. doi: 10.1126/science.1056421. [DOI] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- LeClerc JE, Li B, Payne WL, Cebula TA. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science. 1996;274:1208–1219. doi: 10.1126/science.274.5290.1208. [DOI] [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- Denamur E, Lecointre G, Darlu P, Tenaillon O, Acquaviva C, Sayada C, Sunjevaric I, Rothstein R, Elion J, Taddei F, Radman M, Matic I. Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell. 2000;103:711–721. doi: 10.1016/S0092-8674(00)00175-6. [DOI] [PubMed] [Google Scholar]
- Brown EW, LeClerc JE, Li B, Payne WL, Cebula TA. Phylogenetic evidence for horizontal transfer of mutS alleles among naturally occurring Escherichia coli strains. J Bacteriol. 2001;183:1631–1644. doi: 10.1128/JB.183.5.1631-1644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt RP, Logsdon JM, Jr, Healy B, Dorey MW, Doolittle WF, Embley TM. Microsporidia are related to Fungi: evidence from the largest subunit of RNA polymerase II and other proteins. Proc Natl Acad Sci U S A. 1999;96:580–585. doi: 10.1073/pnas.96.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiller JW, Duffield EC, Hall BD. Amitochondriate amoebae and the evolution of DNA-dependent RNA polymerase II. Proc Natl Acad Sci U S A. 1998;95:11769–11774. doi: 10.1073/pnas.95.20.11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2128. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Karlin S, Campbell A, Mrazek J. Comparative analysis across diverse genomes. Annu Rev Genet. 1998;32:185–225. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- Bissell AF. Weighted cumulative sums for text analysis using word counts. J Statist Soc. 1995;158:525–545. [Google Scholar]
- Pearson WR. Effective protein sequence comparison. Methods Enzymol. 1996;266:227–258. doi: 10.1016/s0076-6879(96)66017-0. [DOI] [PubMed] [Google Scholar]