

Summary
Interleukin (IL)-33 is a key cytokine involved in type 2 immunity and allergic airway diseases. Abundantly expressed in lung epithelial cells, IL-33 plays critical roles in both innate and adaptive immune responses in mucosal organs. In innate immunity, IL-33 and group 2 innate lymphoid cells (ILC2s) provide an essential axis for rapid immune responses and tissue homeostasis. In adaptive immunity, IL-33 interacts with dendritic cells, Th2 cells, follicular T cells, and regulatory T cells, where IL-33 influences the development of chronic airway inflammation and tissue remodeling. The clinical findings that both the IL-33 and ILC2 levels are elevated in patients with allergic airway diseases suggest that IL-33 plays an important role in the pathogenesis of these diseases. IL-33 and ILC2 may also serve as biomarkers for disease classification and to monitor the progression of diseases. In this article, we reviewed the current knowledge of the biology of IL-33 and discussed the roles of the IL-33 in regulating airway immune responses and allergic airway diseases.
Keywords: IL-33, type 2 immunity, ILC2s, Th2 cells, asthma, rhinosinusitis
Introduction
Allergic airway diseases are thought to be caused by aberrant immune responses against inhaled airborne allergens (1). Exposed to the outside environment, the lung epithelium not only serves as a barrier against pathogens and environmental insults, but also plays an important role in airway immune responses. Indeed, upon exposure to inhaled airborne allergens, lung epithelial cells secrete inflammatory cytokines that activate immune cells involved in type 2 immunity (2). Among various products of epithelial cells, three epithelium-derived cytokines, interleukin (IL)-33, IL-25, and thymic stromal lymphopoietin (TSLP), have been identified as having critical functions in the pathophysiology of allergic airway diseases (3, 4).
IL-33 was first identified in 1999 as the protein DVS27 expressed in canine arteries (5), and later in 2003 as “nuclear factor from high endothelial venules” (NF-HEV) expressed in human endothelial cells (6). IL-33 was rediscovered in 2005 by a computational sequence search, which identified it as an IL-1 family member. At the time, IL-33 was given the official name “IL-33” (7). Conversely, IL-33 receptor, designated IL-1-receptor-like 1 (IL1RL1) and commonly known as suppression of tumorigenicity 2 (ST2), was first reported in 1989 as a serum-responsive protein in murine fibroblasts that shared sequence homology with the IL-1 receptor (8). In 2005, Schmitz et al. identified IL-33 as a ligand for the orphan receptor ST2 (7). IL-33 and ST2 have different cellular distributions. IL-33 is predominantly expressed by tissue cell types, including epithelial cells, fibroblasts, and endothelial cells (9–11). IL-33 expression can be induced in immune cells, such as mast cells and dendritic cells (DCs), in inflammatory conditions (12–14). ST2 is predominantly expressed by immune cells including CD4+ T cells (15), CD8+ T cells (16), mast cells (17), group 2 innate lymphoid cells (ILC2s) (18–20), macrophages (21), eosinophils (22), DCs (23), basophils, natural killer (NK) T cells, and NK cells (24, 25). Through binding to the ST2, IL-33 activates target cells to produce and secrete cytokines and growth factors that promote and/or regulate local and systemic immunity.
Since the discovery of IL-33, tremendous progress has been made in understanding its biological functions. Studies have shown that IL-33 is a multifunctional cytokine that plays critical roles in a variety of biological responses, such as the development and regulation of immune responses, maintenance of tissue homeostasis, repair, and remodeling. Evidence suggests that IL-33 is likely involved in pathogenesis of various human diseases. Outstanding reviews have been published recently to describe the biology of IL-33 and its roles in diseases in general (26–28), and readers are asked to refer to them for comprehensive information. The potential importance of IL-33 in allergic airway diseases in humans has been supported by multiple genome-wide association studies (GWAS), which have shown that IL-33 and its receptor ST2 are associated with asthma susceptibility (29–33). Animal models have shown that the IL-33/ST2 pathway plays a critical role in allergen-induced innate and adaptive immune responses and inflammation in the airway (34–38). In this article, we have focused on the roles of IL-33 in airway mucosa and have cited the evidence for the involvement of IL-33 in allergic airway diseases.
Molecular features of IL-33
The human and mouse IL-33 genes are located on chromosome 9p24.1 and chromosome 19qC1, respectively. The human IL-33 gene contains eight exons, with exon one being non-coding and exons 2–8 coding for the IL-33 protein (7, 39, 40). In the mouse, two different promoters have been found to initiate Il33 gene transcription. The resulting IL-33 transcripts, Il33a and Il33b, differ in their 5′-untranslated regions but encode the same protein. The expression of Il33a and Il33b transcripts was cell type- and stimulus-dependent (40, 41). Besides the full-length IL33 mRNA, several IL33 splice variants have been identified in human cells (39–42), which are derived from alternative splicing of some or all IL33 exons 3, 4, or 5.
Human and mouse full-length IL-33 proteins are 270 and 266 amino acids long, respectively, and the two proteins are 55% homologous (7). The IL-33 protein can be divided into three functional domains; nuclear domain, central domain, and IL-1-like cytokine domain (43) (Figure 1A). The nuclear domain (amino acids 1–65 in humans) is encoded by exons 2–3 and contains a chromatin-binding motif (amino acids 40–58) (44). Under basal conditions, the chromatin-binding motif localizes IL-33 protein to the nucleus and tethers IL-33 to chromatin by interacting with the histone H2A-H2B dimer (44). The central domain (amino acids 66–111 in humans) of IL-33 is encoded by exon 4 and contains protease cleavage sites, which are sensitive to neutrophil- and mast cell-derived proteases (45, 46). The IL-1-like cytokine domain (amino acids 112–270 in humans) of IL-33 is encoded by exons 5–8, binds to ST2 on target cells, and mediates the cytokine activities (7, 47).
Figure 1.
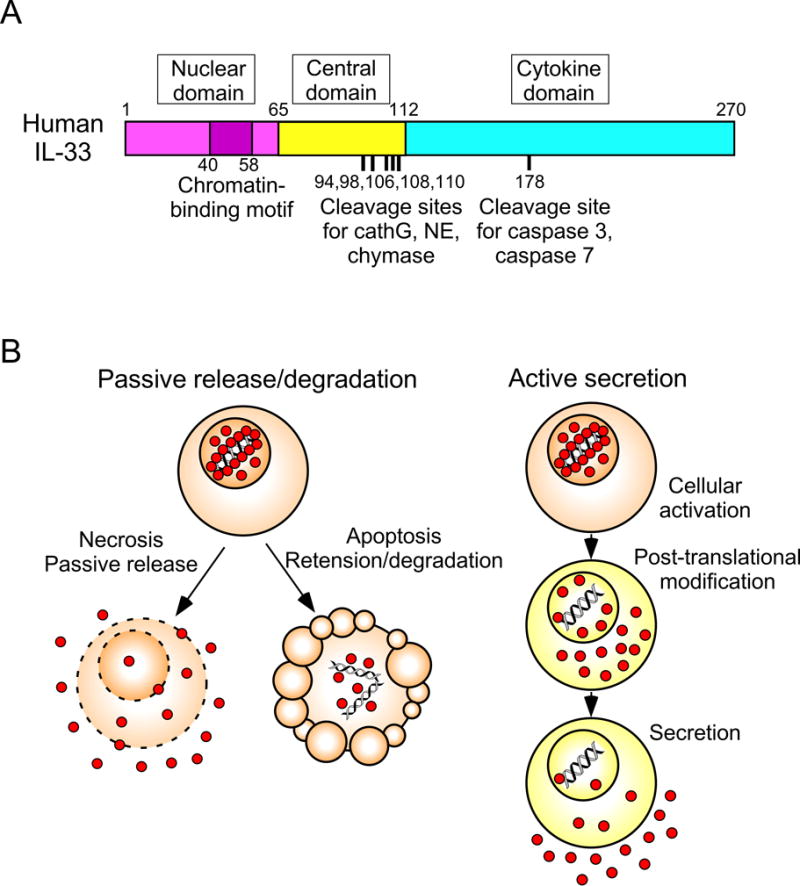
The structure of IL-33 and mechanisms of its extracellular release. Panel A. Human IL-33 protein is composed of two evolutionary conserved domains (the nuclear domain and the cytokine domain) that are separated by the highly divergent central domain. Chromatin-binding motif and cleavage sites for inflammatory and apoptotic proteases are indicated. CathG; cathepsin G, NE; neutrophil elastase. Panel B. Proposed mechanisms for extracellular IL-33 release. IL-33 that are constitutively produced and stored in the nucleus is passively released during necrosis when cells lose integrity of plasma and nuclear membrane. Upon apoptosis, IL-33 is retained within the cells and inactivated by apoptotic proteases, such as caspase 3 and caspase 7. Alternatively, cellular stress that is caused by exposure to enviromental factors, such as proteases and ATP, induces cellular activation and post-translational modification of nuclear IL-33 to remove the nuclear domain and central domain. The processed IL-33 is most likely actively secreted extracellularly by structural cells.
While full-length ~31 kDa IL-33 has cytokine activities (48), processing of IL-33 by cleavage at the central domain and removal of the N-terminal peptides increased its activity by ~30-fold (45, 46). Exogenous proteases that are derived from inflammatory cells, such as neutrophil cathepsin G, neutrophil elastase, and mast cell serine proteases, are capable of processing IL-33 (45, 46) (Figure 1), while it is still unknown whether and which endogenous protease(s) have a similar capacity. In addition, caspases that are active during cellular apoptosis, such as caspase 3, caspase 7, and likely caspase 1, regulate IL-33 activity. Indeed, the IL-33 protein contains a consensus caspase cleavage site in its cytokine domain (Figure 1). In contrast to the neutrophil- and mast cell-derived proteases, caspases inactivate IL-33 by cleaving it into biologically-inactive fragments (43, 48).
Expression of IL-33
Under basal conditions, IL33 mRNA and protein are constitutively and abundantly expressed in many tissues in mice and humans (6, 7, 9). Therefore, IL-33 has been considered as an “alarmin” cytokine that is stored and released quickly in response to cellular damage or tissue injury (9). Specifically in the airway, bronchial epithelial cells and endothelial cells of high endothelial venules are the major source of IL-33 in humans (9, 49). In contrast, in mice, IL-33 is not constitutively expressed by endothelial cells while its expression can be induced in endothelial cells under chronic inflammation conditions (10, 50). Furthermore, mouse IL-33 is mainly expressed by lung alveolar type II pneumocytes, whereas human IL-33 is expressed by bronchial epithelial cells (38, 51). These species differences may need to be taken into consideration to translate the results of mouse models to humans. IL-33 is also constitutively expressed by mouse and human platelets (52).
The abundant basal IL-33 expression in tissue cells is further upregulated by inflammation or tissue stress. For example, during intestinal nematode infections, lung influenza infections, and airway exposure to cigarette smoke, cysteine proteases, uric acid, and allergens all increase IL-33 expression in the mouse lung (36, 38, 51, 53, 54). In humans, increased IL-33 expression has been observed in nasal or lung epithelium of patients with allergic asthma, allergic rhinitis, and chronic obstructive pulmonary disease (COPD) (49, 55–57). Furthermore, inflammation can also induce IL-33 expression in bone marrow-derived cells, such as macrophages, DCs, B cells, and mast cells (12, 13, 38, 53, 58, 59). However, the functional importance of immune cell-derived IL-33 in allergic immune responses is not fully understood. Indeed, a recent study showed that IL-33 produced by tissue cells, but not that produced by immune cells, was essential for allergen-driven airway inflammation (60).
Processing and release of IL-33
The cytokine activity of IL-33 is regulated by its cellular location and biochemical modification. Under basal conditions, IL-33 protein is stored in the cell nucleus as a ~31 kDa full-length protein (61). This nuclear localization of IL-33 appears to be critical to prevent detrimental effects of this cytokine on the host. For example, when the chromatin-binding domain of IL-33 (Figure 1A) was selectively eliminated in an IL-33 knock-in murine model, IL-33 was constitutively released extracellularly, resulting in ST2-dependent lethal eosinophilic and neutrophilic inflammation of multiple organs in the animal (62).
IL-33 lacks a classical signal sequence to direct the cytokine to the endoplasmic reticulum-Golgi secretory pathway. While the molecular mechanisms that make nuclear IL-33 available to the extracellular milieu are not fully understood, the current knowledge suggests two mechanisms; passive release and active secretion (Figure 1B). Upon infection with microbes or tissue damage, full-length IL-33 was passively released from the nucleus of necrotic cells into the extracellular space (43, 48). Thus, IL-33 has been considered a danger- or damage-associated molecular patterns (DAMPs) component that initiates immune responses in response to cellular injury (63, 64). IL-33 can also be secreted from living cells when cells encounter the factors that cause cellular stress or minor (and repairable) injury. For example, in normal human bronchial airway epithelial (NHBE) cells, exposure to allergen extracts from fungus Alternaria or cockroach or uric acid induced the translocation of IL-33 from the nucleus to the cytoplasm and subsequent extracellular release of the protein without apparent signs of cell death (54, 65). IL-33 secretion by airway epithelial cells induced by extracts of natural allergens has been observed by several investigators in both in vitro and in vivo models (35, 66, 67). Stimulation of human tracheobronchial epithelial cells with exogenous ATP also induced non-cytotoxic release of IL-33 (55); ATP has been considered a classical DAMP that is released by stressed cells (4). Finally, the application of biomechanical strain to human fibroblasts induced IL-33 secretion in the absence of cellular necrosis (68).
The signaling pathway that mediates active IL-33 secretion is currently unknown, although several key factors have been identified. IL-33 release from NHBE cells that are exposed to fungal allergens involved acute extracellular accumulation of ATP, autocrine and paracrine activation of P2 purinergic receptors, and subsequent increase in intracellular Ca2+ concentrations (65). The administration of Ca2+ chelators, cellular treatment with P2 purinergic receptor antagonists, or genetic knockdown of these receptors inhibited Alternaria-induced IL-33 release. Likewise, the inhibition of P2 purinergic receptors reduced Alternaria-induced IL-33 release as reported in other studies (35, 67). In addition, in airway epithelial cells exposed to Alternaria or house dust mite (HDM) extracts, NADPH oxidase dual oxidase 1 (DUOX1) has been shown to be activated as a result of the engagement of P2 purinergic receptors and mediated IL-33 secretion (67). Nucleotide-binding domain leucine-rich repeat-containing protein (NLRP)3-inflammasome may also played a role, because knockdown of NLRP3 attenuated IL-33 secretion induced by the adjuvant aluminum hydroxide (alum) by macrophages (69); however, it still remains to be determined whether this observation can be applied to more physiological stimuli, such as allergens. Collectively, these findings suggested that intracellular calcium and P2 purinergic receptors were likely to be key players in the signaling cascade involved in IL-33 secretion from airway epithelial cells.
It remains unknown whether IL-33 is released as a ~31 kDa full-length form or a ~20 kDa processed form (Figure 1A). Several forms of IL-33 are detected in the mucosal tissues at resting conditions and during inflammation. In naïve mice, two forms of IL-33 were detected in lung homogenates; a full-length form and a processed form that migrated around the 35 kDa and 20 kDa range on SDS-PAGE gels, respectively (70). The expression of both forms of IL-33 was upregulated by intranasal administration of chitin, a component of common airborne allergens (70). Similarly, the expression of both the full-length and processed forms of IL-33 was upregulated in the mouse lung during chronic airway inflammation (71). Upon Alternaria exposure in mice, IL-33 was released into the bronchoalveolar lavage (BAL) as a 19-kDa protein (72). Further studies will be necessary to elucidate whether this processed IL-33 is generated by the actions of endogenous proteases expressed by epithelial cells themselves or by exogenous proteases secreted from inflammatory cells (e.g., neutrophils) in the surrounding tissues. A question also remains whether the biological activities of IL-33 are enhanced or decreased by processing in vivo. It should also be noted that the activity of IL-33 can be regulated by biochemical modifications. When oxidized, IL-33 undergoes a rapid conformational change involving formation of two disulfide bonds that switches IL-33 from an active form to an inactive form (72). Thus, proteolytic cleavage and oxidation likely play key roles to regulate IL-33 activity.
Biological functions of IL-33
In principle, IL-33 can work intracellularly as a transcription regulator or extracellularly as a cytokine. Although IL-33 is expressed constitutively and the protein is localized in the nucleus, its nuclear function is not well understood. IL-33 has been shown to regulate chromatin compaction (44) and NF-κB transcriptional activity in transfected cells (73). However, when nuclear IL-33 was knocked down in primary human endothelial cells, these cells displayed unaltered proteomes and NF-kB expression as compared to that of the wild-type cells, suggesting that endogenous nuclear IL-33 does not play a significant role in gene transcription (74). Furthermore, the mice lacking IL-33 gene were fertile and did not appear to show gross abnormalities at the resting condition.
The functions of extracellular IL-33 as a cytokine have been extensively studied (Figure 2). While tissue cells, such as epithelial cells, are a major source of IL-33, these cells also express IL-33 receptor ST2 and respond to IL-33. For example, IL-33 has been shown to stimulate CXCL8/IL-8 production by goblet cells (75). In bronchial epithelial cells and endothelial cells, IL-33 has been shown to induce IL-8 and IL-17F expression and to be involved in rhinovirus-induced CXCL10 production (76–78). Thus, IL-33 might act in a self-amplification loop in these tissue-resident cells.
Figure 2.
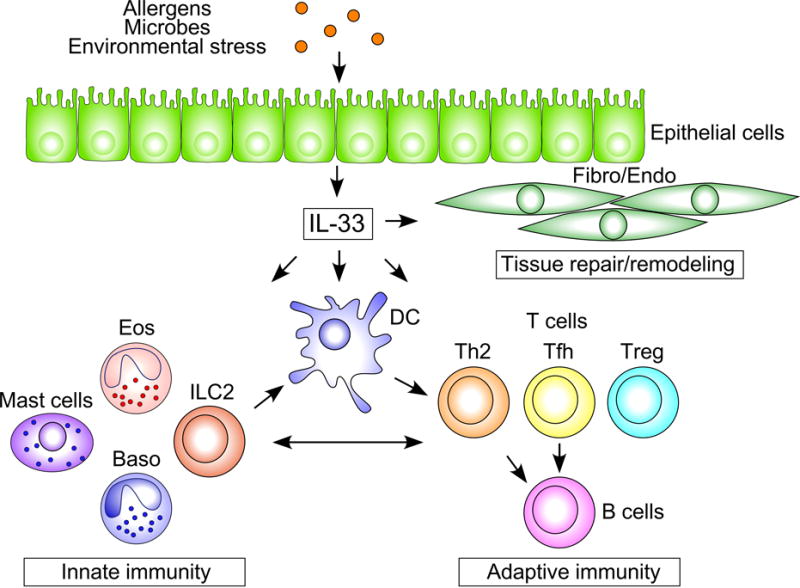
The roles of epithelial IL-33 in mucosal immune responses. Exposure to allergens, microbes and perhaps environmetal stress induces IL-33 release from airway epithelial cells. IL-33 activates the innate immune cells, including ILC2s, basophils, eosinophils, and mast cells to drive type 2 inflammation. IL-33 also activates DCs and CD4+ T cells and drives proliferation and differentiation of Th2, Tfh and Treg cells and production of antibody by B cells. Note that a bidirectional cross-talk exists betewen the cells involved in the innate immunity and adaptive immunity. Chronic activation of these immune responses results in changes in structural cells, such as epithelial cells, fibroblasts and endotherial cells, and promotes tissue repiar and remodeling. Fibro; fibroblasts, Endo; endothelial cells, Eos; eosinophils, and Baso; basophils.
Many immune cells that are involved in innate immunity express ST2, including mast cells (17), group 2 innate lymphoid cells (ILC2s) (18, 20), macrophages (21), DCs (23), eosinophils (22), basophils, NK cells (24, 25), and NK T cells (24, 25). In addition, the cells involved in adaptive immunity express ST2, including both CD4+ T cells (15) and CD8+ T cells (16). The best characterized function of IL-33 is the initiation and development of innate and adaptive type 2 immune responses that are mediated by ILC2s and CD4+ T cells, while the role of IL-33 in CD8+ T cell-mediated immune responses has also been reported (16). Furthermore, certain subsets of regulatory T (Treg) cells expressed ST2 (79–81), and IL-33 may modulate the functions of this cell type. The following two sections describe how IL-33 works with these cell types to regulate innate and adaptive immunity in the airway (Figure 2).
IL-33 in innate immunity
The predominant IL-33 expression in the epithelial barrier positions IL-33 as an important cytokine for the initiation of immune responses against invading pathogens or other environmental insults. In the airway, IL-33 responds quickly to allergen exposure. For example, when mice were exposed to the fungal allergen Alternaria or cysteine proteases (papain and bromelain), IL-33 was rapidly released into the airway lumen within 1 hour, and IL-33 mRNA transcription in the lungs was upregulated within 6 hours, (34, 54, 66). The critical role of IL-33 in innate allergic airway responses has been demonstrated by various studies that utilized ST2- or IL-33-deficient mice. Exposure to papain, Alternaria, influenza virus, and hookworm induced acute airway inflammation that was significantly dependent on IL-33 or ST2 (34, 53, 82, 83). The rapid IL-33-driven immune responses occurred even in the absence of adaptive immunity. For example, airway administration of IL-33 induced type 2 cytokine production, airway eosinophilia and airway hyper-responsiveness in Rag−/− mice that were deficient in T and B cells (34, 84). Importantly, the primary target of IL-33 in the airway was found to be lung ILC2s (34, 85) that were enriched in mucosal tissues and barrier surfaces (86). Although ILC2 numbers are low in the airway of naïve mice, lung ILC2s proliferate and produce a large quantity of type 2 cytokines, such as IL-5 and IL-13, in response to IL-33 (34, 85). Likewise, the importance of the IL-33/ILC2 axis in innate-type allergic airway inflammation has been shown in a number of experimental models (11, 34, 85, 87).
In addition to ILC2s, IL-33 can also regulate other innate immune cells involved in allergic airway responses, including macrophages, eosinophils, mast cells, and basophils. IL-33 stimulation in vitro leads to the activation and production of inflammatory mediators, cytokines, and chemokines by these cell types (22, 25, 88–90). For example, IL-33 induced superoxide production and degranulation more potently than IL-5 in human eosinophils (22). IL-33 administration into mouse airways led to IL-13-dependent polarization of alternatively-activated macrophages (AAMs) that contributed to airway inflammation (57). IL-33-stimulated mast cells showed increased histamine release that may play an important role in the early phase of allergic rhinitis (91). Furthermore, IL-33-stimulated mast cells also induced Th17 differentiation in eosinophil-deficient mice that led to neutrophil-dominant airway inflammation (92). Thus, IL-33 that is released quickly by airway epithelium upon exposure to allergens and other respiratory insults likely initiates type 2 innate immunity in the airways through interacting with ILC2s as well as other innate immune cells.
IL-33 in adaptive immunity
The roles of the IL-33/ST2 pathway in allergen-induced type 2 adaptive immune responses have been demonstrated by using several experimental models (36, 54, 83, 91). Generally, IL-33 regulates adaptive immunity by two pathways; by activating innate immune cells, such as DCs and ILCs, and by directly activating ST2-expressing adaptive immune cells. For example, DCs play important roles in T cell activation and differentiation. Although ST2 surface expression is relatively low in DCs, stimulation with IL-33 induces the production of a number of cytokines and chemokines, such as IL-6, TNF, IL-1β, and CCL17, by bone marrow-derived DCs and upregulates the expression of class II MHC and co-stimulatory molecules (23, 93). Furthermore, when exposed to IL-33, these DCs facilitate the proliferation and differentiation of naïve CD4+ T cells to preferentially produce IL-5 and IL-13 (23). Conversely, allergen-induced maturation and migration of DCs was attenuated in ST2-deficient mice (93).
IL-33-activated ILC2s may also be involved in proliferation and differentiation of CD4+ T cells, as depletion of ILC2s in mice severely impaired the adaptive type 2 immune responses induced by exposure to papain (94). In this model, ILC2s secreted IL-13 that in turn induced the migration of lung DCs into the draining lymph nodes to prime naïve T cells (94). IL-33-activated lung ILC2s can promote CD4+ T cell functions by presenting antigen to T cells and by stimulating CD4+ T cell proliferation and Th2 cytokine production in a contact-dependent manner (94–96). The adoptive transfer of both ILC2s and CD4+ T cells, but not each cell population alone, into IL-7R-deficient mice that lacked both ILC2s and CD4+ T cells, induced a robust airway inflammation and antigen-specific type 2 immune responses (95). Furthermore, IL-33-activated ILC2s enhanced B cell proliferation and promoted antibody production, including IgM, IgG1, IgA, and IgE, in vitro (97). Consistent with this data, the papain-induced IgE response was attenuated in ILC2-deficient mice (94).
ST2 was originally found to be expressed by highly-differentiated Th2-type CD4+ T cells (15). Naïve CD4+ T cells expressed limited numbers of ST2 on their surfaces. Upon activation, however, ST2 expression on CD4+ T cells was upregulated, thus rendering these cells highly responsive to IL-33. When naïve CD4+ T cells were cultured with IL-33 plus T cell receptor (TCR) agonists or agonists that activated the STAT5 pathway (e.g., TSLP), IL-33 polarized CD4+ T cells to produce IL-5 and IL-13 (98). Furthermore, in highly-differentiated ST2+ Th2 cells, IL-33 alone was sufficient to induce IL-5 and IL-13, but not IL-4 production, even in the absence of in vitro TCR stimulation (99). The results from mouse models also supported the roles for IL-33 in mediating the differentiation and activation of Th2-type CD4+ T cells. Airway exposure to innocuous antigens, such as OVA and keyhole limpet hemocyanin, generally induced immunological tolerance to the antigens (100). When mice were exposed to the same antigens together with IL-33, they developed robust and long-lasting Th2-type immune responses and asthma-like lung pathology, including airway eosinophilia, mucus hyperplasia, and airway hyper-reactivity (101). Although many of the in vitro studies failed to detect IL-4 production by CD4+ T cells that were cultured with IL-33 (98, 99), airway exposure to IL-33 with antigen in vivo rapidly induced the expression of IL-4 in CD4+ T cells in the draining lymph nodes. This endogenous IL-4 appeared to be critical in the Th2-type differentiation of the antigen-specific CD4+ T cells because depletion of IL-4 led to the development of Th17-type CD4+ T cells (101).
A hallmark of allergic airway diseases is the presence of an antigen-specific IgE antibody. Conventionally, Th2-type CD4+ T cells have been considered to play critical roles in allergic immune responses, including type 2 cytokine production, airway eosinophilia, mucus hyperplasia, and IgE antibody production (102, 103). Follicular helper T (Tfh) cells are distinguished from other CD4+ T cells by their selective role in orchestrating germinal center responses and in promoting the development of memory B cells and long-lived plasma cells (104, 105). Recent findings suggested that IL-33 was also involved in the development of Tfh cells (106). Indeed, airway administration of IL-33 together with OVA antigen induced IL-4 expression in two distinct CD4+ T cell subsets, CD4+ST2+CXCR5− Th2 cells and CD4+ST2−CXCR5+ Tfh cells, in lung draining lymph nodes (106). Adoptive transfer experiments showed that Th2 cells mediated eosinophilic airway inflammation and transient production of antigen-specific IgE antibody, whereas the Tfh cells mediated sustained production of IgE. Interestingly, the induction of Tfh cells required lower doses of IL-33 as compared to those needed for the induction of Th2 cells. Collectively, these findings indicated that IL-33 induced different types of effector CD4+ T cells and influenced various aspects of allergic immune responses.
IL-33 may also be involved in the regulation and homeostasis of immune responses. Treg cells are critically involved in immune homeostasis and tolerance to innocuous antigens (107–109). Under basal conditions, constitutive ST2 expression was observed in subsets of CD4+Foxp3+ Treg cells in various tissues, including those in the lung and gastrointestinal tract (79–81, 110). Interestingly, these ST2+ Treg cells also expressed the canonical Th2 transcription factor GATA3. When exposed to IL-33, ST2+ Treg cells proliferated and upregulated GATA3 and ST2 expression (80, 81, 110). IL-33-stimulated GATA3+ Treg cells, but not GATA3− Treg cells, expressed Il5 and Il13 mRNA, and were capable of producing IL-5 and IL-13 proteins (110). Furthermore, using an in vitro co-culture assay, IL-33 was shown to impair the ability of Foxp3+ Treg cells to suppress the proliferation of effector CD4+ T cells. In contrast, in certain settings, IL-33 stabilized Foxp3 expression in ST2+ Treg cells and promoted their suppressive functions (79–81). IL-33 also indirectly regulated Treg cells through activating ILC2s, DCs, or mast cells; these cells produced IL-2 that subsequently promoted Treg cell expansion and function (79, 111, 112). Thus, IL-33 may promote or regulate CD4+ T cell-mediated immune responses depending on their conditions and/or tissue environment. Future studies will be necessary to elucidate the mechanisms of how IL-33 dictates the direction of these immune responses.
IL-33 and chronic airway inflammation
Human asthma is generally characterized by chronic airway inflammation, airway hyper-reactivity, and tissue remodeling (113). While IL-33 is likely involved in the initiation of innate and adaptive type 2 immunity, emerging data also suggest that IL-33 plays an important role in chronic and persistent airway inflammation. Allergic patients with asthma are often sensitized to multiple allergens (114). To mimic this, mouse models have been developed in which mice are exposed to the extracts of several airborne allergens, such as Alternaria, HDM, ragweed, and Aspergillus, for a prolonged period of up to 12 weeks (36, 71). In these models, IL-33 expression in the lung was dramatically increased during the chronic phase of allergen exposure, and was accompanied by increased numbers of CD4+ T cells and ILC2s in the lung. ST2-deficient mice or the administration of anti-IL-33 antibody caused attenuated allergen-induced immune responses in these “chronic” models (36, 71), suggesting that increased IL-33 expression in the lung may play a key role in maintaining the chronic airway response. Furthermore, in these models, ILC2-derived IL-13 appeared to play roles in the upregulation of IL-33 in airway epithelium (71). Importantly, a separate study showed that IL-33-activated and primed ILC2s persisted in the draining lymph nodes for several weeks even in the absence of subsequent exposure to IL-33 (115), further suggesting that IL-33 signaling may have a long-term effect on innate and perhaps adaptive immune responses.
After chronic exposure to IL-33, the phenotype of Th2-type CD4+ T cells was further modified beyond conventional Th2 cells. After several rounds of exposure to IL-33, both mouse and human CD4+ T cells developed into a “memory” CD4+ Th2 cell type that was highly pathological and preferentially produce IL-5 over other Th2-type cytokines through the p38 MAP kinase pathway (116). Conversely, Il33−/− mice showed impaired development of IL-5-producing CD4+ memory Th2 cells and attenuated eosinophilic airway inflammation (116). In humans, IL-5-producing memory Th2 cells appeared to play critical roles in driving the pathology of allergic airway inflammation that was typically observed in patients with eosinophilic chronic rhinosinusitis (CRS) (116, 117). In addition, besides targeting ILC2s and Th2 cells, IL-33 may promote persistent type 2 inflammation by activating basophils and mast cells, as shown by the presence of increased basophils and mast cells in the induced sputum and airway biopsy specimens from patients with asthma (42, 118).
IL-33 and airway remodeling
Chronic airway inflammation is often accompanied by airway remodeling that is characterized by increased airway wall thickness and smooth muscle mass, abnormal composition of the extracellular matrix, and increased vascularity (119, 120). While many cell types and inflammatory mediators likely contribute to this pathological process (119), recent studies suggested that IL-33 may play a significant role. For example, the administration of anti-ST2 antibody or genetic manipulation of ST2 in mouse models of fungus- or HDM-induced airway inflammation attenuated airway remodeling (121, 122). Furthermore, ST2 deficiency or anti-IL-33 antibody treatment diminished bleomycin-induced lung fibrosis (123). IL-33 may regulate airway remodeling by stimulating airway fibroblasts directly to secret collagen, or by engaging with AAMs and ILC2s (122, 123). In a COPD model, IL-33 was required for the expression of Il13 and mucin in lung epithelial cells, two genes implicated in tissue remodeling (55). IL-33 may also engage Treg cells to mediate airway remodeling. Recent studies in animal models showed that Treg cells were essential to repair physical damage of certain tissues (124, 125). Furthermore, IL-33 induced the production of amphiregulin by Treg cells and ILC2s that was essential for lung tissue repair after influenza virus infection (124, 126). Thus, airway remodeling can be considered to occur as a result of extensive inflammation and as a part of tissue repair, which are not mutually exclusive.
IL-33 in human airway diseases
As described above, the use of animal models and in vitro studies have suggested that IL-33 may play a key role in immunity, inflammation, and homeostasis of mucosal tissues. IL-33 may be important for allergic airway diseases in humans. Indeed, the IL33 and ST2/IL1RL1 genes were found to be associated with asthma and allergic rhinitis in several GWAS studies that included thousands of human subjects from diverse ethnic groups (29–33, 127). Below, we have discussed the studies that investigated the potential roles for IL-33 and ILC2s in human airway diseases, including asthma, allergic rhinitis, and CRS.
Asthma
Examinations of airway specimens from patients with asthma showed increased IL-33 mRNA expression in cells from induced sputum (128) and in airway epithelial cells from bronchial biopsy specimens (49). Interestingly, the expression of an IL33 spliced variant lacking exons 3 and 4 was strongly associated with airway type 2 inflammation in patients with asthma (42). As discussed above, exon 3 and exon 4 encode the nuclear localization domain and central domain, respectively, suggesting that processing of IL-33 is implicated in disease pathogenesis in these patients. Likewise, elevated levels of IL-33 protein have been found in several specimens from patients with asthma, including serum/plasma (71, 128), induced sputum (128), brochoalveolar lavage (BAL) fluids (49, 71), airway epithelial cells (49, 57), and submucosal inflammatory cells (122); the levels of IL-33 generally correlated with disease severity in these studies. In addition, fungal sensitization in children with severe asthma was associated with increased IL-33 levels in BAL fluids and airway submucosal tissues as compared to those children who did not show evidence of fungal sensitization (129). Similarly to IL-33, serum and plasma levels of soluble ST2 were increased in patients with asthma (128, 130).
Since the discovery of ILC2s in mice, ILC2s have been identified in peripheral blood and mucosal tissues of humans (131). The numbers of IL-33-responsive ILC2s were increased in peripheral blood and BAL fluids in subjects with allergic asthma as compared to those of healthy control subjects or those with allergic rhinitis (71, 132, 133). Moreover, the ILC2s from patients with asthma appeared to be more sensitive to IL-33 when compared to those from control subjects (132). The number of ILC2s in blood was associated with the magnitudes of tissue eosinophilia (134) and clinically unstable asthma (133). Furthermore, IL-33 and type 2 cytokines were induced during a rhinovirus-induced asthma exacerbation (135), suggesting that virus-induced IL-33 and IL-33-responsive T cells and ILC2s provide key mechanistic links between viral infection and exacerbation of asthma. Altogether, these findings suggested that IL-33 may be involved in the pathophysiology of human asthma, and justifies the need for pharmacological and biological agents that can control the levels of IL-33 as a potential therapeutic approach for patients with asthma.
Allergic rhinitis
ST2 mRNA, ST2 protein, and IL-33 mRNA expression are consistently increased in the nasal epithelium of patients with allergic rhinitis as compared to that of healthy control subjects (56, 91). However, there are conflicting reports regarding the levels of IL-33 protein. One study showed reduced levels of IL-33 (91) while another showed increased IL-33 in nasal epithelial cells (56). While the reason(s) for this discrepancy is not clear, the differences in the assay systems used to detect IL-33 protein and the potentially different forms (e.g., full-length vs. processed) of IL-33 in biological specimens may offer some explanation. Indeed, using immunohistochemistry, IL-33 was shown to be present only in the nuclei of nasal epithelial cells from healthy subject, while IL-33 staining was observed in both the nuclei and cytoplasm of the samples from patients with allergic rhinitis (56). IL-33 levels in serum/plasma have consistently been found to be increased in patients with allergic rhinitis (56, 127, 136).
While peripheral blood ILC2 numbers in allergic rhinitis were not different from those of control subjects under stable conditions (e.g., out of allergy season) (132), airway allergen exposure in patients with allergic rhinitis increased the numbers of blood ILC2s (137). Blood ILC2 numbers also were significantly increased in patients with seasonal allergic rhinitis during grass pollen season (138). Interestingly, ILC2s were increased in allergic rhinitis patients who were sensitive to HDM, but not to those sensitive to mug wort (139), suggesting that the biochemical nature of the allergens may have affected the levels of ILC2s and perhaps of IL-33. Thus, within the patients who showed clinical symptoms of allergic rhinitis, some heterogeneity existed in the immunopathogenesis of the disease.
Chronic rhinosinusitis
CRS is one of the most common chronic diseases in adults in the United States, and it produces significant morbidity and cost to the health care system (140). CRS represents >12 weeks of chronic inflammation in paranasal sinuses, and can be divided into two subtypes, CRS without nasal polyps (CRSsNP) and CRS with nasal polyps (CRSwNP) (141). CRSsNP and CRSwNP show distinct inflammatory endotypes with CRSwNP being associated with increased levels of tissue IL-5, tissue eosinophilia, and concomitant asthma (142). IL-33 was upregulated in nasal polyp tissues from patients with CRSwNP as compared with those of CRSsNP or healthy control subjects (143–146). Furthermore, IL-33 mRNA expression in nasal biopsy specimens was higher in CRSwNP patients, in particular those who were resistant to treatment (147). Similarly, ILC2 numbers in sinus mucosal biopsies were increased in patients with CRSwNP and were associated with tissue and blood eosinophilia (131, 148–150). The levels of ILC2s correlated with the severity of nasal symptoms (148) and were reduced following systemic corticosteroid treatment (150). Furthermore, IL-33-responsive and highly pathological memory CD4+ T cells were enriched in nasal polyp tissues from patients with eosinophilic CRS (115).
Conclusions and perspectives
As summarized in Figure 2, there is accumulating evidence to suggest that IL-33 plays important roles in innate and adaptive immunity in mucosal organs as well as in the homeostasis and repair of the airway. In innate immunity, IL-33 activates ILC2s, basophils, eosinophils, and mast cells to drive the early immune response against allergens and other environmental insults. In adaptive immunity, IL-33 regulates the functions of DCs and drives and influences the development of Th2, Tfh, and Treg cells. While robust type 2 immunity and airway inflammation are distinct features of the immunological effects of IL-33, IL-33 may also be involved in tissue homeostasis at the resting condition, and could help to repair mucosal tissues after injury. Furthermore, correlative evidence has been increasing to suggest the roles for IL-33 and their targets, such as ILC2 and memory Th2 cells, in chronic airway diseases in humans.
However, a number of questions still remain. For example, many aspects of the biology of IL-33 are not well understood, including its stability, metabolism, and the biological activity of any processed products that might be generated from intracellular or extracellular proteolytic enzymes. Furthermore, the fates of different forms of IL-33 are unknown, although post-translational modification and redox state might affect its activity. Regarding the cellular source of IL-33, in addition to tissue-resident cells, IL-33 can be induced in bone marrow-derived cells, such as DCs and mast cells, and our knowledge is limited regarding whether and how these sources of IL-33 contribute to the immune responses and disease processes.
One of the distinct characteristics of IL-33 is its involvement in chronic and persistent airway inflammation; however, the mechanisms appears to be complex and involve several cell types. Because IL-33 may also be involved in the regulation of Treg cells and tissue homeostasis and repair, it will be critical to identify the pathological and potentially beneficial roles of IL-33. Accumulating clinical evidence suggests that IL-33 and ILC2s may serve as biomarkers to define the subtypes of allergic airway diseases. However, more studies are needed to validate the usefulness of these biomarkers in large populations with different ethnic backgrounds, and to analyze how age, sex, and other variables affect these biomarkers. Finally, the human studies reported thus far are correlative, and it is therefore critically important to know the cause-effect relationships that IL-33 may play in various airway diseases. The biological and pharmacological agents that block the activities of IL-33 will likely be helpful in this regard. As the field of IL-33 is advancing and expanding quickly, one can expect that new knowledge will be gained in the coming years to answer many of these questions.
Acknowledgments
The authors thank Ms. LuRaye S. Eischens for secretarial help. This work was supported by grants from the National Institutes of Health (R01 AI71106, R01 HL117823, R01 AI128729) and the Mayo Foundation.
Footnotes
Conflict of Interest
The authors identify no conflict of interest.
References
- 1.Cookson W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat Rev Immunol. 2004;4:978–988. doi: 10.1038/nri1500. [DOI] [PubMed] [Google Scholar]
- 2.Lambrecht BN, Hammad H. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol. 2014;134:499–507. doi: 10.1016/j.jaci.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 3.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity. 2015;43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Onda H, Kasuya H, Takakura K, et al. Identification of genes differentially expressed in canine vasospastic cerebral arteries after subarachnoid hemorrhage. J Cerebr Blood F Met. 1999;19:1279–1288. doi: 10.1097/00004647-199911000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Baekkevold ES, Roussigne M, Yamanaka T, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. American journal of pathology. 2003;163:69–79. doi: 10.1016/S0002-9440(10)63631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Tominaga S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. 1989;258:301–304. doi: 10.1016/0014-5793(89)81679-5. [DOI] [PubMed] [Google Scholar]
- 9.Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PloS one. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pichery M, Mirey E, Mercier P, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. 2012;188:3488–3495. doi: 10.4049/jimmunol.1101977. [DOI] [PubMed] [Google Scholar]
- 11.Barlow JL, Peel S, Fox J, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. Journal of allergy and clinical immunology. 2013;132:933–941. doi: 10.1016/j.jaci.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Hsu CL, Bryce PJ. Inducible IL-33 expression by mast cells is regulated by a calcium-dependent pathway. J Immunol. 2012;189:3421–3429. doi: 10.4049/jimmunol.1201224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardman CS, Panova V, McKenzie AN. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol. 2013;43:488–498. doi: 10.1002/eji.201242863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams JW, Tjota MY, Clay BS, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Comm. 2013;4:2990. doi: 10.1038/ncomms3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu D, Chan WL, Leung BP, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. Journal of experimental medicine. 1998;187:787–794. doi: 10.1084/jem.187.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonilla WV, Frohlich A, Senn K, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science. 2012;335:984–989. doi: 10.1126/science.1215418. [DOI] [PubMed] [Google Scholar]
- 17.Moritz DR, Rodewald HR, Gheyselinck J, Klemenz R. The IL-1 receptor-related T1 antigen is expressed on immature and mature mast cells and on fetal blood mast cell progenitors. J Immunol. 1998;161:4866–4874. [PubMed] [Google Scholar]
- 18.Moro K, Yamada T, Tanabe M, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 19.Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price AE, Liang HE, Sullivan BM, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–379. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 22.Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. Journal of allergy and clinical immunology. 2008;121:1484–1490. doi: 10.1016/j.jaci.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol. 2009;123:1047–1054. doi: 10.1016/j.jaci.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:1526–1534. doi: 10.1182/blood-2008-05-157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 26.Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 2015;42:1005–1019. doi: 10.1016/j.immuni.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17:122–131. doi: 10.1038/ni.3370. [DOI] [PubMed] [Google Scholar]
- 28.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 29.Bonnelykke K, Sleiman P, Nielsen K, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46:51–55. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 30.Gudbjartsson DF, Bjornsdottir US, Halapi E, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41:342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 31.Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savenije OE, Mahachie JM, Granell R, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. 2014;134:170–177. doi: 10.1016/j.jaci.2013.12.1080. [DOI] [PubMed] [Google Scholar]
- 33.Torgerson DG, Ampleford EJ, Chiu GY, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43:887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–1513. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snelgrove RJ, Gregory LG, Peiro T, et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134:583–592 e586. doi: 10.1016/j.jaci.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iijima K, Kobayashi T, Hara K, et al. IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol. 2014;193:1549–1559. doi: 10.4049/jimmunol.1302984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willart MA, Deswarte K, Pouliot P, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. 2012;209:1505–1517. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yasuda K, Muto T, Kawagoe T, et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A. 2012;109:3451–3456. doi: 10.1073/pnas.1201042109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuda H, Komine M, Karakawa M, Etoh T, Tominaga S, Ohtsuki M. Novel splice variants of IL-33: differential expression in normal and transformed cells. J Invest Dermatol. 2012;132:2661–2664. doi: 10.1038/jid.2012.180. [DOI] [PubMed] [Google Scholar]
- 40.Polumuri SK, Jayakar GG, Shirey KA, et al. Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. J Immunol. 2012;189:50–60. doi: 10.4049/jimmunol.1003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Talabot-Ayer D, Calo N, Vigne S, Lamacchia C, Gabay C, Palmer G. The mouse interleukin (Il)33 gene is expressed in a cell type- and stimulus-dependent manner from two alternative promoters. J Leukoc Biol. 2012;91:119–125. doi: 10.1189/jlb.0811425. [DOI] [PubMed] [Google Scholar]
- 42.Gordon ED, Simpson LJ, Rios CL, et al. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc Natl Acad Sci U S A. 2016;113:8765–8770. doi: 10.1073/pnas.1601914113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–37. doi: 10.1016/j.coi.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 44.Roussel L, Erard M, Cayrol C, Girard JP. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO reports. 2008;9:1006–1012. doi: 10.1038/embor.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lefrancais E, Roga S, Gautier V, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A. 2012;109:1673–1678. doi: 10.1073/pnas.1115884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lefrancais E, Duval A, Mirey E, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U S A. 2014;111:15502–15507. doi: 10.1073/pnas.1410700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lingel A, Weiss TM, Niebuhr M, et al. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors–insight into heterotrimeric IL-1 signaling complexes. Structure. 2009;17:1398–1410. doi: 10.1016/j.str.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Prefontaine D, Nadigel J, Chouiali F, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 50.Shinoda K, Hirahara K, Linuma T, et al. Thy1+IL−7+ lymphatic endothelial cells in iBALT provide a survival niche for memory T-helper cells in allergic airway inflammation. PNAS. 2016;113:E2842–2851. doi: 10.1073/pnas.1512600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kearley J, Silver JS, Sanden C, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity. 2015;42:566–579. doi: 10.1016/j.immuni.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 52.Takeda T, Unno H, Morita H, et al. Platelets constitutively express IL-33 protein and modulate eosinophilic airway inflammation. J Allergy Clin Immunol. 2016;138:1395–1403 e1396. doi: 10.1016/j.jaci.2016.01.032. [DOI] [PubMed] [Google Scholar]
- 53.Chang YJ, Kim HY, Albacker LA, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hara K, Iijima K, Elias MK, et al. Airway uric acid is a sensor of inhaled protease allergens and initiates type 2 immune responses in respiratory mucosa. J Immunol. 2014;192:4032–4042. doi: 10.4049/jimmunol.1400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Byers DE, Alexander-Brett J, Patel AC, et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. 2013;123:3967–3982. doi: 10.1172/JCI65570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamekura R, Kojima T, Takano K, Go M, Sawada N, Himi T. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin Exp Allergy. 2012;42:218–228. doi: 10.1111/j.1365-2222.2011.03867.x. [DOI] [PubMed] [Google Scholar]
- 57.Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 58.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PloS one. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nile CJ, Barksby E, Jitprasertwong P, Preshaw PM, Taylor JJ. Expression and regulation of interleukin-33 in human monocytes. Immunology. 2010;130:172–180. doi: 10.1111/j.1365-2567.2009.03221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakanishi W, Yamaguchi S, Matsuda A, et al. IL-33, but not IL-25, is crucial for the development of house dust mite antigen-induced allergic rhinitis. PloS one. 2013;8:e78099. doi: 10.1371/journal.pone.0078099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carriere V, Roussel L, Ortega N, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bessa J, Meyer CA, de Vera Mudry MC, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. 2014;55:33–41. doi: 10.1016/j.jaut.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 63.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 64.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186:4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doherty TA, Khorram N, Chang JE, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol-Lung Cell Mol Physiol. 2012;303:L577–588. doi: 10.1152/ajplung.00174.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hristova M, Habibovic A, Veith C, et al. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol. 2016;137:1545–1556 e1511. doi: 10.1016/j.jaci.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. 2012;287:6941–6948. doi: 10.1074/jbc.M111.298703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal immunol. 2016;9:275–286. doi: 10.1038/mi.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christianson CA, Goplen NP, Zafar I, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136:59–68 e14. doi: 10.1016/j.jaci.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen ES, Scott IC, Majithiya JB, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Comm. 2015;6:8327. doi: 10.1038/ncomms9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ali S, Mohs A, Thomas M, et al. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J Immunol. 2011;187:1609–1616. doi: 10.4049/jimmunol.1003080. [DOI] [PubMed] [Google Scholar]
- 74.Gautier V, Cayrol C, Farache D, et al. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci Rep. 2016;6:34255. doi: 10.1038/srep34255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanabe T, Shimokawaji T, Kanoh S, Rubin BK. IL-33 stimulates CXCL8/IL-8 secretion in goblet cells but not normally differentiated airway cells. Clin Exp Allergy. 2014;44:540–552. doi: 10.1111/cea.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fujita J, Kawaguchi M, Kokubu F, et al. Interleukin-33 induces interleukin-17F in bronchial epithelial cells. Allergy. 2012;67:744–750. doi: 10.1111/j.1398-9995.2012.02825.x. [DOI] [PubMed] [Google Scholar]
- 77.Ganesan S, Pham D, Jing Y, et al. TLR2 Activation Limits Rhinovirus-Stimulated CXCL-10 by Attenuating IRAK-1-Dependent IL-33 Receptor Signaling in Human Bronchial Epithelial Cells. J Immunol. 2016;197:2409–2420. doi: 10.4049/jimmunol.1502702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yagami A, Orihara K, Morita H, et al. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol. 2010;185:5743–5750. doi: 10.4049/jimmunol.0903818. [DOI] [PubMed] [Google Scholar]
- 79.Molofsky AB, Van Gool F, Liang HE, et al. Interleukin-33 and Interferon-gamma Counter-Regulate Group 2 Innate Lymphoid Cell Activation during Immune Perturbation. Immunity. 2015;43:161–174. doi: 10.1016/j.immuni.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schiering C, Krausgruber T, Chomka A, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vasanthakumar A, Moro K, Xin A, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. 2015;16:276–285. doi: 10.1038/ni.3085. [DOI] [PubMed] [Google Scholar]
- 82.Hung LY, Lewkowich IP, Dawson LA, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci U S A. 2013;110:282–287. doi: 10.1073/pnas.1206587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oboki K, Ohno T, Kajiwara N, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kondo Y, Yoshimoto T, Yasuda K, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 85.Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity. 2012;37:463–474. doi: 10.1016/j.immuni.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 86.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 87.Kamijo S, Takeda H, Tokura T, et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J Immunol. 2013;190:4489–4499. doi: 10.4049/jimmunol.1201212. [DOI] [PubMed] [Google Scholar]
- 88.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–2054. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 89.Suzukawa M, Iikura M, Koketsu R, et al. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J Immunol. 2008;181:5981–5989. doi: 10.4049/jimmunol.181.9.5981. [DOI] [PubMed] [Google Scholar]
- 90.Yang Z, Grinchuk V, Urban JF, et al. Macrophages as IL-25/IL-33-responsive cells play an important role in the induction of type 2 immunity. PloS one. 2013;8:e59441. doi: 10.1371/journal.pone.0059441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haenuki Y, Matsushita K, Futatsugi-Yumikura S, et al. A critical role of IL-33 in experimental allergic rhinitis. J Allergy Clin Immunol. 2012;130:184–194 e111. doi: 10.1016/j.jaci.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 92.Cho KA, Suh JW, Sohn JH, et al. IL-33 induces Th17-mediated airway inflammation via mast cells in ovalbumin-challenged mice. Am J Physiol-Lung Cell Mol Physiol. 2012;302:L429–440. doi: 10.1152/ajplung.00252.2011. [DOI] [PubMed] [Google Scholar]
- 93.Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol. 2011;41:1675–1686. doi: 10.1002/eji.201041033. [DOI] [PubMed] [Google Scholar]
- 94.Halim TY, Steer CA, Matha L, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40:425–435. doi: 10.1016/j.immuni.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy. 2014;69:1300–1307. doi: 10.1111/all.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mirchandani AS, Besnard AG, Yip E, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol. 2014;192:2442–2448. doi: 10.4049/jimmunol.1300974. [DOI] [PubMed] [Google Scholar]
- 97.Drake LY, Iijima K, Bartemes K, Kita H. Group 2 Innate Lymphoid Cells Promote an Early Antibody Response to a Respiratory Antigen in Mice. J Immunol. 2016;197:1335–1342. doi: 10.4049/jimmunol.1502669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kurowska-Stolarska M, Kewin P, Murphy G, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–4790. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 99.Guo L, Wei G, Zhu J, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A. 2009;106:13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001;2:725–731. doi: 10.1038/90667. [DOI] [PubMed] [Google Scholar]
- 101.Kobayashi T, Iijima K, Checkel JL, Kita H. IL-1 family cytokines drive Th2 and Th17 cells to innocuous airborne antigens. Am J Respir Cell Mol Biol. 2013;49:989–998. doi: 10.1165/rcmb.2012-0444OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402:B18–23. doi: 10.1038/35037014. [DOI] [PubMed] [Google Scholar]
- 103.Akdis CA. Therapies for allergic inflammation: refining strategies to induce tolerance. Nat Med. 2012;18:736–749. doi: 10.1038/nm.2754. [DOI] [PubMed] [Google Scholar]
- 104.King C. New insights into the differentiation and function of T follicular helper cells. Nat Rev Immunol. 2009;9:757–766. doi: 10.1038/nri2644. [DOI] [PubMed] [Google Scholar]
- 105.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 106.Kobayashi T, Iijima K, Dent AL, Kita H. Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol. 2017;139:300–313 e307. doi: 10.1016/j.jaci.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Josefowicz SZ, Niec RE, Kim HY, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duan W, So T, Croft M. Antagonism of airway tolerance by endotoxin/lipopolysaccharide through promoting OX40L and suppressing antigen-specific Foxp3+ T regulatory cells. J Immunol. 2008;181:8650–8659. doi: 10.4049/jimmunol.181.12.8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Soroosh P, Doherty TA, Duane W, et al. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med. 2013;210:775–788. doi: 10.1084/jem.20121849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T (Treg) cells and impairs established immunological tolerance in the lungs. J Allergy Clin Immunol. 2017 doi: 10.1016/j.jaci.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Matta BM, Lott JM, Mathews LR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. 2014;193:4010–4020. doi: 10.4049/jimmunol.1400481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morita H, Arae K, Unno H, et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity. 2015;43:175–186. doi: 10.1016/j.immuni.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 114.Simpson A, Tan VY, Winn J, et al. Beyond atopy: multiple patterns of sensitization in relation to asthma in a birth cohort study. Am J Respir Crit Care Med. 2010;181:1200–1206. doi: 10.1164/rccm.200907-1101OC. [DOI] [PubMed] [Google Scholar]
- 115.Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity. 2016;45:198–208. doi: 10.1016/j.immuni.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 116.Endo Y, Hirahara K, Linuma T, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity. 2015;42:294–308. doi: 10.1016/j.immuni.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 117.Prussin C, Lee J, Foster B. Eosinophilic gastrointestinal disease and peanut allergy are alternatively associated with IL-5+ and IL-5(-) T(H)2 responses. J Allergy Clin Immunol. 2009;124:1326–1332 e1326. doi: 10.1016/j.jaci.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dougherty RH, Sidhu SS, Raman K, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol. 2010;125:1046–1053 e1048. doi: 10.1016/j.jaci.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Homer RJ, Elias JA. Airway remodeling in asthma: therapeutic implications of mechanisms. Physiology. 2005;20:28–35. doi: 10.1152/physiol.00035.2004. [DOI] [PubMed] [Google Scholar]
- 120.Pascual RM, Peters SP. Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. J Allergy Clin Immunol. 2005;116:477–486. doi: 10.1016/j.jaci.2005.07.011. quiz 487. [DOI] [PubMed] [Google Scholar]
- 121.Ramaprakash H, Shibata T, Duffy KE, et al. Targeting ST2L potentiates CpG-mediated therapeutic effects in a chronic fungal asthma model. Am J Pathol. 2011;179:104–115. doi: 10.1016/j.ajpath.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saglani S, Lui S, Ullmann N, et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J Allergy Clin Immunol. 2013;132:676–685 e613. doi: 10.1016/j.jaci.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li D, Guabiraba R, Besnard AG, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–1432 e1411. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arpaia N, Green JA, Moltedo B, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kuswanto W, Burzyn D, Panduro M, et al. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity. 2016;44:355–367. doi: 10.1016/j.immuni.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sakashita M, Yoshimoto T, Hirota T, et al. Association of serum interleukin-33 level and the interleukin-33 genetic variant with Japanese cedar pollinosis. Clin Exp Allergy. 2008;38:1875–1881. doi: 10.1111/j.1365-2222.2008.03114.x. [DOI] [PubMed] [Google Scholar]
- 128.Hamzaoui A, Berraies A, Kaabachi W, Haifa M, Ammar J, Kamel H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J Asthma. 2013;50:803–809. doi: 10.3109/02770903.2013.816317. [DOI] [PubMed] [Google Scholar]
- 129.Castanhinha S, Sherburn R, Walker S, et al. Pediatric severe asthma with fungal sensitization is mediated by steroid-resistant IL-33. J Allergy Clin Immunol. 2015;136:312–322 e317. doi: 10.1016/j.jaci.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oshikawa K, Kuroiwa K, Tago K, et al. Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am J Respir Crit Care Med. 2001;164:277–281. doi: 10.1164/ajrccm.164.2.2008120. [DOI] [PubMed] [Google Scholar]
- 131.Mjosberg JM, Trifari S, Crellin NK, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 132.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134:671–678 e674. doi: 10.1016/j.jaci.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jia Y, Fang X, Zhu X, et al. IL-13+ Type 2 Innate Lymphoid Cells Correlate with Asthma Control Status and Treatment Response. Am J Respir Cell Mol Biol. 2016;55:675–683. doi: 10.1165/rcmb.2016-0099OC. [DOI] [PubMed] [Google Scholar]
- 134.Liu T, Wu J, Zhao J, et al. Type 2 innate lymphoid cells: A novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respir Med. 2015;109:1391–1396. doi: 10.1016/j.rmed.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 135.Jackson DJ, Makrinioti H, Rana BM, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbation in vivo. Am J Respir Crit Care Med. 2014;190:1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vocca L, Di Sano C, Uasuf CG, et al. IL-33/ST2 axis controls Th2/IL-31 and Th17 immune response in allergic airway diseases. Immunobiology. 2015;220:954–963. doi: 10.1016/j.imbio.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 137.Doherty TA, Scott D, Walford HH, et al. Allergen challenge in allergic rhinitis rapidly induces increased peripheral blood type 2 innate lymphoid cells that express CD84. J Allergy Clin Immunol. 2014;133:1203–1205. doi: 10.1016/j.jaci.2013.12.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lao-Araya M, Steveling E, Scadding GW, Durham SR, Shamji MH. Seasonal increases in peripheral innate lymphoid type 2 cells are inhibited by subcutaneous grass pollen immunotherapy. J Allergy Clin Immunol. 2014;134:1193–1195 e1194. doi: 10.1016/j.jaci.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 139.Fan D, Wang X, Wang M, et al. Allergen-Dependent Differences in ILC2s Frequencies in Patients With Allergic Rhinitis. Allergy Asthma Immunol Res. 2016;8:216–222. doi: 10.4168/aair.2016.8.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bhattacharyya N, Orlandi RR, Grebner J, Martinson M. Cost burden of chronic rhinosinusitis: a claims-based study. Otolaryng Head Neck. 2011;144:440–445. doi: 10.1177/0194599810391852. [DOI] [PubMed] [Google Scholar]
- 141.Steinke JW, Borish L. Chronic rhinosinusitis phenotypes. Ann Allergy Asthma Immunol. 2016;117:234–240. doi: 10.1016/j.anai.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tomassen P, Vandeplas G, Van Zele T, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. 2016;137:1449–1456 e1444. doi: 10.1016/j.jaci.2015.12.1324. [DOI] [PubMed] [Google Scholar]
- 143.Shaw JL, Fakhri S, Citardi MJ, et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188:432–439. doi: 10.1164/rccm.201212-2227OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liao B, Cao PP, Zeng M, et al. Interaction of thymic stromal lymphopoietin, IL-33, and their receptors in epithelial cells in eosinophilic chronic rhinosinusitis with nasal polyps. Allergy. 2015;70:1169–1180. doi: 10.1111/all.12667. [DOI] [PubMed] [Google Scholar]
- 145.Lam M, Hull L, Imrie A, et al. Interleukin-25 and interleukin-33 as mediators of eosinophilic inflammation in chronic rhinosinusitis. Am J Rhinol Allergy. 2015;29:175–181. doi: 10.2500/ajra.2015.29.4176. [DOI] [PubMed] [Google Scholar]
- 146.Kim DK, Jin HR, Eun KM, et al. The role of interleukin-33 in chronic rhinosinusitis. Thorax. 2016 doi: 10.1136/thoraxjnl-2016-208772. [DOI] [PubMed] [Google Scholar]
- 147.Reh DD, Wang Y, Ramanathan M, Jr, Lane AP. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy. 2010;24:105–109. doi: 10.2500/ajra.2010.24.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ho J, Bailey M, Zaunders J, et al. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin Exp Allergy. 2015;45:394–403. doi: 10.1111/cea.12462. [DOI] [PubMed] [Google Scholar]
- 149.Miljkovic D, Bassiouni A, Cooksley C, et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy. 2014;69:1154–1161. doi: 10.1111/all.12440. [DOI] [PubMed] [Google Scholar]
- 150.Walford HH, Lund SJ, Baum RE, et al. Increased ILC2s in the eosinophilic nasal polyp endotype are associated with corticosteroid responsiveness. Clin Immunol. 2014;155:126–135. doi: 10.1016/j.clim.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]