

Abstract
C1 domain-containing proteins, such as protein kinase C (PKC), have a central role in cellular signal transduction. Their involvement in many diseases, including cancer, cardiovascular disease, and immunological and neurological disorders has been extensively demonstrated and has prompted a search for small molecules to modulate their activity. By employing a diacylglycerol (DAG)-lactone template, we have been able to develop ultra potent analogs of diacylglycerol with nanomolar binding affinities approaching those of complex natural products such as phorbol esters and bryostatins. One current challenge is the development of selective ligands capable of discriminating between different protein family members. Recently, structure-activity relationship studies have shown that the introduction of an indole ring as a DAG-lactone substituent yielded selective Ras guanine nucleotide-releasing protein (RasGRP1) activators when compared to PKCα and PKCε. In the present work, we examine the effects of ligand selectivity relative to the orientation of the indole ring and the nature of the DAG-lactone template itself. Our results show that the indole ring must be attached to the lactone moiety through the sn-2 position in order to achieve RasGRP1 selectivity.
Graphical Abstract
1. Introduction
The second messenger DAG plays a central role in cell signaling from both receptor tyrosine kinases and G-protein-coupled receptors.1 Upon receptor stimulation, the activation of phospholipase C catalyzes phosphatidylinositol 4,5-bisphosphate hydrolysis, liberating DAG and inositol 1,4,5-triphosphate. The rise in DAG concentration is transduced into downstream signals via activation of seven families of signaling proteins through binding of DAG to their C1 domains, which function as the DAG recognition motif. The most extensively studied proteins containing these domains are members of the PKC family, which belongs to the superfamily of serine/threonine kinases and is known to regulate numerous cellular functions such as cell growth, cell differentiation, metabolism, and apoptosis.2 After the identification of the PKCs, six other families of proteins with homologous, DAG-responsive C1 domains have been identified. The protein kinase D family is involved in Golgi function, proliferation, metastasis and apoptosis.3 The chimaerins act as inhibitors (GAPs, GTPase activating proteins) for the small GTPase Rac and are candidate tumor suppressors.4 The Unc-13 family members are responsible for promoting vesicle priming.5 The DAG kinases terminate DAG signaling by phosphorylating DAG.6 MRCK is a downstream effector of cdc42 involved in filopodia formation, contributing to tumor invasion.7 Finally, the RasGRP family members function as activators (GEFs, GTP Exchange factors) for Ras.8
PKC was first identified as a cellular receptor for the phorbol ester tumor promoters more than 30 years ago.9,10 Today, it is recognized that the involvement of the PKC isoforms in cellular biology extends far beyond its role in tumor promotion.11 Their central role in cellular signal transduction underlies their extensive involvement in many diseases, including cancer, cardiovascular disease, and immunological and neurological disorders. Thus far, the clinical trial results of PKC modulators have been disappointing, largely due to inadequate therapeutic effect and/or unanticipated adverse reactions. Given PKC’s critical roles in both normal physiology and disease, this family of kinases remains an enticing target for drug development. In this context, the development of selective modulators will contribute to the unraveling of the biology of PKC and to the future success in developing drugs for PKC-mediated diseases.11
RasGRP family members function as activators (GEFs, guanine nucleotide exchange factors) for the Ras family of small GTPases and are prominently expressed in blood cells. RasGRP malfunction likely contributes to autoimmunity and may contribute to blood malignancies.12 In addition, a role for RasGRP3 in prostate cancer 13 and melanoma 14 and of RasGRP1 in skin cancer15 has been demonstrated. Furthermore, it has been shown that RasGRPs are targets of the anticancer drug ingenol-3-angelate.16 Consequently, given the important biological roles of RasGRP family members, the discovery of selective agents capable of specifically interacting with their C1 domains could provide exciting lead structures for drug development and biochemical studies.
C1 domains are zinc finger structures of ~50 amino acids. Those that bind phorbol ester / diacylglycerol are termed “typical” whereas those that do not do so are referred to as “atypical”.17 DAG-lactones have provided a powerful platform for probing C1 domain structure-activity relations. They combine impressive structural simplification compared to the structurally complex phorbol esters while still achieving comparable nanomolar potencies.18 A current challenge is the development of ligands possessing selectivity, whether between the different families of C1 domain containing proteins or between isoforms within a single family, while retaining high potency for their selective target. A key previous finding using a combinatorial chemical approach was that DAG-lactones substituted with sn-1 and sn-2 aromatic rings displayed much higher binding affinities for RasGRP1/3 than for PKCα and other PKC isozymes.14
Building on these findings, exploration of heteroaryl moieties at the sn-2 position yielded the DAG-indololactone 1 as the most selective and potent compound in the series for the activation of RasGRP3.20 This compound showed a 22-fold more potent binding affinity for RasGRP3 as compared to PKCα and exhibited subnanomolar affinities. Starting with this lead DAG-indololactone 1, we synthesized a family of regioisomers that probed the influence on potency and selectivity of the position of the linkage between the indole ring and the lactone moiety.16 All compounds were potent and selective activators of RasGRP1 and RasGRP3 when compared to PKCα and revealed that the orientation of the 1-methyl-1H-indole ring on the DAG-lactone plays a critical role in selectivity, with the most selective compound having the linkage at position 3 of the indole ring (1).
In the present study, we have synthesized and evaluated a new family of DAG-indololactones in which the 1-methyl-1H-indole ring is attached through the sn-1 ester position rather than in the sn-2 position and we have then varied the nature of the substituent in the sn-2 position (Figure 1). As in previous studies, DAG-indololactones were synthesized as racemates but bearing in mind that the R isomer would be twice as potent.22 The results presented here show that both binding affinity and selectivity are decreased when the heterocyclic ring is present at the sn-1 position.
Figure 1.

Structures of parent sn-2 indololactone 1 and target sn-1 indololactones.
2. Results
2.1. Chemistry
The synthesis of the target compounds was accomplished through a well established procedure utilizing a sequential alkylation-elimination and acylation steps.17 Previously, we presented the synthesis of a family of regioisomers of lead compound 1 that differ in the position of the linkage between the 1-methyl-1H-indole ring and the DAG-lactone moiety at the sn-2 position.16 During the course of that work it became evident that the stability of indole ring towards deprotection conditions depended on its substitution pattern. From previous work we knew that 3-substituted indole rings were stable to the deprotection chemistry used for silyl ethers, so, we decided to synthesize the simple sn-1 indololactone derivative 2 starting with the asymmetrically protected DAG-lactone 9.24
Aldol condensation with acetone followed by mesylation and elimination of the aldol product gave 10 in 62% yield (Scheme 1). The benzyl ether group was selectively removed with BCl3 in 82% yield giving pure compound 11 after column chromatography purification. Acylation employing 1-methyl-1H-indole-3-carbonyl chloride and subsequent treatment with triethylamine trihydrofluoride to remove the tert-butyldiphenylsilyl ether afforded the expected lactone 2.
Scheme 1.

Synthesis of indololactone 2.
Reagents and conditions: (a) 1. (CH3)2CO, LiHMDS, THF, −78 °C, 4 h; 2. (i) CH3SO2Cl, Et3N, 0 °C, 2 h; (ii) DBU, CH2Cl2, room temp, 24 h, 62%; (b) BCl3, CH2Cl2, −78 °C, 2 h, 82%; (c) 1-methyl-1H-indole-3-carbonyl chloride, Et3N, DMAP, room temp, 24 h, 85%; (d) HF.Et3N, THF, reflux, 24 h, 78%.
During scale up of the synthesis of lead compound 1, we learned that 3-substituted indole rings were also stable during Lewis Acid treatment required for benzyl removal (unpublished results). So, for the preparation of target compounds 3, 5, 6 and 8 we employed lactone 13 that was available in higher amounts (Scheme 2). In most cases, the alkylation step generated mixtures of the E/Z-isomers; the only exception was the aromatic aldehyde that afforded exclusively E-16. As with previously synthesized DAG-lactones, the geometry of the exocyclic double bond was assigned by 1H NMR. The vinyl proton corresponding to the E-isomer appears as a characteristic multiplet that is further downfield compared to that of the corresponding Z-isomer.23 After separation of the E/Z-isomers by column chromatography, 14 (E-isomer) and 15 (individual regioisomers E and Z) were converted to the corresponding DAG-lactones 3, 5 and 6, respectively. Selective deprotection of the TBDMS group with TBAF, acylation of the free primary hydroxyl group, and final deprotection of the second primary hydroxyl group were carried out with very good yields. In the case of the aromatic derivative 16, the treatment with TBAF led to the desired monoalcohol 20 but with a high degree of isomerization, presumably due to the basic nature of the reagent. The isomerization was confirmed by HPLC analysis and two-dimensional NMR experiments (see Supplementary Data). As the isomers of 20 were not chromatographically separable, we continued the DAG-lactone synthesis with the mixture. Thus, after acylation and benzyl ether removal compound 8 was obtained as an E/Z mixture.
Scheme 2.

Synthesis of indololactones 3–8.
Reagents and conditions: (a) 1. RCHO, LiHMDS, THF, −78 °C, 2–4 h; 2. (i) CH3SO2Cl, Et3N, 0 °C, 5 h; (ii) DBU, CH2Cl2, room temp, 24 h; (b) TBAF, THF, room temp, 30 min; (c) CAN, CH3CN/H2O, 0 °C, 20 min; (d) 1-methyl-1H-indole-3-carbonyl chloride, Et3N, DMAP, room temp, 24 h; (e) BCl3, CH2Cl2, −78 °C, 1 h.
At this point we had several examples of compatibility between lactones bearing the 3-substituted indole moiety and typical conditions employed for benzyl group cleavage (BCl3, −78 °C, CH2Cl2). Consequently, in order to avoid double bond isomerization we prepared compound 7 starting with known lactone 2525 protected with benzyl and p-methoxyphenyl (PMP) groups and obtained excellent results. Compound 4 was synthesized following the same strategy.
2.2. Binding of ligands to PKCα, PKCε and RasGRP1
The binding affinities of compounds 1–8 for PKCα, PKCε and RasGRP1 were determined in vitro by competition with bound [20-3H]phorbol 12,13-dibutyrate (PDBU) in the presence of 100 μg/ml phosphatidylserine as previously described.26 For characterization, we examined PKCα, PKCε, and RasGRP1. We selected PKCα and PKCε because these isoforms had behaved differently from one another in a previous study, in which our lead DAG-indololactone showed marked selectivity for RasGRP1/3, compared to PKCα, with intermediate selectivity relative to PKCε.27 We limited our comparison to RasGRP1, since both RasGRP1 and RasGRP3 closely resembled one another in binding affinity for aryl-DAG lactones27 and for the DAG-indololactones in our previous study.21 In general, none of the compounds were appreciably more potent than parent compound 1 for any of the studied proteins (Table 1). Although all compounds showed equal or stronger affinity for RasGRP1 than for PKCα, the obtained values of selectivity, ranging from 1 to 6.5-fold, indicated that none of the synthesized DAG-indololactones improved the selectivity of the parent compound 1 (22-fold in parallel measurements, Table 1). Likewise, compound 1 remained the most selective for PKCε with a selectivity of 53, compared to values of 0.45 to 5.8 for compounds 2–8. In terms of potencies, compounds 6 and 8 were the most potent for RasGRP1 but 56-fold and 39-fold less potent than the phorbol ester PDBu and compound 1, respectively.
Table 1.
Binding selectivity of DAG-lactones 1–8 for PKCα and PKCε versus RasGRP1
| [3H]PDBub | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|---|
| PKCα | 0.28 ± 0.02 | 12.9 ± 1.1 | 698 ± 57 | 106 ± 16 | 504 ± 49 | 73 ± 12 | 83.1 ± 6.3 | 257 ± 38 | 116 ± 18 |
| PKCε | 0.22 ± 0.05 | 31.0 ± 1.6 | 301 ± 31 | 59.6 ± 9.5 | 295 ± 41 | 27.3 ± 7.9 | 55 ± 10 | 119 ± 14 | 131 ± 38 |
| RasGRP1 | 0.40 ± 0.03 | 0.58 ± 0.02 | 666 ± 13 | 63.3 ± 8.3 | 133 ± 12 | 31.3 ± 4.9 | 22.6 ± 3.1 | 40 ± 11 | 22.6 ± 1.8 |
| PKCα/RasGRP1 | --- | 22 | 1.08 | 1.7 | 3.8 | 2.3 | 3.7 | 6.5 | 5.1 |
| PKCε/RasGRP1 | --- | 53 | 0.45 | 0.94 | 2.2 | 0.87 | 2.4 | 3.0 | 5.8 |
| PKCα/PKCε | --- | 0.42 | 2.3 | 1.8 | 1.7 | 2.7 | 1.5 | 2.2 | 0.89 |
| ClogPc | --- | 5.0 | 2.6 | 6.4 | 5.2 | 6.6 | 6.6 | 7.0 | 7.0 |
Values (nM) represent mean ± SEM of the Kd or Ki from at least triplicate independent experiments.
[Kd(nM)].
The Clog P values were calculated using ChemBioDraw Ultra, version 12.0.2.
We have observed in previous studies18 that the binding affinity or Ki for the DAG-lactones is correlated with their lipophilicity (logP). This is because binding to the C1 domain requires ligand partitioning into the phosphatidylserine lipid bilayer (or the cell membrane in vivo) and the formation of a ternary complex between C1 domain, ligand, and the membrane. As expected from its lower lipophilicity, compound 2 was the least potent of the sn-1 indololactones studied here, and there is a general trend toward increasing affinity with increased logP (Table 1). The direction and orientation of the aliphatic chains relative to the lactone ring is also important.18 This can be seen here in the striking difference between compounds 1 and 4, where the 1-methyl-1H-indole ring and the branched alkyl group are reciprocally substituted. Similarly, compound 8, bearing a mixture of 1:1 E/Z isomers at the sn-2 aromatic group, presented approximately a 2-fold difference in Ki values for PKCα and RasGRP1 compared to the pure E isomer (7).
2.3. Modeling
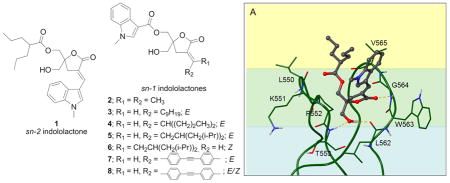
The parent compound 1 and the reciprocally substituted compound 4 were docked into the C1 domain of RasGRP1. The available crystal structure of RasGRP128 is relatively low resolution (3 Å) and the C1 domain has no ligand bound. We compared this structure to that of the phorbol-bound PKCδ C1b domain29 to determine whether the binding site would be suitable for docking, and found that while the backbone trace of the binding site loops could be aligned quite closely, residue Q568 at the bottom of the binding site was in a conformation that would clash with a bound ligand. The side chain amide orientation in this glutamine residue also appeared to be mis-assigned, since when flipped it could form three hydrogen bonds to surrounding residues (Figure 2). Before docking, we therefore adjusted the conformation of Q568 to an orientation similar to that seen in the equivalent residue (Q257) in the phorbol-bound C1b domain of PKCδ.29 Both compounds adopted the expected binding mode for DAG-lactones, in which hydrogen bonds are formed between the C1 domain backbone and the lactone carbonyl oxygen as well as the hydroxymethyl group. The overall docking scores and the calculated hydrogen bonding energy were virtually identical between the two compounds due to this similarity in binding mode. The indole group does not appear to form any strong intermolecular interactions with binding site residues in either docked structure (Figure 3). To explain the difference in binding affinity between compounds 1 and 4, therefore, we consider a model of the ternary complex between C1 domain, ligand, and the cell membrane, which as discussed above is the relevant structure for understanding DAG-lactone activities. Although the exact depth of penetration and orientation of the RasGRP1 C1 domain relative to the membrane bilayer is not known experimentally, we can develop a reasonable estimate based on the location of hydrophobic residues in the binding site loops. The positioning of the indole group relative to the bilayer differs significantly between compounds 1 and 4. In compound 1 (Figure 3A), the indole is predicted to be located in the interfacial region of the bilayer whereas when compound 4 is bound (Figure 3B), the indole ring is directed more deeply into the bilayer to interact with the hydrophobic lipid acyl chains.
Figure 2.

Comparison of the C1 domain binding sites of PKCδ C1b with bound phorbol (1PTR),29 shown in light blue, and RasGRP1 (4L9M),28 shown in black. The original position of residue Gln 568 in the RasGRP1 structure is colored white, and its adjusted position, as used for docking, is in black. Hydrogen bonds formed by this residue to the backbone are shown as dashed green lines.
Figure 3.

Model of the RasGRP1 C1 domain relative to the lipid bilayer. The colored background represents the location of bilayer regions: blue – polar region of charged lipid headgroups and water; green – interfacial region with lipid glycerol backbone and ester groups; yellow – hydrophobic regions with lipid acyl chains. A) Docked complex of compound 1. B) Docked complex of compound 4. Hydrogen bonds to the C1 domain backbone are shown as dashed yellow lines.
3. Discussion
As a starting point, compound 2 was selected as the simplest indololactone bearing the 1-methyl-1H-indole-3-carbonyl group at the sn-1 position of the DAG-lactone template. Initially, because of our concern with the stability of the indole ring to the reaction conditions required for final hydroxyl group deprotection, we decided to use a starting lactone scaffold containing at least one silyl protecting group, e.g., the TBDPS or TBDMS ether. However, for aromatic derivative 7 this approach generated an E/Z mixture of the deprotected compound upon removal of the silyl ether from the intermediate 16 with TBAF. As an alternative, we employed the available paramethoxyphenyl- and benzyl-protected lactone 25 as starting material (Scheme 2). From a chemical point of view, this approach seemed feasible since the most reactive 3-position of the indole ring was blocked, and its derivatives should have sufficient stability in the presence of the Lewis acids required for benzyl group removal.20 Indeed, this proved to be the case and the single E-isomer 7 was obtained in very good yield after final deprotection by BCl3 treatment. Thus there is no need to use silylated lactones such as 9 and 13 as the starting material. We therefore proceeded to use lactone 25 as the appropriate starting material for preparation of our family of target indololactones.
A clear finding of our study was that the exchange of the heterocyclic indole from the sn-2 position, as in the parent compound 1, to the sn-1 position decreased the affinity of the lactones to the proteins. Comparing compounds 1 and 4, which possess the identical indole group and alkyl chain arrayed in opposite orientations on the DAG-lactone, we observe 40-fold and 229-fold weaker affinities for PKCα and RasGRP1, respectively, when the indole group is in the sn-1 position. This result is consistent with a proposed binding model in which the DAG-lactone binds to the C1 domain with its acyl chain oriented toward the inner lipid core of the membrane and its R-alkylidene chain projected along the membrane surface in the interfacial region (Figure 2).29 This binding model is also supported by observed differences in affinity for other pairs of compounds with functionalized phenyl groups alternatively in either the sn-1 or sn-2 positions of the DAG-lactone template where again, more polar substituents such as phenol rings showed stronger C1 domain binding when located in the sn-2 position.19,29
It is well known that indole-bearing tryptophan residues are preferentially located in membrane protein structures where they can partition into the membrane-water interface.30 Furthermore, in C1 domains themselves, the conserved tryptophan residue in the binding site (located at position 563 in RasGRP1), has been shown to be important for ligand binding and membrane translocation in PKC θ and PKCα,32,33 and is predicted in our model to be located in the interfacial region upon membrane insertion of the C1 domain. It has been demonstrated computationally that isolated indole moieties also retain an energetic preference for the interfacial region of the lipid bilayer over the hydrophobic center.34 In the case of the 1-methyl-1H-indole substituent analyzed here, we believe that the contrasting behavior displayed by structural isomers 1 and 4 points to differences in the membrane interactions the two compounds can form once bound to the C1 domain in the ternary complex.
4. Conclusions
Indololactones bearing an indole ring at the sn-2 position were previously shown to possess potent affinity and selectivity for RasGRP1 versus PKCα. Here, we sought to compare their behavior when the indole ring was shifted to the sn-1 position. We developed a synthetic strategy for preparation of indololactones bearing the indole ring at the sn-1 position and assessed their binding affinities for PKCα, PKCε, and RasGRP1. We conclude that locating the heteroaromatic ring at the sn-2 position affords superior binding affinity as well as enhanced selectivity for RasGRP1. This preference probably arises from more favorable interactions of the indole ring in the interfacial region of cellular membranes. In summary, at the current time, compound 1 still remains the most selective RasGRP activator and a valuable tool for activating this class of C1 domain-containing proteins.
Experimental section
General procedures
All chemical reagents were commercially available. [20-3H]phorbol 12,13-dibutyrate (PDBu) was obtained from Perkin-Elmer, Waltham, MA. Melting points were determined on an Electrothermal IA9000series digital melting point apparatus and are uncorrected. Column chromatography was performed on a Teledyne Isco CombiFlash Companion instrument under gradient elution conditions with RediSep disposable flash columns. Analytical TLC was performed on Merck silica gel 254F plates. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX 400 instrument at 400 and 100 MHz, respectively. Spectra are referenced to the solvent in which they were run (7.26 ppm for CDCl3). High resolution, positive ion, electrospray ionization (ESI) mass spectra were obtained for all compounds. These analyses were carried out on a Thermo-Fisher LTQ-XL Orbitrap hybrid LC/MS/MS system equipped with an in-line diode-array UV detector and operated at a resolution of 30,000 (FWHM). Sample introduction into the mass spectrometer was accomplished by either flow-injection analysis (FIA) or by gradient LC/MS analysis; the choice of sample introduction mode was based on compound structure and estimated sample hydrophobicity. For FIA mode a sample solution was injected directly into the mass spectrometer using 1:1 CH3CN/H2O containing 0.1% HCOOH at a flow rate of 200 μl/min. Selected compounds, including indololactone target compounds 2–8, were analyzed by LC/MS to confirm identity and assess homogeneity. Separations were carried out on a narrow-bore (100 X 2.1 mm), small-particle (3.5-μm), Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column (12.5 X 2.1 mm) that was eluted with a 20-min combination of linear gradient and isocratic elution using mobile phases varying from 2 – 90% of CH3CN/H2O containing 0.1% HCOOH at a flow rate of 250 μl/min. Both the total-ion chromatogram (TIC) and the UV-chromatogram generated by this LC/MS analysis were used to confirm compound purity. For LC/MS mode, the chromatographic conditions were capable of separating E/Z isomers. For both FIA and LC/MS high resolution MS analyses, the resulting accurate mass measurement of a molecular species ([M+H]+, [M+Na]+ or M+NH4]+) was then used to determine a unique elemental composition for each particular compound. When appropriate, 1H and 13C NMR data were used to set elemental constraints for this calculation.
Chemistry
General Procedure A. Aldol condensation followed by olefination
Standard Alkylation Procedure
A solution of lactone (1 equiv) in THF (5 mL/mmol) at −78 °C was treated dropwise with LiHMDS (1.5 equiv, 1 M in THF). After the mixture was stirred at −78 °C for 1 h, a solution of R2CHO (1.5 equiv) in THF (1 mL/mmol) was added and stirring continued for 2–3 h more at −78 °C. The reaction was quenched by slow addition of a saturated aqueous solution of NH4Cl, and allowed to warm to room temperature. The layers were separated, and the aqueous layer was extracted with Et2O (3×). The combined organic phases were washed with H2O (1×) and brine (1×), dried over MgSO4, and concentrated in vacuo. Purification by silica gel flash column chromatography (gradient 0–20% EtOAc/hexanes) gave a mixture of diastereomers, which were used directly in the next step.
Standard Mesylation-Olefination Procedure
A solution of the alkylation product in dichloromethane (10 mL/mmol) at 0 °C was treated with methanesulfonyl chloride (2 eq) and triethylamine (4 eq) and then stirred at room temperature for 2–5 h. DBU (5 equiv) was added at 0 °C, and the resulting solution was stirred overnight at ambient temperature. The reaction mixture was treated with a saturated solution of NH4Cl (10 mL/mmol) and extracted with CH2Cl2 (3 ×). The combined organics were washed with H2O (2 ×) and brine (1 ×), dried (Na2SO4), and concentrated in vacuo. Purification by silica gel flash column chromatography (gradient 0–20% EtOAc/hexanes) gave 10, 14, 15, 16 and 27.
5-(benzyloxymethyl)-5-((tert-butyldiphenylsilyloxy)methyl)-3-(propan-2-ylidene)dihydrofuran-2(3H)-one (10)
Starting from 9 (1.38 g, 2.90 mmol) and following general procedure A, 10 was isolated as a colorless oil (926 mg, 62% yield): 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 7.1 Hz, 4 H, Ph), 7.44-7.26 (m, 11 H, Ph), 4.54 (s, 2 H, OCH2Ph), 3.72 (AB q, J = 10.7 Hz, 2 H, CH2OSi), 3.55 (AB q, J = 10.1 Hz, 2 H, CH2OBn), 2.76 (m, 2 H, H-4a,b), 2.25 (s, 3 H, CH3), 1.83 (s, 3 H, CH3), 1.01 (s, 9 H, (CH3)3CSi); 13C NMR (50 MHz, CDCl3): δ 169.6, 149.2, 137.7, 135.6, 135.6, 132.9, 132.7, 129.8, 128.3, 127.7, 127.6, 127.6, 120.3, 82.5, 73.6, 72.0, 66.5, 32.6, 26.6, 24.4, 19.8, 19.2. IR (neat) 1748 cm−1; HRMS (ESI) m/z calcd. for C32H42O4NSi (M+NH4)+: 532.2878, found: 532.2873. Elem. Anal. Calcd. for C32H38O4Si: C, 74.67; H, 7.44. Found: C, 74.57; H, 7.23.
(E) and (Z)-5-(benzyloxymethyl)-5-((tert-butyl(methoxy)(phenyl)silyloxy)methyl)-3-decylidenedihydrofuran-2(3H)-one (14)
Starting from 13 (489 mg, 1.40 mmol) and following general procedure A, 14 were isolated as a colorless oil, 7:3 mixture of E- and Z-isomers respectively (341 mg, 50% yield).
14E: 1H NMR (400 MHz, CDCl3) δ 7.36-7.26 (m, 5 H, Ph), 6.70-6.64 (m, 1 H, C=CH), 4.56 (s, 2 H, OCH2Ph), 3.69 (s, 2 H, CH2OSi), 3.55 (AB q, J = 10.3 Hz, 2 H, CH2OBn), 2.78-2.64 (AB q, J = 16.8 Hz, 2 H, H-4a,b), 2.18-2.10 (m, 2 H, CH2CH2(CH2)6CH3), 1.50-1.40 (m, 2 H, CH2CH2(CH2)6CH3), 1.26 (m, 12 H, CH2CH2(CH2)6CH3), 0.88 (t, J = 6.8 Hz, 3 H, CH2CH2(CH2)6CH3), 0.85 (s, 9 H, (CH3)3CSi,), 0.04 (s, 6 H, Si(CH3)2); 13C NMR (50 MHz, CDCl3) δ 170.5, 140.1, 137.8, 128.4, 127.7, 127.6, 127.1, 84.4, 73.7, 72.0, 65.6, 31.8, 30.1, 29.9, 29.5, 29.4, 29.3, 29.3, 28.1, 25.7, 22.6, 18.1, 14.1, −5.6; IR (neat) 2926, 1760, 1682 cm−1; HRMS (ESI) m/z calcd. for C29H49O4Si (M+H)+: 489.3395, found 489.3383.
14Z: 1H NMR (400 MHz, CDCl3) δ 7.36-7.27 (m, 5 H, Ph), 6.16-6.10 (m, 1 H, C=CH), 4.56 (s, 2 H, OCH2Ph), 3.67 (s, 2 H, CH2OSi), 3.53 (AB q, J = 10.3 Hz, 2 H, CH2OBn), 2.72-2.84 (m, 2 H, H-4a,b), 2.72-2.64 (m, 2 H, CH2(CH2)7CH3), 1.45-1.22 (m, 14 H, CH2(CH2)7CH3), 0.90-0.84 (m, 12 H, (CH3)3CSi, CH2(CH2)7CH3), 0.04 (s, 6 H, Si(CH3)2); 13C NMR (50 MHz, CDCl3) δ 169.3, 143.7, 137.8, 128.4, 127.7, 127.6, 125.0, 83.6, 73.7, 72.0, 65.5, 33.2, 31.9, 29.5, 29.5, 29.3, 29.1, 27.6, 25.7, 22.7, 18.1, 14.1, −5.6, −5.6; IR (neat) 2925, 1756, 1671 cm−1; HRMS (ESI) m/z calcd. for C29H49O4Si (M+H)+: 489.3395, found 489.3376.
(E) and (Z)-5-(benzyloxymethyl)-5-((tert-butyldimethylsilyloxy)methyl)-3-(3-isobutyl-5-methylhexylidene)dihydrofuran-2(3H)-one (15)
Starting from 13 (950 mg, 2.7 mmol) and following general procedure A, 15E and 15Z were isolated as a colorless oil, 6:4 mixture of E- and Z-isomers respectively (298 mg, 22% yield).
15E: 1H NMR (400 MHz, CDCl3): δ 7.37-7.25 (m, 5 H, Ph), 6.75-6.67 (m, 1 H, C=CH), 4.56 (s, 2 H, OCH2Ph), 3.69 (s, 2 H, CH2OSi), 3.55 (AB q, J = 10.2 Hz, 2 H, CH2OBn), 2.71 (AB q, J = 16.9 Hz, 2 H, H-4a,b), 2.10 (t, J = 6.4 Hz, 2 H, C=CHCH2), 1.73-1.55 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.09 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.86 (m, 21 H, (CH3)3CSi, CH(CH2(CH(CH3)2)2), 0.04 (s, 6 H, Si(CH3)2); 13C NMR (50 MHz, CDCl3) δ 170.3, 139.0, 137.7, 128.4, 128.0, 127.7, 127.6, 84.3, 73.7, 72.0, 65.5, 43.8, 34.6, 32.8, 30.0, 25.7, 25.2, 22.9, 22.6, 18.1, −5.6; IR (neat): 2953, 1760, 1681 cm−1; HRMS (ESI) m/z calcd. for C30H51O4Si (M+H)+: 503.3551, found 503.3541.
15Z: 1H NMR (400 MHz, CDCl3): δ 7.36-7.26 (m, 5 H, Ph), 6.14 (t, J = 7.3 Hz, 1 H, C=CH), 4.56 (s, 2 H, OCH2Ph), 3.70-3.63 (m, 2 H, CH2OSi), 3.56-3.50 (m, 2 H, CH2OBn), 2.80 (AB q, J = 16.2 Hz, 2 H, H-4a,b), 2.68-2.62 (m, 2 H, C=CHCH2), 1.69-1.56 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.08 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.86 (m, 21 H, (CH3)3CSi, CH(CH2(CH(CH3)2)2), 0.04 (s, 6 H, Si(CH3)2); 13C NMR (50 MHz, CDCl3) δ 169.2, 142.6, 137.8, 128.3, 127.6, 127.5, 125.8, 83.5, 73.6, 71.8, 65.3, 43.90, 43.9, 33.2, 33.2, 31.9, 25.7, 25.1, 25.1, 23.0, 22.7, 18.1, −5.6, −5.6; IR (neat): 2953, 1756, 1671 cm−1; HRMS (ESI) m/z calcd. for C30H51O4Si (M+H)+: 503.3551, found 503.3537.
(E)-5-(benzyloxymethyl)-5-((tert-butyldimethylsilyloxy)methyl)-3-(4-(ptolylethynyl) benzylidene)dihydrofuran-2(3H)-one (16)
Starting from 13 (501 mg, 1.43 mmol) and following general procedure A, 16 was obtained as a white solid (196 mg, 25% yield): 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 8.1 Hz, 2 H, Ph), 7.46-7.39 (m, 5 H, Ph, C=CH), 7.32-7.23 (m, 5 H, Ph), 7.14 (d, J = 7.8 Hz, 2 H, Ph), 4.55 (s, 2 H, OCH2Ph), 3.75-3.69 (m, 2 H, CH2OSi), 3.61-3.56 (m, 1 H, CH2OBn), 3.07 (AB q, J = 17.7 Hz, 2 H, H-4a,b), 2.35 (s, 3 H, CH3Ar), 0.79 (s, 9 H, (CH3)3Si), 0.00 (s, 6 H, CH3Si); 13C NMR (50 MHz, CDCl3) δ 171.5, 138.8, 137.6, 134.8, 134.4, 131.8, 131.6, 129.8, 129.2, 128.4, 127.8, 127.7, 126.4, 124.7, 119.8, 91.9, 88.4, 84.7, 73.7, 71.8, 65.7, 32.5, 25.6, 21.5, 18.1, −5.6; IR (neat) 3031, 2927, 2855, 1742, 1652 cm−1; HRMS (ESI) m/z calcd. for C35H41O4Si (M+H)+: 553.2769, found 553.2753; Mp: 128–131 °C.
(E)-5-(benzyloxymethyl)-5-((4-methoxyphenoxy)methyl)-3-(2-propylpentylidene) dihydrofuran-2(3H)-one (26)
A solution of lactone 25 (398 mg, 1.16 mmol) was treated following general procedure A, using potassium tert-butoxide (3 equiv) in place of DBU and stirring solution at room temperature for 20 min in the final step, to give 26 as a colorless oil, one single diastereomer (163 mg, 31% yield): 1H NMR (400 MHz, CDCl3) δ 7.33-7.26 (m, 5 H, Ph), 6.81 (s, 4 H, Ph), 6.51 (d, J = 10.8 Hz, 1 H, C=CH), 4.58 (s, 2 H, OCH2Ph), 4.03 (AB q, J = 9.7 Hz, 2 H, CH2OPhOCH3), 3.76 (s, 3 H, OCH3), 3.67 (AB q, J = 10.0 Hz, 2 H, CH2OBn), 2.85 (AB q, J = 16.2 Hz, 2 H, H-4a,b), 2.28-2.17 (m, 1 H, CH((CH2)2CH3)2), 1.48-1.15 (m, 8 H, CH((CH2)2CH3)2), 0.88-0.82 (m, 6 H, CH((CH2)2CH3)2); 13C NMR (50 MHz, CDCl3) δ 170.2, 154.3, 152.5, 145.9, 137.6, 128.4, 127.8, 127.6, 126.1, 115.6, 114.6, 82.8, 73.7, 72.1, 70.7, 55.7, 40.8, 37.1, 37.1, 30.8, 20.6, 20.6, 14.2, 14.2; IR (neat) 2926, 1760, 1682 cm−1; HRMS (ESI) m/z calcd. for C28H37O5 (M+H)+: 453.2636, found 453.2628.
(E)-5-(benzyloxymethyl)-5-((4-methoxyphenoxy)methyl)-3-(4-(ptolylethynyl) benzylidene)dihydrofuran-2(3H)-one (27)
Starting from 25 (440 mg, 1.28 mmol) and following general procedure A, 27 was obtained as a white solid, one single diastereomer (328 mg, 47% yield): 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.4 Hz, 2 H, Ph), 7.55 (s, 1 H, C=CH), 7.48-7.42 (m, 4 H, Ph), 7.32-7.26 (m, 5 H, Ph), 7.16 (d, J = 7.9 Hz, 2 H, Ph), 6.80 (s, 4 H, Ph), 4.59 (s, 2 H, OCH2Ph), 4.08 (AB q, J = 9.7 Hz, 2 H, CH2OPhOCH3), 3.74 (s, 3 H, CH3OAr), 3.71 (AB q, J = 10 Hz, 2 H, CH2OBn), 3.23 (AB q, J = 17.8 Hz, 2 H, H-4a,b), 2.37 (s, 3 H, CH3Ar); 13C NMR (50 MHz, CDCl3) δ 171.1, 154.4, 152.4, 138.9, 137.4, 135.8, 134.1, 131.9, 131.6, 129.9, 129.2, 128.4, 127.8, 127.6, 125.2, 125.0, 119.7, 115.7, 114.6, 92.1, 88.3, 83.1, 73.7, 71.9, 70.7, 55.7, 32.9, 21.5; IR (neat) 3031, 2918, 2866, 1747, 1659 cm−1; HRMS (ESI) m/z calcd. for C36H33O5 (M+H)+: 545.2323, found 545.2307; Mp: 88°C.
General Procedure B. TBDMS deprotection
To a stirred solution of 15E, 15Z or 16 (1 equiv) in anhydrous THF (30 mL/mmol) tetrabutylammonium fluoride (TBAF, 1.0 M solution in THF, 1.5 equiv) was added. The reaction mixture was stirred at room temperature for 15 min. The crude solution was concentrated in vacuo and purified by silica gel flash column chromatography (gradient 0–40% EtOAc/hexanes) to obtain 18, 19, or 20.
(E)-5-(benzyloxymethyl)-5-(hydroxymethyl)-3-(3-isobutyl-5-methylhexylidene)dihydrofuran-2(3H)-one (18)
Starting from 15E (130 mg, 0.26 mmol) and following general procedure B, 18 was obtained as a colorless oil (79 mg, 79% yield): 1H NMR (400 MHz, CDCl3) δ 7.37-7.26 (m, 5 H, Ph), 6.79-6.73 (m, 1 H, C=CH), 4.56 (s, 2 H, OCH2Ph), 3.76 (dd, J = 11.7, 5.6 Hz, 1H, CHaHOH), 3.67 (dd, J = 11.6, 4.9 Hz, 1H, CHHbOH), 3.57 (AB q, J = 10.0 Hz, 2 H, CH2OBn), 2.73 (AB q, 2 H, H-4a,b), 2.11 (m, 3 H, C=CHCH2, OH), 1.74-1.57 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.09 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.86 (m, 12 H, CH(CH2(CH(CH3)2)2); 13C NMR (50 MHz, CDCl3) δ 170.1, 140.4, 137.5, 128.5, 127.9, 127.6, 127.2, 84.0, 73.7, 71.9, 65.6, 43.8, 34.7, 32.8, 30.1, 25.2, 23.0, 22.9, 22.6; IR (neat) 3432, 2953, 2912, 1755, 1677 cm−1; HRMS (ESI) m/z calcd. for C24H37O4 (M+H)+: 389.2686, found 389.2678.
(Z)-5-(benzyloxymethyl)-5-(hydroxymethyl)-3-(3-isobutyl-5-methylhexylidene)dihydrofuran-2(3H)-one (19)
Starting from 15Z (120 mg, 0.24 mmol) and following general procedure B, 19 was obtained as a colorless oil (68 mg, 73% yield): 1H NMR (400 MHz, CDCl3) δ 7.38-7.26 (m, 5 H, Ph), 6.20 (t, J = 7.4 Hz, 1 H, C=CH), 4.56 (s, 2 H, OCH2Ph), 3.70 (AB q, J = 11.6 Hz, 2 H, CH2OH), 3.55 (AB q, J = 10.0 Hz, 2 H, CH2OBn), 2.82 (s, 2 H, H-4a,b), 2.66 (m, 2 H, C=CHCH2), 2.20 (s, 1 H, OH), 1.69-1.56 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.08 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.85 (m, 12 H, CH(CH2(CH(CH3)2)2); 13C NMR (50 MHz, CDCl3) δ 169.1, 144.0, 137.5, 128.4, 127.8, 127.6, 125.1, 83.3, 73.7, 71.8, 65.4, 43.9, 33.4, 33.2, 32.0, 25.1, 23.0, 22.7, 22.7; IR (neat) 3432, 2953, 2908, 1752, 1668 cm−1; HRMS (ESI) m/z calcd. for C24H37O4 (M+H)+: 389.2686, found 389.2673.
5-(benzyloxymethyl)-5-(hydroxymethyl)-3-(4-(p-tolylethynyl)benzylidene) dihydrofuran-2(3H)-one (20)
Starting from 16 (170 mg, 0.30 mmol) and following general procedure B, 20 was obtained as a colorless oil, inseparable 1:1 mixture of E- and Z-isomers (84 mg, 64% yield): 1H NMR (400 MHz, CDCl3) δ 7.58-7.12 (m, 27 H, Ar isomer E, Ar isomer Z, C=CH isomer E), 6.92 (s, 1 H, C=CH isomer Z), 4.55 (s, 2 H, OCH2Ph isomer E), 4.51 (AB q, 2 H, J = 11.3 Hz, OCH2Ph isomer Z), 3.54-3.84 (m, 10 H, CH2OBn, CH2OH, H-4a,b isomer Z), 3.13 (s, 2 H, H-4a,b isomer E), 2.36 (s, 6 H, CH3Ar); 13C NMR (50 MHz, CDCl3) δ 172.4, 171.5, 148.9, 138.9, 138.4, 137.3, 137.2, 137.1, 136.0, 135.4, 134.0, 131.9, 131.6, 131.4, 131.1, 130.7, 130.0, 129.5, 129.2, 129.1, 128.9, 128.5, 128.0, 127.9, 127.7, 125.3, 125.0, 122.0, 120.1, 119.7, 92.1, 89.6, 88.4, 88.4, 84.5, 73.8, 73.7, 71.8, 70.4, 65.3, 63.9, 32.4, 31.7, 22.6, 21.5; IR (neat) 3365, 2920, 2865, 1729, 1652 cm−1; HRMS (ESI) m/z calcd. for C29H27O4 (M+H)+: 439.1904, found 439.1891.
General Procedure C. PMP deprotection
Ceric ammonium nitrate (3 equiv) was added to a solution of 26, 27 (1 equiv) in 80% CH3CN/H2O (v/v, 10 mL/mmol) at 0 °C. The reaction mixture was stirred for 15 min. A solution of 5% Na2S2O3 was added at room temperature and the aqueous phase was extracted with CH2Cl2 (3 ×). The organic phase was dried (Na2SO4) and concentrated in vacuo. Purification by silica gel flash column chromatography (gradient 0–30% EtOAc/hexanes) gave 28, 29.
(E)-5-(benzyloxymethyl)-5-(hydroxymethyl)-3-(2-propylpentylidene)dihydrofuran-2(3H)-one (28)
Starting from 26 (149 mg, 0.33 mmol) and following general procedure C, 28 was obtained as a colorless oil (101 mg, 89% yield): 1H NMR (400 MHz, CDCl3) δ 7.37-7.26 (m, 5 H, Ph), 6.49 (dt, J = 10.7, 2.7 Hz, 1 H, C=CH), 4.55 (d, J = 2.3 Hz, 2 H, OCH2Ph), 3.76 (dd, J = 12.1, 7.1 Hz, 1 H, CHaHOH), 3.68 (dd, J = 12.1, 6.6 Hz, 1 H, CHHbOH), 3.57 (AB q, J = 10.0 Hz, 2 H, CH2OBn), 2.79 (dd, J = 16.9, 2.6 Hz, 1 H, H-4a), 2.71 (dd, J = 16.8, 2.9 Hz, 1H, H-4b), 2.26-2.16 (m, 1 H, CH((CH2)2CH3)2), 2.09 (t, J = 6.8 Hz, 1H, CH2OH), 1.48-1.13 (m, 8 H, CH((CH2)2CH3)2), 0.87 (t, J = 7.2 Hz, 3 H, CH((CH2)2CH3)2), 0.84 (t, J = 7.2 Hz, 3 H, CH((CH2)2CH3)2); 13C NMR (50 MHz, CDCl3) δ 171.0, 147.0, 138.2, 129.1, 128.6, 128.3, 126.8, 84.7, 74.5, 72.7, 66.3, 41.5, 37.8, 30.9, 21.3, 21.2, 14.8; IR (neat) 3437, 2955, 1755, 1737, 1678 cm−1; HRMS (ESI) m/z calcd. for C21H31O4 (M+H)+: 347.2217, found 347.2208.
(E)-5-(benzyloxymethyl)-5-(hydroxymethyl)-3-(4-(p-tolylethynyl)benzylidene) dihydrofuran-2(3H)-one (29)
Starting from 27 (308 mg, 0.56 mmol) and following general procedure C, 29 was obtained as a yellow solid (181 mg, 74% yield): 1H NMR (400 MHz, CDCl3) δ 7.58-7.50 (m, 3 H, Ar, C=CH isomer E), 7.47-7.40 (m, 3 H, Ar), 7.35-7.25 (m, 6 H, Ar ), 7.19-7.12 (m, 2 H, Ar), 4.55 (s, 2 H, OCH2Ph), 3.80 (m, 2 H, CH2OH), 3.63 (AB q, J = 10.0 Hz, 2 H, CH2OBn), 3.12 (s, 2 H, H-4a,b), 2.36 (s, 3 H, CH3Ar), 1,92 (s, 1H, OH).
General Procedure D. BnO Deprotection
A solution of 10, 21, 22, 23, 30, 31 (1 equiv) in anhydrous dichloromethane (20 mL/mmol) at −78°C was treated dropwise with boron trichloride (3 equiv). The reaction was monitored by TLC and quenched upon completion (0.5–2 h) by the slow addition of NaHCO3 and the aqueous phase was extracted with dichloromethane (2 ×). The organic phase was dried (Na2SO4) and concentrated in vacuo. Purification by silica gel flash column chromatography (gradient 0–40% EtOAc/hexanes) gave 11, 3, 4, 5, 6, 7 and 8.
General Procedure E. Acylation
A solution of 11, 17, 18, 19, 20, 28 or 29 (1 equiv) in dichloromethane (12 ml/mmol) was treated with Et3N (3 equiv), 1-methyl-1H-indole-3-carbonyl chloride (1.3 equiv) and a catalytic amount of DMAP (0.1 equiv). The reaction was stirred at room temperature and monitored by TLC, and upon completion (20–27 h) it was concentrated in vacuo. Purification by silica gel flash column chromatography (gradient 0–30% EtOAc/hexanes) gave 12, 21, 22, 23, 30, 31.
5-((tert-butyldiphenylsilyloxy)methyl)-5-(hydroxymethyl)-3-(propan-2-ylidene)dihydrofuran-2(3H)-one (11)
Starting from 10 (229 mg, 0.44 mmol) and following general procedure D, 11 was obtained as an amber oil (154 mg, 82%yield): 1H NMR (400 MHz, CDCl3) δ 7.66-7.61 (m, 4 H, Ph), 7.46-7.36 (m, 6 H, Ph), 3.76-3.60 (m, 4 H, CH2OSi, CH2OH), 2.76 (AB q, J = 16.2 Hz, 2 H, H-4a,b), 2.24 (s, 3 H, CH3), 1.94 (t, J = 6.8 Hz, 1 H, OH), 1.86 (s, 3 H, CH3), 1.03 (s, 9 H, (CH3)3CSi); 13C NMR (50 MHz, CDCl3) δ 169.5, 150.1, 135.6, 135.6, 132.8, 132.6, 129.9, 127.8, 120.1, 83.2, 66.4, 65.4, 31.9, 26.6, 24.5, 19.8, 19.2; IR (neat): 3417, 1746 cm− 1; HRMS (ESI) m/z calcd. for C25H33O4Si (M+H)+: 425.2143, found 425.2124.
(2-((tert-butyldiphenylsilyloxy)methyl)-5-oxo-4-(propan-2-ylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (12)
Starting from 11 (434 mg, 1.02 mmol) and following general procedure E, 12 was obtained as a colorless oil (508 mg, 85% yield): 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 7.7 Hz, 1 H, H-4′), 7.70-7.63 (m, 5 H, Ph, H-2′), 7.44-7.21 (m, 9 H, Ph, H-5′, H-6′, H-7′), 4.47 (AB q, J = 11.7 Hz, 2 H, CH2CO2), 3.86-3.81 (m, 2 H, CH2OSi), 3.80 (m, 3 H, CH3N), 2.84 (AB q, J = 16.3 Hz, 2 H, H-3a,b), 2.23 (s, 3 H, CH3), 1.81 (s, 3 H, CH3), 1.04 (s, 9 H, (CH3)3CSi); 13C NMR (100 MHz, CDCl3): δ 169.3, 164.2, 149.8, 137.2, 135.6, 135.6, 132.7, 132.6, 129.8, 129.8, 127.8, 127.8, 126.5, 122.8, 122.1, 121.5, 119.9, 109.7, 106.1, 81.6, 66.5, 65.0, 33.4, 32.7, 26.6, 24.4, 19.8, 19.2; IR (neat): 1746, 1699 cm−1; HRMS (ESI) m/z calcd. for C35H40NO5Si (M+H)+: 582.2670, found 582.2662.
(E)-(2-(benzyloxymethyl)-4-decylidene-5-oxotetrahydrofuran-2-yl)methyl-1-methyl-1H-indole-3-carboxylate (21)
Starting from 14E (191 mg, 0.37 mmol) and following general procedures B and E, 21 was obtained as a white solid (138 mg, 70% yield): 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.2 Hz, 1 H, H-4′), 7.69 (s, 1 H, H2′), 7.37-7.24 (m, 8 H, Ph, H-5′, H-6′, H-7′), 6.77-6.70 (m, 1 H, C=CH), 4.60 (s, 2 H, OCH2Ph), 4.57 (d, J = 11.9 Hz, 1 H, CO2CHaH), 4.44 (d, J = 11.8 Hz, 1 H, CO2CHHb), 3.83 (s, 3 H, CH3N), 3.68 (AB q, J = 10.1 Hz, 2 H, BnOCH2), 2.84 (AB q, J = 17.4 Hz, 2 H, H-3a,b), 2.15-2.06 (m, 2 H, CH2(CH2)7CH3), 1.39-1.14 (m, 14 H, CH2(CH2)7CH3), 0.87 (t, J = 6.9 Hz, 3 H, CH3); 13C NMR (50 MHz, CDCl3): δ 170.2, 164.1, 141.2, 137.4, 137.2, 135.6, 128.4, 127.8, 127.7, 126.5, 126.3, 122.9, 122.1, 121.5, 109.8, 105.9, 82.8, 73.7, 72.0, 65.2, 33.5, 31.8, 30.6, 30.2, 29.4, 29.3, 29.3, 28.0, 22.6, 14.1; IR (neat): 2926, 2854, 1750, 1688, 1538 cm−1; HRMS (ESI) m/z calcd. for C33H42NO5 (M+H)+: 532.3057, found 532.3045; Mp: 92°C.
(E)-(2-(benzyloxymethyl)-4-(3-isobutyl-5-methylhexylidene)-5-oxotetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (22)
Starting from 18 (74 mg, 0.18 mmol) and following general procedure E, 22 was obtained as a colorless oil (92 mg, 93% yield): 1H NMR (400 MHz, CDCl3) δ 8.08 (d, 1 H, J = 7.3 Hz, H-4′), 7.69 (s, 1 H, H2′), 7.36-7.24 (m, 8 H, Ph, H-5′, H-6′, H-7′), 6.77 (m, 1 H, C=CH), 4.59 (s, 2 H, OCH2Ph), 4.56 (d, 1 H, J = 11.8 Hz, CO2CHaH), 4.44 (d, 1 H, J = 11.8 Hz, CO2CHHb), 3.82 (s, 3 H, CH3N), 3.67 (AB q, J = 10.1 Hz, 2 H, BnOCH2), 2.83 (AB q, J = 17.1 Hz, 2 H, H-3a,b), 2.14-2.01 (m, 2 H, C=CHCH2), 1.68-1.50 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.07-0.98 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.83-0.75 (m, 12 H, CH(CH2(CH(CH3)2)2); 13C NMR (50 MHz, CDCl3) δ 170.0, 164.1, 140.1, 137.4, 137.2, 135.6, 128.4, 127.8, 127.6, 127.1, 126.6, 122.9, 122.1, 121.5, 109.8, 105.9, 82.7, 73.7, 71.9, 65.1, 43.7, 34.6, 33.5, 32.7, 30.8, 25.1, 22.9, 22.8, 22.6, 22.5. IR (neat): 2953, 2912, 2867, 1757, 1701, 1534, 748 cm−1; HRMS (ESI) m/z calcd. for C34H44NO5 (M+H)+: 546.3214, found 546.3199.
(Z)-(2-(benzyloxymethyl)-4-(3-isobutyl-5-methylhexylidene)-5-oxotetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (23)
Starting from 19 (64 mg, 0.16 mmol) and following general procedure E, 23 was obtained as a colorless oil (74 mg, 85% yield): 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 6.8 Hz, 1 H, H-4′), 7.71 (s, 1 H, H-2′), 7.36-7.23 (m, 8 H, Ph, H-5′, H-6′, H-7′), 6.17 (m, 1 H, C=CH), 4.59 (s, 2 H, OCH2Ph), 4.55 (d, J = 11.8 Hz, 1 H, CO2CHaH), 4.43 (d, J = 11.8 Hz, 1 H, CO2CHHb), 3.82 (s, 3 H, CH3N), 3.66 (AB q, J = 10.1 Hz, 2 H, BnOCH2), 2.97 (dd, J = 16.4, 1.9 Hz, 1 H, H-3a), 2.86 (dd, J = 16.4, 1.9 Hz, 1 H, H-3b), 2.81-2.73 (m, 1 H, C=CHCHaH), 2.52-2.43 (m, 1 H, C=CHCHHb), 1.65-1.53 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.07-1.02 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.84-0.80 (m, 12 H, CH(CH2(CH(CH3)2)2); 13C NMR (50 MHz, CDCl3 δ 169.0, 164.2, 143.7, 137.5, 137.2, 135.6, 128.4, 127.8, 127.6, 126.5, 124.9, 122.9, 122.2, 121.6, 109.8, 106.1, 82.0, 73.7, 71.8, 64.9, 43.9, 43.9, 34.1, 33.5, 33.2, 32.0, 25.1, 23.0, 23.0, 22.7, 22.6; IR (neat): 2952, 2909, 2866, 1755, 1702, 1534, 748 cm−1; HRMS (ESI) m/z calcd. for C34H44NO5 (M+H)+: 546.3214, found 546.3197.
(E)-(2-(benzyloxymethyl)-5-oxo-4-(2-propylpentylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (30)
Starting from 28 (90 mg, 0.26 mmol) and following general procedure E, 30 was obtained as a colorless oil (129 mg, 98% yield): 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.4 Hz, 1 H, H-4′), 7.69 (s, 1 H, H-2′), 7.36-7.23 (m, 8 H, Ph, H-5′, H-6′, H-7′), 6.50 (m, 1 H, C=CH), 4.59 (s, 2 H, OCH2Ph), 4.58 (d, J = 11.8 Hz, 1 H, CO2CHaH), 4.42 (d, J = 11.8 Hz, 1 H, CO2CHHb), 3.82 (s, 3 H, CH3N), 3.67 (AB q, J = 10.1 Hz, 2 H, BnOCH2), 2.90 (dd, J = 17.0, 2.7 Hz, 1 H, H-3a), 2.78 (dd, J = 17.0, 2,6 Hz, 1 H, H-3b), 2.22-2.11 (m, 1 H, CH((CH2)2CH3)2), 1.45-0.98 (m, 8 H, CH((CH2)2CH3)2), 0.83 (t, J = 7.1 Hz, 3 H, CH((CH2)2CH3)2), 0.61 (t, J = 7.2 Hz, 3 H, CH((CH2)2CH3)2); 13C NMR (50 MHz, CDCl3): δ 170.2, 164.1, 146.0, 137.4, 137.2, 135.6, 128.4, 127.8, 127.7, 126.6, 126.0, 122.9, 122.2, 121.5, 109.7, 105.9, 82.7, 73.7, 72.0, 65.2, 40.7, 37.0, 33.4, 30.8, 20.6, 20.5, 14.2, 14.9; IR (neat): 2955, 2927, 2870, 1755, 1701, 1533, 748 cm−1; HRMS (ESI) m/z calcd. for C31H38NO5 (M+H)+: 504.2744, found 504.2735.
(E)-(2-(benzyloxymethyl)-5-oxo-4-(4-(p-tolylethynyl)benzylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (31)
Starting from 27 (308 mg, 0.56 mmol) and following general procedures C and E, 31 was obtained as a white solid (127 mg, 38% yield): 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.9 Hz, 1 H, H-4′), 7.66 (s, 1 H, H-2′), 7.15-7.56 (m, 17 H, Ar, H-5′, H-6′, H-7′, C=CH), 4.61 (d, J = 2.3 Hz, 2 H, OCH2Ph), 4.57 (d, J = 3.6 Hz, 2 H, CO2CH2), 3.79 (s, 3 H, CH3N), 3.73 (AB q, J = 10.1 Hz, 1 H, BnOCH2), 3.29 (dd, J = 17.9, 2.7 Hz, 1 H, H-3a), 3.21 (dd, J = 2.7, 17.9 Hz, 1 H, H-3b), 2.38 (s, 3 H, CH3Ar); 13C NMR (50 MHz, CDCl3): δ 171.1, 164.1, 139.0, 137.3, 137.2, 135.8, 135.7, 134.0, 131.9, 131.6, 129.9, 129.2, 128.5, 127.9, 127.9, 126.5, 125.2, 125.1, 122.9, 122.1, 121.5, 119.7, 109.8, 105.9, 92.1, 88.3, 83.0, 73.8, 71.9, 65.1, 33.5, 33.1, 21.5; IR (neat) 3117, 2919, 1742, 1691 cm−1; HRMS (ESI) m/z calcd. for C39H34NO5 (M+H)+: 596.2431, found 596.2415; Mp: 207–209°C.
(2-(hydroxymethyl)-5-oxo-4-(propan-2-ylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (2)
A solution of compound 12 (351 mg, 0.60 mmol) in anhydrous THF (12 mL) was treated dropwise with triethylamine hydrofluoride (10 eq). The reaction mixture was stirred at 70°C for 7 h. Volatiles were removed and the residue was purified by silica gel flash column chromatography to yield pure final DAG-indololactone 2 as a white solid (160 mg, 78% yield): 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 6.6 Hz, 1 H, H-4′), 7.75 (s, 1 H, H-2′), 7.34-7.23 (m, 3 H, H-5′, H-6′, H-7′), 4.51 (d, J = 11.9 Hz, 1 H, CO2CHaH), 4.41 (d, J = 11.9 Hz, 1 H, CO2CHHb), 3.78 (s, 3 H, CH3N), 3.77-3.73 (m, 2 H, CH2OH), 3.16 (t, J = 6.8 Hz, 1 H, OH), 2.92 (d, J = 16.5 Hz, 1 H, H-3a), 2.76 (d, J = 16.5 Hz, 1 H, H-3b), 2.21 (s, 3 H, CH3), 1.82 (s, 3 H, CH3); 13C NMR (100 MHz, CDCl3): δ 169.5, 164.8, 151.6, 137.3, 136.0, 126.5, 123.0, 122.2, 121.5, 119.3, 110, 105.9, 82.0, 65.0, 64.8, 33.5, 32.4, 24.6, 20.0; IR (neat) 3425, 1705, 1661 cm1; HRMS (ESI) m/z calcd. for C19H22NO5 (M+H)+: 344.1492, found 344.1482; Mp: 140–141°C.
(E)-(4-decylidene-2-(hydroxymethyl)-5-oxotetrahydrofuran-2-yl)methyl 1-methyl-1Hindole-3-carboxylate (3)
Starting from 21 (134 mg, 0.25 mmol) and following general procedure D, 3 was obtained as a white solid (72 mg, 65% yield): 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.1 Hz, 1 H, H-4′), 7.77 (s, 1 H, H-2′), 7.38-7.26 (m, 3 H, H-5′, H-6′, H-7′), 6.78 (t, J = 7.5 Hz, 1 H, C=CH), 4.58 (d, J = 12.0 Hz, 1 H, CO2CHaH), 4.43 (d, J = 12.0 Hz, 1 H, CO2CHHb), 3.84 (s, 3 H, CH3N), 3.75 (d, J = 6.8 Hz, 2 H, CH2OH), 2.92 (d, J = 17.2 Hz, 1 H, H-3a), 2.75 (d, J = 17.2 Hz, 1 H, H-3b), 2.50 (t, J = 6.9 Hz, 1 H, OH), 2.19-2.11 (m, 2 H, CH2CH2(CH2)6CH3), 1.45-1.35 (m, 2 H, CH2CH2(CH2)6CH3), 1.30-1.17 (m, 12 H, CH2CH2(CH2)6CH3), 0.87 (t, J = 6.8 Hz, 3 H, CH2CH2(CH2)6CH3); 13C NMR (100 MHz, CDCl3) δ 170.4, 164.8, 142.3, 137.4, 136.0, 126.7, 128.1, 123.2, 122.4, 121.6, 110.0, 105.8, 83.8, 64.7, 64.6, 33.6, 31.9, 30.4, 30.1, 29.5, 29.5, 29.4, 29.4, 28.2, 22.8, 14.2; IR (neat) 3497, 2919, 1753, 1686 cm−1; HRMS (ESI) m/z calcd. for C26H36NO5 (M+H)+: 442.2588, found 442.2583; Mp 95–96°C.
(E)-(2-(hydroxymethyl)-5-oxo-4-(2-propylpentylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (4)
Starting from 30 (118 mg, 0.23 mmol) and following general procedure D, 4 was obtained as a colorless oil (86 mg, 90% yield): 1H NMR (400 MHz, CDCl3) δ 8.13-8.09 (m, 1 H, H-4′), 7.77 (s, 1 H, H-2′), 7.37-7.26 (m, 3 H, H-5′, H-6′, H-7′), 6.55 (dt, J = 10.6, 2.7 Hz, 1 H, C=CH), 4.55 (d, J = 12 Hz, 1 H, CO2CHaH), 4.46 (d, J = 12 Hz, 1 H, CO2CHHb), 3.84 (s, 3 H, CH3N), 3.74 (d, J = 7.0 Hz, 2 H, CH2OH), 2.90 (dd, J = 17.0, 2.7 Hz, 1 H, H-3a), 2.75 (dd, J = 17.0, 2.7 Hz, 1 H, H-3b), 2.45 (t, J = 7.0 Hz, 1 H, OH), 2.25-2.15 (m, 1 H, CH((CH2)2CH3)2), 1.47-1.01 (m, 8 H, CH((CH2)2CH3)2), 0.87 (t, J = 7.1 Hz, 3 H, CH((CH2)2CH3)2), 0.67 (t, J = 7.2 Hz, 3 H, CH((CH2)2CH3)2); 13C NMR (100 MHz, CDCl3) δ 170.4, 164.8, 147.1, 137.4, 136.0, 126.7, 125.7, 123.2, 122.4, 121.6, 110.0, 105.7, 83.8, 64.7, 64.7, 41.0, 37.1, 37.1, 33.6, 30.4, 20.7, 20.6, 14.3, 14.1. IR (neat) 3498, 2929, 1741, 1675 cm−1; HRMS (ESI) m/z calcd. for C24H32NO5 (M+H)+: 414.2275, found 414.2272.
(E)-(2-(hydroxymethyl)-4-(3-isobutyl-5-methylhexylidene)-5-oxotetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (5)
Starting from 22 (91 mg, 0.16 mmol) and following general procedure D, 5 was obtained as a colorless oil (57 mg, 78% yield): 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 7.0 Hz, 1 H, H-4′), 7.77 (s, 1 H, H-2′), 7.36-7.25 (m, 3 H, H-5′, H-6′, H-7′), 6.80 (t, 1 H, J = 7.3 Hz, C=CH), 4.57 (d, J = 12.0 Hz, 1 H, CO2CHaH), 4.43 (d, J = 12.0 Hz, 1 H, CO2CHHb), 3.83 (s, 3 H, CH3N), 3.75 (s, 2 H, CH2OH), 2.91 (d, J = 17.1 Hz, 1 H, H-3a), 2.73 (d, J = 17.1 Hz, 1 H, H-3b), 2.26 (s, 1 H, OH), 2.13-2.07 (m, 2 H, C=CHCH2), 1.71-1.63 (m, 1 H, CH(CH2(CH(CH3)2)2), 1.62-1.51 (m, 2 H, CH(CH2(CH(CH3)2)2), 1.11-0.99 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.84-0.78 (m, 12 H, CH(CH2(CH(CH3)2)2); 13C NMR (100 MHz, CDCl3) δ 170.0, 164.7, 141.1, 137.2, 135.9, 126.7, 126.5, 123.1, 122.3, 121.5, 109.9, 105.6, 83.6, 64.5, 43.8, 34.7, 33.5, 32.7, 30.3, 25.2, 22.9, 22.9, 22.6, 22.5; IR (neat) 3452, 2954, 1748, 1679, 1532, 739 cm−1; HRMS (ESI) m/z calcd. for C27H38NO5 (M+H)+: 456.2744, found 456.2742.
(Z)-(2-(hydroxymethyl)-4-(3-isobutyl-5-methylhexylidene)-5-oxotetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (6)
Starting from 23 (67 mg, 0.12 mmol) and following general procedure D, 6 was obtained as a colorless oil (39 mg, 70% yield): 1H NMR (400 MHz, CDCl3) δ 8.13-8.09 (m, 1 H, H-4′), 7.79 (s, 1 H, H-2′), 7.38-7.26 (m, 3 H, H-5′, H-6′, H-7′), 6.24 (t, 1 H, J = 7.5 Hz, C=CH), 4.57 (d, J = 12.0 Hz, 1 H, CO2CHaH), 4.42 (d, J = 12.0 Hz, 1 H, CO2CHHb), 3.84 (s, 3 H, CH3N), 3.73 (s, 2 H, CH2OH), 3.00 (dd, J = 16.4, 1.8 Hz, 1 H, H-3a), 2.82 (dd, J = 16.4, 1.8 Hz, 1 H, H-3b), 2.78-2.72 (m, 1 H, C=CHCHaH), 2.56-2.47 (m, 2 H, C=CHCHHb, OH), 1.64-1.55 (m, 3 H, CH(CH2(CH(CH3)2)2), 1.09-1.03 (m, 4 H, CH(CH2(CH(CH3)2)2), 0.86-0.81 (m, 12 H, CH(CH2(CH(CH3)2)2); 13C NMR (100 MHz, CDCl3) δ 169.0, 164.8, 144.7, 137.2, 135.9, 126.5, 124.5, 123.1, 122.3, 121.5, 109.9, 105.7, 82.8, 64.3, 64.3, 43.9, 33.5, 33.5, 33.2, 32.1, 25.1, 23.0, 23.0, 22.7, 22.6; IR (neat) 3421, 2952, 1749, 1701, 1536, 738 cm−1; HRMS (ESI) m/z calcd. for C27H38NO5 (M+H)+: 456.2744, found 456.2742.
(E)-(2-(hydroxymethyl)-5-oxo-4-(4-(p-tolylethynyl)benzylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (7)
Starting from 31 (65 mg, 0.11 mmol) and following general procedure D, 7 was obtained as a white solid (44 mg, 80% yield): 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.8 Hz, 1 H, H-4′), 7.75 (s, 1 H, H-2′), 7.57-7.21 (m, 10 H, Ph, H-5′, H-6′, H-7′, C=CH), 7.17 (d, J = 7.9 Hz, 2 H, Ph), 4.68 (d, J = 12.1 Hz, 1 H, CO2CHaH), 4.45 (d, J = 12.1 Hz, 1 H, CO2CHHb), 3.82 (s, 5 H, CH3N, CH2OH), 3.33 (dd, J = 17.8, 2.7 Hz, 1 H, H-3a), 3.16 (dd, J = 17.8, 2.6 Hz, 1 H, H-3b), 2.54 (t, J = 6.8 Hz, 1 H, OH), 2.38 (s, 3 H, CH3Ar); 13C NMR (100 MHz, CDCl3/MeOD) δ 172.0, 164.6, 138.8, 137.1, 136.1, 135.9, 133.7, 131.8, 131.5, 129.9, 129.1, 126.3, 125.1, 122.9, 122.1, 121.2, 119.5, 109.8, 105.5, 92.1, 88.1, 84.5, 64.8, 64.3, 33.3, 32.2, 21.3; IR (neat) 3118, 1741, 1690 cm−1; HRMS (ESI) m/z calcd. for C32H28NO5 (M+H)+: 506.1962, found 506.1956; Mp: 218–220°C.
(2-(hydroxymethyl)-5-oxo-4-(4-(p-tolylethynyl)benzylidene)tetrahydrofuran-2-yl)methyl 1-methyl-1H-indole-3-carboxylate (8)
Starting from 20 (65 mg, 0.15 mmol) and following general procedures E and D, 8 was obtained as an inseparable mixture of diastereomeric products (37 mg, 50% yield): 1H NMR (400 MHz, CDCl3) δ 8.09-8.06 (s, 2 H, H-4′), 7.75 (s, 1 H, H-2′ isomer E), 7.61 (s, 1 H, H-2′ isomer Z), 7.56-7.00 (m, 24 H, Ph, H-5′, H-6′, H-7′, C=CH), 4.68 (d, J = 12.1 Hz, 2 H, CO2CHaH), 4.55 (d, J = 12.0 Hz, 1 H, CO2CHHb isomer Z), 4.45 (d, J = 12.1 Hz, 1 H, CO2CHHb isomer E), 3.82 (s, 10 H, CH3N, CH2OH), 3.55 (s, 2 H, H-3a,b isomer Z), 3.34 (dd, J = 17.9, 2.8 Hz, 1 H, H-3a isomer E), 3.16 (dd, J = 17.9, 2.7 Hz, 1 H, H-3b isomer E), 2.61 (t, J = 6.9 Hz, 1 H, OH isomer E), 2.47 (t, J = 6.9 Hz, 1 H, OH isomer Z), 2.38 (m, 6 H, CH3Ar); 13C NMR (100 MHz, CDCl3/MeOD): δ 173.1, 172.1, 164.7, 164.4, 148.1, 138.9, 138.4, 137.2, 136.82, 136.0, 133.8, 131.8, 131.4, 131.3, 131.2, 129.9, 129.1, 128.6, 126.3, 125.3, 125.1, 122.9, 122.0, 121.1, 110.1, 109.9, 105.4, 105.2, 92.1, 89.0, 88.1, 84.6, 64.9, 64.2, 63.2, 61.7, 33.2, 32.2, 31.5, 21.2; IR (neat) 3446, 1739, 1690 cm−1; HRMS (ESI) m/z calcd. for C32H28NO5 (M+H)+: 506.1962, found 506.1956.
Analysis of Inhibition of [3H]PDBU Binding
Enzyme-ligand interactions were analyzed by competition with [3H]PDBu binding for the enzymes PKCα, PKCε, and RasGRP1 in the presence of phosphatidylserine (100 μg/mL, porcine brain phosphatidylserine (Avanti)) as described previously.24 To optimize stability, binding was performed at 37 °C. Each competition curve in each experiment consisted of six concentrations of the indololactone with triplicate measurements at each concentration. The IC50 values were determined by least-squares fitting of the theoretical sigmoidal competition curve to the binding data. Ki values were calculated from the IC50 values after correction for competition by the [3H]PDBu and represent the mean ± SE of at least three independent experiments.
Molecular Modeling
Structures for compounds 1 and 4 were docked into the C1 domain of the RasGRP1 crystal structure 4L9M.28 In order to allow ligand docking into the binding site, the position of residue Gln 568 was adjusted by flipping the amide group and rotating the χ2 angle into an orientation similar to that seen in the phorbol-bound C1b domain of PKCδ.29 Docking was done using GOLD version 5.2.235 with standard genetic algorithm search efficiency and the GoldScore scoring function. The binding site was defined by atoms within a 10.0 Ǻ radius of the Nε atom in Gln 568. Default torsion distributions were used for ligand flexibility, with ring corners allowed to flip. Constraints were included to prioritize poses with hydrogen bonds to the backbone of Thr 553, Leu 562, and Gly 564.
Supplementary Material
Acknowledgments
The research was supported in part by The National Institute of Industrial Technology (INTI), Consejo Nacional de Investigación Científica y Tecnológica (CONICET), in part through the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Project Z1A BC 005270), and in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Abbreviations
- BCl3
boron trichloride
- CAN
ceric ammonium nitrate
- DAG
diacylglycerol
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DMAP
4-dimethylaminopyridine
- GEFs
guanine nucleotide exchange factors
- LiHMDS
lithium hexamethyldisilazide
- MRCKs
myotonic dystrophy kinase-related Cdc42-binding kinases (MRCKs)
- PDBu
[20-3H]phorbol 12,13-dibutyrate
- PKC
protein kinase C
- PKD
protein kinase D
- PMP
p-methoxyphenyl
- RasGRP
Ras guanine nucleotide-releasing protein
- TBAF
tetra-n-butylammonium fluoride
- TBDMS
tert-butyldimethylsilyl
- TBDPS
tert-butyldiphenylsilyl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kazanietz MG. Mol Pharmacol. 2002;61:759–67. doi: 10.1124/mol.61.4.759. [DOI] [PubMed] [Google Scholar]
- 2.Newton AC. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 3.Wang QJ. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Bruinsma SP, Baranski TJ. Cell Cycle. 2007;6:2440–2444. doi: 10.4161/cc.6.20.4786. [DOI] [PubMed] [Google Scholar]
- 5.Silinsky EM, Searl TJ. Br J Pharmacol. 2003;138:1191–1201. doi: 10.1038/sj.bjp.0705213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topham MK. J Cell Biochem. 2006;97:474–484. doi: 10.1002/jcb.20704. [DOI] [PubMed] [Google Scholar]
- 7.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 8.Stone JC. Biochem Soc Trans. 2006;34:858–861. doi: 10.1042/BST0340858. [DOI] [PubMed] [Google Scholar]
- 9.Kikkawas U, Takai Y, Tanaka Y, Miyake R, Nishizuka Y. J Biol Chem. 1983;258:11442–11445. [PubMed] [Google Scholar]
- 10.Leach KL, James ML, Blumberg PM. Proc Natl Acad Sci U S A. 1983;80:4208–4212. doi: 10.1073/pnas.80.14.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mochly-Rosen D, Das K, Grimes K. Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stone JC. Genes and Cancer. 2011;2:320–334. doi: 10.1177/1947601911408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang D, Kedei N, Li L, Tao J, Velasquez JF, Michalowski AM, Tóth BI, Marincsák R, Varga A, Bíró T, Yuspa SH, Blumberg PM. Cancer Res. 2010;70:7905–7917. doi: 10.1158/0008-5472.CAN-09-4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang D, Tao J, Li L, Kedei N, Tóth ZE, Czap A, Velasquez JF, Mihova D, Michalowski AM, Yuspa SH, Blumberg PM. Oncogene. 2011;30:4590–4600. doi: 10.1038/onc.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma A, Fonseca LL, Rajani C, Yanagida JK, Endo Y, Cline JM, Stone JC, Ji J, Ramos JW, Lorenzo PS. Carcinogenesis. 2014;35:1084–1091. doi: 10.1093/carcin/bgu016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song X, Lopez-Campistrous A, Sun L, Dower Na, Kedei N, Yang J, Kelsey JS, Lewin NE, Esch TE, Blumberg PM, Stone JC. PLoS One. 2013;8:e72331. doi: 10.1371/journal.pone.0072331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Protein Sci. 1997;6:477–480. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marquez VE, Blumberg PM. Acc Chem Res. 2003;36:434–443. doi: 10.1021/ar020124b. [DOI] [PubMed] [Google Scholar]
- 19.Duan D, Sigano DM, Kelley JA, Lai CC, Lewin NE, Kedei N, Peach ML, Lee J, Abeyweera TP, Rotenberg SA, Kim H, Kim YH, El Kazzouli S, Chung J-U, Young HA, Young MR, Baker A, Colburn NH, Haimovitz-Friedman A, Truman J-P, Parrish DA, Deschamps JR, Perry NA, Surawski RJ, Blumberg PM, Marquez VE. J Med Chem. 2008;51:5198–5220. doi: 10.1021/jm8001907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Kazzouli S, Lewin NE, Blumberg PM, Marquez VE. J Med Chem. 2008;51:5371–5386. doi: 10.1021/jm800380b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia LC, Donadío LG, Mann E, Kolusheva S, Kedei N, Lewin NE, Hill CS, Kelsey JS, Yang J, Esch TE, Santos M, Peach ML, Kelley Ja, Blumberg PM, Jelinek R, Marquez VE, Comin MJ. Bioorg Med Chem. 2014;22:3123–3140. doi: 10.1016/j.bmc.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang J-H, Siddiqui Ma, Sigano DM, Krajewski K, Lewin NE, Pu Y, Blumberg PM, Lee J, Marquez VE. Org Lett. 2004;6:2413–2416. doi: 10.1021/ol0492041. [DOI] [PubMed] [Google Scholar]
- 23.Nacro K, Bienfait B, Lee J, Han KC, Kang JH, Benzaria S, Lewin NE, Bhattacharyya DK, Blumberg PM, Marquez VE. J Med Chem. 2000;43:921–944. doi: 10.1021/jm9904607. [DOI] [PubMed] [Google Scholar]
- 24.Gal N, Kolusheva S, Kedei N, Telek A, Naeem TA, Lewin NE, Lim L, Mannan P, Garfield SH, El Kazzouli S, Sigano DM, Marquez VE, Blumberg PM, Jelinek R. ChemBioChem. 2011;12:2331–2340. doi: 10.1002/cbic.201100246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J. Arch Pharm Res. 1998;21:452–457. doi: 10.1007/BF02974642. [DOI] [PubMed] [Google Scholar]
- 26.Lewin NE, Blumberg PM. In: Protein Kinase C Protocols. Newton AC, editor. Humana Press; Totowa, NJ: 2003. pp. 129–156. [Google Scholar]
- 27.Pu Y, Perry NA, Yang D, Lewin NE, Kedei N, Braun DC, Choi SH, Blumberg PM, Garfield SH, Stone JC, Duan D, Marquez VE. J Biol Chem. 2005;280:27329–27338. doi: 10.1074/jbc.M414132200. [DOI] [PubMed] [Google Scholar]
- 28.Iwig JS, Vercoulen Y, Das R, Barros T, Limnander A, Che Y, Pelton JG, Wemmer DE, Roose JP, Kuriyan J. Elife. 2013;2:e00813. doi: 10.7554/eLife.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 30.Kang J, Benzaria S, Sigano DM, Lewin NE, Pu Y, Peach ML, Blumberg PM, Marquez VE. J Med Chem. 2006;49:3185–3203. doi: 10.1021/jm060011o. [DOI] [PubMed] [Google Scholar]
- 31.Yau WM, Wimley WC, Gawrisch K, White SH. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 32.Rahman GM, Shanker S, Lewin NE, Kedei N, Hill CS, Prasad BVV, Blumberg PM, Das J. Biochem J. 2013;451:33–44. doi: 10.1042/BJ20121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart MD, Morgan B, Massi F, Igumenova TI. J Mol Biol. 2011;408:949–970. doi: 10.1016/j.jmb.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norman KE, Nymeyer H. Biophys J. 2006;91:2046–2054. doi: 10.1529/biophysj.105.080275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones G, Willett P, Glen RC, Leach AR, Taylor R, Uk KBR. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.