

Abstract
Episodic memories initially require rapid synaptic plasticity within the hippocampus for their formation and are gradually consolidated in neocortical networks for permanent storage. However, the engrams and circuits that support neocortical memory consolidation remain unknown. We found that neocortical prefrontal memory engram cells, critical for remote contextual fear memory, were rapidly generated during initial learning via inputs from both hippocampal-entorhinal cortex and basolateral amygdala. After their generation, the prefrontal engram cells, with support from hippocampal memory engram cells, became functionally mature with time. Whereas hippocampal engram cells gradually became silent with time, engram cells in the basolateral amygdala, which were necessary for fear memory, are maintained. Our data provide new insights into the functional reorganization of engrams and circuits underlying systems consolidation of memory.
Memories are thought to be initially stored within the hippocampal-entorhinal cortex (HPC-EC) (recent memory) and over-time are slowly consolidated within the neocortex for permanent storage (remote memory) (1–7). Systems memory consolidation models suggest that the interaction between the HPC-EC and the neocortex during and after an experience is crucial (8–12). Experimentally, prolonged inhibition of hippocampal or neocortical networks during the consolidation period produces deficits in remote memory formation (13–15). However, little is known regarding specific neural circuit mechanisms underlying the formation and maturation of neocortical memories through interactions with the HPC-EC network. By employing activity-dependent cell labeling technology (16–18) combined with viral vector–based transgenic, anatomical (19, 20), and optogenetic strategies (19, 21) for circuit-specific manipulations and in vivo calcium imaging (22), we investigated the nature and dynamics of neocortical and subcortical memory engram cells (a population of neurons that are activated by learning, have enduring cellular changes, and are reactivated by a part of the original stimuli for recall (18)) and their circuits for systems consolidation of memory.
We first traced entorhinal projections to frontal cortical structures (the medial prefrontal cortex (PFC), caudal anterior cingulate cortex (cACC), retrosplenial cortex (RSC)) involved in contextual fear memory, and the basolateral amygdala (BLA), with injections of the retrograde tracer cholera toxin subunit B (CTB)–Alexa555 into these regions (fig. S1). CTB injections resulted in labeling in the medial entorhinal cortex (MEC) specifically in cells in layer Va (Fig. 1A–D, H and fig. S1A–D), indicating that MEC-Va cells have extensive projections to the neocortex and BLA (23). We then sought to inhibit these specific projections by bilaterally injecting adeno-associated virus 8 (AAV8)–calcium/calmodulin-dependent protein kinase II (CaMKII):eArchT–enhanced yellow fluorescent protein (eYFP) in the deep layers of the MEC in wild-type (WT) mice with bilaterally implanted optic fibers above the PFC, cACC, or RSC (Fig. 1E, J, and fig. S2G). Expression of eArchT-eYFP was abundant in MEC-Va terminals located in the PFC, cACC and RSC (Fig. 1B, I, and fig. S2D). These mice were then subjected to contextual fear conditioning (CFC) while we delivered green light bilaterally to the different cortical areas receiving MEC-Va projections during either the conditioning period (Day-1) (fig. S2E) or recall test period (Day-2, Day-8, Day-15 and Day-22) (fig. S2F). Axon terminal inhibition with optogenetics of MEC-Va cells within the PFC during Day-1 of CFC disrupted memory at Day-15 and Day-22, but not at Day-2 or Day-8 (Fig. 1F). Terminal inhibition during memory recall tests did not affect memory retrieval (Fig. 1G). Finally, terminal inhibition in the cACC or RSC during CFC or recall had no effect on memory throughout these periods (Fig. 1J–L and fig. S2G–I).
Fig. 1. MECVa input to PFC during conditioning is crucial for generation of PFC engram cells.
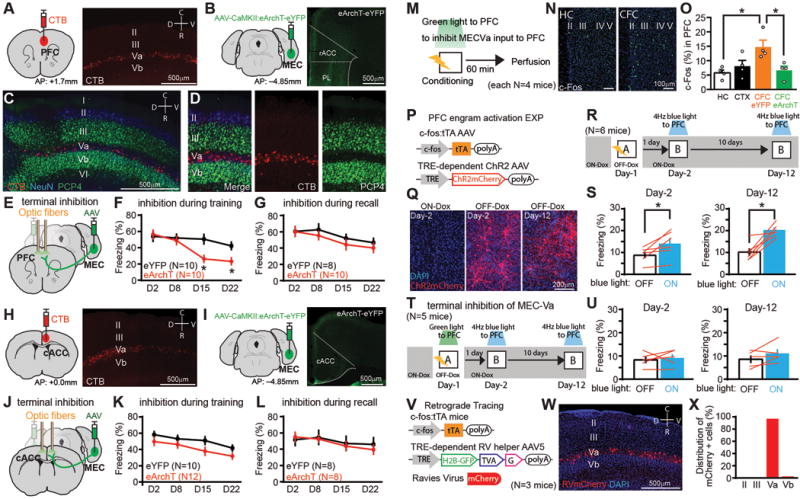
(A) CTB injection into PFC. Sagittal section of MEC with CTB-labeled cells (red) (B) Coronal sections of PFC with MECVa axons expressing eYFP (green). rACC; rostral ACC, PL; prelimbic cortex. (C, D) Sagittal section of MEC with CTB-labeled cells (red) and immunostained with anti-PCP4 (green) and anti-NeuN (blue). PCP4 is a marker for layer III and Vb cells in MEC. CTB injection into BLA. (E, J) Viral injections and optic fiber implantations. (F, G) Time courses of freezing during recall tests. Green light was shone into the PFC during conditioning (F) or testing (G). N presents number of animals. (H) CTB injection into caudal ACC (cACC). Sagittal section of MEC with CTB-labeled cells (red). (I) Coronal sections of ACC with MECVa axons (green). (K, L) Time courses of freezing during recall tests. Green light was shone into the ACC during conditioning (K) or testing (L). (M) Experimental schedule. (N) Coronal section of PFC with anti-c-Fos (green). (O) Percentages of c-Fos+ cells in PFC of homecage (HC), context exposure (CTX), CFC with eYFP, and CFC with eArchT group. (P) Virus-mediated engram cell labelling with ChR2. (Q) Coronal section of PFC with ChR2-mCherry (red). (R, T) Experimental schedule. (S, U) Averaged freezing for light-off and light-on epochs. (V) Retrograde trans-synaptic labeling with activity-dependent cell labeling. (W) Sagittal section of MEC with rabies virus–specific mCherry (red). (X) Distribution of mCherry+ cells in the MEC (n=212 mCherry+ cells, n presents number of cells.). *P < 0.05 by unpaired t-test compared to eYFP (F, G, K, L), one-way ANOVA with Tukey-Kramer test (O) and paired t-test (S, U). Error bars mean ± s.e.m.
The above results suggest that MEC-Va input into the PFC during CFC is crucial for the eventual formation of remote memory. This hypothesis was supported by several findings. First, CFC increased the number of c-Fos+ cells in the PFC compared to that of homecage mice (Fig. 1M–O), whereas context only exposure did not increase c-Fos activity in the PFC (Fig. 1O). Second, optogenetic terminal inhibition of MEC-Va projections within the PFC during CFC inhibited the observed increase of c-Fos+ cells in the PFC (Fig. 1O). Finally, we identified CFC engram cells in the PFC. We targeted injections of AAV9-c-fos:tTA and AAV9-TRE:channelrhodopsin-2 (ChR2)-mCherry (Fig. 1P, Q) and optic fibres to the PFC of WT mice and labeled the PFC cells activated by CFC with ChR2 while the mouse was off doxycycline (OFF-Dox) (Fig. 1R). Blue light stimulation at 4 Hz, but not the conventional 20 Hz, of ChR2-mCherry-expressing cells in the PFC induced freezing behavior on Day-2 and Day-12 in an unconditioned context (Fig. 1S, fig. S3) compared with freezing under the blue light-off condition. This blue light-induced freezing was prevented when MEC-Va fibers in the PFC were inhibited during CFC on Day-1 (Fig. 1T, U, fig. S4). Using trans-synaptic retrograde tracing combined with the activity-dependent cell labelling, we confirmed that the PFC engram cells generated by CFC receive monosynaptic input from MEC-Va cells (Fig. 1V–X, fig. S5).
To examine whether PFC engram cells are also reactivated by the conditioned context (rather than by blue light) at a recent and remote time point, we targeted injections of AAV9-TRE: human histone H2B-green fluorescent protein (H2B-GFP) to the PFC of c-fos:tTA transgenic mice (Fig. 2A). The mice underwent CFC on Day-1 and then re-exposed to the conditioned (Context-A) or an unconditioned (Context-B) context on Day-2 or Day-13 (Fig. 2B). Cells activated by CFC were labeled with H2B-GFP and the cells activated by the context test with a c-Fos antibody, and we calculated the proportion of double labeled cells (Fig. 2A–B, fig. S6B). H2B-GFP+ cells (PFC engram cells) were preferentially reactivated in Context-A on Day-13 but not Day-2, compared to the H2B-GFP negative cells (Fig. 2C). There was no difference in c-Fos expression between H2B-GFP positive and negative cells when mice were tested in Context-B (Fig. 2C). We also found that the spine density of the PFC engram cells on Day-12 was significantly higher than on Day-2 (Fig. 2D, E, fig. S7), in line with previous findings of a positive correlation between dendritic spine density on memory engram cells and memory expression triggered by natural recall cues (24–26).
Fig. 2. PFC engram cells mature with time.
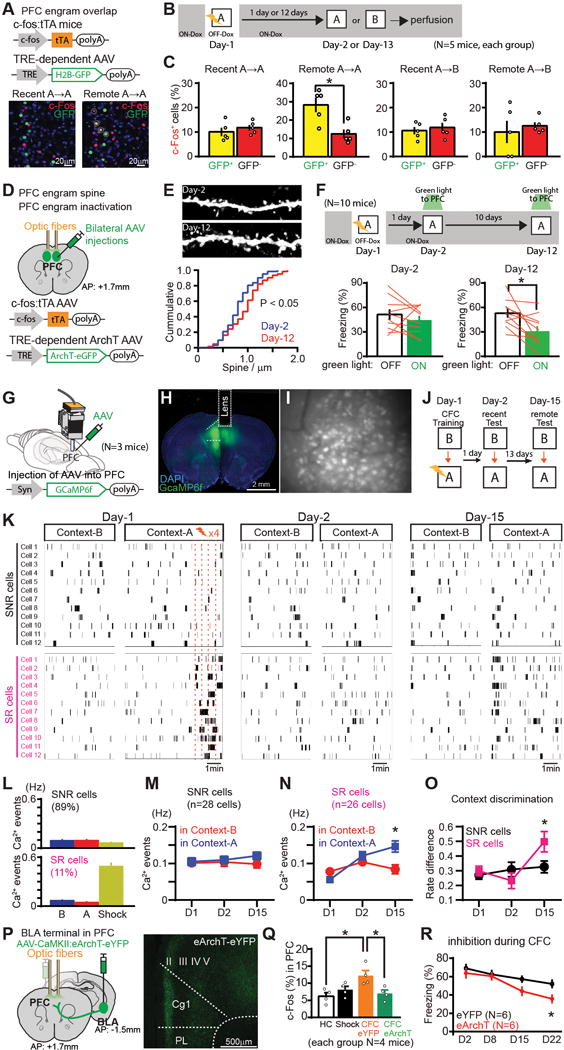
(A) PFC engram cell labelling with H2B-GFP. Coronal sections of PFC with H2B-GFP (green), anti-c-Fos (red). Marked cells are double-positive. (B) Experimental schedule. (C) Percentages of c-Fos+ cells in H2B-GFP+ and H2B-GFP− cells in PFC. (D) PFC engram cell labelling with ArchT. (E) Images showing dendritic spines from PFC engram cells. Cumulative probability of the spine density of PFC engram. (F) Experimental schedule. Averaged freezing for light-off and light-on epoch during recall test. (G, H) Viral injections and GRIN lens implantation. (I) Stacked image acquired through the microendoscope over 10 mins of imaging in PFC. (J) Experimental schedule. (K) Raster plots of Ca2+ event in shock non-responding (SNR) cells and shock responding (SR) cells in PFC (showing 12 example cells). (L-N) Average Ca2+ event frequency of SNR cells and SR cells on Day-1, Day-2 and Day-3. (O) Average rate difference index of Ca2+ activity. (P) Viral injections and optic fiber implantations. Coronal sections of PFC visualizing BLA axons (green). (Q) Percentages of c-Fos+ cells in PFC of homecage (HC), shock only (Shock), CFC with eYFP, and CFC with eArchT, groups. (R) Time courses of freezing during recall tests. *P < 0.05 by unpaired t-test (C, O, R), KS test (E), paired t-test (F, M, N), one-way ANOVA with Tukey-Kramer test (Q). Error bars mean ± s.e.m.
To test whether PFC engram cells are necessary for memory recall by natural cues, we bilaterally targeted injections of AAV9-c-fos:tTA and AAV9-TRE:ArchT-eGFP (Fig. 2D, F) and optic fibres to the PFC of WT mice and labeled the PFC engram cells that were activated by CFC with ArchT while the mice were OFF-Dox (Fig. 2F). Cell body inhibition of the PFC engram cells by green light during retrieval did not affect recent memory (Day-2), however at the remote time-point (Day-12) memory retrieval was disrupted compared to the green light-off condition (Fig. 2F).
To further investigate the characteristics of PFC engram cells we monitored transient calcium (Ca2+) events in PFC cells in vivo. WT mice were injected with AAV5-Syn:GCaMP6f into the PFC and implanted with a micro GRIN lens targeting the PFC (Fig. 2G–I, fig. S8) (22, 27). On Day-1 mice were first exposed to Context-B followed by CFC in Context-A. Mice were then re-exposed to both contexts in the same order on Day-2 and Day-15 (Fig. 2J). The averaged frequency of Ca2+ events in PFC cells did not significantly change in either a time- or context-dependent manner (fig. S9B). However a small but significant difference was revealed in the cumulative distribution curves of a rate difference index (assessing context selectivity; see Methods) between Day-1 conditioning and Day-15 recall, and between Day-2 recall and Day-15 recall (fig. S9C). PFC cells did not appear to discriminate between the two contexts on Day-1 prior to footshock presentation (Fig. 2K, L). However, after footshock presentation, about 11% of cells showed a significant increase in Ca2+ transients (shock responding cells; SR cells) (Fig. 2K, L). The remaining ~89% of PFC cells did not respond to the shocks (shock non-responding cells; SNR cells). The SR cells were less active than SNR cells during exposure to Context-B and Context-A on Day-1 prior to footshock presentation (Fig. 2L–N, fig. S9D). During recall, the transient Ca2+ activity of SR cells in Context-A (conditioned context) was significantly higher compared to that in Context-B on Day-15 but not on Day-1 or Day-2, whereas the frequency of Ca2+ transient events in SNR cells remained constant, irrespective of context (Fig. 2M, N). This produced a significant rate difference index of Ca2+ activity for the conditioned context between the SR and SNR cells on Day-15 but not on Day-1 (excluding the shock delivery period) or Day-2 (Fig. 2O). These results combined with c-Fos activation data (Fig. 1M–O) suggest that the SR cells may be the PFC memory engram cells, given that the generation of the PFC engram cells requires both context exposure and footshocks.
Our calcium imaging data suggest that footshock stimulus input into the PFC is crucial for the generation of PFC engram cells. Because the BLA integrates footshock information arriving from the thalamus (28) and projects to the PFC (fig. S5I, S10), we optogenetically inhibited the pathway from BLA to PFC during CFC (Fig. 2P). Optogenetic inhibition of BLA terminals in the PFC during CFC disrupted the generation of PFC engram cells (Fig. 2Q). The terminal inhibition during CFC also inhibited remote memory formation (Fig. 2R).
To test whether the HPC engram cells play a crucial role in the functional maturation of PFC engram cells during the system consolidation process, we bilaterally targeted injection of AAV9-TRE:tetanus toxin light chain (TeTX) or AAV9-TRE:eYFP (as a control) to the hippocampal dentate gyrus (DG) of c-fos:tTA transgenic mice (Fig. 3A). When the mice were subjected to CFC, DG engram cells were labeled with TeTX. DG engram cell labeling with TeTX caused a robust inhibition of DG engram cell output, as revealed by greatly reduced immunoreactivity of vesicle-associated membrane protein 2 (VAMP2), which is essential for activity-dependent neurotransmitter release from presynaptic terminals (13), within the stratum lucidum (s.l.) in hippocampal CA3 in OFF-Dox mice compared to ON-Dox mice (Fig. 3B, C). Optogenetic activation of DG engram cells with ChR2 failed to produce the increase in c-Fos+ cells in CA3 of TeTX-expressing mice that was observed in control mice compared to home cage controls (Fig. 3D). TeTX expression in HPC engram cells inhibited the reactivation of PFC engram cells during exposure to the conditioned context 12 days after CFC compared to the eYFP control group (Fig. 3E, F). TeTX expression also blocked the increase of dendritic spine density of PFC engram cells compared to the eYFP group (Fig. 3G). In vivo calcium imaging revealed that TeTX expression in HPC engram cells after CFC blocked the increase of the context discrimination index in SR cells in the PFC (Fig. 3H, fig. S11).
Fig. 3. HPC engram cells support the maturation of PFC engram cells, while HPC engram cells become silent with time.
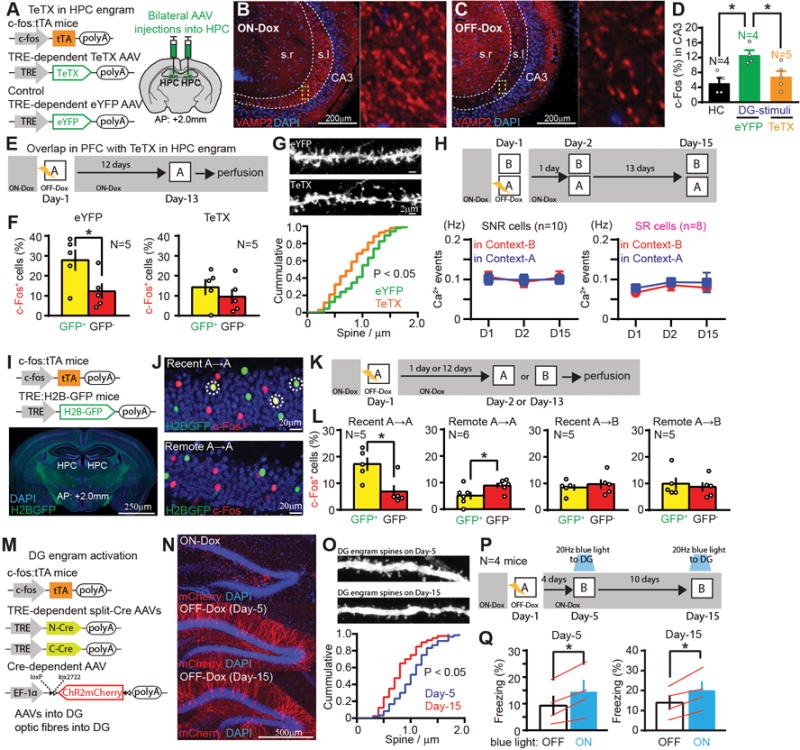
(A) DG engram cell labelling with TeTX. (B, C) Sagittal sections of HPC with anti-VAMP2 (red). (D) Percentages of c-Fos+ cells in hippocampal CA3 of homecage, blue-light-ON mice with eYFP or with TeTX. (E) Experimental schedule. (F) Percentages of c-Fos+ cells in H2B-GFP+ and H2B-GFP− cells in the PFC of eYFP- and TeTX-expressing mice. (G) Images showing dendritic spines from PFC engram. Cumulative probability of the spine density of PFC engram of eYFP- and TeTX-expressing mice. (H) Experimental schedule, average Ca2+ event frequency of SNR cells and SR cells under TeTX-expressing condition. (I) Transgenic strategy of DG engram cell labelling with H2B-GFP. (J) Coronal sections of DG with H2B-GFP (green), anti-c-Fos (red). Marked cells are double-positive. (K, P) Experimental schedule. (L) Percentages of c-Fos+ cells in H2B-GFP+ and H2B-GFP− cells in the DG. (M) Long-term DG engram cell labeling with ChR2. (N) Coronal sections of DG with ChR2-mCherry (red). (O) Images showing dendritic spines from DG engram. Cumulative probability of the spine density of DG engram. (Q) Averaged freezing by blue light stimulation for light-off and light-on epochs. *P < 0.05 by one-way ANOVA with Tukey-Kramer test (D), unpaired t-test (F, L), KS test (G, O), paired t-test (H, Q). Error bars mean ± s.e.m.
To investigate the post-consolidation fate of HPC engram cells we crossed c-fos:tTA TG mice with TRE:H2B-GFP TG mice (29), subjected them to CFC, and then re-exposed them to the conditioned (Context-A) or an unconditioned (Context-B) context on Day-2 or Day-13 (Fig. 3I–K). DG engram cells were preferentially reactivated in Context-A on Day-2 but not Day-13, compared to the non-engram cells (Fig. 3L). No difference was observed in the activation of DG engram and non-engram cells by Context-B (Fig. 3L). We were unable to maintain labeled DG engram cells with ChR2 beyond 12 days with injection of AAV9-TRE:ChR2-mCherry. To extend this technical limit we targeted injections of AAV1,5,8,9-TRE:CCre, AAV1,5,8,9-TRE:NCre and AAV5-EF1a:ChR2-mCherry to the DG of c-fos:tTA transgenic mice (Fig. 3M). We could thus extend viable labelling by a few days (Fig. 3N). The spine density of DG engram cells on Day-15 was significantly reduced compared to Day-5 (Fig. 3O, fig. S12). On both Day-5 and Day-15, optogenetic activation of DG engram cells induced freezing behaviour (Fig. 3P, Q).
We finally investigated the role of MEC-Va projections to the BLA in recent and remote memory (Fig. 4A, fig. S2A). Inhibition of MEC-Va terminals in the BLA during CFC disrupted contextual fear memory formation. Retrieval was impaired at all time-points tested (Fig. 4B). When terminal inhibition was restricted to retrieval, recent memory tested on Day-2 and Day-8 was impaired, but remote memory retrieval on Day-15 and Day-22 was unaffected (Fig. 3C). In contrast, inhibition of PFC engram cell terminals in the BLA did not impair memory retrieval on Day-2 but did impair memory retrieval on Day-12 (Fig. 4D, E). To investigate whether the BLA fear memory engram cells formed on Day-1 are maintained and used for PFC engram-dependent remote memory recall, we subjected the double transgenic mice (Fig. 4F, I) to CFC and re-exposed them to the conditioned context at recent or remote time-points (Fig. 4G). BLA engram cells were reactivated equally well by the conditioned context at both recent and remote time-points (Fig. 4H). Similarly, BLA cells activated by recent recall were reactivated again equally well by re-exposure to the conditioned context at recent and remote time-points (Fig. 4J, K).
Fig. 4. BLA engram cells are maintained throughout consolidation but with a switch of the recall circuit.
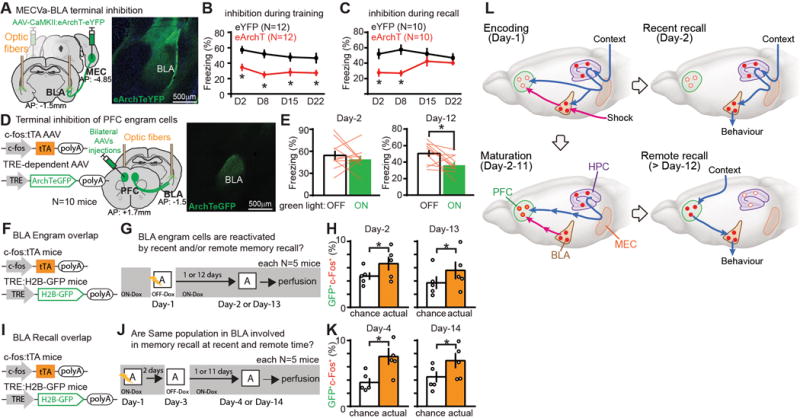
(A) Viral injections and optic fiber implantations. Coronal sections of BLA with MECVa axons expressing eYFP (green). (B, C) Time courses of freezing during recall tests. Green light was shone into the BLA during conditioning (B) or testing periods (C). (D) Viral injections and optic fiber implantations. Coronal sections of BLA visualizing axons of PFC engram cells (green). (E) Averaged freezing for green light-off and light-on epochs during recall test. (F, I) BLA engram cell labelling with H2B-GFP. (G, J) Experimental schedules. (H, K) Percentages of double labeling with c-Fos+ and H2B-GFP+ in the BLA compared to the calculated chance level. (L) A new model for systems consolidation of memory. *P < 0.05 by unpaired t-test (B, C, H, K) or by paired t-test (E). Error bars mean ± s.e.m.
Here, we found that PFC memory engram cells for CFC were rapidly formed during Day-1 training by virtue of inputs from both MEC-Va and BLA, but they were not retrievable with natural recall cues. The immature PFC engram cells functionally, structurally, and physiologically matured during the subsequent few weeks and this process required inputs from HPC engram cells presumably through MEC-Va. In contrast to their formation on Day-1, retrieval of the PFC engram at a remote time did not require MEC-Va input. HPC engram cells formed during training became silent with time; they are not retrieved on Day-14 by natural recall cues but are still re-activatable optogenetically for recall. However, fear memory BLA engrams formed during training are functionally maintained even after the consolidation-mediated switch in recall circuits (Fig. 4L).
Our model (Fig. 4L) introduces the concept that the prefrontal memory engram is already generated, albeit in an immature form, on Day-1 of training via inputs from both HPC-EC and BLA (Fig. 1). The standard model (1, 2, 4, 6, 7, 11) hypothesized that remote memory is formed in the cortex by a slow transfer of hippocampal memory. In contrast, in our study the role of the hippocampus in cortical memory is for the rapid generation of immature engram cells in the PFC during training and for the subsequent functional maturation of these preexisting engram cells (Fig. 3). The immature PFC engram may correspond to the cortical “tagging” speculated in an earlier study (14). Whereas in a previous study the BLA is crucial for both recent and remote fear memory expression (30), our results demonstrated an overlapping set of BLA engram cells for both recent and remote fear memory retrieval which were quickly formed during training (Fig. 4). However, the source of input into the BLA engrams for retrieval shifts from MEC-Va at recent time-points to the PFC engram at remote time-points (Fig. 4L). The route through which contextual stimuli activate the mature PFC engram is unknown. Most likely the information processed in a variety of sensory cortices reaches the PFC via the thalamus (31). Supporting this idea, PFC engram cells receive monosynaptic input from both the medial-dorsal and anteromedial thalamus (fig. S5).
Our finding of the lasting hippocampal engrams (Fig. 3Q) is consistent with multiple trace theory (5, 11). However, at the post-consolidation stage, the hippocampal engrams were not activatable by natural recall cues, but with optogenetic stimulation. A similar state of hippocampal engrams has previously been observed in anisomycin-induced amnesia (24) and in mouse models of early Alzheimer’s disease (26), and the early (Day-2) PFC engram cells showed a similar property (Fig. 1S, 2C). Although we did not determine how long after encoding this “silent state” of the hippocampal engram lasts, we speculate that the hippocampal engram will eventually lose the original memory information (29, 32, 33). Alternatively, the silent engram cells may still participate in the successful remote recall of discrete episodic details (5, 11).
As in previous studies (18, 20, 29), we observed that training resulted in wide-spread neuronal activation in the neocortex, including the ACC and RSC. However, whereas the activation of PFC neurons is crucial for formation of remote memory, MEC-Va input into the cACC or RSC is dispensable for this process. For remote memory, the PFC may thus have a distinctive role compared to other neocortical areas in integrating multiple sensory information stored in various cortical areas (11). Finally, our data show that the remote memory expressed by the PFC engram is conditioned-context specific, suggesting that it is episodic-like.
Supplementary Material
One Sentence Summary.
We discovered the neocortical remote memory engram cells, and the neural circuit mechanisms responsible for their generation and maturation.
Acknowledgments
We thank F. Bushard, J. Martin, T. Ryan, J. Yamamoto, C. Sun, W. Yu, S. Huang, M. Ragion, A. Arons, X. Zhou, C. Ragion, A. Moffa, L. Brenner, A. Hamalian, and D. King, for help with experiments; and all members of the Tonegawa laboratory for their support. We thank Ian Wickersham for providing rabies virus, Yasuyuki Shima and Sacha B Nelson for providing TRE3G split Cre AAV. All data necessary to understand and assess the conclusions of this research are available in the supplementary materials. This work was supported by the RIKEN Brain Science Institute, the Howard Hughes Medical Institute and the JPB Foundation (to S.T.). AAV9-c-fos:tTA, AAV9-TRE:ChR2mCherry, AAV9-TRE:ArchTeGFP, AAV9-TRE:TeTX, AAV9-TRE:eYFP were developed at MIT by the group of S.T.; virus plasmids are available through a material transfer agreement.
Footnotes
References and Notes
- 1.Marr D. Philos Trans R Soc Lond B Biol Sci. 1971 Jul 1;262:23. doi: 10.1098/rstb.1971.0078. [DOI] [PubMed] [Google Scholar]
- 2.Squire LR. Science. 1986 Jun 27;232:1612. doi: 10.1126/science.3086978. [DOI] [PubMed] [Google Scholar]
- 3.Kim JJ, Fanselow MS. Science. 1992 May 1;256:675. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 4.McClelland JL, McNaughton BL, O’Reilly RC. Psychol Rev. 1995 Jul;102:419. doi: 10.1037/0033-295X.102.3.419. [DOI] [PubMed] [Google Scholar]
- 5.Nadel L, Moscovitch M. Curr Opin Neurobiol. 1997 Apr;7:217. doi: 10.1016/s0959-4388(97)80010-4. [DOI] [PubMed] [Google Scholar]
- 6.Tse D, et al. Science. 2007 Apr 06;316:76. doi: 10.1126/science.1135935. [DOI] [PubMed] [Google Scholar]
- 7.McClelland JL. J Exp Psychol Gen. 2013 Nov;142:1190. doi: 10.1037/a0033812. [DOI] [PubMed] [Google Scholar]
- 8.Buzsaki G. Cereb Cortex. 1996 Mar-Apr;6:81. doi: 10.1093/cercor/6.2.81. [DOI] [PubMed] [Google Scholar]
- 9.Siapas AG, Wilson MA. Neuron. 1998 Nov;21:1123. doi: 10.1016/s0896-6273(00)80629-7. [DOI] [PubMed] [Google Scholar]
- 10.Wiltgen BJ, Brown RA, Talton LE, Silva AJ. Neuron. 2004 Sep 30;44:101. doi: 10.1016/j.neuron.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Frankland PW, Bontempi B. Nat Rev Neurosci. 2005 Feb;6:119. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- 12.Preston AR, Eichenbaum H. Curr Biol. 2013 Sep 9;23:R764. doi: 10.1016/j.cub.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakashiba T, Buhl DL, McHugh TJ, Tonegawa S. Neuron. 2009 Jun 25;62:781. doi: 10.1016/j.neuron.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesburgueres E, et al. Science. 2011 Feb 18;331:924. [Google Scholar]
- 15.Zelikowsky M, Bissiere S, Fanselow MS. J Neurosci. 2012 Mar 07;32:3393. doi: 10.1523/JNEUROSCI.4339-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reijmers LG, Perkins BL, Matsuo N, Mayford M. Science. 2007 Aug 31;317:1230. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, et al. Nature. 2012 Mar 22;484:381. doi: 10.1038/nature11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tonegawa S, Liu X, Ramirez S, Redondo R. Neuron. 2015 Sep 2;87:918. doi: 10.1016/j.neuron.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Kitamura T, et al. Science. 2014 Feb 21;343:896. doi: 10.1126/science.1244634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye L, et al. Cell. 2016 Jun 16;165:1776. [Google Scholar]
- 21.Deisseroth K. Nat Neurosci. 2015 Sep;18:1213. doi: 10.1038/nn.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziv Y, et al. Nat Neurosci. 2013 Mar;16:264. doi: 10.1038/nn.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surmeli G, et al. Neuron. 2015 Dec 2;88:1040. doi: 10.1016/j.neuron.2015.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S. Science. 2015 May 29;348:1007. doi: 10.1126/science.aaa5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi-Takagi A, et al. Nature. 2015 Sep 17;525:333. doi: 10.1038/nature15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy DS, et al. Nature. 2016 Mar 24;531:508. doi: 10.1038/nature17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitamura T, et al. Neuron. 2015 Sep 23;87:1317. doi: 10.1016/j.neuron.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pellman BA, Kim JJ. Trends Neurosci. 2016 Jun;39:420. doi: 10.1016/j.tins.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tayler KK, Tanaka KZ, Reijmers LG, Wiltgen BJ. Curr Biol. 2013 Jan 21;23:99. doi: 10.1016/j.cub.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 30.Maren S, Aharonov G, Fanselow MS. Behav Neurosci. 1996 Aug;110:718. doi: 10.1037//0735-7044.110.4.718. [DOI] [PubMed] [Google Scholar]
- 31.Do-Monte FH, Quinones-Laracuente K, Quirk GJ. Nature. 2015 Mar 26;519:460. doi: 10.1038/nature14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denny CA, et al. Neuron. 2014 Jul 02;83:189. doi: 10.1016/j.neuron.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitamura T, et al. Cell. 2009 Nov 13;139:814. doi: 10.1016/j.cell.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 34.Tumbar T, et al. Science. 2004 Jan 16;303:359. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okuyama T, Kitamura T, Roy DS, Itohara S, Tonegawa S. Science. 2016 Sep 30;353:1536. doi: 10.1126/science.aaf7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyamichi K, et al. Nature. 2011 Apr 14;472:191. doi: 10.1038/nature09714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramirez S, et al. Science. 2013 Jul 26;341:387. doi: 10.1126/science.1239073. [DOI] [PubMed] [Google Scholar]
- 38.Ramirez S, et al. Nature. 2015 Jun 18;522:335. doi: 10.1038/nature14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu CR, et al. Neuron. 2004 May 27;42:553. doi: 10.1016/s0896-6273(04)00224-7. [DOI] [PubMed] [Google Scholar]
- 40.Hirrlinger J, et al. PLoS One. 2009;4:e4286. doi: 10.1371/journal.pone.0004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shima Y, et al. Elife. 2016 Mar 21;5:e13503. doi: 10.7554/eLife.13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickersham IR, Finke S, Conzelmann KK, Callaway EM. Nat Methods. 2007 Jan;4:47. doi: 10.1038/NMETH999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohara K, et al. Nat Neurosci. 2014 Feb;17:269. doi: 10.1038/nn.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goosens KA, Maren S. Learn Mem. 2001 May-Jun;8:148. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Pignatelli M, Xu S, Itohara S, Tonegawa S. Nat Neurosci. 2016 Oct 17; doi: 10.1038/nn.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitamura T, Macdonald CJ, Tonegawa S. Learn Mem. 2015 Sep;22:438. doi: 10.1101/lm.038687.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokoyama M, Matsuo N. Front Behav Neurosci. 2016;10:218. doi: 10.3389/fnbeh.2016.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Redondo RL, et al. Nature. 2014 Sep 18;513:426. doi: 10.1038/nature13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Restivo L, Vetere G, Bontempi B, Ammassari-Teule M. J Neurosci. 2009 Jun 24;29:8206. doi: 10.1523/JNEUROSCI.0966-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinto L, Dan Y. Neuron. 2015 Jul 15;87:437. doi: 10.1016/j.neuron.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun C, et al. Proc Natl Acad Sci U S A. 2015 Jul 28;112:9466. doi: 10.1073/pnas.1511668112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.