

Abstract
Ordinary differential equations (ODEs) are a popular approach to quantitatively model molecular networks based on biological knowledge. However, such knowledge is typically restricted. Wrongly modelled biological mechanisms as well as relevant external influence factors that are not included into the model are likely to manifest in major discrepancies between model predictions and experimental data. Finding the exact reasons for such observed discrepancies can be quite challenging in practice. In order to address this issue, we suggest a Bayesian approach to estimate hidden influences in ODE-based models. The method can distinguish between exogenous and endogenous hidden influences. Thus, we can detect wrongly specified as well as missed molecular interactions in the model. We demonstrate the performance of our Bayesian dynamic elastic-net with several ordinary differential equation models from the literature, such as human JAK–STAT signalling, information processing at the erythropoietin receptor, isomerization of liquid α-Pinene, G protein cycling in yeast and UV-B triggered signalling in plants. Moreover, we investigate a set of commonly known network motifs and a gene-regulatory network. Altogether our method supports the modeller in an algorithmic manner to identify possible sources of errors in ODE-based models on the basis of experimental data.
Keywords: ordinary differential equations, systems biology, dynamic elastic-net, modelling
1. Introduction
Mathematical models of biological systems become more and more complex and contribute important insights into various biological processes [1–7]. Since biological systems are naturally open, formulating mathematical models and specifying their boundaries is a highly non-trivial task [8,9]. Consequently, most researchers in systems biology are faced with the still unsolved issue to find a compromise between model complexity and the limited amount of knowledge, data and time [9–11]. Researchers in other fields including earth and environmental sciences are facing similar challenges [12]. In many cases, missed and unknown external influences as well as erroneous interactions in a model could lead to severely misleading results [7].
Current research is mostly focused on inference of perturbation effects and model selection [13–15]. Although, perturbation experiments are labour and cost intensive, which raises the need for a careful prioritization strategy [14–17]. On the other hand, statistical model selection and related methods require a strong knowledge about the system and its alternatives which is rarely given in practice [7,18]. Thus model selection can be very difficult, specifically if nothing is known about missing variables and their possible mechanisms.
In most situations, researchers have partial knowledge and preliminary hypotheses about their system, which needs to be integrated into a restricted but still predictive and experimentally validatable model [19]. Even if the biological system is partially known and the data are given for almost all molecular species, it is not clear how to deal with insufficient predictions [7]. Often this ends in trial-and-error approaches and does not ensure that the selected model reflects the reality rather than just fitting the given dataset. The question is, how to detect so far unknown molecules and their interactions in a more data driven manner. This could guide the modeller towards points in the given model, where the model is likely erroneous. In a second step, the modeller can then try to link these erroneous points to known mechanisms.
Recently, we proposed the dynamic elastic-net (DEN) as a more principled method to address this issue [20]. The DEN aims for estimating the dynamics of hidden influence variables in ordinary differential equations (ODE)-based systems via a penalized estimation procedure resembling elastic-net regression [21]. While our previous method was tested successfully on several applications, such as the erythropoietin receptor (EpoR)-dependent signalling network [19], it has still several shortcomings, which we address in this paper. More specifically, DEN is not a probabilistic approach and thus does not fully address the unavoidable uncertainty about estimates. DEN does not answer the question, whether estimated hidden influences could be attributed to missed or wrongly modelled interactions among the known molecular species. Here we introduce the Bayesian DEN (BDEN) as a new and fully probabilistic approach, which deals with all these aspects. In contrast with DEN, our new BDEN method does not require pre-specified hyper-parameters. We illustrate the predictive power of BDEN compared with DEN in several real biological models and test cases. The BDEN thus provides a systematic Bayesian computational method to identify target nodes and reconstruct the corresponding error signal including detection of missing and wrong molecular interactions within the assumed model. The method works for ODE-based systems even with uncertain knowledge and noisy data. In contrast with approaches based on point estimates the Bayesian framework incorporates the given uncertainty and circumvents numerical pitfalls which frequently arise from optimization methods [22,23].
2. Material and methods
2.1. Motivation
We assume the modelling process to start with an initial, potentially incomplete or partially misspecified nominal model including all known but not necessarily observable molecules [24]. Figure 1 illustrates the general idea. In most situations, the real system differs from the initially modelled nominal system, which is reflected by an insufficient fit to the given data caused by (i) hidden influences and (ii) erroneous molecular interactions. Exogenous hidden influences could, for example, be stimulatory (e.g. phosphorylation) or inhibitory (e.g. de-phosphorylation) events affecting the modelled system from outside. In addition, there could exist stimulatory or inhibitory influences within the system, which are not included in the model due to lack of knowledge, i.e. missing molecular interactions. Similarly, wrongly included molecular interactions could exist. In biochemical systems, molecular interactions are based on biochemical reactions, e.g. phosphorylation and binding events.
Figure 1.

Illustration of the Bayesian approach to estimating hidden influence variables in ODE-based models. Here, a fictional interaction network is shown. (a) True system (black) with two inputs and one fictive observable measurement as a combination of two nodes (box). (b) In reality, the available knowledge represented by an a priori model (blue) does not necessarily cover the whole system but only a part of the true system. Hence the nominal model leads to an unsatisfactory fit with the observable measurement. This may caused by exogenous influences or by misspecified molecular interactions (i.e. missing or wrong edges in the interaction network). (c) Our approach aims for estimating these hidden influences (red) and the directly involved molecular species. (d) Some of the estimated hidden influences may correspond to missing or wrong molecular interactions within the system. Hence, in a last step, our method tries to further distinguish between intrinsic and exogenous hidden influences. We therefore identify erroneous parts of the nominal ODE system and give detailed hints for their correction.
Owing to the fact that biological systems are open, the number of potential erroneous nodes (e.g. proteins or other molecules) within the nominal model is huge [9]. Without further knowledge, independent error terms have to be assigned to each node. If the respective node is in reality not directly targeted by a hidden influence, the hidden input takes the value zero. Only nodes directly affected by hidden influences have non-zero errors. Wrongly modelled or missing interactions between two nodes can be represented by two error terms, one for each of the respective nodes, which will be correlated over time. We exploited this idea to detect missing or erroneous interactions in a given ODE-based nominal model.
2.2. Approach
We assume the dynamical model
| 2.1a |
| 2.1b |
| 2.1c |
Here, 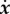 denotes the time derivative of the state vector
denotes the time derivative of the state vector 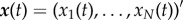 with initial value η. The not necessarily linear function f represents the nominal model, which describes the current assumptions about the dynamic interactions between the state variables and in addition the effect of the known input function
with initial value η. The not necessarily linear function f represents the nominal model, which describes the current assumptions about the dynamic interactions between the state variables and in addition the effect of the known input function 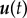 .
.
No model of a biological system can ever be totally complete and comprehensive. Therefore, we add the hidden influences 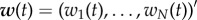 to the nominal model function f. The additive dynamic hidden influence w(t) subsumes missing or wrong interactions between the state variables as well as exogenous influences caused by crosstalk with other biological processes (see electronic supplementary material, §3). Of course, w(t) is unknown and we aim to estimate these hidden influences from the data [20]. Notably, the hidden influence w(t) is not restricted to be constant or linear and thus can be any arbitrary function of time.
to the nominal model function f. The additive dynamic hidden influence w(t) subsumes missing or wrong interactions between the state variables as well as exogenous influences caused by crosstalk with other biological processes (see electronic supplementary material, §3). Of course, w(t) is unknown and we aim to estimate these hidden influences from the data [20]. Notably, the hidden influence w(t) is not restricted to be constant or linear and thus can be any arbitrary function of time.
Often it is impossible to measure all components of the state vector x, e.g. concentrations of reacting substances (due to technical limitations, e.g. non-availability of phospho-protein specific antibodies). The map from the state to the measurable output 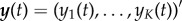 , with K not necessarily equal to N, is given by the measurement function h, which we assume to be known (equation (2.1b)). In addition, we assume white Gaussian measurement noise
, with K not necessarily equal to N, is given by the measurement function h, which we assume to be known (equation (2.1b)). In addition, we assume white Gaussian measurement noise 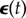 with expectation zero and a noise covariance matrix
with expectation zero and a noise covariance matrix  , see below.
, see below.
In practice, the data are given as measurements 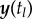 at discrete time points tl with l ∈ {1, …, T}. We will use the notation yk,l = yk(tl) for the measured output k ∈ {1, …, K} at measurement time tl and the analogous notation for the other variables, i.e. xi(tl) = xi,l and
at discrete time points tl with l ∈ {1, …, T}. We will use the notation yk,l = yk(tl) for the measured output k ∈ {1, …, K} at measurement time tl and the analogous notation for the other variables, i.e. xi(tl) = xi,l and 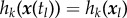 . For sake of simplicity in the following, we denote the matrix of observed measurements by
. For sake of simplicity in the following, we denote the matrix of observed measurements by 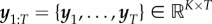 and the corresponding state and hidden influence matrices by
and the corresponding state and hidden influence matrices by  and
and  .
.
From now on, we are interested in hidden influences at discrete time points. Under this assumption equation (2.1) can be rewritten as
| 2.2a |
| 2.2b |
| 2.2c |
Consequently, we obtain a first-order Markov process over the state variables x. The function 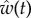 is obtained by fitting a cubic smoothing spline through each of the N discrete time series of hidden influence signals wi,1:T [25].
is obtained by fitting a cubic smoothing spline through each of the N discrete time series of hidden influence signals wi,1:T [25].
The assumption of Gaussian measurement noise can, if necessary, approximately be fulfilled after a variance-stabilizing transformation [26]. In addition, we assume uncorrelated measurement noise and thus Ξl = diag(ξ21,l, …, ξ2K,l).
2.3. Marginal likelihood of the data
The likelihood of the observed data
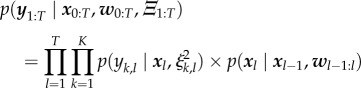 |
2.3 |
can be factorized due to the independence of the measurement noise with respect to time and observables. Note that 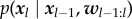 is defined by equation (2.2a). In addition,
is defined by equation (2.2a). In addition,  and
and 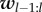 are conditionally independent from Ξ1:T.
are conditionally independent from Ξ1:T.
Since typically the number of replicate measurements per time point is small, the empirical variance is not a reliable estimator of the true measurement noise. Therefore, we impose an inverse gamma prior on the variance of the measurement noise
| 2.4 |
The marginal likelihood of the data are obtained by marginalizing over the variance of the measurement noise variable
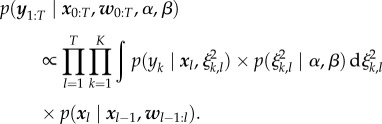 |
2.5 |
This integral can analytically be calculated to yield [27]
 |
2.6 |
A detailed derivation of equation (2.6) is provided in electronic supplementary material, §4.
According to Bayes' theorem, the posterior density over the hidden input signals 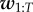 with initial value
with initial value  is given by
is given by
| 2.7 |
Using equation (2.6), we can directly draw samples from the posterior density of the hidden influence. For this purpose, we propose a Bayesian elastic-net prior as detailed in the following sections.
2.4. Smoothness and sparsity via a Bayesian elastic-net prior
The hidden input signals 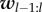 can be understood as the statistical residuals of the nominal system, and every deviation of observations from the nominal system could thus be explained by non-zero components in
can be understood as the statistical residuals of the nominal system, and every deviation of observations from the nominal system could thus be explained by non-zero components in 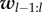 . However, we are only interested in hidden input signals, which are far stronger than measurement noise. Therefore, we assume that the hidden input signal is smooth and sparse. Sparsity corresponds to the a priori belief that only a small subset of state variables is truly affected by unknown external or internal input signals. In addition, we assume the hidden input signal to be smooth over time. Smoothness and sparsity are encoded by a prior distribution inspired by the Bayesian elastic-net, which is here combined with a first-order Markov process over
. However, we are only interested in hidden input signals, which are far stronger than measurement noise. Therefore, we assume that the hidden input signal is smooth and sparse. Sparsity corresponds to the a priori belief that only a small subset of state variables is truly affected by unknown external or internal input signals. In addition, we assume the hidden input signal to be smooth over time. Smoothness and sparsity are encoded by a prior distribution inspired by the Bayesian elastic-net, which is here combined with a first-order Markov process over 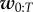 [28,29]. Overall our proposed approach is thus a hierarchical graphical model shown in figure 2. Details can be found in electronic supplementary material, §5.
[28,29]. Overall our proposed approach is thus a hierarchical graphical model shown in figure 2. Details can be found in electronic supplementary material, §5.
Figure 2.

Representation of the proposed Bayesian dynamic elastic-net approach as a probabilistic graphical model. The hidden influences wl form a Markov chain over all time points l = 1, …, T and are directly dependent on the shared parameters 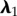 and λ2. Since the outcome of one integration step represents the initial value for the next integration step, the system state variables
and λ2. Since the outcome of one integration step represents the initial value for the next integration step, the system state variables  are also successively dependent.
are also successively dependent.
Briefly, the Bayesian elastic-net defines a conditional Gaussian prior over each wi,l|wi,l−1 (i = 1, …, N, l = 1, …, T). The scale of the variance of the Gaussian prior is a strongly decaying and smooth distribution peaking at zero, which depends on parameters λ2, 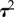 and σ2. The parameter
and σ2. The parameter 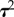 is itself given by an exponential distribution (one for each component of vector
is itself given by an exponential distribution (one for each component of vector 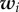 ) with parameters
) with parameters 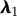 . In consequence, sparsity is dependent on the parameter vector
. In consequence, sparsity is dependent on the parameter vector 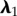 , whereas smoothness is mainly controlled by λ2 [28,30]. These parameters are drawn from hyper-priors, which can be set in a non-informative manner or with respect to prior knowledge about the degree of shrinkage and smoothness of the hidden influences [31]. We refer the reader to electronic supplementary material, §§5 and 7, for details.
, whereas smoothness is mainly controlled by λ2 [28,30]. These parameters are drawn from hyper-priors, which can be set in a non-informative manner or with respect to prior knowledge about the degree of shrinkage and smoothness of the hidden influences [31]. We refer the reader to electronic supplementary material, §§5 and 7, for details.
2.5. Estimating hidden influences from data
To estimate the hidden input and the parameters in the hierarchical model, we devise a Metropolis–Hastings algorithm with Gibbs updates of the Bayesian elastic-net hyper-parameters. The algorithm proceeds sequentially between the different time points 1, …, T by drawing different samples at each supporting point. At sampling step s and time point l, a random component wi,l is selected of the hidden input vector (a node in the network) at the previous time point, which is modified by a sample from a univariate Gaussian transition kernel π [32]. The resulting vector 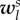 is accepted with probability
is accepted with probability
 |
2.8 |
where 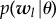 is the Bayesian elastic-net prior over the hidden influences conditioned by hyper-parameters
is the Bayesian elastic-net prior over the hidden influences conditioned by hyper-parameters  (for details, see electronic supplementary material, §§5 and 7). Because of the Gaussian measurement errors, the discrepancy between data component yk,l and the corresponding model output
(for details, see electronic supplementary material, §§5 and 7). Because of the Gaussian measurement errors, the discrepancy between data component yk,l and the corresponding model output 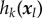 in equation (2.6) is given by the quadratic error
in equation (2.6) is given by the quadratic error 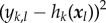 . Note that
. Note that 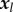 is obtained by numerically integrating the ODE system from time point l − 1 using
is obtained by numerically integrating the ODE system from time point l − 1 using 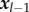 as initial value according to equation (2.2a). Code for the sampling algorithm is provided in electronic supplementary material, §6.
as initial value according to equation (2.2a). Code for the sampling algorithm is provided in electronic supplementary material, §6.
2.6. Estimating endogenous hidden influences: missing and wrong reactions
After having estimated hidden influences on state components in the nominal ODE system, the question arises whether these hidden variables could in fact correspond to missing or wrongly specified interactions within the nominal system. A simple strategy, which we followed here, is to rank all state variables in the nominal system by their temporal correlation with the estimated hidden influences. The essential idea is that in case of a wrong or missing reaction the estimated hidden time courses should ‘compensate’ erroneous predictions by the nominal system (figure 3). In general, wrong or missing interactions can either have an increasing (stimulatory) or decreasing (inhibitory) influence on the target nodes. This results in a negative hidden influence with 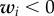 in the case of inhibition and a positive hidden influence with
in the case of inhibition and a positive hidden influence with 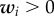 in the case of stimulatory events. More specifically, we distinguish several cases which are listed in table 1 and further illustrated in figure 3. Briefly, the idea is that an modelled stimulation between two state variables x1, x4 in the ODE system yields two error signals w1 (influencing x1) and w4 (influencing x4), which are anticorrelated. This is because w1 and w4 capture the unmodelled dynamics of the ODE system. Similarly, an unmodelled inhibition yields w1,
in the case of stimulatory events. More specifically, we distinguish several cases which are listed in table 1 and further illustrated in figure 3. Briefly, the idea is that an modelled stimulation between two state variables x1, x4 in the ODE system yields two error signals w1 (influencing x1) and w4 (influencing x4), which are anticorrelated. This is because w1 and w4 capture the unmodelled dynamics of the ODE system. Similarly, an unmodelled inhibition yields w1, 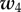 and a positive correlation of w1 and w4. A wrongly modelled stimulation results in
and a positive correlation of w1 and w4. A wrongly modelled stimulation results in 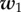 and
and 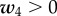 which are anticorrelated. A wrongly modelled inhibition yields w1,
which are anticorrelated. A wrongly modelled inhibition yields w1, 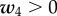 and a positive correlation.
and a positive correlation.
Figure 3.

Illustration of hidden exogenous and endogenous influences by an arbitrary example system. (a) Hidden exogenous influence. No significant temporal correlation between x4 and w1 is expected. (b) Hidden endogenous influence as a missing stimulatory interaction (arrow) from x4 to x1. Here, hidden influences w4 and w1 are highly negatively correlated. This is caused by a missing stimulating effect of x4 on x1. The decrease of x4 is correlated with an increase of w1. (c) Hidden endogenous influence as a missing inhibitory interaction. Here, hidden influences w4 and w1 have a strong positive correlation and compensate a missing inhibitory effect of x4 on x1. The increase of x4 is correlated with an decrease of w1. (d) Hidden endogenous influence as erroneous stimulation. Here, hidden influences w4 and w1 have a strong negative correlation. The increase of x4 goes along with a decrease of w1. (e) Hidden endogenous influence as an erroneous inhibition. The hidden influences w1 and w4 are concordant and correlate strongly with the state component x4.
Table 1.
An endogenous hidden influence is expected to yield different types of (cross-)correlations with other hidden influences (Corr. HI) and state variable dynamics (Corr. State), depending on whether a molecular interaction is missing or wrongly specified in the nominal system. Missing and wrong molecular interactions can be further distinguished depending on whether the true molecular interaction is of stimulatory (stim.) or inhibitory (inh.) nature. Furthermore, molecular interactions between two species can either be modelled in the correlation between the target hidden influence dynamics and the related estimated model variables (Corr. State). In case of endogenous hidden influences, these correlations are expected to be either strongly positive (+) or strongly negative (−) if they reflect true molecular interactions. Figure 3 illustrates all four cases in detail.
| event | Corr. HI | Corr. State | |
|---|---|---|---|
| missing | stim. | − | + |
| inh. | + | − | |
| wrong | stim. | − | − |
| inh. | + | + |
As exemplified in figure 3, due to differences in the expected correlations, our analysis should also allow for distinguishing missing inhibitory versus stimulating effects of missing monotonous interactions.
Different measures exist to capture the strength of correlation between time courses. Apart from the Pearson correlation, we here used the the cross-correlation coefficient
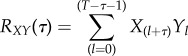 |
2.9 |
to quantify temporal associations between two time series X and Y [33]. The cross-correlation RXY(τ) depends on the time lag τ which was chosen as argmaxτ[RXY (τ)].
3. Results
3.1. Tested mathematical models
The EpoR-induced JAK–STAT signalling pathway mediates a rapid signal transduction from the receptor to the nucleus related to cell proliferation and differentiation [19]. This pathway involves a rapid nucleocytoplasmic cycling of the signal transducer and activator of transcription 5 (STAT5) molecules which is not directly measurable [19].
The G protein cycling model quantitatively characterizes the heterotrimeric G protein activation and deactivation in yeast [34]. This model serves as a fully observed but complex test case where all states are measured.
In contrast, the model of the UV-B signalling in plants systematically links several signalling events induced by UV-B light to a comprehensive informational signalling pathway [35]. Only combinations of small amounts of the involved molecules are accessible and thus it serves as a complex and not fully observed test case.
Network motifs are thought to represent building blocks of larger biological systems [36]. It is thus informative to test BDEN with respect to these motifs to better understand the possible dependency of the performance of our models on different basic network topologies.
The dynamic EpoR model reflects the information processing at EpoR including turnover, recycling and mobilization of EpoR after stimulation with erythropoietin (Epo) at the cell membrane [37]. Consequently, it details the first part of the JAK–STAT signalling pathway. Only combinations of Epo concentrations in the medium, on the surface and in the cells are accessible and thus it represents a complex model with limited experimental data [37].
By contrast, the thermal isomerization of α-Pinene (αP) in the liquid phase has the purpose to investigate the applicability of BDEN to small compound reaction networks [38]. The model details the racemization of αP and its simultaneous isomerization to dipentene (dP) and allo-ocimene (aO).
To further investigate the utility of BDEN for complex systems, we used a gene-regulatory network composed of six genes and related proteins obtained from the DREAM6 challenge [39]. Further details about the described models are given in electronic supplementary material, §8.
3.2. Simulation study: testing the performance of Bayesian DEN
We first compared the performances of BDEN as well as our old DEN approach to correctly predict the location of single hidden influence for the JAK–STAT signalling model, the heterotrimeric G protein cycling, the U-VB signalling in plants and the aforementioned network motifs [19,34–36]. This was done on the basis of simulated data for each system. Details about the simulations are given in electronic supplementary material. For BDEN, we computed for each 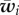 (where
(where 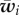 denotes the posterior mean taken over MCMC samples) the area under the predicted hidden influence curve by trapezoidal numerical integration [40]. For DEN, we applied the same method based on the provided point estimates of hidden influence curves. The area under the predicted hidden influence curve was compared against the simulated existence and non-existence of a hidden signal at that node. Consequently, we were able to compute an area under ROC (AUC) value and a corresponding Brier score (BS), i.e. the squared difference between the prediction score and the Boolean indicator of a true hidden influence [41,42]. Table 2 and electronic supplementary material, table S1, show a favourable overall performance of our new method for different levels of measurement noise and simulated errors of kinetic parameter estimates.
denotes the posterior mean taken over MCMC samples) the area under the predicted hidden influence curve by trapezoidal numerical integration [40]. For DEN, we applied the same method based on the provided point estimates of hidden influence curves. The area under the predicted hidden influence curve was compared against the simulated existence and non-existence of a hidden signal at that node. Consequently, we were able to compute an area under ROC (AUC) value and a corresponding Brier score (BS), i.e. the squared difference between the prediction score and the Boolean indicator of a true hidden influence [41,42]. Table 2 and electronic supplementary material, table S1, show a favourable overall performance of our new method for different levels of measurement noise and simulated errors of kinetic parameter estimates.
Table 2.
Performance of BDEN and DEN regarding the dependence on measurement noise (median). The median absolute deviation for the AUC (ROC) and Brier score (BS) are given in brackets.
| model | noise level | method | AUC | BS |
|---|---|---|---|---|
| JAK–STAT | 2.5% | BDEN | 0.90 (0.15) | 0.11 (0.11) |
| DEN | 0.60 (0.40) | 0.16 (0.06) | ||
| 7.5% | BDEN | 0.83 (0.18) | 0.21 (0.19) | |
| DEN | 0.43 (0.29) | 0.30 (0.14) | ||
| 12.5% | BDEN | 0.75 (0.25) | 0.26 (0.16) | |
| DEN | 0.42 (0.31) | 0.41 (0.12) | ||
| G protein | 2.5% | BDEN | 0.99 (0.02) | 0.04 (0.03) |
| DEN | 1.00 (0.00) | 0.09 (0.02) | ||
| 7.5% | BDEN | 0.88 (0.13) | 0.17 (0.09) | |
| DEN | 0.80 (0.13) | 0.16 (0.09) | ||
| 12.5% | BDEN | 0.80 (0.16) | 0.22 (0.10) | |
| DEN | 0.71 (0.16) | 0.20 (0.11) | ||
| UV-B | 2.5% | BDEN | 0.91 (0.11) | 0.19 (0.06) |
| DEN | 0.80 (0.19) | 0.22 (0.08) | ||
| 7.5% | BDEN | 0.88 (0.14) | 0.19 (0.04) | |
| DEN | 0.80 (0.19) | 0.20 (0.06) | ||
| 12.5% | BDEN | 0.81 (0.15) | 0.19 (0.05) | |
| DEN | 0.71 (0.15) | 0.19 (0.05) | ||
| motifs | 2.5% | BDEN | 1.00 (0.00) | 0.00 (0.00) |
| DEN | 0.90 (0.14) | 0.11 (0.09) | ||
| 7.5% | BDEN | 1.00 (0.00) | 0.00 (0.00) | |
| DEN | 0.81 (0.19) | 0.19 (0.04) | ||
| 12.5% | BDEN | 1.00 (0.00) | 0.00 (0.00) | |
| DEN | 0.80 (0.15) | 0.19 (0.05) |
Next, we investigated the performance of BDEN to correctly detect more than one hidden influence. We used the comparatively large G protein cycle model for this purpose. Results can be found in electronic supplementary material, table S2. In this simulation, we randomly added hidden influences for up to 50% of the nodes and still observed a good prediction performance.
In a similar manner, we investigated the performance of BDEN to detect wrong and missing interactions (table 3). We simulated wrong model specifications of the heterotrimeric G protein cycling, the UV-B signalling in plants and the synthetic JAK–STAT signalling by randomly removing and adding interactions. As described above, the quantitative predictions of BDEN are given in terms of (cross-)correlations. By comparing these correlation values against the true existence and non-existence of a particular interaction, we were able to compute an AUC value. Notably, missing and wrong interaction detection is only possible with our new BDEN approach. Again we archived a very good performance for all systems under investigation. On average, 80% of the missing interactions are correctly detected by BDEN. Among the correctly identified missing interactions, on average 90% were correctly classified as ‘stimulating’ and ‘inhibiting’, respectively (electronic supplementary material, table S3). Details regarding the dependency on the measurement noise are given in table 3 and results in dependency of deviance of the parameter estimates are given in electronic supplementary material, tables S4 and S5.
Table 3.
Performance of BDEN to detect wrong and missing interactions depending on the measurement noise (median) evaluated for the JAK–STAT (JS), G protein (GP) and UV-B network. The median absolute deviation for the AUC (ROC) is given in brackets.
| model | noise level | AUC | |
|---|---|---|---|
| missing interaction | JS | 2.5% | 1.00 (0.00) |
| 7.5% | 0.83 (0.28) | ||
| 12.5% | 0.80 (0.32) | ||
| GP | 2.5% | 0.81 (0.19) | |
| 7.5% | 0.78 (0.22) | ||
| 12.5% | 0.62 (0.47) | ||
| UV-B | 2.5% | 1.00 (0.00) | |
| 7.5% | 0.91 (0.11) | ||
| 12.5% | 0.76 (0.16) | ||
| wrong interaction | JS | 2.5% | 1.00 (0.00) |
| 7.5% | 0.87 (0.32) | ||
| 12.5% | 0.80 (0.23) | ||
| GP | 2.5% | 1.00 (0.00) | |
| 7.5% | 0.81 (0.32) | ||
| 12.5% | 0.73 (0.40) | ||
| UV-B | 2.5% | 0.81 (0.20) | |
| 7.5% | 0.70 (0.24) | ||
| 12.5% | 0.68 (0.30) |
3.3. Examples with real data
In the following, we further illustrate the results obtained with BDEN for the JAK–STAT signalling model, the information processing at EpoR and the isomerization of αP using experimental data.
3.3.1. JAK–STAT signalling
The JAK–STAT signalling pathway model (§3.1) consists of four molecular species.
Unbound STAT5 molecules become phosphorylated (STAT5p) catalysed by the erythropoietin receptor. Two STAT5p molecules can form a dimer (STAT5di) and thus are able to enter the nucleus (STAT5n). Only the amount of phosphorylated STAT5 molecules, the total amount of STAT5 and the erythropoietin receptor are directly accessible. Experimental measurements are available at 16 time points [19].
Figure 4 illustrates the application of our method when ignoring the back-translocation of STAT5n into the cytoplasm, which was hypothesized by the authors [19]. After parameter fitting the nominal system is not in sufficient agreement with the data. Introducing hidden influence terms 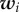 leads to good agreement with the observations. Our method clearly localized two hidden influences
leads to good agreement with the observations. Our method clearly localized two hidden influences 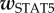 and
and 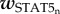 at STAT5 and STAT5n. Subsequent analysis shows a high positive (cross-)correlation of
at STAT5 and STAT5n. Subsequent analysis shows a high positive (cross-)correlation of 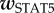 with STAT5n and a negative one with
with STAT5n and a negative one with 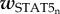 . Exactly the opposite pattern can be observed for
. Exactly the opposite pattern can be observed for 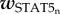 . According to our above explained procedure we thus predict a stimulatory influence of
. According to our above explained procedure we thus predict a stimulatory influence of 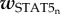 on STAT5. This is in agreement with the claimed nucleocytoplasmic cycling of the phosphorylated STAT5 dimer [19,43].
on STAT5. This is in agreement with the claimed nucleocytoplasmic cycling of the phosphorylated STAT5 dimer [19,43].
Figure 4.

Reconstructing the model error for the JAK–STAT signalling pathway [19]. (a) Nominal model of the JAK–STAT signalling (blue) including its nucleocytoplasmic cycling (red). (b,c) Measurements (black) for phosphorylated cytoplasmic STAT5 and total cytoplasmic STAT5 and posterior BDEN predictions including the 95% credible intervals (red) based on the nominal system (blue). (d) Estimated hidden inputs (posterior means) and 95% credible intervals. There is a clear input located at STAT5 and STAT5 in the nucleus. (e) Estimated time series posterior means for all modelled variables including 95% credible intervals. (f) Estimated correlations (Corr) and cross-correlations (xCorr) of the estimated hidden input 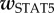 located at STAT5 with all estimated state variables. (g) Estimated correlations and cross-correlations of
located at STAT5 with all estimated state variables. (g) Estimated correlations and cross-correlations of 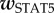 with all remaining hidden influences. Here
with all remaining hidden influences. Here 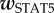 is clearly correlated to the hidden input located at STAT5n and in addition it is correlated to the time series of STAT5n. High cross-correlation is a necessary but not sufficient condition.
is clearly correlated to the hidden input located at STAT5n and in addition it is correlated to the time series of STAT5n. High cross-correlation is a necessary but not sufficient condition.
3.3.2. EpoR model
The complex core model of the EpoR regulation via receptor mobilization, turnover and recycling involves six species and eight time points. Here the ligand Epo binds to EpoR on the surface and builds a ligand–receptor complex (Epo–EpoR). In consequence, Epo–EpoR triggers the phoshorylation of the cytoplasmic EpoR and thus induces the JAK–STAT signalling pathway [37]. Several mechanisms affect the amount of active EpoR. This model covers the ligand-induced receptor endocytosis and thus the internalization of the ligand-bound receptor (Epo–EpoRi), receptor recycling and degradation of the internalized ligand-bound receptor. Location-dependent degradation results in degraded Epo in cytoplasm (dEpoi) and in medium (dEpoe).
As a test case for BDEN, we wrongly specified a receptor-induced feedback on the amount of available Epo. In consequence, we expect to detect this wrong interaction. As shown in figure 5, BDEN allows to correctly localize and characterize this erroneous interaction in the nominal model.
Figure 5.

Reconstructing the hidden influence in a model of the EpoR regulation [37]. (a) Reaction graph of the model (E = Epo, ER = EpoR, EER = Epo − EpoR, EERi = Epo − EpoRi, dEi = dEpoi, dEe = dEpoe). The red arrow is a wrong reaction. (b,c,d) Output measurements (black) compared with posterior BDEN predictions (red) including 95% credible intervals and the nominal model (blue). (e) Estimates of the hidden influences (posterior mean) including 95% credible intervals. (f) Estimated correlations (Corr) and cross-correlations (xCorr) of the hidden influence related to wE with all estimated state variables. A clear correlation with EpoR is observable. (g) Estimated correlations (Corr) and cross-correlations (xCorr) of the hidden influence related to wE with all estimated remaining hidden influences. Again wE is clearly correlated with wER. In consequence, the direct interaction between ER and E is correctly classified as wrong and has to be removed.
3.3.3. αP isomerization
The model of the dynamic isomerization of αP is composed of four molecular species. Measurements of αP, dP, aO and the dimer (Di) are available [38]. After heating, αP reacts either to dP or builds a dimer by reacting with aO. Furthermore, Di can react to aO.
To test BDEN the dimerization step was wrongly replaced with a simple reaction involving only aO. In consequence the interaction between αP and dP is completely independent from the interaction between aO and Di. The erroneous nominal system can be corrected by using BDEN as illustrated in figure 6. BDEN is able to correctly locate and add the falsely removed reaction.
Figure 6.

Reconstructing the hidden influence in a model of the dynamic αP isomerization [38]. (a) Reaction graph. The red arrow indicates a missing reaction. (b,c,d,e) Output measurements (black) compared with the posterior BDEN predictions (red) including 95% credible intervals and the nominal model (blue). (f) Estimates of the hidden influences (posterior mean) including 95% credible intervals. (g) Estimated relative cross-correlations (xCorr) of the hidden influence of wDi to all estimated remaining hidden influences and state variables. BDEN is able to correctly detect the missing reaction.
3.4. Further examples with simulated data
In the following, we further illustrate the results obtained with BDEN for the G protein cycle in yeast, the UV-B signalling model and a generic gene-regulatory network using simulated data (2.5% noise level).
3.4.1. G protein cycle in yeast
The heterotrimeric G protein cycle in yeast involves six species which are directly observable and coupled by several types of kinetics (for details see electronic supplementary material, §8) [34]. Experimental data at 8 time points were simulated by adding Gaussian distributed noise to the predicted values of the observable variables. We assumed a noise intensity of 2.5% relative to the mean of the related time series for each observable variable. The nominal system was generated by adding one additional ‘wrong’ interaction (between the ligand–receptor complex (LR) and the G proteinα-inactive (GPαi)). Electronic supplementary material, figure S1, illustrates the ability of BDEN to localize and recover the wrong interaction within the nominal system.
3.4.2. UV-B Signalling
As a more complex example, we simulated seven data points of the photomorphogenic UV-B signalling in plants [35]. The model of the photomorphogenic UV-B signalling in the model plant Arabidopsis thaliana consists of 11 species coupled by several different kinetic rate expressions and five observable variables as a combination of seven different species (for details see electronic supplementary material, §8). As the nominal system, we used the literature given model and included a missing link by removing one interaction which influences two different species. Observed data were simulated by adding Gaussian distributed noise to the predicted values of the observable variables. We assumed a noise intensity of 2.5% with respect to the mean of the related time series for each observable variable. As shown in electronic supplementary material, figure S2, the BDEN is able to detect the missing molecular interaction and correctly identifies the corresponding proteins.
3.4.3. DREAM6 Challenge Network
To investigate the applicability of BDEN to gene-regulatory networks we took a model from the DREAM6 challenge [39]. The model consists of six genes and six proteins coupled by mass action and hill kinetics. In this model, all proteins and one mRNA species are assumed to be directly observable (for details see electronic supplementary material, §8) [39]. As the nominal system, we used the provided model and included one inhibitory mechanism. Observed data were simulated at five time points by adding Gaussian distributed noise to the predicted values of observable variables according to the original challenge [39]. BDEN is able to detect and correct the spurious interaction, as illustrated in electronic supplementary material, figure S3.
4. Conclusion
Mathematical modellers in systems biology are frequently confronted with incomplete knowledge and limited understanding of a complex biochemical system [9–11]. Consequently, there is a non-negligible chance that relevant molecular species are missed or interactions are misspecified [8,9]. Our proposed method addresses this issue by adopting a Bayesian framework which allows for inferring hidden influence variables, as well as estimating missing and wrong molecular interactions. It has been successfully validated in several real models as well as common network motifs. This was done with simulated as well as experimental data. Owing to the fully Bayesian formulation all model parameters are sampled. Furthermore, the Bayesian approach allows to assign confidence levels to predictions. Besides these general features of a fully probabilistic framework our newly proposed BDEN method seems to be more stable and more robust because within the Bayesian framework, we average over a large number of parameters and do not rely on stiff integration methods.
A unique feature of our new approach is the distinction between exogenous and endogenous hidden influences in the biological system, allowing for the detection of missing and misspecified equations in the ODE system. Altogether we thus see our BDEN method as a further step towards a better automated and more objective revision of ODE-based models in systems biology and possibly other fields, such as pharmacokinetics, earth science, robotics and engineering [12,20]. In that context, we emphasize again that BDEN is not designed to learn ODE systems purely from data and should thus not be confused with network reverse engineering methods [44]. Much more, the utility of BDEN is to ease identification of sources of errors in mechanism-based mathematical models.
Supplementary Material
Data accessibility
The source code (Matlab) with several examples is available online at http://www.abi.bit.uni-bonn.de/index.php?id=17.
Authors' contributions
B.E. performed the simulations; B.E., M.K. and H.F developed the method; B.E. and H.F interpreted the data; B.E., M.K. and H.F drafted the manuscript; M.K. and H.F. designed the research. All authors reviewed the manuscript and gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
B.E. was supported by the Research Training Group 1873, Deutsche Forschungsgemeinschaft (DFG).
References
- 1.Li C. et al 2010. BioModels database: an enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 4, 1–14. ( 10.1186/1752-0509-4-92) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunawardena J. 2014. Models in biology: ‘accurate descriptions of our pathetic thinking’. BMC Biol. 12, 29 ( 10.1186/1741-7007-12-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shlomi T, Cabili MN, Herrgård MJ, Palsson BØ, Ruppin E. 2008. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 26, 1003–1010. ( 10.1038/nbt.1487) [DOI] [PubMed] [Google Scholar]
- 4.Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. 2014. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 7, 501–501 ( 10.1038/msb.2011.35) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cvijovic M. et al. 2014. Bridging the gaps in systems biology. Mol. Genet. Genomics 289, 727–734. ( 10.1007/s00438-014-0843-3) [DOI] [PubMed] [Google Scholar]
- 6.Kuepfer L, Peter M, Sauer U, Stelling J. 2007. Ensemble modeling for analysis of cell signaling dynamics. Nat. Biotechnol. 25, 1001–1006. ( 10.1038/nbt1330) [DOI] [PubMed] [Google Scholar]
- 7.Azeloglu EU, Iyengar R. 2015. Good practices for building dynamical models in systems biology. Sci. Signal 8, fs8 ( 10.1126/scisignal.aab0880) [DOI] [PubMed] [Google Scholar]
- 8.Bertalanffy Lv. 1950. The theory of open systems in physics and biology. Sciences 111, 23–29. ( 10.1126/science.111.2872.23) [DOI] [PubMed] [Google Scholar]
- 9.Babtie AC, Kirk P, Stumpf MPH. 2014. Topological sensitivity analysis for systems biology. Proc. Natl Acad. Sci. USA 111, 18507–18512. ( 10.1073/pnas.1414026112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barzel B, Liu YY, Barabási AL. 2015. Constructing minimal models for complex system dynamics. Nat. Commun. 6, 7186 ( 10.1038/ncomms8186) [DOI] [PubMed] [Google Scholar]
- 11.Gao J, Liu YY, D'Souza RM, Barabási AL. 2014. Target control of complex networks. Nat. Commun. 5, 5415 ( 10.1038/ncomms6415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abarbanel H. 2013. Predicting the future understanding complex systems. New York, NY: Springer. [Google Scholar]
- 13.Toni T, Stumpf MPH. 2010. Simulation-based model selection for dynamical systems in systems and population biology. Bioinformatics 26, 104–110. ( 10.1093/bioinformatics/btp619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirk P, Thorne T, Stumpf MP. 2013. Model selection in systems and synthetic biology. Curr. Opin. Biotechnol. 24, 767–774. ( 10.1016/j.copbio.2013.03.012) [DOI] [PubMed] [Google Scholar]
- 15.Xu TR. et al. 2010. Inferring signaling pathway topologies from multiple perturbation measurements of specific biochemical species. Sci. Signal 3, ra20 ( 10.1126/scisignal.2000517) [DOI] [PubMed] [Google Scholar]
- 16.Sunnaker M, Zamora-Sillero E, López García de Lomana A, Rudroff F, Sauer U, Stelling J, Wagner A. 2014. Topological augmentation to infer hidden processes in biological systems. Bioinformatics 30, 221–227. ( 10.1093/bioinformatics/btt638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vyshemirsky V, Girolami MA. 2008. Bayesian ranking of biochemical system models. Bioinformatics 24, 833–839. ( 10.1093/bioinformatics/btm607) [DOI] [PubMed] [Google Scholar]
- 18.Ollier E, Heritier F, Bonnet C, Hodin S, Beauchesne B, Molliex S, Delavenne X. 2015. Population pharmacokinetic model of free and total ropivacaine after transversus abdominis plane nerve block in patients undergoing liver resection. Br. J. Clin. Pharmacol. 80, 67–74. ( 10.1111/bcp.12582) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swameye I, Müller TG, Timmer J, Sandra O, Klingmüller U. 2003. Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by databased modeling. Proc. Natl Acad. Sci. USA 100, 1028–1033. ( 10.1073/pnas.0237333100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelhardt B, Fröhlich H, Kschischo M. 2016. Learning (from) the errors of a systems biology model. Sci. Rep. 6, 20772 ( 10.1038/srep20772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou H, Hastie T. 2005. Regularization and variable selection via the Elastic Net. J. R. Stat. Soc. B 67, 301–320. ( 10.1111/j.1467-9868.2005.00503.x) [DOI] [Google Scholar]
- 22.Betts JT. 2009. Practical methods for optimal control and estimation using nonlinear programming, 2nd edn Philadelphia, PA: Society for Industrial and Applied Mathematics. [Google Scholar]
- 23.Rao AV. 2009. A survey of numerical methods for optimal control. In AAS/AIAA Astrodynamics Specialist Conf., Pittsburgh, PA, August, AAS Paper 09-334. [Google Scholar]
- 24.Liu YY, Slotine JJ, Barábasi AL. 2013. Observability of complex systems. Proc. Natl Acad. Sci. USA 110, 2460–2465. ( 10.1073/pnas.1215508110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fritsch FN, Carlson RE. 1980. Monotone piecewise cubic interpolation. SIAM J. Numer. Anal. 17, 238–246. ( 10.1137/0717021) [DOI] [Google Scholar]
- 26.Atkinson AC. 1987. Plots, transformations, and regression: an introduction to graphical methods of diagnostic regression analysis, Reprint edn New York, NY: Oxford University Press. [Google Scholar]
- 27.Zacher B, Abnaof K, Gade S, Younesi E, Tresch A, Fröhlich H. 2012. Joint Bayesian inference of condition-specific miRNA and transcription factor activities from combined gene and microRNA expression data. Bioinformatics 28, 1714–1720. ( 10.1093/bioinformatics/bts257) [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Lin N. 2010. The Bayesian elastic net. Bayesian Anal. 5, 151–170. ( 10.1214/10-BA506) [DOI] [Google Scholar]
- 29.Friedman N, Murphy K, Russell S. 1998. Learning the structure of dynamic probabilistic networks. In Proc. of the Fourteenth Conf. on Uncertainty in Artificial Intelligence. UAI'98, pp. 139–147. San Francisco, CA: Morgan Kaufmann Publishers Inc. [Google Scholar]
- 30.Zou H, Zhang HH. 2009. On the adaptive elastic-net with a diverging number of parameters. Ann. Stat. 37, 1733–1751. ( 10.1214/08-AOS625) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyung M, Gill J, Ghosh M, Casella G. 2010. Penalized regression, standard errors, and Bayesian lassos. Bayesian Anal. 5, 369–411. ( 10.1214/10-BA607) [DOI] [Google Scholar]
- 32.Brooks SP. 1998. Markov chain Monte Carlo method and its application. J. R. Stat. Soc. D 47, 69–100. ( 10.1111/1467-9884.00117) [DOI] [Google Scholar]
- 33.Petre S, Moses R. 2005. Spectral analysis of signals. Upper Saddle River, NJ: Prentice Hall. [Google Scholar]
- 34.Yi TM, Kitano H, Simon MI. 2003. A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc. Natl Acad. Sci. USA 100, 10 764–10 769. ( 10.1073/pnas.1834247100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouyang X, Huang X, Jin X, Chen Z, Yang P, Ge H, Deng XW. 2014. Coordinated photomorphogenic UV-B signaling network captured by mathematical modeling. Proc. Natl Acad. Sci. USA 111, 11 539–11 544. ( 10.1073/pnas.1412050111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milo R. 2002. Network motifs: simple building blocks of complex networks. Sciences 298, 824–827. (doi:0.1126/science.298.5594.824) [DOI] [PubMed] [Google Scholar]
- 37.Becker V, Schilling M, Bachmann J, Baumann U, Raue A, Maiwald T, Timmer J, Klingmüller U. 2010. Covering a broad dynamic range: information processing at the erythropoietin receptor. Sciences 328, 1404–1408. ( 10.1126/science.1184913) [DOI] [PubMed] [Google Scholar]
- 38.Fuguitt RE, Hawkins JE. 1947. Rate of the thermal isomerization of α-Pinene in the liquid phase. J. Am. Chem. Soc. 69, 319–322. ( 10.1021/ja01194a047) [DOI] [Google Scholar]
- 39.Meyer P. et al. 2014. Network topology and parameter estimation: from experimental design methods to gene regulatory network kinetics using a community based approach. BMC Syst. Biol. 8, 13 ( 10.1186/1752-0509-8-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atkinson KE. 1989. An introduction to numerical analysis, 2nd edn New York, NY: Wiley. [Google Scholar]
- 41.Fawcett T. 2006. An introduction to ROC analysis. Pattern Recognit. Lett. 27, 861–874. ( 10.1016/j.patrec.2005.10.010) [DOI] [Google Scholar]
- 42.Murphy AH. 1973. A new vector partition of the probability score. J. Appl. Meteorol. 12, 595–600. ( 10.1175/1520-0450(1973)012%3C0595:ANVPOT%3E2.0.CO;2) [DOI] [Google Scholar]
- 43.Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmüller U, Timmer J. 2009. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics 25, 1923–1929. ( 10.1093/bioinformatics/btp358) [DOI] [PubMed] [Google Scholar]
- 44.Sachs K, Perez O, Pe'er D, Lauffenburger DA, Nolan GP. 2005. Causal protein-signaling networks derived from multiparameter single-cell data. Sciences 308, 523–529. ( 10.1126/science.1105809) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code (Matlab) with several examples is available online at http://www.abi.bit.uni-bonn.de/index.php?id=17.