

Summary
Mitochondria perform central functions in cellular bioenergetics, metabolism, and signaling, and their dysfunction has been linked to numerous diseases. The available studies cover only part of the mitochondrial proteome, and a separation of core mitochondrial proteins from associated fractions has not been achieved. We developed an integrative experimental approach to define the proteome of east mitochondria. We classified > 3,300 proteins of mitochondria and mitochondria-associated fractions and defined 901 high-confidence mitochondrial proteins, expanding the set of mitochondrial proteins by 82. Our analysis includes protein abundance under fermentable and nonfermentable growth, submitochondrial localization, single-protein experiments, and subcellular classification of mitochondria-associated fractions. We identified mitochondrial interactors of respiratory chain supercomplexes, ATP synthase, AAA proteases, the mitochondrial contact site and cristae organizing system (MICOS), and the coenzyme Q biosynthesis cluster, as well as mitochondrial proteins with dual cellular localization. The integrative proteome provides a high-confidence source for the characterization of physiological and pathophysiological functions of mitochondria and their integration into the cellular environment.
Keywords: mitochondria, quantitative MS, protein-protein interaction, subcellular localization, SILAC, protein copy numbers, submitochondrial localization, dual localization
Graphical Abstract
Highlights
-
•
Classification of > 3,300 proteins of mitochondria and associated fractions
-
•
High-confidence mitochondrial proteome with absolute quantification and topology
-
•
Interactors of oxidative phosphorylation complexes and cristae organizing system
-
•
Identification of system linking respiratory chain and AAA quality control
Morgenstern et al. describe an integrative organelle proteomics analysis to define the mitochondrial proteome in baker’s yeast. The study provides a quantitative footprint of the proteome and its dynamics under different conditions. The results expand the set of proteins assigned to the mitochondria and provide a resource for future mitochondrial research.
Introduction
Mitochondria are essential for the viability of eukaryotic cells. They are crucial for numerous cellular functions, including oxidative phosphorylation, metabolism of amino acids and lipids, biosynthesis of heme and iron-sulfur (Fe/S) clusters, and programmed cell death (Nunnari and Suomalainen, 2012). Mitochondria are extensively connected with other cellular compartments. This includes the transport of numerous metabolites and ions between cytosol and mitochondria, the import of precursor proteins from the cytosol, the formation of contact sites with the endoplasmic reticulum (ER) and additional organelles, as well as the interaction of mitochondria with the cytoskeleton (Eisenberg-Bord et al., 2016, Klecker et al., 2014, Labbé et al., 2014, Neupert and Herrmann, 2007, Wiedemann and Pfanner, 2017).
Initial proteomic studies led to the identification of a considerable portion of the mitochondrial proteome (Mootha et al., 2003, Ohlmeier et al., 2004, Pflieger et al., 2002, Prokisch et al., 2004, Sickmann et al., 2003, Taylor et al., 2003). This led to a boost of studies on mitochondrial functions under physiological and pathophysiological conditions, including the identification and functional characterization of further mitochondrial protein import pathways, of machineries required for maintaining mitochondrial membrane architecture, of proteins involved in the biosynthesis of Fe/S clusters, and of enzymes required for lipid biosynthesis (Lill, 2009, Neupert and Herrmann, 2007, van der Laan et al., 2016, Wiedemann and Pfanner, 2017). The effective combination of genetic, cell biological, and biochemical methods in the yeast Saccharomyces cerevisiae provided powerful approaches for analyzing protein functions in vivo and in vitro. Further studies expanded the mitochondrial proteome of both yeast and mammals (Itzhak et al., 2016, Müller et al., 2016, Pagliarini et al., 2008, Rao et al., 2017, Reinders et al., 2006, Renvoisé et al., 2014, Stefely et al., 2016). In addition to direct proteomic studies of purified mitochondria (mentioned above) and mitochondrial subcompartments (Hung et al., 2014, Rhee et al., 2013, Vögtle et al., 2012, Zahedi et al., 2006), data mining, prediction programs for protein targeting, and literature entries were used to construct databases. Furthermore, numerous proteins were assigned to the mitochondrial compartment by genome-wide high-throughput localization studies using fluorescent tags (Kumar et al., 2002, Huh et al., 2003, Stadler et al., 2013, Yofe et al., 2016).
However, our knowledge about the mitochondrial proteome is far from being complete and the validity of mitochondrial entries in the widely used databases vary considerably for several reasons. (1) The high sensitivity of mass spectrometry (MS)-based approaches leads to the identification of even minor contaminations of organelle preparations, and thus the numerous lists of identified proteins contain a mixture of authentic mitochondrial proteins, loosely attached proteins, and contaminants. (2) High-throughput tagging studies yield important information about the global distribution of proteins but can lead to a misassignment of individual proteins to cellular compartments when they are not combined with single-protein analysis. (3) Prediction programs of protein targeting to distinct cell organelles and literature mining can provide important information, yet their use for databases without validating individual entries by experimental approaches can be misleading.
We developed an integrative experimental approach for organelle proteomics, generating a comprehensive proteome of mitochondria and associated fractions of baker’s yeast. We combined high-sensitivity MS with quantitative analysis of protein abundance under different growth conditions, enrichment of proteins in distinct fractions, and subcellular classification of mitochondria-associated fractions. Many identified mitochondrial proteins were subjected to single-protein studies for in organello protein transport, interaction analysis, and subcellular and submitochondrial fractionation to define their localization and functional context, revealing partner proteins of key mitochondrial processes in respiration, quality control, and membrane architecture.
Results
Experimental Approaches toward a Comprehensive Mitochondrial Proteome
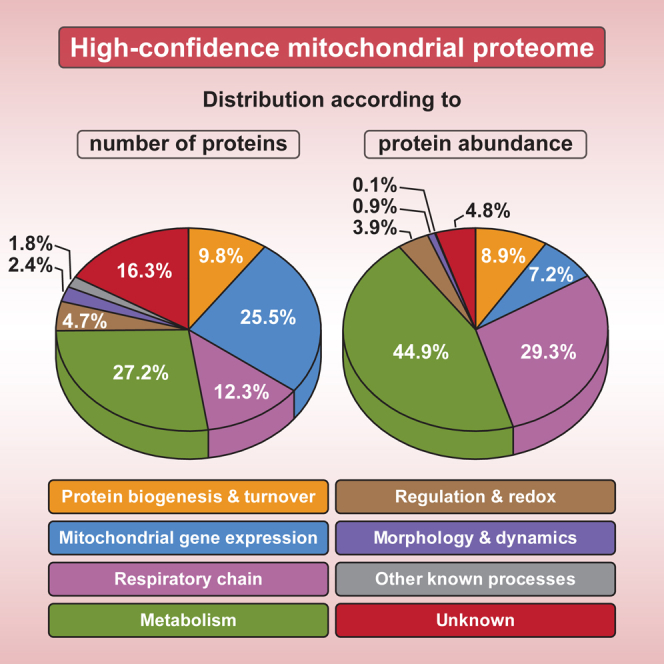
To systematically explore the proteome of yeast mitochondria, we combined subcellular fractionation and biochemical methods with high-resolution proteomics and quantitative MS techniques (Figure 1A). We prepared highly purified mitochondria. The abundance of marker proteins was assessed by immunoblotting and MS (Figure 1B; Tables S1 and S2A). The purified mitochondria were virtually devoid of cytosolic and peroxisomal components, but still contained a fraction of ER proteins (Figure 1B, lanes 4 and 8). Figure 1C summarizes our multifaceted functional proteomic strategy. (1) Crude and purified mitochondria prepared from stable isotope labeling by amino acids in cell culture (SILAC)-yeast cells were analyzed by quantitative MS using a multi-protease digestion and orthogonal separation approach (Figures 1C and S1A). (2) Yeast cells grown under three different conditions were analyzed by MS to define an absolute quantitative blueprint of the mitochondrial proteome (Figures 1C and S1B). (3) We established the mitochondrial localization of identified and poorly characterized mitochondrial candidate proteins by subcellular protein profiling in a dual approach comprising label-free quantitative MS and epitope tagging combined with immunoblotting or microscopy (Figure 1C). (4) We studied submitochondrial protein localization using a gel-enhanced MS-based proteome profiling approach (Figure S1C) and mitochondrial import assays with radiolabeled precursor proteins (Figure 1C). (5) We delineated the functional protein interaction networks of identified mitochondrial proteins by SILAC-based quantitative affinity purification-MS (q-AP-MS) (Figure 1C). Altogether, we collected data of 913 liquid chromatography-tandem mass spectrometry (LC-MS/MS) runs with a total measurement time of 72 days. Overviews of the functional classification of the high-confidence mitochondrial proteome (901 proteins) and of the 201 mitochondrial proteins identified/verified in this study are shown in Figures 1D and 1E (see also Tables S3 and S4).
Figure 1.

Multi-dimensional MS Approach for Global Profiling and In-Depth Characterization of the Mitochondrial Proteome of S. cerevisiae
(A) Outline of experimental strategy. Total, cell lysate; PMS, post-mitochondrial supernatant; Mito, mitochondria; pMito, gradient-purified mitochondria.
(B) Western blot (lanes 1–4) and MS data (lanes 5–8) for selected subcellular marker proteins. Error bars indicate SEM for n ≥ 3 and the range for n = 2.
(C) Overview of experimental approaches followed to define and characterize mitochondrial proteins.
(D) Overview of the functional classification of the high-confidence mitochondrial proteome. Relative protein quantification was based on the analysis of gradient-purified mitochondria from cells grown on glycerol.
(E) Overview of the functional classification of mitochondrial proteins identified or verified in this study.
Classification of Mitochondria and Mitochondria-Associated Fractions
Our MS-based proteome profiling of crude and highly purified yeast mitochondria resulted in the identification of 3,512 proteins (SD, ±10) on average per biological replicate (n = 4), of which 1,024 (SD, ±1.4) were annotated as mitochondrial proteins in the Saccharomyces Genome Database (SGD) (Cherry et al., 2012) (Figure S2A; Tables S2B and S2C). To obtain a high coverage of protein sequences by identified peptides, we used five different proteases, leading to a 1.6-fold average increase of sequence coverage (1.5-fold for mitochondrial proteins) in comparison to the use of trypsin alone (Figure S2B; Table S2B; exemplified with the TIM23 presequence translocase of the inner membrane, Figure S2C). The overall sequence coverage for proteins with mitochondrial annotation was 64% compared to 45% for all identified proteins. Mitochondrial annotation of proteins in the SGD includes a broad range of approaches from detailed single-protein localization and functional studies to high-throughput analyses and putative mitochondrial assignments. To obtain a reference set of mitochondrial proteins, we used 583 mitochondrial proteins, for which a submitochondrial localization was reported in SGD (pointing toward single-protein studies); 520 proteins were assigned to a single submitochondrial compartment. We identified 98% of these reference set proteins (Figure 2A), indicating a high coverage of well-established mitochondrial proteins by our MS analysis.
Figure 2.

Quantitative Deep Proteome Analysis of Yeast Mitochondria by SILAC-MS
(A) Coverage of proteins of distinct submitochondrial localization. Proteins identified in pure/crude mitochondria were categorized as outer membrane (OM), intermembrane space (IMS), inner membrane (IM), or matrix protein based on GO annotations. Shown is the number of proteins assigned to a given term and (in parentheses) the total number of proteins with this annotation. Mito, sum of all proteins in OM, IMS, IM, and matrix.
(B) Protein composition of crude versus gradient-purified mitochondria. Shown are mean percentages of intensity-based absolute quantification (iBAQ) values (n = 4). Loc., localized; PM, plasma membrane; SGD, Saccharomyces Genome Database.
(C) Ratio-intensity plots of quantified proteins (n = 4). Proteins exclusively localized to mitochondria (left), to mitochondria and other organelles (middle), and selected proteins with multiple subcellular localization (right) are highlighted.
(D) Distribution of proteins grouped into distinct classes by statistical data analysis (left panel) and selected GO cellular component terms significantly enriched in each class (right panel) with the number of proteins assigned. Cyto. ribo., cytosolic ribosome; Cytosk., cytoskeleton; Nucl. chr./lum., nuclear chromosome/lumen.
(E and F) Ratio-intensity plots as shown in (D) highlighting mitochondrial subcompartment annotations (E) and subunits of the TOM, TIM23-PAM, MICOS, and ERMES complex as well as mitochondrial ribosomal proteins and proteins involved in mitochondrial dynamics (F). Coloring reflects the class a protein was assigned to.
With a number of 3,539 proteins identified in total across all replicates (based on 125,648 identified peptide sequences), this analysis considerably exceeds previous studies on the yeast mitochondrial proteome (Figures S2D and S2E; Tables S1 and S2B) (Ohlmeier et al., 2004, Prokisch et al., 2004, Reinders et al., 2006, Vögtle et al., 2012, Zahedi et al., 2006); however, due to the high sensitivity of MS, a large number of non-mitochondrial proteins were also identified. We thus searched for criteria to distinguish the core mitochondrial proteome from associated and contaminating proteins. Non-mitochondrial proteins were markedly decreased in abundance, but not in number of identified proteins, in purified mitochondria in comparison with crude mitochondria (Figure 2B; Tables S1 and S2B). For relative quantification, we calculated SILAC ratios of 3,453 proteins and established ratio-intensity plots of 3,365 proteins reliably quantified in all four biological replicates (Figures 2C, S2F, and S2G; Table S2B). Proteins with an exclusive mitochondrial localization exhibited a narrow distribution centered at a median log2 SILAC ratio of purified to crude mitochondria of 0.3 (Figures 2C, left plot, and S2H), whereas mitochondrial proteins with dual or multiple localizations (Yogev and Pines, 2011) displayed a broader distribution (Figure 2C, middle and right plots). Using a statistical approach, we classified proteins based on their mean log2 ratios, leading to a subdivision of the large dataset into four distinct classes (Figures 2D and S2H; Table S2B). All four classes contained mitochondrial proteins, but to a considerably different extent. Using gene ontology (GO) enrichment analysis (SGD), class 1 (864 proteins) contained a strong enrichment of the term mitochondrion including all four submitochondrial compartments, whereas this term was underrepresented in classes 2–4 (Table S5A). Class 2 features a set of nuclear proteins (105), whereas a larger population of nuclear proteins (784) are part of class 3 along with proteins of various subcellular compartments including the ER, Golgi, and the plasma membrane. GO enrichment analysis for class 4 showed that purified mitochondrial fractions are mostly devoid of vacuolar and cytosolic proteins including cytosolic ribosomes (Figure 2D; see also Figures 1B and 2B).
The proteins of the mitochondrial reference set with a defined submitochondrial localization were preferentially found in class 1 and to a lesser degree in class 3 (Figure 2E). As an example, the subunits of the TIM23 complex and the presequence translocase-associated motor (PAM), the mitochondrial contact site and cristae organizing system (MICOS), and the mitochondrial ribosome featured highly consistent mean log2 ratios centered at 0.3 within class 1 (Figure 2F). The translocase of the outer membrane (TOM) equally qualified for class 1 but is slightly shifted to lower mean log2 ratios compared to TIM23-PAM, MICOS, and the mitochondrial ribosome. The ER-mitochondria encounter structure (ERMES) revealed a remarkable separation with three subunits present in class 1 and two in class 3. This dual distribution is consistent with data from previous work showing that Mdm10, Mdm34, and Gem1 are anchored in the mitochondrial outer membrane, whereas Mmm1 localizes to the ER membrane and Mdm12 is bridging Mmm1 and Mdm34/Mdm10 at the cytosolic side (Ellenrieder et al., 2016, Kornmann et al., 2009). Various proteins controlling mitochondrial fusion and fission were also present in class 1, albeit with a broader distribution (Figure 2F). Dnm1 was considerably depleted in highly purified mitochondria in agreement with its distribution between cytosol and mitochondria (Labbé et al., 2014).
For the definition of a high-confidence mitochondrial proteome, we applied stringent criteria and included the following: class 1 proteins with a sequence coverage of > 20% (SD, < 0.75; Figure S2I); experimentally validated mitochondrial proteins via import of radiolabeled precursors into mitochondria, subcellular fractionation, or fluorescence microscopy; manually curated mitochondrial proteins from single-protein studies; and proteins of dual localization, for which a presence in the mitochondrial proteome was demonstrated by experimental analysis/manual curation as outlined below. In total, this stringent mitochondrial proteome contains 901 proteins (Figure 1D; Tables S1 and S3).
Assessment of Copy Numbers of Mitochondrial Proteins
At present, there is no consistent picture about the absolute copy numbers of mitochondrial and mitochondria-associated proteins. Several studies using glucose-grown yeast cells reported different cellular copy numbers for the same proteins (Tables S1 and S2D) (Chong et al., 2015, Ghaemmaghami et al., 2003, Kulak et al., 2014). To obtain a quantitative understanding of the mitochondrial proteome and its changes under different growth conditions, we grew yeast cells on fermentable (glucose), alternative sugar (galactose), and non-fermentable (glycerol) carbon sources. For each condition, we analyzed biological triplicates using triple-SILAC labeling and high-pH reversed-phase peptide fractionation for proteome-wide quantification by MS. The calculated SILAC ratios of more than 4,000 proteins were reproducible between biological replicates and carbon sources with Pearson correlation coefficients between 0.78 and 0.9 (Figure S3A; Table S2E). We extracted MS intensities and used the proteomic ruler method (Wiśniewski et al., 2014) to estimate copy numbers. Altogether, we report 12,111 copy numbers for 4,039 yeast proteins and thus significantly expand absolute quantitative information on yeast proteins (Figure S3B; Table S2D). A comparison of our glucose-specific copy number data with reported copy numbers showed the best correlation with the MS approach of Kulak et al. (2014), whereas the correlation was lower with quantification data obtained by single-cell imaging (Chong et al., 2015) or a tagging approach (Ghaemmaghami et al., 2003). Our analysis was not biased against low abundant proteins. For both non-mitochondrial and mitochondrial proteins, we observed a large dynamic range of up to six orders of magnitude, varying from proteins with less than 10 to more than 1,000,000 copies per cell (Figure S3C). The majority of proteins (∼70%) were present in the range of 100 to 10,000 copies per cell, whereas approximately the same number of proteins exhibit copy numbers below or above the main distribution. A total of 1,576 proteins exhibited a significant change in abundance (ANOVA, p value ≥ 0.05, n = 3) in yeast grown on galactose or glycerol in relation to glucose and were grouped to 14 clusters by k-means clustering (Figure S3D; Table S2D). We revealed a significant overrepresentation of mitochondrial terms in all three GO domains for proteins in cluster C02–C06 that were considerably more abundant in galactose- and glycerol-grown yeast (Figure S3E; Tables S5B–S5D). We independently corroborated carbon source-dependent effects on the expression levels of mitochondrial proteins by direct comparison of immunoblotting data using antibodies specifically directed against mitochondrial proteins and global MS-based copy number estimation (Figure S3F; Table S2F).
Our analysis leads to a three-carbon source-specific absolute quantitative blueprint of the yeast mitochondrial proteome (Figures 3A and S3G; Tables S1 and S2D); representative examples are shown in Figure 3B. The protein import machineries of the outer membrane (TOM, SAM), inner membrane (TIM23-PAM, TIM22), and intermembrane space (mitochondrial disulfide relay system Mia40-Erv1) (Neupert and Herrmann, 2007, Wiedemann and Pfanner, 2017) constitute a rather static part of the mitochondrial proteome. The TOM complex exhibited a high abundance, highlighting its role as main protein entry gate. Mitochondrial proteins with functions in Fe/S cluster biogenesis and iron homeostasis (Lill, 2009) were mostly low abundant and constant in expression, consistent with these functions being essential and steady functions of mitochondria under all conditions (Figures 3B and S3G). The carbon source-dependent (dynamic) part of the mitochondrial proteome largely consists of the highly abundant oxidative phosphorylation protein network (respiratory complexes II–IV, ATP synthase, and proteins of the tricarboxylic acid [TCA] cycle) (Murphy et al., 2015, Ohlmeier et al., 2004, Paulo et al., 2016); the metabolite channel Por1/VDAC and its interaction partners OM14 and OM45 (Lauffer et al., 2012), which are the most abundant proteins in the outer membrane (Zahedi et al., 2006); and the highly abundant carrier proteins of the inner membrane such as the phosphate carrier Mir1 and the adenine nucleotide translocators Pet9 and Aac1 (Palmieri et al., 2006). Por2, the paralog of Por1, was found to be five orders of magnitude lower expressed than Por1, and expression of Pic2 was two orders of magnitude lower than Mir1, to which it is functionally redundant. Mitochondrial ribosomes were of medium abundance yet showed a growth condition-dependent expression as expected (Figure S3G). The MICOS complex also exhibited a carbon source-dependent expression pattern in agreement with its dynamic role in the maintenance of inner membrane architecture (van der Laan et al., 2016).
Figure 3.

Proteome-wide Absolute Quantification of Carbon Source-Dependent Protein Expression
(A) Overview of the functional classification of 661 high-confidence class 1 mitochondrial proteins according to copy numbers per cell determined for three different carbon sources. The areas of the pie charts (ii), (iii), and (iv) are directly proportional to the determined copy numbers of the mitochondrial proteins.
(B) Proteins of selected mitochondrial complexes, functions, or pathways and their carbon source-dependent copy numbers per cell. Colors indicate absolute mean copy numbers per cell and carbon source (n = 3). Asterisks, low sequence coverage or low quantification accuracy can lead to an underestimation of the copy number of components such as for Atp6 and Tim17. Fe/S, iron–sulfur cluster biosynthesis; FoF1, ATP synthase; TCA, tricarboxylic acid cycle; II/III/IV, complexes II/III/IV of the respiratory chain.
Identification of Mitochondrial Proteins and Submitochondrial Localization
Our high-confidence mitochondrial proteome includes 82 proteins that were previously not demonstrated to localize to mitochondria (Table S4A). The proteome also includes 119 proteins, for which a mitochondrial annotation was inferred from high-throughput studies without further verification (Table S4B). In addition to the presence in class 1 using stringent criteria (Figures 4A and 4B), we employed distinct experimental assays to demonstrate a mitochondrial localization for this group. A total of 58 proteins was selected for this in-depth analysis (shown in bold in Table S4). (1) Precursor proteins were synthesized in a cell-free system, labeled with [35S]methionine and imported into isolated mitochondria; proteolytic processing and/or translocation to a protease-protected location in dependence on the mitochondrial membrane potential (Δψ) were taken as specific criteria for mitochondrial import. This includes the respiratory chain interactors Rci37 and Rci50, the coenzyme Q cluster protein Coq21, and the mitochondrial glutaredoxin-like protein Mgp12 (Figure 4C). (2) Proteins were tagged with a C-terminal green fluorescent protein (GFP), and the subcellular localization was analyzed by fluorescence microscopy. The GFP signal showed a typical elongated mitochondrial network for the selected proteins (Figure 4D). For comparison, we show the localization of the recently identified mini-ER open reading frame (ORF) Meo1 (Yofe et al., 2016). (3) We used N-terminal GFP tag libraries with native promoters (Figure S4A) or with constitutive promoter (Figures S4B and S4C; Tom5 as reference) (Yofe et al., 2016) to show mitochondrial localization, including the transmembrane protein (TMEM) family members Tmh11 and Tmh18 as well as Rci37 and Rci50. (4) We individually tagged 38 proteins with C-terminal protein A or hemagglutinin tags and performed biochemical subcellular fractionation (Figures 4E, S4D, and S4E). (5) Subcellular fractionations of untagged yeast cells were subjected to MS analysis, yielding an additional dataset to corroborate the high-confidence mitochondrial proteome (Figures 4E, S5A, and S5B; Table S2A). Our analysis included the mitochondrial localization of Rso55 (related to spastic paraplegia with optic atrophy and neuropathy SPG55) (Figures 4C, S4A, and S4D), a homolog of the human gene C12orf65 that was previously linked to spastic paraplegia SPG55 and associated with mitochondrial translation defects (Perocchi et al., 2006, Shimazaki et al., 2012).
Figure 4.

Biochemical, Fluorescence Microscopy, and MS-Based Subcellular Localization Analysis of Mitochondrial Proteins
(A and B) Ratio-intensity plots, as shown in Figures 2C–2F, highlighting proteins from Tables S4A (A) and S4B (B).
(C) Protein import assays with radiolabeled mitochondrial precursor proteins. +/−Δψ, mitochondria with or without membrane potential; Prot. K, proteinase K; p, precursor; m, mature form.
(D) Images of yeast cells expressing GFP-tagged proteins analyzed by fluorescence microscopy. cER/nER, cortical/nuclear ER. Scale bar, 5 μm.
(E) Subcellular protein profiling. Yeast strains were subjected to subcellular fractionation followed by SDS-PAGE and immunoblotting (lanes 1–4) or quantitative MS analysis (n = 4) (lanes 5–8). Shown are data for selected marker proteins (upper panel) and proteins that were only listed as ORF in the SGD and named in this study (shown in red, lower panel). Y axes of bar charts, mean of normalized MS intensities; error bars indicate SEM for n ≥ 3 and the range for n = 2. PNS, post-nuclear supernatant; PMS, post-mitochondrial supernatant; pMito, gradient-purified mitochondria.
See also Figures S4 and S5, and Tables S1 and S2A.
To determine the suborganellar localization, we combined protease accessibility assays, in which gradient-purified mitochondria were differentially treated with detergents and proteases, with SILAC-MS (Figure S1C; Tables S2G and S2H). Incubation of intact mitochondria with proteinase K and trypsin (Figure 5A, sample 1 [S1]) leads to degradation of outer membrane proteins Tom22 and Tom20 (Figure 5B, lane 2), whereas proteins of the other mitochondrial subcompartments remain unaffected. Protease treatment of mitoplasts (Figure 5A, sample 2 [S2]), generated by rupturing the outer membrane with low concentrations of digitonin, leads to proteolytic degradation of intermembrane space-exposed proteins such as Pam18 and Mic12 (Figure 5B, lane 3). Matrix-exposed proteins like Mdm38 and Tim44 are only accessible to proteases upon lysis of both mitochondrial membranes by Triton X-100 (Figure 5A, sample 3 [S3]; Figure 5B, lane 4). SILAC-MS analysis of S1–S3 mixed with intact mitochondria (n = 3; Figure S6A) led to signature plots for proteins of different mitochondrial subcompartments (Figure 5B, lanes 5–7). Class 1 proteins were subjected to clustering analysis, revealing three clusters specific for outer membrane proteins, intermembrane space-exposed proteins, or matrix-exposed proteins (Figure 5C; Table S2G). Principal-component analysis of proteins in these clusters and further proteins meeting the criteria for the signature plots visualizes the data in a two-dimensional plot, showing the separation into three distinct clusters specific for individual subcompartments in good agreement with GO cellular component annotations (Figure 5D). Using the clustering analysis and signature plots, we established the submitochondrial localization of 219 proteins, for which the assignment to mitochondrial subcompartments had not been achieved previously. A total of 21 proteins were located at the outer membrane, 50 proteins were exposed to the intermembrane space, and 148 proteins were exposed to the matrix (Figure 5D, right plot; Tables S2G and S5E). To control the MS-based submitochondrial protein localization, we prepared mitochondria and mitoplasts from yeast strains expressing representative protein A-tagged class 1 proteins, treated both fractions with proteinase K, and analyzed them by immunoblotting. The biochemical data were in very good agreement with the MS data and confirmed the submitochondrial assignment of the selected proteins (Figures 5E, 5F, S6B, and S6C; Tables S2G and S6). Comparison of our suborganellar data with previous studies targeting the outer membrane and intermembrane space proteomes (Zahedi et al., 2006, Vögtle et al., 2012) showed substantial overlap with proteins of our high-confidence mitochondrial proteome (Figure S6D).
Figure 5.

Profiling of Suborganellar Localization and Membrane Topology of Mitochondrial Proteins
(A and B) Protease accessibility assay. Gradient-purified mitochondria (M, S1), mitoplasts (S2), and mitochondrial Triton X-100 (TX-100) lysates (S3) were treated with proteases (proteinase K and trypsin) as indicated. (B) Samples were analyzed by SDS-PAGE and immunoblotting using antisera against marker proteins of the mitochondrial outer membrane (OM), and intermembrane space (IMS)- and matrix-exposed proteins (lanes 1–4). For submitochondrial profiling, SILAC-labeled untreated mitochondria (M) and S1, S2, or S3 were mixed and analyzed by MS (n = 3) (lanes 5–7). Y axes, mean S/M protein ratios; error bars indicate SEM for n = 3 and the range for n = 2; dotted lines, S/M ratios of 0.25. IM, inner membrane.
(C) Top, k-means clustering performed based on S/M ratios of proteins (n = 3) with a ratio ≤1.1 that were present in class 1 in pure/crude experiments and showed decreasing ratios with increasing accessibility of the proteases (i.e., S1/M ≥ S2/M > S3/M). Bottom, selected GO terms significantly enriched in each cluster.
(D) Principal-component (PC) analysis of log2-transformed mean S/M ratios of the proteins present in the clusters depicted in (C) and further proteins meeting the criteria for signature plots. Shown are all proteins of C1–C3 (left), proteins with previous GO annotations (middle), and proteins without previous GO annotation that were assigned to mitochondrial subcompartments based on our experimental data (right). PC 1–3 account for 45.7%, 35.3%, and 19.0% of the variability in the data. Arrows point to the direction of increasing values for the different experimental conditions.
(E and F) Mitochondrial sublocalization and membrane protein topology. Intact mitochondria (lanes 1 and 2) or mitoplasts (lanes 3 and 4) were treated with proteinase K (Prot. K) where indicated. Marker proteins of the mitochondrial outer (Tom70) and inner membrane (Tim23) shown in (E) belong to the fractionation of Mco8ProtA (Figure S6). Bar charts (lanes 5–7) show the corresponding S/M protein ratios. Peptide plots illustrate the topology for Tom70, Tim23 (both summarized in adjacent cartoons), and selected proteins of the high-confidence mitochondrial proteome. Plotted are S/M ratios of tryptic peptides identified in submitochondrial profiling experiments. Transmembrane segments (TMHMM prediction or according to Alder et al. [2008] for Tim23) are indicated in blue (OM) or green (IM). Y axes of bar charts and peptide plots, mean S/M ratio; dotted lines indicate S/M ratios of 0.25; error bars indicate SEM for n = 3 and the range for n = 2.
To gain insight into the membrane topology of authentic (untagged) mitochondrial proteins, we established peptide plots based on S/M SILAC ratios of the submitochondrial profiling analysis (Figures 5E and 5F). We first demonstrated the feasibility of the approach by analyzing two proteins with known topology. Tom70 contains a single transmembrane domain (TMD) near the N terminus (amino acids [aa] 12–29) (Li and Shore, 1992). A high S1/M ratio of the peptide N-terminal to the TMD indicated a protection from degradation when intact mitochondria were treated with proteases, whereas the low S1/M ratios of peptides C-terminal to the TMD confirmed that this part was accessible to the proteases and thus facing the cytosol (Figure 5E; Table S2I). Tim23 contains four TMDs and an N-terminal hydrophilic domain in the intermembrane space (Alder et al., 2008). When mitoplasts were subjected to protease treatment (S2), the peptides N-terminal to the first TMD and the peptide stretching across the fourth TMD exhibited low S2/M ratios, confirming that N and C termini were exposed to the intermembrane space (Figure 5E; Table S2I).
We then applied this approach to proteins with unknown topology, using TMHMM predictions to reveal putative TMDs (Figures 5E and 5F; Table S2I). Our analysis indicates that the single-spanning proteins Pth2, a peptidyl-tRNA hydrolase, and Tcd2, tRNA threonylcarbamoyladenosine dehydratase, are located in the outer membrane with a short N-terminal portion residing in the intermembrane space and the major C-terminal portion exposed to the cytosol. For Scm4, an outer membrane protein with four predicted TMDs (Becker et al., 2011), peptide ratios suggest that both protein termini are located in the cytosol. The inner membrane proteins Aim11, Iai11, and Rci37, all predicted to have two TMDs, project both termini into the intermembrane space, whereas the single-spanning proteins Min8, Rci50, and Dpc7 show a matrix-intermembrane space orientation of their N and C termini, respectively. Information about S/M peptide ratios determined for all proteins with TMHMM prediction identified in this experiment are provided in Table S2I.
Mitochondrial Proteins with Dual Localization
Studies by Pines and colleagues demonstrated a dual cellular localization of several yeast mitochondrial proteins (Yogev and Pines, 2011). To identify further dual-localized mitochondrial proteins, we analyzed the high-confidence class 1 mitochondrial proteome by stringent criteria outlined in Table S7, including the presence of significant amounts of a protein in both the post-mitochondrial supernatant (PMS/total ratio > 0.5) and the mitochondrial fraction in the cellular fractionation. These criteria led to a list of 57 proteins (Table S7).
We tested the validity of our selection approach by two means. (1) We performed a literature analysis: 30 of the 57 proteins were previously assigned to mitochondria by manual annotation (single-protein analysis). A total of 15 of these proteins was shown to possess a dual/multiple cellular localization (summarized in SGD): the copper chaperone Ccs1, which is located in the mitochondrial intermembrane space, cytosol, and nucleus; the cytosolic and mitochondrial glutathione oxidoreductase Glr1, the glycerol-3-phosphate dehydrogenase Gpd2, the protease Prd1, the flavohemoglobin protein Yhb1, the kynurenine aminotransferase Bna3, and Nif3, a protein of unknown function; the nuclear and mitochondrial DNA ligase Cdc9 and the 5′-flap endonuclease Rad27; and the cytosolic/nuclear mitochondrial tRNA metabolism-related proteins Dia4, Hts1, Mod5, Pus4, Trm5, and Vas1. (ii) We selected 11 of the further proteins from Table S7 for experimental analysis (Figures 6A and 6B). Eight candidates were previously annotated to the nucleus or cytosol, but not to mitochondria, including the tRNA metabolism-related enzymes Deg1, Smm1, and Sua5; Ymr087w, now named Pdl32 for protein of dual localization of 32 kDa; the glutathione transferase Ecm4; Meu1, which catalyzes the initial step in the methionine salvage pathway; the putative nitroreductase Hbn1; and acyl-protein thioesterase 1 (Ylr118c) (Duncan and Gilman, 2002), now named Tml25 for 25-kDa acyl-protein thioesterase with multiple localizations (Figure 6C, upper panel). Two candidates were previously localized to peroxisomes by single-protein studies and linked to mitochondria only by high-throughput analysis: the peroxisomal matrix protein Pxp2 and acyl-CoA thioesterase Tes1 (Jones et al., 1999, Nötzel et al., 2016). The thiouridine modification enzyme Tum1 was linked to the cytoplasm and to mitochondria by high-throughput analysis (Figure 6C, upper panel). For an experimental analysis, we individually tagged nine candidate proteins and controlled the proper subcellular fractionation of each resulting yeast strain manually, demonstrating a dual localization for each protein, including the presence of Pdl32 and Tml25 in both cytosol and mitochondria (Figure 6A). Seven candidate proteins were efficiently synthesized and labeled with [35S]methionine in a cell-free system, and thus their Δψ-dependent import into mitochondria could be analyzed: each protein was imported in a Δψ-dependent manner, but not processed (Figure 6B). Submitochondrial analysis by swelling followed by protease treatment demonstrated that each of these hydrophilic proteins was translocated to the mitochondrial matrix (Figure 6B). Taken together, these experiments fully support a dual/multiple localization of the 11 candidate proteins (Figure 6C, lower panel).
Figure 6.

Mitochondrial Proteins with Dual Localization
(A) Yeast strains expressing proteins with a protein A tag previously reported to localize to the cytosol, nucleus, or peroxisome were subjected to subcellular fractionation as described in Figure 4E.
(B) 35S-labeled proteins were incubated with isolated wild-type mitochondria in the presence (+Δψ) or absence (−Δψ) of a membrane potential. Where indicated, mitochondria were subjected to hypoosmotic swelling and/or proteinase K (Prot. K) treatment.
(C) Schematic representation of the previously reported subcellular localization of selected proteins (upper panel). In this study, all proteins were additionally found in mitochondria by quantitative MS (Tables S4A and S4B) and single-protein analysis (lower panel). ‡/∗, subcellular localization reported from high-throughput studies/manual curation (SGD).
We conclude that literature analysis and biochemical characterization demonstrate a dual localization of 26 proteins selected from the list of 57 putatively dual-localized mitochondrial proteins (Table S7), providing strong evidence for the validity of the approach.
Interaction Networks of Identified Proteins
To obtain functional information about identified or poorly characterized mitochondrial proteins (Tables S4A and S4B), we analyzed the organization of selected proteins in protein complexes and networks (summarized in Figures 7 and S7, and Table S2J). For an overview, mitochondria containing tagged proteins or imported 35S-labeled proteins were lysed with digitonin and analyzed by blue native electrophoresis, revealing distinct high-molecular-weight complexes (Figures 7A and S7A). Δψ-dependent formation of protein complexes demonstrates that the precursor proteins were translocated into or across the inner membrane to form a complex (shown here for Dpa10, Mco14, Dpi29, and Dpi34), whereas Δψ-independent complex formation typically points to an outer membrane/intermembrane space location (Min6, in agreement with its localization to the outer membrane [Figure 5D; Table S6]).
Figure 7.

Mitochondrial Protein Interaction Networks
(A) Mitochondria isolated from indicated yeast strains were analyzed by blue native gel electrophoresis using 3%–13% (lanes 1–13) or 6%–16.5% (lanes 14–16) discontinuous polyacrylamide gels.
(B) Coq21 interaction network identified by SILAC q-AP-MS (n = 2).
(C–E) Rcf3, Rci37, and Rci50 interaction networks identified by SILAC q-AP-MS (n = 2 each). In addition, IgG chromatography eluates were analyzed by SDS-PAGE and immunoblotting using the indicated antisera. Load = 5% (C and D) or 0.4% (E); eluate = 100%. (D, lanes 5–14) 35S-labeled Yil077c was imported into mitochondria isolated from wild-type (WT) or yil077cΔ strains for the indicated periods at 25°C and subsequently treated with proteinase K. Where indicated (−Δψ), the membrane potential was dissipated prior to import reactions. Samples were solubilized with 1% digitonin and analyzed by blue native gel electrophoresis and digital autoradiography.
To define interaction partners of the selected proteins, we performed SILAC labeling of wild-type yeast and yeast strains containing protein A-tagged proteins with a tobacco etch virus (TEV) cleavage site. After lysis with digitonin, interacting proteins were identified by q-AP-MS. In addition, interaction partners were analyzed by affinity purification and immunoblotting using specific antibodies. (1) Ybr230w-a, which was imported into the mitochondrial matrix in a Δψ-dependent manner (Figure 4C), specifically co-purified the coenzyme Q biosynthesis enzymes Coq4, Coq5, Coq6, Coq9, Coq11, and Cat5/Coq7 (Figure 7B), and was thus named Coq21. (2) The mitochondrial class 1 protein of 10 kDa (Mco10) is an interaction partner of the F1Fo-ATP synthase, revealed by the specific enrichment of ten ATP synthase subunits (Figure S7B). (3) Ybl059w interacted with the altered inheritance rate of mitochondria protein Aim11, the genetic interactor of prohibitins Gep7, and the high-confidence class 1 protein Mtc3 (Figure S7C, left panel). The reverse pulldown via Aim11 similarly co-purified Ybl059w, Gep7, and Mtc3 (Figure S7C, right panel). Both pulldowns led to the co-purification of three subunits of respiratory complex IV (Cox2, Cox6, Cox20), the mitochondrial peculiar membrane protein Mpm1, and numerous subunits of the F1Fo-ATP synthase. Due to the specific interaction with Aim11, Ybl059w was named Iai11 (interactor of Aim11). The topology of Aim11 and Iai11 in the inner membrane was determined in Figure 5F. We conclude that Aim11 and Iai11 are core components of an inner membrane complex that interacts with a set of complex IV components and the F1Fo-ATP synthase. (4) Tmh11 (TMEM14 homolog of 11 kDa) contains conserved GxxxG transmembrane segment interaction motifs in two of its predicted membrane anchors (Figure S7D, alignment). 35S-labeled Tmh11 was imported into isolated mitochondria in a Δψ-dependent manner and assembled into a high-molecular-weight complex in the megadalton range (Figure S7D, left panel). Tmh11 predominantly interacted with other GxxxG motif-containing mitochondrial inner membrane proteins such as the MICOS subunit Mic10, the F1Fo-ATP synthase dimerization subunit e (Tim11), and Fmp10 (with unknown function) (Figure S7D, middle panel). The matrix-facing subunits Sdh1 and Cor1 of respiratory complexes II and III also interacted with Tmh11. Remarkably, Tmh11 only co-purified Mic10, but not other MICOS subunits (Figure S7D, right panel), suggesting that Tmh11 interacts with an inner membrane pool of Mic10 that is distinct from the MICOS complex. (5) During the preparation of this manuscript, Ybr255c-a was identified as Rcf3, a homolog of the respiratory supercomplex factor Rcf2 (Römpler et al., 2016). Our co-purification analysis agrees with the interaction of Rcf3 with Rcf1, Rcf2, and respiratory complexes III and IV (Römpler et al., 2016). In addition, we observed that Rcf3 co-purified all six subunits Mic10, Mic12, Mic19, Mic26, Mic27, and Mic60 of the MICOS complex (Figure 7C, SILAC and immunoblotting) (van der Laan et al., 2016). Rcf3 may thus function as mediator between respiratory chain III–IV supercomplexes and MICOS. (6) Yil077c assembled into large blue native gel complexes that are characteristic for III–IV supercomplexes (Figures 7A and 7D, right panel), in agreement with the co-purification of Yil077c with Rcf3 (Figure 7C) (Römpler et al., 2016). Tagged Yil077c, named Rci37 (respiratory chain interacting protein of 37 kDa), co-purified subunits of complexes III and IV and additionally the subunits Afg3 and Yta12 of the inner membrane m-AAA protease (Rugarli and Langer, 2012), followed by the prohibitin complex (Figure 7D). (7) Ykl133c co-purified subunits of respiratory complexes III and IV and was named Rci50 (respiratory chain interacting protein of 50 kDa). Rci50 shows sequence similarity to the i-AAA adaptor Mgr3 (Dunn et al., 2008). SILAC and immunoblotting analysis indeed revealed that Rci50 co-purified the i-AAA protease Yme1 (Figure 7E) (Rugarli and Langer, 2012). Thus, Rci37 and Rci50 are interaction partners of the two inner membrane AAA proteases: Rci37 of the matrix-exposed m-AAA protease and Rci50 of the intermembrane space-exposed i-AAA protease.
Discussion
We developed an extensive protein resource of mitochondria and mitochondria-associated fractions in baker’s yeast (Table S1). Approaches were established to classify > 3,300 proteins into a high-confidence mitochondrial proteome (Figure 1D; Table S3) and three distinct mitochondria-associated fractions. The resource includes the absolute copy numbers of yeast proteins under different growth conditions, substantially expanding previous studies on the absolute quantification of the mitochondrial proteome (Figures 3 and S3; Tables S1 and S2) (Chong et al., 2015, Ghaemmaghami et al., 2003, Kulak et al., 2014). We characterized the subcellular and submitochondrial localization of proteins, defined an assay for determining the membrane topology of mitochondrial proteins, and identified many mitochondrial proteins with dual subcellular location (Figures 2, 4, 5, 6, S2, and S4–S6; Tables S2, S6, and S7).
Our high-confidence mitochondrial proteome of 901 proteins includes 82 proteins that were not assigned to mitochondria previously and 119 proteins with a previous ambiguous mitochondrial localization, providing a rich source for the identification and characterization of mitochondrial functions (Figure 1E; Tables S1, S3, and S4). As proof-of-principle for the validity of the proteome and its impact for the analysis of mitochondrial functions, we performed single-protein analysis of 58 identified/ambiguous proteins and demonstrated their bona fide mitochondrial localization, including subfractionation, microscopy, and import of precursor proteins into mitochondria (Table S6). Interaction analysis of selected proteins under native conditions revealed the high potential of the proteome for the definition of interactors of mitochondrial machineries (Figures 7 and S7; Table S2J). We identified an inner membrane complex, formed by Aim11-Gep7-Iai11-Mtc3, that provides a link between the F1Fo-ATP synthase and a subset of cytochrome c oxidase (respiratory complex IV) subunits. Tmh11 interacts with oligomerization-promoting subunits of MICOS and the F1Fo-ATP synthase (Arnold et al., 1998, Barbot et al., 2015, Bohnert et al., 2015, Rabl et al., 2009), pointing to an interaction of Tmh11 with a special pool of the MICOS core component Mic10. We found that Rcf3 associates not only with respiratory chain supercomplexes (Römpler et al., 2016) but also with the entire MICOS complex, indicating a role of Rcf3 in linking respiratory chain and the cristae organizing system. Mco10 was found as partner protein of the F1Fo-ATP synthase, and Coq21 was identified as component of the coenzyme Q biosynthesis cluster. We demonstrate that Rso55, a homolog of human C12orf65 that has been linked to spastic paraplegia with optic atrophy and neuropathy SPG55 (Perocchi et al., 2006, Shimazaki et al., 2012), is a high-confidence mitochondrial protein. A further striking example of our proteome is the observation that Rci37 and Rci50 provide a connection between respiratory chain supercomplexes and the AAA proteases of the inner membrane (Rugarli and Langer, 2012, Tatsuta and Langer, 2009). Rci37 interacts with III–IV supercomplexes and the m-AAA protease, which degrades matrix-exposed proteins, whereas Rci50 provides a link between III–IV supercomplexes and the i-AAA protease, which degrades intermembrane space-exposed proteins. These findings suggest that Rci37 and Rci50 provide a system to link respiratory chain complexes to the AAA quality control system on both sides of the mitochondrial inner membrane.
Further intriguing examples of this resource are derived from the list of dual-localized proteins, including the metalloprotease Ste23, the mitochondrial aspartyl-tRNA synthetase Msd1, the cysteinyl-tRNA synthetase Ynl247w, the folic acid biosynthesis enzyme Fol1, and the dihydrofolate synthetase Fol3 (Table S7). The presence of the mitochondria-ER-cortex-anchor Mdm36 (Klecker et al., 2014, Lackner et al., 2013) among the dual-localized proteins as well as the striking distribution of ERMES components between class 1 and class 3 of the proteome (Figure 2F) reveal the potential of this resource for the characterization of proteins that are involved in the formation of contact sites between mitochondria and other cellular membranes.
Taken together, our comprehensive analysis of mitochondria and associated fractions provides a high-confidence resource for the definition of mitochondrial functions, interaction networks, and integration of mitochondria into the cellular context.
Experimental Procedures
Further details and an outline of resources used in this work can be found in Supplemental Experimental Procedures.
Yeast Strains
All yeast strains used in this study are derived from S. cerevisiae BY4741, BY4742, or YPH499. For further details, see Supplemental Experimental Procedures.
Statistical Methods
Proteins quantified in the crude/pure mitochondrial dataset were classified based on mean log2 ratios using equivalence and t tests (p value < 0.01). Error bars in figures represent means ± SEM. For further details, see Supplemental Experimental Procedures.
Author Contributions
M.M., S.B.S., P.L., S.D., U.W., P.H., R.F., and C.S. performed the experiments and analyzed the data (together with C.D.P., F.D., M.G., M.B., M.v.d.L., M.S., S.O., N.P., N.W., and B.W.). B.W., N.W., N.P., and S.O. designed and supervised the project. M.M., S.B.S., P.L., C.D.P., S.D., U.W., P.H., R.F., C.S., S.O., and N.W. prepared the figures and tables. B.W., N.W., N.P., and S.O. wrote the manuscript (together with M.M., S.B.S., C.D.P., and C.S., and input of the other authors). All authors discussed results from the experiments and commented on the manuscript.
Acknowledgments
We thank M. Opalińska, Ł. Opaliński, E. Fitzke, L. Martens, and A. Argentini for discussion and experimental advice, and B. Knapp for technical assistance in LC-MS analyses. Work included in this study has also been performed in partial fulfillment of the requirements for the doctoral theses of M.M., P.L., C.D.P., and S.D., the bachelor’s thesis of P.H., and the diploma theses of R.F. and C.S. at the University of Freiburg. This work was supported by the European Research Council (ERC) Consolidator Grants 646604 and 648235, the Excellence Initiative of the German federal and state governments (EXC 294, BIOSS; GSC-4, Spemann Graduate School), Deutsche Forschungsgemeinschaft (PF 202/8-1 and WA 1598/5-1), and Sonderforschungsbereiche 746, 1140, and 1190.
Published: June 27, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.06.014.
Contributor Information
Nils Wiedemann, Email: nils.wiedemann@biochemie.uni-freiburg.de.
Bettina Warscheid, Email: bettina.warscheid@biologie.uni-freiburg.de.
Accession Numbers
The accession numbers for the MS raw data and result files reported in this study are ProteomeXchange Consortium: PXD006128 (subcellular profiling), PXD006151 (pure/crude dataset), PXD006146 (absolute protein quantification under different growth conditions), PXD006128 (submitochondrial profiling), and PXD006147 (q-AP-MS data) (Vizcaíno et al., 2016).
Supplemental Information
(A) Related to Figures 1, 4, and S5. Result of subcellular profiling experiments. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with the identifier PXD006128. (B and C) Related to Figures 2, 4, S2, and S5. Summary (B) and MaxQuant results (C) of the SILAC-based quantitative mass spectrometry analysis of gradient-purified versus crude mitochondria. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006151. (D–F) Related to Figures 3 and S3. Summary (D) and MaxQuant results (E) of proteome-wide absolute quantification of proteins under fermentable and nonfermentable growth conditions based on the 'Proteomic Ruler' approach. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006146. Quantification of the corresponding immunoblots of Figure S3F (F). (G–I) Related to Figures 5 and S6. Summary (G) and MaxQuant results (H) of SILACMS-based submitochondrial profiling experiments as well as corresponding peptide data providing information about the topology of membrane proteins (I). Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006128. (J) Related to Figures 7 and S7. Result of SILAC-based q-AP-MS experiments of mitochondrial protein complexes. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006147.
The high confidence mitochondrial proteome includes: class 1 proteins with a sequence coverage of > 20% (SD < 0.75; Figure S2I); mitochondrial proteins experimentally validated via import of radiolabeled precursors into mitochondria, subcellular fractionation or fluorescence microscopy; manually curated mitochondrial proteins from single-protein studies; and proteins of dual localization, for which a presence in the mitochondrial proteome was demonstrated by experimental analysis/manual curation.
(A) Related to Figure 2. GO term analysis for the domain 'cellular component' (GOCC) for proteins of class 1 - class 4 defined in pure-versus-crude mitochondria experiments. (B–D) Related to Figure S3. GO term analysis for the domains 'cellular component' (GOCC; B), 'biological process' (GOBP; C) and 'molecular function' (GOMF; D) for proteins with significant changed in protein copy numbers in cells grown on different carbon sources. (E) Related to Figure 5. GO term analysis for the domain 'cellular component' (GOCC) for proteins of class 1 as determined in pure-versus-crude mitochondria experiments that had S/M ratios for all experimental conditions (i.e., S1–S3) in ≥ two replicates.
References
- Alder N.N., Jensen R.E., Johnson A.E. Fluorescence mapping of mitochondrial TIM23 complex reveals a water-facing, substrate-interacting helix surface. Cell. 2008;134:439–450. doi: 10.1016/j.cell.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Arnold I., Pfeiffer K., Neupert W., Stuart R.A., Schägger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J. 1998;17:7170–7178. doi: 10.1093/emboj/17.24.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbot M., Jans D.C., Schulz C., Denkert N., Kroppen B., Hoppert M., Jakobs S., Meinecke M. Mic10 oligomerizes to bend mitochondrial inner membranes at cristae junctions. Cell Metab. 2015;21:756–763. doi: 10.1016/j.cmet.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Becker T., Wenz L.-S., Krüger V., Lehmann W., Müller J.M., Goroncy L., Zufall N., Lithgow T., Guiard B., Chacinska A. The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J. Cell Biol. 2011;194:387–395. doi: 10.1083/jcb.201102044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert M., Zerbes R.M., Davies K.M., Mühleip A.W., Rampelt H., Horvath S.E., Boenke T., Kram A., Perschil I., Veenhuis M. Central role of Mic10 in the mitochondrial contact site and cristae organizing system. Cell Metab. 2015;21:747–755. doi: 10.1016/j.cmet.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Y.T., Koh J.L.Y., Friesen H., Duffy S.K., Cox M.J., Moses A., Moffat J., Boone C., Andrews B.J. Yeast proteome dynamics from single cell imaging and automated analysis. Cell. 2015;161:1413–1424. doi: 10.1016/j.cell.2015.04.051. [DOI] [PubMed] [Google Scholar]
- Duncan J.A., Gilman A.G. Characterization of Saccharomyces cerevisiae acyl-protein thioesterase 1, the enzyme responsible for G protein alpha subunit deacylation in vivo. J. Biol. Chem. 2002;277:31740–31752. doi: 10.1074/jbc.M202505200. [DOI] [PubMed] [Google Scholar]
- Dunn C.D., Tamura Y., Sesaki H., Jensen R.E. Mgr3p and Mgr1p are adaptors for the mitochondrial i-AAA protease complex. Mol. Biol. Cell. 2008;19:5387–5397. doi: 10.1091/mbc.E08-01-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Bord M., Shai N., Schuldiner M., Bohnert M. A tether is a tether is a tether: tethering at membrane contact sites. Dev. Cell. 2016;39:395–409. doi: 10.1016/j.devcel.2016.10.022. [DOI] [PubMed] [Google Scholar]
- Ellenrieder L., Opaliński Ł., Becker L., Krüger V., Mirus O., Straub S.P., Ebell K., Flinner N., Stiller S.B., Guiard B. Separating mitochondrial protein assembly and endoplasmic reticulum tethering by selective coupling of Mdm10. Nat. Commun. 2016;7:13021. doi: 10.1038/ncomms13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S., Huh W.-K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Huh W.-K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Hung V., Zou P., Rhee H.W., Udeshi N.D., Cracan V., Svinkina T., Carr S.A., Mootha V.K., Ting A.Y. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell. 2014;55:332–341. doi: 10.1016/j.molcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak D.N., Tyanova S., Cox J., Borner G.H. Global, quantitative and dynamic mapping of protein subcellular localization. eLife. 2016;5:e16950. doi: 10.7554/eLife.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J.M., Nau K., Geraghty M.T., Erdmann R., Gould S.J. Identification of peroxisomal acyl-CoA thioesterases in yeast and humans. J. Biol. Chem. 1999;274:9216–9223. doi: 10.1074/jbc.274.14.9216. [DOI] [PubMed] [Google Scholar]
- Klecker T., Böckler S., Westermann B. Making connections: interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol. 2014;24:537–545. doi: 10.1016/j.tcb.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Kornmann B., Currie E., Collins S.R., Schuldiner M., Nunnari J., Weissman J.S., Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Kumar A., Agarwal S., Heyman J.A., Matson S., Heidtman M., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé K., Murley A., Nunnari J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. [DOI] [PubMed] [Google Scholar]
- Lackner L.L., Ping H., Graef M., Murley A., Nunnari J. Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc. Natl. Acad. Sci. USA. 2013;110:E458–E467. doi: 10.1073/pnas.1215232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer S., Mäbert K., Czupalla C., Pursche T., Hoflack B., Rödel G., Krause-Buchholz U. Saccharomyces cerevisiae porin pore forms complexes with mitochondrial outer membrane proteins Om14p and Om45p. J. Biol. Chem. 2012;287:17447–17458. doi: 10.1074/jbc.M111.328328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.M., Shore G.C. Protein sorting between mitochondrial outer and inner membranes. Insertion of an outer membrane protein into the inner membrane. Biochim. Biophys. Acta. 1992;1106:233–241. doi: 10.1016/0005-2736(92)90001-3. [DOI] [PubMed] [Google Scholar]
- Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Bunkenborg J., Olsen J.V., Hjerrild M., Wiśniewski J.R., Stahl E., Bolouri M.S., Ray H.N., Sihag S., Kamal M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- Müller C.S., Bildl W., Haupt A., Ellenrieder L., Becker T., Hunte C., Fakler B., Schulte U. Cryo-slicing blue native-mass spectrometry (csBN-MS), a novel technology for high resolution complexome profiling. Mol. Cell. Proteomics. 2016;15:669–681. doi: 10.1074/mcp.M115.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J.P., Stepanova E., Everley R.A., Paulo J.A., Gygi S.P. Comprehensive temporal protein dynamics during the diauxic shift in Saccharomyces cerevisiae. Mol. Cell. Proteomics. 2015;14:2454–2465. doi: 10.1074/mcp.M114.045849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert W., Herrmann J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- Nötzel C., Lingner T., Klingenberg H., Thoms S. Identification of new fungal peroxisomal matrix proteins and revision of the PTS1 consensus. Traffic. 2016;17:1110–1124. doi: 10.1111/tra.12426. [DOI] [PubMed] [Google Scholar]
- Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmeier S., Kastaniotis A.J., Hiltunen J.K., Bergmann U. The yeast mitochondrial proteome, a study of fermentative and respiratory growth. J. Biol. Chem. 2004;279:3956–3979. doi: 10.1074/jbc.M310160200. [DOI] [PubMed] [Google Scholar]
- Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.-E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F., Agrimi G., Blanco E., Castegna A., Di Noia M.A., Iacobazzi V., Lasorsa F.M., Marobbio C.M.T., Palmieri L., Scarcia P. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim. Biophys. Acta. 2006;1757:1249–1262. doi: 10.1016/j.bbabio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Paulo J.A., O’Connell J.D., Everley R.A., O’Brien J., Gygi M.A., Gygi S.P. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteomics. 2016;148:85–93. doi: 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F., Jensen L.J., Gagneur J., Ahting U., von Mering C., Bork P., Prokisch H., Steinmetz L.M. Assessing systems properties of yeast mitochondria through an interaction map of the organelle. PLoS Genet. 2006;2:e170. doi: 10.1371/journal.pgen.0020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflieger D., Le Caer J.-P., Lemaire C., Bernard B.A., Dujardin G., Rossier J. Systematic identification of mitochondrial proteins by LC-MS/MS. Anal. Chem. 2002;74:2400–2406. doi: 10.1021/ac011295h. [DOI] [PubMed] [Google Scholar]
- Prokisch H., Scharfe C., Camp D.G., 2nd, Xiao W., David L., Andreoli C., Monroe M.E., Moore R.J., Gritsenko M.A., Kozany C. Integrative analysis of the mitochondrial proteome in yeast. PLoS Biol. 2004;2:e160. doi: 10.1371/journal.pbio.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl R., Soubannier V., Scholz R., Vogel F., Mendl N., Vasiljev-Neumeyer A., Körner C., Jagasia R., Keil T., Baumeister W. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J. Cell Biol. 2009;185:1047–1063. doi: 10.1083/jcb.200811099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R.S.P., Salvato F., Thal B., Eubel H., Thelen J.J., Møller I.M. The proteome of higher plant mitochondria. Mitochondrion. 2017;33:22–37. doi: 10.1016/j.mito.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Reinders J., Zahedi R.P., Pfanner N., Meisinger C., Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 2006;5:1543–1554. doi: 10.1021/pr050477f. [DOI] [PubMed] [Google Scholar]
- Renvoisé M., Bonhomme L., Davanture M., Valot B., Zivy M., Lemaire C. Quantitative variations of the mitochondrial proteome and phosphoproteome during fermentative and respiratory growth in Saccharomyces cerevisiae. J. Proteomics. 2014;106:140–150. doi: 10.1016/j.jprot.2014.04.022. [DOI] [PubMed] [Google Scholar]
- Rhee H.-W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A., Ting A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römpler K., Müller T., Juris L., Wissel M., Vukotic M., Hofmann K., Deckers M. Overlapping role of respiratory supercomplex factor Rcf2 and its N-terminal homolog Rcf3 in Saccharomyces cerevisiae. J. Biol. Chem. 2016;291:23769–23778. doi: 10.1074/jbc.M116.734665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli E.I., Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki H., Takiyama Y., Ishiura H., Sakai C., Matsushima Y., Hatakeyama H., Honda J., Sakoe K., Naoi T., Namekawa M., Japan Spastic Paraplegia Research Consortium (JASPAC) A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55) J. Med. Genet. 2012;49:777–784. doi: 10.1136/jmedgenet-2012-101212. [DOI] [PubMed] [Google Scholar]
- Sickmann A., Reinders J., Wagner Y., Joppich C., Zahedi R., Meyer H.E., Schönfisch B., Perschil I., Chacinska A., Guiard B. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA. 2003;100:13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler C., Rexhepaj E., Singan V.R., Murphy R.F., Pepperkok R., Uhlén M., Simpson J.C., Lundberg E. Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods. 2013;10:315–323. doi: 10.1038/nmeth.2377. [DOI] [PubMed] [Google Scholar]
- Stefely J.A., Kwiecien N.W., Freiberger E.C., Richards A.L., Jochem A., Rush M.J.P., Ulbrich A., Robinson K.P., Hutchins P.D., Veling M.T. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol. 2016;34:1191–1197. doi: 10.1038/nbt.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T., Langer T. AAA proteases in mitochondria: diverse functions of membrane-bound proteolytic machines. Res. Microbiol. 2009;160:711–717. doi: 10.1016/j.resmic.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Taylor S.W., Fahy E., Zhang B., Glenn G.M., Warnock D.E., Wiley S., Murphy A.N., Gaucher S.P., Capaldi R.A., Gibson B.W., Ghosh S.S. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- van der Laan M., Horvath S.E., Pfanner N. Mitochondrial contact site and cristae organizing system. Curr. Opin. Cell Biol. 2016;41:33–42. doi: 10.1016/j.ceb.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J.A., Csordas A., del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(D1):D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vögtle F.N., Burkhart J.M., Rao S., Gerbeth C., Hinrichs J., Martinou J.-C., Chacinska A., Sickmann A., Zahedi R.P., Meisinger C. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteomics. 2012;11:1840–1852. doi: 10.1074/mcp.M112.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N., Pfanner N. Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 2017 doi: 10.1146/annurev-biochem-060815-014352. Published online March 15, 2017. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J.R., Hein M.Y., Cox J., Mann M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics. 2014;13:3497–3506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yofe I., Weill U., Meurer M., Chuartzman S., Zalckvar E., Goldman O., Ben-Dor S., Schütze C., Wiedemann N., Knop M. One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nat. Methods. 2016;13:371–378. doi: 10.1038/nmeth.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev O., Pines O. Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta. 2011;1808:1012–1020. doi: 10.1016/j.bbamem.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Zahedi R.P., Sickmann A., Boehm A.M., Winkler C., Zufall N., Schönfisch B., Guiard B., Pfanner N., Meisinger C. Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol. Biol. Cell. 2006;17:1436–1450. doi: 10.1091/mbc.E05-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Related to Figures 1, 4, and S5. Result of subcellular profiling experiments. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with the identifier PXD006128. (B and C) Related to Figures 2, 4, S2, and S5. Summary (B) and MaxQuant results (C) of the SILAC-based quantitative mass spectrometry analysis of gradient-purified versus crude mitochondria. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006151. (D–F) Related to Figures 3 and S3. Summary (D) and MaxQuant results (E) of proteome-wide absolute quantification of proteins under fermentable and nonfermentable growth conditions based on the 'Proteomic Ruler' approach. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006146. Quantification of the corresponding immunoblots of Figure S3F (F). (G–I) Related to Figures 5 and S6. Summary (G) and MaxQuant results (H) of SILACMS-based submitochondrial profiling experiments as well as corresponding peptide data providing information about the topology of membrane proteins (I). Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006128. (J) Related to Figures 7 and S7. Result of SILAC-based q-AP-MS experiments of mitochondrial protein complexes. Raw MS data and complete MaxQuant results files are available via ProteomeXchange with identifier PXD006147.
The high confidence mitochondrial proteome includes: class 1 proteins with a sequence coverage of > 20% (SD < 0.75; Figure S2I); mitochondrial proteins experimentally validated via import of radiolabeled precursors into mitochondria, subcellular fractionation or fluorescence microscopy; manually curated mitochondrial proteins from single-protein studies; and proteins of dual localization, for which a presence in the mitochondrial proteome was demonstrated by experimental analysis/manual curation.
(A) Related to Figure 2. GO term analysis for the domain 'cellular component' (GOCC) for proteins of class 1 - class 4 defined in pure-versus-crude mitochondria experiments. (B–D) Related to Figure S3. GO term analysis for the domains 'cellular component' (GOCC; B), 'biological process' (GOBP; C) and 'molecular function' (GOMF; D) for proteins with significant changed in protein copy numbers in cells grown on different carbon sources. (E) Related to Figure 5. GO term analysis for the domain 'cellular component' (GOCC) for proteins of class 1 as determined in pure-versus-crude mitochondria experiments that had S/M ratios for all experimental conditions (i.e., S1–S3) in ≥ two replicates.