

Abstract
A key question regarding the signaling mechanism for G protein-coupled receptors (GPCRs) is what triggers agonism versus antagonism. Peptide analogs derived from the chemokine, complement fragment 5 anaphylatoxin (C5a), can act as agonists or antagonists to the C5a receptor, a member of the GPCR family [Gerber, B. O., Meng, E. C., Dotsch, V., Baranski, T. J. & Bourne, H. R. (2001) J. Biol. Chem. 276, 3394–4000]. Recently, we showed that two Cys residues engineered near a proposed binding site in the C5a receptor on transmembrane helices III and VI can selectively and reversibly trap short Cys-containing 3-mer peptides derived from C5a by disulfide bond formation [Buck, E. A., Bourne, H. & Wells, J. A. (November 18, 2004) J. Biol. Chem., 10.1074/jbc.C400500200]. Here, a library of 10,000 compounds, each containing an exchangeable thiol, was screened to identify specific small-molecule mimics that block binding of C5a. Some of the selected compounds acted as agonists and were as potent as the natural C5a ligand, and some acted as antagonists. A residue near these compounds, Ile-116 in helix III, functions as a “gatekeeper” to modulate these effects. A small substitution, Ile-116-Ala, enhanced affinity for some compounds and allowed antagonists to function as agonists; a larger substitution, Ile-116-Trp, decreased affinity and agonism. Thus, subtle changes in either the structure of the ligand or the receptor at the site between helix III, VI, and VII can switch the receptor on or off. This ligand binding and activation site may be similarly positioned in other members of the chemokine receptor family. Selective ligand trapping by reversible disulfide formation may serve to nucleate the development of small-molecule mimics.
Keywords: fragment-based discovery, ligand binding, C5a, chemokine, Tethering
G protein-coupled receptors (GPCRs) are primary responders to a diverse collection of extracellular stimuli (1–3). The receptors relay information to intracellular signal transduction networks that evoke rapid changes in cell metabolism. GPCRs regulate a wide range of physiology, including visual signal transduction, smell, learning and memory, and inflammation among others. This receptor family is one of the top targets for drug discovery, and about half of the top-marketed drugs are thought to act at GPCRs (4). Given the importance of these receptors in biology and medicine, the mechanism by which they are activated by their ligands has remained of intense interest.
One of the most striking features of the GPCRs is how changes in ligand structure can turn it on or off (1). For example, 6-mer peptides derived from analoging the C terminus of complement fragment 5 anaphylatoxin (C5a) can bind and activate the C5a receptor (C5aR), whereas subtle changes at the penultimate residue can inhibit C5aR activity (5–7). Recent studies showed that 3-mer peptides derived from the 6-mer peptides but containing a Cys residue on their N termini can be reversibly trapped by disulfide bond formation to specific Cys residues engineered into the C5aR (8). These trapped peptides retained their respective properties as agonist or antagonist. This work further localized the binding and activation site to a specific transmembrane region that was previously proposed from mutational data and modeling hypotheses (Fig. 1A) (9).
Fig. 1.

Model and functional effects for disulfide trapping of small peptides. (A) Model based on coordinates provided by H. Bourne of the peptide (Phe-Lys-Pro-dCha-Trp-dArg) (sticks) bound to C5aR (ribbons). The Gly-262-Cys anchor site (blue) and residue Ile-116 (cyan) are highlighted. (B) Effects of various concentrations of CysCha peptide (Cys-dCha-Cha-dArg) on the receptor activation of Gly-262-Cys (○) or Ile-116-Ala/Gly-262-Cys (•). (C) Effects of various concentrations of CysTrp peptide (Cys-dCha-Trp-dArg) on the receptor activation of Gly-262-Cys (○) or Ile-116-Ala/Gly-262-Cys (•). (D) Effects of various concentrations of the CysTrp peptide on C5a ligand binding (0.1 nM) for Gly-262-Cys (○) or Ile-116-Ala/Gly-262-Cys (•). Results shown are a representative of three or more independent experiments.
Here, specific small-molecule functional agonists and antagonists were identified at these same receptor Cys sites by screening a library of 10,000 small molecules, each of which contains an exchangeable thiol. Mutation of a receptor amino acid, Ile-116-Ala on transmembrane helix III, converts the Cys-trapped antagonist peptide to an agonist, as was seen for binding of the larger peptide from which it was derived (5). The Ile-116-Ala mutant can also switch these small molecules from acting as antagonists to agonists. Thus, subtle structural changes in these selectively trapped small molecules or the receptor can switch the complex on or off. These studies serve to further define the location and sensitivity of ligand binding and activation site in the C5aR by using an approach that may be useful to other GPCRs. The specific trapping of small organic compounds may also suggest the possibilities for small-molecule drug discovery.
Experimental Procedures
Materials. Human recombinant C5a was from Sigma. 125I-labeled C5a and [3H]myo-inositol were from Perkin–Elmer. All peptides were from the Tufts University Core Facility.
Transfection of COS-7 Cells and Preparation of Membranes. COS-7 cells were transfected with a plasmid carrying the cDNA for C5aR by using the FuGENE 6 transfection reagent (Roche). COS-7 cells were cultured to 80% confluence before transfection and harvested 2 days [for inositol(1,4,5)trisphosphate (IP3) accumulation] or 3 days (for preparation of membranes) after transfection. For binding studies, membranes were prepared as described in ref. 8.
Measurement of 125I-C5a Binding and IP3 Accumulation. Binding studies were conducted on membranes derived from COS-7 cells that were transfected with the C5aR. The procedure for measuring binding has been described in ref. 8. For measurement of IP3 accumulation, COS-7 cells are cultured and cotransfected with Gα16 and C5aR in 24-well culture dishes. Twenty-four hours after transfection, cell monolayers were washed one time with PBS, and growth media was exchanged with inositol-free DMEM containing 10% dialyzed FBS and 100 μM [3H]myoinositol. Cells were assayed for IP3 accumulation 24 h after labeling as described in ref. 8.
Small-Molecule Library Screening. Solutions containing a 50 μM concentration of each thiol-containing compound were pooled in groups of 10 and incubated with membranes derived from COS-7 cells transfected with C5aR in binding buffer containing 10 mM 2-mercaptoethanol (2-ME) for 2 h. 125I-C5a at a final concentration of 0.1 nM was added and incubated for an additional 2 h before filtration as described (8). Positive pools were identified as those that inhibited C5a ligand binding by at least 15% to membranes containing the Cys receptor mutants (Phe-93-Cys, Pro-113-Cys, Gly-262-Cys, or Leu-117-Cys) but not to membranes containing the wild-type receptor. For each positive pool, discrete compounds were then tested individually to deconvolute the specific compound responsible for inhibition of ligand binding. The identity of all inhibitory compounds was verified by mass spectrometry and structures shown in Fig. 6, which is published as supporting information on the PNAS web site.
Results
Functional Consequences for Trapped Agonist and Antagonist Peptides. Ile-116 in the C5aR has been implicated from Ala-scanning mutational studies to be part the signaling mechanism (5, 9). In particular, the Ile-116-Ala mutation converts a 6-mer antagonist peptide [Phe-Lys-Pro-cyclohexylalanine (Cha)-Trp-d-enantiomer of Arg (dArg)] to an agonist (5). Ile-116 is positioned between transmembrane helices III and VII, in the vicinity of trapped Cys-containing, 3-mer peptides (Fig. 1 A) (8). We tested the effect of the Ile-116-Ala mutation on the binding and functional effects of the trapped Cys-containing, 3-mer peptides. This mutation lowered the EC50 for activation of Ile-116-Ala/Gly-262-Cys by >2-fold relative to Gly-262-Cys and increased the maximal stimulation by ≈2-fold for the trapping of the agonist peptide [Cys-d-enantiomer of Cha (dCha)-Cha-dArg] (Fig. 1B). Moreover, the double mutant was fully activated by the antagonist peptide (Cys-dCha-Trp-dArg) (Fig. 1C), and it improved the ability to trap this peptide by ≈8-fold (Fig. 1D). The double mutant does not affect binding or activation by the C5a ligand (data not shown), indicating that these effects are induced by capturing the peptides. The fact that similar functional effects were seen for the disulfide-trapped 3-mers or the larger free-standing 6-mer peptides indicates that disulfide bonding does not distort the functional consequences for the trapped peptides but only compensates for the reduced noncovalent binding energy of the 3-mers relative to the 6-mers.
Use of Tethering to Trap Smaller-Molecular Mimics. It was of interest to see whether these same Cys anchor sites could be used to trap low-molecular-weight compounds and whether they could modulate receptor activity. This approach would facilitate a more thorough exploration of the structural requirements for agonism and antagonism. A 10,000-member library of thiol-containing compounds (10) was screened for disulfide trapping at the Cys sites in the C5aR that were previously tested with the Cys peptides (8) (Fig. 2A). The compounds from this library have been used extensively for Tethering small organic molecules to nucleate the ligand discovery process at natural and engineered residues in proteins (10–12). Each of the compounds was reacted with the four single-Cys mutants (Phe-93-Cys, Leu-117-Cys, Phe-113-Cys, Gly-262-Cys) in the C5aR near the proposed peptide binding site (Fig. 2A). Each of the Cys mutant receptors had expression levels, C5a-binding affinities, and signaling efficacies that were similar to the wild-type receptor, suggesting that the Cys residues by themselves had little or no effect on the folding and function of the receptor (8). The disulfide-containing compounds in pools of 10 (50 μM each) were incubated with cell membranes containing the variant C5aR under highly reducing conditions (10 mM 2-ME/500 μM total disulfide-containing compound). The initial set of compounds were selected for their ability to inhibit C5a binding by >15%, as described in Materials and Methods.
Fig. 2.

Model and functional effects for disulfide trapping of small molecules. (A) Ribbon diagram of a model of the C5aR. Sites that served as Cys anchors are indicated. (B) Representative example of one thiol-containing compound identified in the screen for Pro-113-Cys. The effect of various concentrations of 2-ME (β-ME) on the inhibition of C5a ligand binding by a 50 μM concentration of compound (cmpd). (C) Effects of a representative panel of compounds at 150 μM identified in screening Gly-262-Cys on C5aR basal activity. Also indicated is the maximal stimulation by the C5a ligand and cystamine. (D) Effect of various compounds 10, 11, and 12 (at 150 μM) on the dose–response for C5a-stimulated receptor activity. Structures of compounds are shown in Fig. 6.
Disulfide trapping was performed in highly reducing conditions to promote thiol–disulfide exchange and thus ensure that the selection of active compounds was driven by their intrinsic binding properties and not thiol reactivity. The noncovalent binding affinities of the selected compounds identified from Tethering are typically in the 0.1–5 mM range (11, 12). Thus, these compounds, like the 3-mer peptides used in the previous study (8), would not be readily identified were it not for the additional free energy of forming a reversible disulfide bond in the thiol–disulfide exchange buffer. A compound was classified as a specific “hit” if it satisfied three criteria: (i) it was shown to be selective for the Cys mutant forms of the receptor, (ii) it had no effect on the wild-type version of the receptor, and (iii) its effect could be reversed by a reducing agent.
After screening ≈10,000 compounds at each of the four Cys sites, we recovered 65, 36, and 24 hit compounds that were specific for the Pro-113-Cys, Gly-262-Cys and Lys-117-Cys mutants, respectively (Table 1). None were found from Phe-93-Cys mutant. Examples of the screening results are shown in Fig. 2B for two such hit compounds bound to the Pro-113-Cys mutant. The best compounds at a concentration of 50 μM inhibited binding of C5a to the receptor ranging from 50% to 80%. The inhibition was fully reversed by 2-ME in a dose-dependent manner. The fact that neither compound inhibited binding of C5a to the wild-type receptor nor to the other Cys mutants (data not shown) suggests that they exert their effects by binding to a specific site.
Table 1. Screening of thiol-containing small molecules by disulfide trapping onto wild type or various Cys mutants in the C5aR.
| Mutants | Compounds, n | Binding inhibitors, n | Agonists, n | Antagonists, n |
|---|---|---|---|---|
| Wild type | 10,143 | 0 | N/A | N/A |
| Phe-93-Cys | 10,143 | 0 | N/A | N/A |
| Lys-117-Cys | 10,023 | 24 | ND | ND |
| Pro-113-Cys | 10,118 | 65 | 10 | 13 |
| Gly-262-Cys | 10,023 | 36 | 11 | 9 |
See Experimental Procedures for further details. ND, not determined; N/A, not applicable.
Approximately 90% of the hits were specific to one of the Cys mutants and only ≈10% showed cross-reactivity to two of the Cys mutants. Such cross-reactivity has been seen for other systems studied by Tethering (12). This result likely reflects the flexibility of the compound to access the same binding site from two neighboring thiols as has been directly observed by crystallographic analysis in several other proteins (12).
The two most hit-rich sites, Pro-113-Cys and Gly-262-Cys, were the same sites that could capture the Cys-containing 3-mer peptides (8). The most potent small molecules that bound by means of Pro-113-Cys or Gly-262-Cys were functionally analyzed to determine whether they acted as agonists by stimulating IP3 accumulation or as antagonists by blocking C5a-stimulated IP3 accumulation in transformed COS-7 cells. The compounds split into roughly even groups of agonists or antagonists (Table 1). Some of the agonist compounds were capable of activating the receptor almost as well as the natural C5a ligand (Fig. 2C). Other compounds had no effect on receptor basal activity but could inhibit activation by the C5a ligand to various extents (Fig. 2D). Thus, depending on their structure, the trapped small molecules could act as agonists or antagonists like the peptides.
The structure activity relationships were analyzed for the most potent agonist, compound 6, a bi-aryl pyrolidine-containing compound (Fig. 3A). When trapped onto the Gly-262-Cys mutant, this compound stimulated receptor activity to >80% the extent achieved by the noncovalently bound C5a or the 6-mer peptide (Cha-Cha) (Fig. 3B). Compound 6 contains a single chiral center, and its enantiomer (compound 14) required 10-fold less 2-ME to reverse its inhibition (Fig. 3C) and was significantly weaker in activating the receptor (Fig. 3D). Some compounds with similar apparent binding affinities, as indicated by their susceptibility to reduction by 2-ME, had different effects on receptor activation. For example, compounds 6 and 15 had identical 2-ME50 values of 1.3 mM (Fig. 3E), but compound 6 behaved as a full agonist and compound 15 behaved as a receptor antagonist (Fig. 3F). These compounds share a common d-Pro amino acid scaffold but contain different substituents, a 2-phenethyl phenyl in the case of the agonist compound 6 and a 3-cyclohexyl propyl in the case of the antagonist compound 15 (Fig. 3A).
Fig. 3.

Structures of select compounds and their effect on the function of the C5aR. (A) Structures of two compounds, 6 and 15, identified in screening Gly-262-Cys. (B) Effects of various concentrations of the Cha-Cha peptide (Phe-Lys-Pro-dCha-Cha-dArg) and the compound (cmpd) 6 on C5aR basal activity. (C) Effect of various concentrations of 2-ME on inhibition of C5a ligand binding by 50 μM of compound 6 or its enantiomer, compound 14. (D) Effect of a 150 μM concentration of compounds 6 and 14 on C5aR basal activity. (E) Effect of various concentrations of 2-ME on the inhibition of C5a ligand binding by a 50 μM concentration of compounds 6 and 15. (F) Effect of compound 15 on C5aR basal activity and C5a-stimulated receptor activity. Results are representative of three or more experiments.
A series of selected compounds containing various substituents extending from the common d-Pro amino acid scaffold of compounds 6 and 15 was evaluated for effects on ligand binding and receptor activation (Fig. 4A). More than half of the compounds that exhibited agonism contained the 2-phenethyl phenyl fragment. All compounds in this series at a concentration of 50 μM could inhibit C5a binding by at least 30%. However, none was as effective in activating the receptor as compound 6 with the 2-phenethyl phenyl group, and the presence of at least two aromatic rings in this substituent fragment was required for at least some activity (Fig. 4B). Collectively, this data indicates that the 2-phenethyl phenyl fragment engages in interactions with the receptor that trigger the receptor activation switch.
Fig. 4.

Structure–activity relationship for various selected compounds on the function of the C5aR. (A) Structures of compounds containing varying R substituent groups extending from the common d-Pro amino acid scaffold. The percentage of inhibition of C5a binding to Gly-262-Cys by a 50 μM concentration of compound is indicated. (B) Effect of a 150 μM concentration of compounds (cmpd) on C5aR basal activity. Results shown are representative of three or more independent experiments.
In the receptor model, Ile-116 is located ≈10 Å from the Gly-262-Cys anchor site (Fig. 5A) and is close enough to make van der Waals contact with the selected small molecules. To test whether the Ile-116-Ala mutation might switch the behavior of the antagonist small-molecule compounds, we analyzed the compound activities on Ile-116-Ala/Gly-262-Cys relative to the Gly-262-Cys. Even though the Ile-116-Ala mutation had no effect on the maximal extent of stimulation by the C5a ligand or the compound 6 agonist, it reversed the activity of many antagonist compounds (Fig. 5B). In particular, the activity of compound 24 on Ile-116-Ala/Gly-262-Cys was comparable with that found for compound 6 and the natural C5a ligand on Gly-262-Cys or the double mutant. This gain in efficacy was accompanied by a >80-fold gain in apparent affinity as measured by a compound titration and inhibition of C5a binding (Table 2). A rough correlation was observed between the gain of agonism and the fold increase in apparent affinity for other compounds (Table 2). A similar trend was observed for the compound 6 agonist, where a 3- to 4-fold increase in apparent affinity was accompanied by an increase in efficacy from 82% to >100% of that observed for the natural C5a ligand (Table 2).
Fig. 5.

Effect of the Ile-116-Ala mutant on activities of various compounds. (A) Ribbon diagram of the C5aR showing the transmembrane III–VI–VII helical triad. The distance between positions 262 and 116 is indicated. (B) Effect of a 150 μM concentration of compound on Gly-262-Cys and Ile-116-Ala/Gly-262-Cys basal activity. Structures of compounds are shown in Fig. 6 and partially listed in Table 2.
Table 2. Effect of compound structure on the ratio of IC50 and maximal stimulation of C5a Gly-262-Cys with or without the Ile-11 G-Ala mutation.
| Compound | IC50(Gly-262-Cys)/IC50(Ile-116-Ala-Gly-262-Cys)* | Maximal stimulation, % | |
|---|---|---|---|
| 24 | 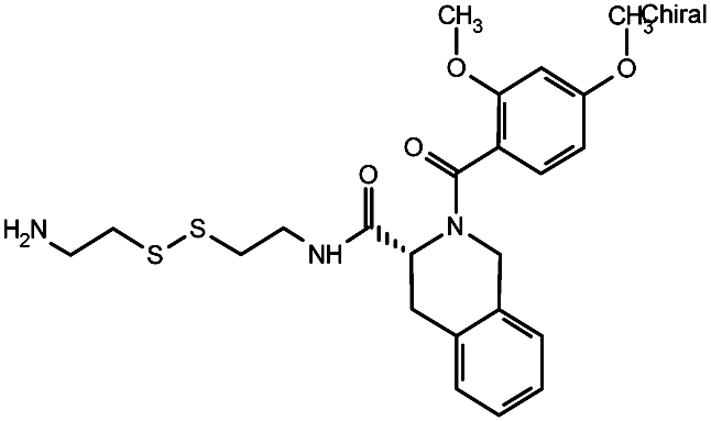 |
82 | 75 |
| 13 |  |
6 | 35 |
| 12 |  |
1.6 | 22 |
| 23 |  |
2.4 | 25 |
| 6 | 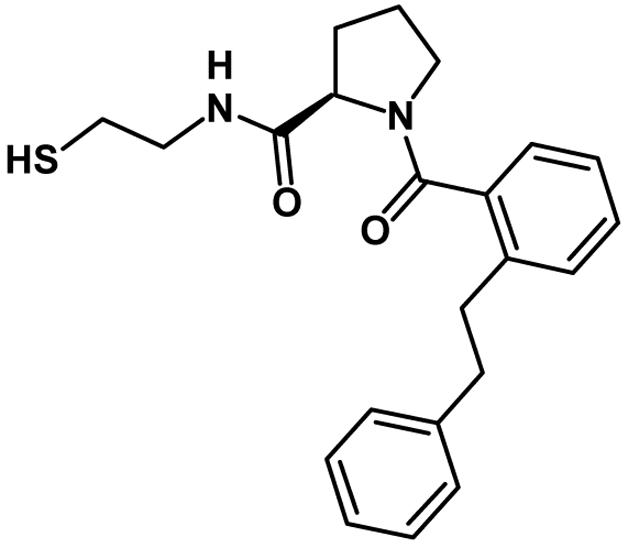 |
3.2 | 107 |
Ratio of IC50(Gly-262-Cys) to IC50(Ile-116-Ala-Gly-262-Cys).
Because the Ile-116-Ala mutation rendered various ligands with tighter receptor binding and gains in agonism, we wondered what effect increasing the size of the side chain at this position might have on the ability of the receptor to respond to agonist and antagonist ligands. Mutation of Ile-116 to Phe had little effect on binding affinity or signaling by the C5a ligand (Table 3). However, the Ile-116-Phe quenched the gain in agonism for the antagonist compounds seen in the Ile-116-Ala mutant. For example, the efficacy of agonist compound 6 was reduced by ≈40% for the Ile-116-Phe double mutant (Table 3). The results were more striking for the larger Ile-116-Trp/Gly-262-Cys double mutant. Although the Ile-116-Trp substitution has little effect on C5a ligand binding affinity, this mutation causes >50% reduction in the ability to signal (Table 3). The Ile-116-Trp mutation results in a 10-fold decrease in the apparent affinity for compound 6 and can no longer can be fully activated (Table 3). Therefore, receptor position 116 is critical in regulating how the receptor will respond to various ligands.
Table 3. Effect of side chain size at position 116 in the Gly-262-Cys C5aR upon binding and signaling by C5a or disulfide-trapped compound 6.
| C5a
|
Compound 6
|
||||
|---|---|---|---|---|---|
| Amino acid C5aR-116 | Van der Waals volume, Å3 | Binding IC50, nM | Activation, % | Binding IC50, μM | Activation, % |
| Ala | 67 | 0.15 | 100 | 1.4 | 107 |
| Ile (wild type) | 124 | 0.17 | 100 | 4.4 | 90 |
| Phe | 135 | 0.27 | 90 | 5.1 | 58 |
| Trp | 163 | 0.30 | 42 | 44.5 | 29 |
Discussion
How ligands bind and activate GPCRs is poorly understood at the molecular level. In the absence of high-resolution structural data, ligand trapping combined with mutational studies can begin to localize binding sites and facilitate determining the functional requirements for binding and activation. The paradigm for this approach was provided by retinal binding to rhodopsin, in which the cofactor trapped by Schiff's base formation to the protein localizes the molecular switch to a specific region of the GPCR. Retinal analogs have been very useful for understanding how small changes in structure can affect the signaling process (13). Schwartz and coworkers (14, 15) expanded this notion to engineered metal chelation traps. These researchers introduced two His residues between helices III and VII to trap copper or zinc metal chelate complexes in the β2 adrenergic receptor. The ligands were found to activate the receptor and provided an approach to identify site-directed ligands. The Cys traps described here provide additional utility in that single Cys mutations suffice, and the number of compounds in the thiol-containing libraries are larger, allowing greater diversity to be screened.
Trapping ligands by reversible disulfide formation is the basis for Tethering, a fragment-based drug discovery tool in which small molecules containing thiols are allowed to undergo thiol– disulfide exchange with natural or engineered thiols on the protein (10–12). Such fragments have been advanced by medicinal chemistry to noncovalent inhibitors. Structural studies with multiple targets have shown these molecules to bind to the target in the same manner as the original disulfide trapped analogs (11, 12). Tethering has been applied to proteins to probe molecular determinants in known binding sites, and binding is detected directly by mass spectrometry of the purified protein (12). Here, the Tethering approach was used to confirm a proposed binding site, and binding was detected by functional analysis of the protein, because direct mass spectrometry was hindered by the low expression levels and the membrane nature of the receptor.
The data strongly support that the functional effects observed are caused by the binding of specific functional sites of interest and not by artifacts. Disulfide trapping occurred for truncated Cys-containing peptides derived from larger analogs known to bind noncovalently and activate the receptor (8). These peptides were trapped onto specific Cys residues and not the wild-type protein, and their functional effects paralleled the noncovalent parents from which they were derived. Similarly, the trapping data for the small molecules strongly support that they exert their functional effects by specific binding interactions and not by artifacts. The hit rate from the 10,000-compound library ranged from 0% to 0.6% depending on the Cys mutant, and there was little cross-reactivity among the hits at specific Cys residues on the C5aR. In fact, the two most hit-rich Cys residues were the same ones that trapped the Cys-containing peptides. Among these hits, there were agonists and antagonists, as seen for the two peptides. One of the trapped small molecules was nearly as potent an agonist as C5a, and, to our knowledge, this is the first report of a small-molecule full-agonist for the C5aR. The functional effects showed sharp structure-activity relationships for the small molecules. Small changes, such as switching a stereocenter or subtle changes in the structure of the trapped compound, could either greatly reduce binding or switch a compound from being an agonist to an antagonist. Specifically, we identified a phenethyl phenyl moity that was important for receptor agonism. None of the antagonist compounds contained this functional group, and changes made to this structural fragment decreased the ability of the agonists to trigger receptor activation. All these effects were reversible by reduction or elimination of the disulfide bond, suggesting the functional effects were site-specifically generated from the engineered Cys residue in the C5aR.
The data presented here and in ref. 8 strongly support that a functionally critical binding site exists between transmembrane helices III, VI, and VII, about three helical rungs down into the receptor from the extracellular side. Not only is the structure activity relationships for the ligand sharp, but the same is true for the receptor. The size of the side-chain at position 116 plays a crucial role in governing whether a ligand behaves as an agonist or antagonist. The van der Waals volume of the amino acid side chain at the 116 position is roughly related to the affinity of ligands for this transmembrane site, (Table 3). Small side-chains, such as Ala, permit more promiscuous signaling, because it was observed that more ligands could bind more tightly and activate the receptor. As the side-chain size increased from Ile to Phe to Trp, there was a systematic reduction in the degree of stimulation by agonists and in the number of trapped ligands that could serve as agonists. Indeed, for the Trp substitution, even C5a lost its ability to signal. Thus, Ile-116, which sits three rungs down from helix III, somehow serves as a gate-keeper in modulating the ease with which the receptor can be activated.
It is possible that this binding site is similarly disposed on other chemokine receptors and perhaps other GPCRs. In fact, the retinal binding site is located in a similar position in rhodopsin. The counterion for the Schiff base of rhodopsin is located one helical turn below the 116 position of C5aR. Rhodopsin is activated when light triggers the isomerization of 11-cis retinal, breaking this transmembrane constraint (1, 16, 17). Therefore, a gatekeeper on transmembrane helix III may be a general mode by which agonism or antagonism is evoked by various ligands. Our studies demonstrate how structural changes that modulate agonism or antagonism can be made at the level of either the ligand or the receptor. This understanding will likely be of significance for the rational design of agonists and antagonists for specific GPCRs. Given that many GPCRs can be expressed in a functionally active form, it is possible that they could be similarly studied with disulfide-trapped ligands. Moreover, such small molecules could be useful for nucleating the drug discovery process to find small-molecule mimics either as agonists or antagonists.
Supplementary Material
Acknowledgments
We thank Dr. Henry Bourne (University of California, San Francisco) for providing the clone of the C5aR and the coordinates for the receptor model and for critically reading the manuscript and our colleagues at Sunesis Pharmaceuticals for generous support and encouragement.
Abbreviations: C5a, complement fragment 5 anaphylatoxin; C5aR, C5a receptor; Cha, cyclohexylalanine; dCha, the d-enantiomer of Cha; dArg, the d-enantiomer of Arg; GPCR, G protein-coupled receptor; 2-ME, 2-mercaptoethanol; IP3, inositol(1,4,5)trisphosphate.
References
- 1.Gether, U. (2000) Endocr. Rev. 21, 90–113. [DOI] [PubMed] [Google Scholar]
- 2.Gether, U. & Kobilka, B. K. (1998) J. Biol. Chem. 273, 17979–17982. [DOI] [PubMed] [Google Scholar]
- 3.Bockaert, J. & Pin, J. P. (1999) EMBO J. 18, 1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurrath, M. (2001) Curr. Med. Chem. 8, 1605–1648. [DOI] [PubMed] [Google Scholar]
- 5.Gerber, B. O., Meng, E. C., Dotsch, V., Baranski, T. J. & Bourne, H. R. (2001) J. Biol. Chem. 276, 3394–3400. [DOI] [PubMed] [Google Scholar]
- 6.DeMartino, J. A., Van Riper, G., Siciliano, S. J., Molineaux, C. J., Konteatis, Z. D., Rosen, H. & Springer, M. S. (1994) J. Biol. Chem. 269, 14446–14450. [PubMed] [Google Scholar]
- 7.Finch, A. M., Wong, A. K., Paczkowski, N. J., Wadi, S. K., Craik, D. J., Fairlie, D. P. & Taylor, S. M. (1999) J. Med. Chem. 42, 1965–1974. [DOI] [PubMed] [Google Scholar]
- 8.Buck, E. A., Bourne, H. & Wells, J. A. (November 18, 2004) J. Biol. Chem., 10.1074/jbc.C400500200. [DOI]
- 9.Baranski, T. J., Herzmark, P., Lichtarge, O., Gerber, B. O., Trueheart, J., Meng, E. C., Iiri, T., Sheikh, S. P. & Bourne, H. R. (1999) J. Biol. Chem. 274, 15757–15765. [DOI] [PubMed] [Google Scholar]
- 10.Erlanson, D. A., Braisted, A. C., Raphael, D. R., Randal, M., Stroud, R. M., Gordon, E. M. & Wells, J. A. (2000) Proc. Natl. Acad. Sci. USA 97, 9367–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkin, M. R. & Wells, J. A. (2004) Nat. Rev. Drug Discovery 3, 301–317. [DOI] [PubMed] [Google Scholar]
- 12.Erlanson, D. A., Wells, J. A. & Braisted, A. C. (2004) Annu. Rev. Biophys. Biomol. Struct. 33, 199–223. [DOI] [PubMed] [Google Scholar]
- 13.Govardhan, C. P. & Oprian, D. D. (1994) J. Biol. Chem. 269, 6524–6527. [PubMed] [Google Scholar]
- 14.Elling, C. E., Thirstrup, K., Holst, B. & Schwartz, T. W. (1999) Proc. Natl. Acad. Sci. USA 96, 12322–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holst, B., Elling, C. E. & Schwartz, T. W. (2000) Mol. Pharmacol. 58, 263–270. [DOI] [PubMed] [Google Scholar]
- 16.Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada, T., Stenkamp, R. E., et al. (2000) Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- 17.Cohen, G. B., Oprian, D. D. & Robinson, P. R. (1992) Biochemistry 31, 12592–12601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.