

Abstract
To determine whether Förster resonance energy transfer (FRET) measurements can provide quantitative distance information in single-molecule fluorescence experiments on polypeptides, we measured FRET efficiency distributions for donor and acceptor dyes attached to the ends of freely diffusing polyproline molecules of various lengths. The observed mean FRET efficiencies agree with those determined from ensemble lifetime measurements but differ considerably from the values expected from Förster theory, with polyproline treated as a rigid rod. At donor–acceptor distances much less than the Förster radius R0, the observed efficiencies are lower than predicted, whereas at distances comparable to and greater than R0, they are much higher. Two possible contributions to the former are incomplete orientational averaging during the donor lifetime and, because of the large size of the dyes, breakdown of the point-dipole approximation assumed in Förster theory. End-to-end distance distributions and correlation times obtained from Langevin molecular dynamics simulations suggest that the differences for the longer polyproline peptides can be explained by chain bending, which considerably shortens the donor–acceptor distances.
Keywords: Förster resonance energy transfer, molecular dynamics, polypeptide, FRET
Almost 40 years ago, Förster resonance energy transfer (FRET) was introduced in classic experiments by Stryer and Haugland (1) as a “spectroscopic ruler” to measure distances in macromolecules. Since then it has been used to address a wide range of biological questions (2–5). More recently, renewed interest has come from the realization that FRET can be used for obtaining distance information in experiments on single biomolecules (6, 7), with a considerable body of work on proteins and polypeptides (8–24). However, it is well known from ensemble experiments that determination of distances from FRET can be complicated by dynamical effects as well as photophysical, photochemical, and instrumental factors (25). Are there additional complications in single-molecule experiments on polypeptides and proteins? To investigate this question, we studied FRET between dyes attached to the N and C termini of polyproline of various lengths. Polyproline, assumed to be a rigid rod, was used as a spacer by Stryer and Haugland to show that the rate of FRET depends on the inverse sixth power of the donor–acceptor distance, as predicted by Förster theory (26).
FRET of individual dye-labeled polyproline molecules freely diffusing in solution was investigated by using a confocal fluorescence microscope setup (13). If a molecule diffuses into the volume illuminated by the focused laser beam, the donor dye is excited. Depending on the distance to the acceptor, a certain rate of energy transfer results, which determines the FRET efficiency, calculated from the fraction of photons emitted by the acceptor. To test the accuracy of the single-molecule results, we also determined FRET efficiencies from ensemble measurements of donor lifetimes in the presence and absence of acceptor by using time-correlated single-photon counting.
Because the FRET efficiencies for the longer peptides were found to be considerably higher than those expected for polyproline treated as a rigid rod, we carried out Langevin molecular dynamics simulations of polyproline of varying lengths. The calculations show that the longer peptides are quite flexible, with end-to-end distance distributions and correlation times that can account for the observed FRET efficiencies.
Materials and Methods
Peptide Preparation. Polyproline peptides of defined length, containing 6, 9, 11, 12, 13, 15, 20, 23, 27, 33 and 40 Pro residues, respectively, were synthesized by using standard FastMoc chemistry with a model 433A peptide synthesizer (Applied Biosystems) on f luorenylmethoxycarbonyl-Cys(Xan)-Wang resin (Peptides International). We included an amino-terminal glycine and a carboxyl-terminal cysteine residue, which react through their amino and sulfhydryl groups, respectively, with succinimide esters and maleimide derivatives of the dyes. After cleavage, the raw material was purified by reversed-phase HPLC on a Vydac (Columbia, MD) C4 column (214TP1022) at a flow rate of 5 ml/min over 45 min by using a linear gradient from 0.1% trifluoroacetic acid in 10% acetonitrile/90% water to 0.1% trifluoroacetic acid in 90% acetonitrile/10% water. Fractions containing the pure peptide as confirmed by electrospray ionization mass spectroscopy were lyophilized, dissolved in buffer, and labeled with Alexa Fluor 488 maleimide (27) at 4°C for 12 h under the conditions recommended by Molecular Probes (Eugene, OR). Singly labeled peptide was purified on a Superdex peptide HR 10/30 size-exclusion chromatography column (Amersham Pharmacia Biosciences) in 100 mM sodium carbonate buffer/0.001% Tween 20, pH 8.3, concentrated to ≈1 mM and labeled by addition of a 20% molar surplus of Alexa Fluor 594 succinimidyl ester and incubation at 20°C for 1 h. Doubly labeled peptide was purified by a second size-exclusion chromatography run, frozen in liquid nitrogen, and stored at –85°C.
Confocal Fluorescence Microscope. Observations of single-molecule fluorescence were made by using a custom confocal microscope as described (13). A 1.4-numerical aperture, 100× microscope objective (Nikon CFN Plan Apo 85025) was coupled with immersion oil to one face of a sample cuvette, consisting of two fused silica coverslips (Esco R425025) separated by 180-μm glass spacers. Light from the 488-nm line of an argon ion laser (Lexel 95-5, Cambridge Laser Laboratories, Fremont, CA) was made circularly polarized by using a single-mode fiber polarization controller (Thorlabs FPC560, Newton, NJ) and filtered (CVI F03-488.0-4-1.00, Albuquerque, NM) to remove unwanted luminescence from the laser-tube discharge and optical fiber. The sample was illuminated by using a dichroic mirror (Omega Optical 505DRLP, Brattleboro, VT) and the objective in a standard epifluorescence arrangement. Sample fluorescence, transmitted by the dichroic mirror, passed through a 100-μm-diameter pinhole in the image plane of the objective. After additional filtering (Omega Optical 495AELP) to remove scattered laser light, the fluorescence was separated into donor and acceptor components by using a second dichroic mirror (Omega Optical 560DCLP) and two final filters (Omega Optical 525AF45 for the donor and Omega Optical 600ALP for the acceptor). Each component was focused onto a photon-counting avalanche photodiode (Perkin–Elmer Optoelectronics SPCM-AQR-15). Output pulses, corresponding to individual detections, from the photodiode modules were collected in 1-ms intervals by a pair of multichannel scalers (EG&G Princeton Applied Research 914P).
Single-Molecule Measurements. For single-molecule experiments, samples of labeled peptides were diluted to a concentration of 35 pM in 50 mM sodium phosphate, adjusted to pH 7.0, containing 0.001% Tween 20 to prevent surface adhesion of the polypeptides. Ensemble experiments were performed in a Spex Fluorolog 2 under identical buffer conditions at peptide concentrations of 10 nM. R0 = 5.4 nm for this dye pair in water was calculated from the measured overlap integral of the normalized donor-emission and acceptor-absorption spectra (with a peak extinction coefficient, supplied by Molecular Probes, of 7.8 × 104 M–1·cm–1), a donor fluorescence quantum yield of 0.5, and a refractive index of 1.33. Even for higher values of the donor fluorescence quantum yield, R0 would not increase beyond 6 nm.
Time-Correlated Single-Photon Counting. Fluorescence lifetime and anisotropy decay measurements were performed with an FL920 f luorometer (Edinburgh Instruments, Livingston, U.K.). A frequency-doubled titanium sapphire laser system (Tsunami 3960, Spectra-Physics) set to λex = 435 nm was used as the excitation light source. The excitation pulse width was determined to be 100 fs by using an autocorrelator (Pulse Check, APE, Berlin), and the laser repetition rate of 80 MHz was reduced to 1 MHz with a pulse picker (Pulse Select, APE). Fluorescence decays were measured with a multichannel plate (ELDY EM1-132/300 MCP-PMT, Europhoton, Berlin). The complete detection system has an instrumental response time of ≈100 ps. For anisotropy decay experiments, emission with vertical and horizontal polarization with respect to the vertically polarized excitation laser beam was measured by using a polarization filter. For data analysis, the commercial software package provided with the FL920 fluorescence spectrometer was used. The experimental fluorescence decay data were iteratively deconvolved from the instrument response function based on a Marquardt algorithm and fit to a single exponential. No attempt was made to extract end-to-end distributions or diffusion coefficients from a more detailed analysis of the decay curves (4). Analysis of anisotropy decay measurements was performed by calculating the anisotropy from the vertical and horizontal polarization intensity decay curves after tail matching.
Molecular Dynamics Simulations. Langevin molecular dynamics simulations were performed for each of seven polypeptides of the form Gly-Pron-Cys, where n = 10, 15, 20, 25, 30, 35, or 40. The starting conformation of each peptide was obtained by appending extended Gly and Cys residues to polyproline modeled in a type II helix by using the angular parameters derived from the crystal structure (28). All simulations were performed with the EEF1 implicit-solvent energy function (29, 30) implemented in the program charmm (31). The EEF1 effective energy function involves a polar-hydrogen description of the peptide solute and a solvation free-energy model. The EEF1 potential has been used successfully to discriminate native protein folds from misfolded decoy structures (30) and in structure-prediction simulations of three peptides of known structure (32). After minimization of the EEF1 energy to a root-mean-square gradient of <0.005 kcal/mol per Å (1 cal = 4.18 J), the dynamics of each peptide were simulated at 298 K with a collision frequency of 50 per ps applied to all heavy (nonhydrogen) atoms. The shake algorithm was applied to the four bonds involving hydrogen atoms (in the terminal residues), and a 2-fs time step was used. Atomic coordinates were saved every 2 ps. Between 1.5 and 4.2 μs of dynamics were analyzed for each peptide.
Results
Energy transfer from the green-fluorescing donor dye (Alexa Fluor 488) at the carboxyl terminus to the red-fluorescing acceptor dye (Alexa Fluor 594) at the amino terminus was measured by determining the fraction of acceptor photons in bursts from individual molecules containing ≥100 detected photons and constructing histograms of FRET efficiencies [E ≡ nA/(nA + nD), where E is the transfer efficiency and nD and nA are the numbers of counts in the donor and acceptor channels, respectively] calculated from the individual bursts. Fig. 1 shows the results for a subset of the polyproline peptides investigated. As expected, the mean transfer efficiencies decrease with increasing peptide length. The histograms show a maximum at transfer efficiencies ranging from ≈0.95 for the six-residue peptide to 0.2 for the 40-residue peptide (Figs. 1 and 2). The additional peak at a transfer efficiency close to zero is thought to be caused by molecules lacking an active acceptor chromophore and was not included in the calculation of mean transfer efficiencies. The peak near zero efficiency increases with increasing laser intensity and with the size of the molecule, whereas in previous experiments (16) it was found to be virtually absent when the solution was flowed. These observations suggest that the acceptor chromophore undergoes a photochemical change before entering the confocal volume to a form that does not accept excitation energy from the donor. Nevertheless, comparison of the FRET efficiency in the first and second half of individual bursts shows no difference, indicating that acceptor photobleaching does not influence the results.
Fig. 1.

Single-molecule FRET efficiency measurements on polyproline peptides. (A) Molecular model of a polyproline peptide used in this study. The acceptor and donor chromophore are linked to the chain by amino- and carboxyl-terminal glycine and cysteine residues, respectively. The conformation of the proline 20-mer is based on the crystal structure (28); the linkers and dyes were placed in arbitrary orientations. (B) Transfer efficiency histograms obtained from confocal single-molecule measurements on polyproline peptides of various lengths.
Fig. 2.

Mean transfer efficiencies from single-molecule measurements (filled red circles) and ensemble time-correlated single-photon measurements (open red circles) as a function of the contour lengths of the peptides (Gly-Pron-Cys) assuming the geometry of polyproline found in the crystal structure (28) in comparison to the dependences calculated for a rod-like spacer assuming isotropic averaging (solid curve) or a random, but static angular distribution of the dipoles of donor and acceptor (dashed curve) (A) and in comparison to the dependences calculated for the different dynamic regimes according to Eqs. 4 (dark-gray squares), 5 (black squares), and 6 (light-gray squares) using the normalized end-to-end distance distributions from the molecular dynamics simulations of the peptides (Fig. 3) (B). The corresponding lines are empirical fits of the data to the equation E = 1/[1 + (r/C)4], where C is the fit parameter. (Inset) The growing deviation of the transfer efficiency measured in the single-molecule experiment (Em) from the prediction of Eq. 1 (EF) as the peptide contour length is increased beyond R0.
Surprisingly, even for peptides with >30 residues, a large amount of transfer was observed. Assuming the same rigid geometry of polyproline as observed in the crystal structure (28), these peptide lengths would correspond to end-to-end distances of >10 nm, and the resulting transfer efficiencies calculated for the dye pair used would be <3%, which would be inseparable from the peak near zero efficiency. A comparison with the Förster curve assuming a fixed distance r between the dyes,
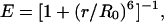 |
[1] |
illustrates the dramatically increased FRET efficiencies for long polyproline peptides (Fig. 2 A), where R0 = 5.4 nm is the Förster radius (the distance of 50% excitation energy transfer) for this dye pair calculated assuming complete orientational averaging of the dye transition dipoles (25). This discrepancy raises two questions. Are the single-molecule FRET efficiencies accurate? Is it valid to assume that polyproline is a rigid rod for the longer peptides?
A potential complication in single-molecule experiments arises from differences in the photon-collection efficiencies of the two detection channels corresponding to donor and acceptor emission. This effect can be corrected for by introducing into the equation used to calculate the transfer efficiency from the experimental data a factor γ that accounts for differences in the detection efficiencies of the emission channels and fluorescence quantum yields of the dyes (33), i.e., E = nA/(nA + γnD); γ must be determined for the particular experimental setup and pair of chromophores. For the instrument and dyes used here, we previously compared single-molecule and ensemble FRET efficiencies measured with a calibrated spectrofluorimeter to show that γ ≈1 (13). To further test this approximation, we performed time-correlated single-photon counting measurements on bulk samples of labeled polyproline to determine FRET efficiencies from the fluorescence lifetime of the donor chromophore as a function of peptide length. The FRET efficiency is given by [1 – (τDA/τD)], where τDA and τD are the donor lifetimes in the presence and absence of acceptor, respectively. As shown in Fig. 2, the efficiencies from the lifetime measurements are in good agreement with those from the single-molecule intensity measurements, eliminating the possibility of the high efficiencies arising from γ < 1.
For the shortest peptides, a significantly lower transfer efficiency than expected is observed experimentally. Polarization measurements point to a possible reason for decreased energy transfer. For the shortest polyproline peptides, an increase in the steady-state residual anisotropy is observed for the donor, from 0.05 for the longest peptides to 0.11 for the hexaproline peptide: the decrease in the lifetime of the excited state of the donor caused by the very rapid energy transfer to the acceptor no longer permits complete orientational averaging of the chromophores. Consequently, the value of 2/3 for the orientation factor κ2 derived assuming rapid orientational averaging, and most commonly used for the analysis of FRET experiments in solution, is not applicable. As a limiting case, we calculated the distance dependence of the mean transfer efficiency 〈E〉 for dyes with a fixed separation and random but static relative transition dipole orientations from the isotropic probability density p(κ2) using††
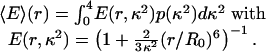 |
[2] |
The result shows a decreased FRET efficiency for small distances (Fig. 2 A), providing an upper bound on the influence of a lack of orientational averaging.
The larger discrepancy with Eq. 1 is observed for the longer polyprolines (Fig. 2 A). To investigate the influence of the polypeptide bending, which would bring the dyes closer together, providing an explanation for the higher FRET efficiency of the long polyprolines, we performed Langevin molecular dynamics simulations. Trajectories of several microseconds were computed for a range of polypeptide lengths. The simulations included the glycine and cysteine residues at the termini but not the dyes. The starting structure was a polyproline type II helix modeled according to the crystal structure (28). The simulations reveal considerable bending of the peptides, especially for the higher oligomers. Because of the strong distance dependence of Förster transfer, this bending will make a significant contribution to an increase in transfer efficiency between the dyes on the termini, particularly for the longer peptides. The ends of the polypeptide containing 40 proline residues, which has a contour length of ≈12 nm, can get to within ≈6 nm in the simulation (Fig. 3), corresponding to an increase in the (instantaneous) transfer rate by a factor of ≈26.
Fig. 3.

End-to-end distance distributions obtained from molecular dynamics simulations of peptides containing 10, 15, 20, 25, 30, 35, or 40 proline residues plus terminal glycine and cysteine residues (black lines) and the least-squares fits to a worm-like chain model (red lines) using Eq. 3. (Inset) The values for lc (filled circles) and lp (open circles) resulting from the fits are shown for the different peptide lengths.
The end-to-end (Gly nitrogen atom to Cys sulfur atom) distance distributions obtained from the simulations are very similar to those of a worm-like chain. To obtain an estimate of the persistence length, the distributions were fit with the equation of Thirumalai and Ha (34), which provides a good approximation for the end-to-end distance distribution P(r) of a semiflexible chain,
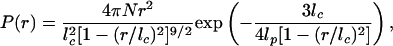 |
[3] |
where r is the end-to-end distance, lc and lp are the contour length and the persistence length of the chain, respectively, and N is a normalization constant. The fits of the worm-like chain model to the data give a persistence length of 4.4 ± 0.9 nm (Fig. 3). The apparently smaller persistence lengths for the shortest peptides may be due to the larger deviations of the fits at the short distance ends of the distributions.
A quantitative determination of the influence of chain bending on the mean FRET efficiency also requires consideration of the time scale of the end-to-end distance fluctuations and the donor fluorescence lifetime (35, 36). Fig. 4 summarizes the relevant times. The typical duration of a photon burst in our experiments is ≈1 ms, much longer than the donor fluorescence lifetime or the end-to-end distance correlation time, resulting in complete conformational averaging during the observation time for an individual molecule. The fluorescence lifetimes of the donor, as determined by time-correlated single-photon counting measurements, range from ≈0.4 to 3.6 ns for the different labeled peptides. The reorientational correlation time of the donor chromophore is ≈0.3 ns as determined from the anisotropy decay of donor-labeled peptide in the absence of transfer. Chain dynamics were quantified from the molecular dynamics simulations by calculating the autocorrelation functions of the end-to-end distance fluctuations and fitting them with single-exponential decays. The resulting relaxation times are between ≈0.2 ns for the proline decamer to ≈10 ns for the longest peptides (Fig. 4). Chain dynamics and fluorescence decay are in a very similar range for all peptides, and for the shortest chains, the donor fluorescence lifetime approaches the reorientational correlation time, as expected from the corresponding increase in residual anisotropy mentioned above. Therefore, we should consider all three physically plausible limits for the possible averaging regimes:
Fig. 4.

Time scales of the dynamic processes involved. Indicated are the rotational correlation time of the donor chromophore (blue dashed line), the relaxation time of the autocorrelation function of the end-to-end distance fluctuations calculated from the simulations (green filled circles), and the fluorescence lifetime of the donor dye (red circles) as a function of the number of proline residues in the chain. The typical duration of a photon burst is ≈1 ms. (Left Inset) The end-to-end distance autocorrelation function for the polyproline 20-mer (green) fit to a single-exponential decay (black). (Right Inset) The anisotropy decay of the donor chromophore (blue) fit to a double-exponential decay (black). The faster component of the decay (0.3 ns), which contains 70% of the amplitude, was assigned to the rotational correlation time of the dye (48), and the slower component (0.8 ns) was assigned to the rotational correlation time of the entire molecule.
- If the rotational correlation time τc of the chromophores is small relative to the fluorescence lifetime τf of the donor and the dynamics of the polyproline chain (with relaxation time τp) are slow relative to τf,
where P(r) is the normalized interdye distance distribution and a is the distance of closest approach of the dyes.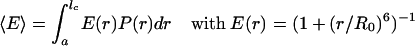
[4] - If τc ≪ τf and τp ≪ τf,
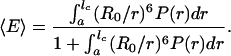
[5] If τc ≫ τf and τp ≫ τf,
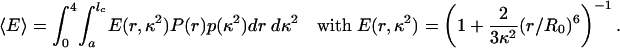 |
[6] |
In all cases we used the normalized end-to-end distance distributions from the molecular dynamics simulations, P(r) (Fig. 3) and the theoretical isotropic probability density p(κ2) to calculate the mean transfer efficiencies for the polyproline peptides studied by simulation.†† The results are plotted in Fig. 2B and illustrate that the flexibility of the chains results in markedly increased transfer efficiencies compared to those calculated for rod-like molecules (Fig. 2 A). Moreover, they demonstrate the strong influence of the different dynamic regimes. Considering that τc ≪ τf and τp ≈ τf for the longer oligomers, we expect the corresponding experimental results to be in the range of the values calculated with Eqs. 4 and 5. The static limit (Eq. 6) will only be relevant for the shortest peptides. All in all, chain flexibility and dynamics account well for the experimentally observed mean transfer efficiencies.‡‡
Discussion
To determine whether FRET measurements can provide quantitative distance information in single-molecule fluorescence experiments, we have measured the FRET efficiency for donor and acceptor dyes attached to the ends of freely diffusing polyproline molecules of various lengths. Polyproline, the stiffest homooligopeptide (37), forms a type II trans helix with a pitch of 0.31 nm per residue in aqueous solution (28), and its conformation does not change after addition of denaturants (13). However, our experimentally observed interdye-distance dependence of the mean FRET efficiency differs greatly from the predictions of the simplest Förster formula, Eq. 1, assuming a perfectly rigid rod for the polyproline spacer and fast, isotropic rotational averaging of the chromophores (Fig. 2 A). For peptides with a length much shorter than the size of the Förster radius, too low a transfer efficiency is observed; for longer peptides, the transfer efficiency is substantially higher than expected for a rod-like behavior of polyproline. The differences are not a result of inaccuracies in the single-molecule measurements, because the mean efficiencies have been confirmed with ensemble lifetime measurements.
Two factors may contribute to the deviations for the smallest oligomers. One factor is the lack of orientational averaging during the donor lifetime, as indicated by the decay of the anisotropy (Fig. 4 Inset), so that the average angular factor κ2 < 2/3. Our calculations (Eqs. 2 and 6) provide an upper bound on this effect, and the experimental data are well within the expected range (Fig. 2). The second factor is the breakdown of the point-dipole approximation of Förster theory, which requires that the size of the donor and acceptor be small compared to their intermolecular separation. In the present case, the lengths of both donor (0.7 nm) and acceptor (1.2 nm) dyes are not small compared to the length of the 10-residue polyproline spacer of ≈3 nm. The point-dipole approximation leads to the important property of the theory that all the parameters can be calculated directly from experimental measurables; if this is not true, the rate must be calculated from a detailed quantum mechanical treatment of the Coulomb interaction between the charge distributions (38–40), which is not nearly as reliable and could lead to either a faster or slower rate of excitation energy transfer (i.e., either a higher or lower FRET efficiency than predicted by using the point-dipole approximation).
For the long peptides, Langevin molecular dynamics simulations show that the difference can be explained by the flexibility of polyproline. The decay of the autocorrelation function indicates that the end-to-end distance distribution is sampled completely within the 1-ms collection time of photons from individual molecules. By considering the averaging regimes relevant for the reorientation of the dyes and the fluctuations of the end-to-end distances of the chains, the main components contributing to the enhancement of the mean FRET efficiencies are accounted for (Fig. 2B). A more complete analysis would include the donor and acceptor dyes and their linkers in simulations with explicit solvent and a direct calculation of the FRET efficiency, as was done, for example, in the case of Förster transfer of excitation energy from tryptophan to heme, calculated by using an all-atom molecular dynamics trajectory of myoglobin (41).
Polyproline peptides were first used in the context of FRET in the work of Stryer and Haugland (1), who experimentally observed the distance dependence of the transfer efficiency predicted by Förster (26), assuming polyproline to be a rigid rod. For the longer polyprolines used in the present work, however, the end-to-end distance distributions obtained from Langevin molecular dynamics simulations are not those of a rigid rod but are much more like the distributions of a worm-like chain (42). To extract an apparent persistence length, the end-to-end distributions were fit with the model function of Thirumalai and Ha (34), Eq. 3 (Fig. 3). The persistence length obtained from the fits is 4.4 ± 0.9 nm, significantly less than the textbook value of 22 nm (37, 42), indicating greater flexibility than previously estimated by assuming a fixed value of the backbone dihedral angle φ (37). However, in the range of the small peptide lengths used by Stryer and Haugland (up to the dodecamer), polyproline can be approximated by a rigid rod.§§
Interestingly, since the experiments of Haugland and Stryer, there have been very few studies of the distance dependence of the FRET efficiency and no rigorous determination of the accuracy of Förster theory at distances for which the point-dipole approximation should apply. In the only previous single-molecule study of the distance dependence, Deniz et al. (43) used B-form, double-stranded DNA as a spacer between large donor and acceptor dyes. By using the reported R0 = 5.3 nm and an assumed correction factor γ = 1, the measured FRET efficiency was also found to be much higher than the predictions of Eq. 1, although flexibility is expected to play a very small role because the persistence length of DNA is ≈10-fold larger than the R0 of the donor–acceptor dye pair. However, a very recent reinvestigation found excellent agreement between measured and predicted FRET efficiencies, by including dye flexibility, by using alternating laser excitation of the donor and acceptor to eliminate the zero-efficiency peak (44), by determining a more accurate value for the correction factor γ, and by redetermining R0 from spectroscopic data to be 6.9 nm (N. K. Lee, A. N. Kapanidis, Y. Wang, X. Michalet, J. Mukhopadhyay, R. H. Ebright, and S. Weiss, personal communication).
Polyproline has been used as a spectroscopic reference for single-molecule fluorescence experiments (13), and because of the large number of potential photochemical complications in single-molecule studies on biomolecular dynamics, we expect labeled polyproline peptides to become an important standard for calibrating measurements and for testing and refining new experimental methods. For single-molecule FRET experiments on proteins, it is desirable to use a polypeptide-based reference molecule, because the type of attachment chemistry and the characteristics of the immediate molecular environment can influence the photophysical properties of fluorophores (45, 46). Single-molecule measurements have the advantage of providing distributions, and they can be used to separate subpopulations and to investigate their dynamics individually. Although the chromophores necessary for these studies are relatively large, our results show that, in principle, there are no additional complications in single-molecule experiments other than those arising from photodestruction due to multiple excitations at the high laser intensities used. As in ensemble FRET experiments, consideration of the dynamics is critical for extracting structural information.
Acknowledgments
We thank Attila Szabo and Irina Gopich for many helpful discussions and developing a comprehensive theoretical description of the single-molecule free-diffusion FRET experiment (36). We also thank Hans-Gerd Löhmannsröben for access to time-correlated single-photon counting instrumentation; Ming Yau for assistance with peptide synthesis; Bernard Brooks and Richard Pastor for advice on the Langevin simulations; Milan Hodoscek for implementing the random number generator used in the program charmm; Yong-Sok Lee and Sergio Hassan for discussion; Jay Knutson for help in lifetime measurements in the initial phases of this work; and Shimon Weiss for a preprint on the length dependence of FRET efficiencies in DNA and sharing other results. This study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). Financial support came from the Deutsche Forschungsgemeinschaft and the VolkswagenStiftung.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: FRET, Förster resonance energy transfer.
Footnotes
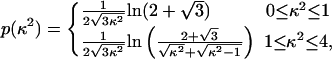 |
We should mention that in the simulations of the 30- and 35-mer, sudden transitions to long-lived kinked structures were observed but were excluded from our analysis (Figs. 3 and 4). We suspect that these structures are artifacts that reflect our use of an implicit-solvent model and will require additional investigation with explicit solvent. Such structures would increase the calculated mean FRET efficiency, bringing the calculated values in even closer agreement with the observed. They might also contribute to the yet-to-be-understood observed excess width of the FRET efficiency distribution (Fig. 1B) over that expected from pure shot noise (36).
Stryer and Haugland (1) also fit their data by using an R0 of 3.4 nm instead of the value of 2.7 nm obtained from the measured spectroscopic parameters. The difference between their experimental data and the curve predicted from Eq. 1 with R0 = 2.7 nm can be accounted for largely by assuming the dye linkers to be flexible instead of stiff extensions of the chain.
References
- 1.Stryer, L. & Haugland, R. P. (1967) Proc. Natl. Acad. Sci. USA 58, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy (Kluwer Academic/Plenum, New York).
- 3.Stryer, L. (1978) Annu. Rev. Biochem. 47, 819–846. [DOI] [PubMed] [Google Scholar]
- 4.Haas, E., Katchalskikatzir, E. & Steinberg, I. Z. (1978) Biopolymers 17, 11–31. [Google Scholar]
- 5.Selvin, P. R. (2000) Nat. Struct. Biol. 7, 730–734. [DOI] [PubMed] [Google Scholar]
- 6.Michalet, X., Kapanidis, A. N., Laurence, T., Pinaud, F., Doose, S., Pflughoefft, M. & Weiss, S. (2003) Annu. Rev. Biophys. Biomol. Struct. 32, 161–182. [DOI] [PubMed] [Google Scholar]
- 7.Ha, T., Enderle, T., Ogletree, D. F., Chemla, D. S., Selvin, P. R. & Weiss, S. (1996) Proc. Natl. Acad. Sci. USA 93, 6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia, Y. W., Talaga, D. S., Lau, W. L., Lu, H. S. M., DeGrado, W. F. & Hochstrasser, R. M. (1999) Chem. Phys. 247, 69–83. [Google Scholar]
- 9.Ishii, Y., Yoshida, T., Funatsu, T., Wazawa, T. & Yanagida, T. (1999) Chem. Phys. 247, 163–173. [Google Scholar]
- 10.Deniz, A. A., Laurence, T. A., Beligere, G. S., Dahan, M., Martin, A. B., Chemla, D. S., Dawson, P. E., Schultz, P. G. & Weiss, S. (2000) Proc. Natl. Acad. Sci. USA 97, 5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talaga, D. S., Lau, W. L., Roder, H., Tang, J. Y., Jia, Y. W., DeGrado, W. F. & Hochstrasser, R. M. (2000) Proc. Natl. Acad. Sci. USA 97, 13021–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ha, T. (2001) Methods 25, 78–86. [DOI] [PubMed] [Google Scholar]
- 13.Schuler, B., Lipman, E. A. & Eaton, W. A. (2002) Nature 419, 743–747. [DOI] [PubMed] [Google Scholar]
- 14.Heyduk, T. (2002) Curr. Opin. Biotechnol. 13, 292–296. [DOI] [PubMed] [Google Scholar]
- 15.Borsch, M., Diez, M., Zimmermann, B., Reuter, R. & Gräber, P. (2002) FEBS Lett. 527, 147–152. [DOI] [PubMed] [Google Scholar]
- 16.Lipman, E. A., Schuler, B., Bakajin, O. & Eaton, W. A. (2003) Science 301, 1233–1235. [DOI] [PubMed] [Google Scholar]
- 17.Rhoades, E., Gussakovsky, E. & Haran, G. (2003) Proc. Natl. Acad. Sci. USA 100, 3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berney, C. & Danuser, G. (2003) Biophys. J. 84, 3992–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen, V. T., Kamio, Y. & Higuchi, H. (2003) EMBO J. 22, 4968–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margittai, M., Widengren, J., Schweinberger, E., Schröder, G. F., Felekyan, S., Haustein, E., König, M., Fasshauer, D., Grubmüller, H., Jahn, R., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 15516–15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasnik, I., Myong, S., Cheng, W., Lohman, T. M. & Ha, T. (2004) J. Mol. Biol. 336, 395–408. [DOI] [PubMed] [Google Scholar]
- 22.Klostermeier, D., Sears, P., Wong, C. H., Millar, D. P. & Williamson, J. R. (2004) Nucleic Acids Res. 32, 2707–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slaughter, B. D., Allen, M. W., Unruh, J. R., Urbauer, R. J. B. & Johnson, C. K. (2004) J. Phys. Chem. B 108, 10388–10397. [Google Scholar]
- 24.Rhoades, E., Cohan, M., Schuler, B. & Haran, G. (2004) J. Am. Chem. Soc. 126, 14686–14687. [DOI] [PubMed] [Google Scholar]
- 25.Van Der Meer, B. W., Coker, G., III, & Chen, S. Y. S. (1994) Resonance Energy Transfer: Theory and Data (VCH, New York).
- 26.Förster, T. (1948) Ann. Phys. 6, 55–75. [Google Scholar]
- 27.Panchuk-Voloshina, N., Haugland, R. P., Bishop-Stewart, J., Bhalgat, M. K., Millard, P. J., Mao, F. & Leung, W. Y. (1999) J. Histochem. Cytochem. 47, 1179–1188. [DOI] [PubMed] [Google Scholar]
- 28.Cowan, P. M. & McGavin, S. (1955) Nature 176, 501–503. [DOI] [PubMed] [Google Scholar]
- 29.Lazaridis, T. & Karplus, M. (1999) Proteins 35, 133–152. [DOI] [PubMed] [Google Scholar]
- 30.Lazaridis, T. & Karplus, M. (1999) J. Mol. Biol. 288, 477–487. [DOI] [PubMed] [Google Scholar]
- 31.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187–217. [Google Scholar]
- 32.Steinbach, P. J. (2004) Proteins 57, 665–677. [DOI] [PubMed] [Google Scholar]
- 33.Deniz, A. A., Laurence, T. A., Dahan, M., Chemla, D. S., Schultz, P. G. & Weiss, S. (2001) Annu. Rev. Phys. Chem. 52, 233–253. [DOI] [PubMed] [Google Scholar]
- 34.Thirumalai, D. & Ha, B. Y. (1988) in Theoretical and Mathematical Models in Polymer Research, ed. Grosberg, A. (Academia, New York), pp. 1–35.
- 35.Gopich, I. V. & Szabo, A. (2003) J. Phys. Chem. B 107, 5058–5063. [Google Scholar]
- 36.Gopich, I. V. & Szabo, A. (2005) J. Chem. Phys. 122, 014707-1–014707-18. [Google Scholar]
- 37.Schimmel, P. R. & Flory, P. J. (1967) Proc. Natl. Acad. Sci. USA 58, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scholes, G. D. (2003) Annu. Rev. Phys. Chem. 54, 57–87. [DOI] [PubMed] [Google Scholar]
- 39.Wong, K. F., Bagchi, B. & Rossky, P. J. (2004) J. Phys. Chem. 108, 5752–5763. [Google Scholar]
- 40.Scholes, G. D. & Fleming, G. R. (2005) Adv. Chem. Phys., in press.
- 41.Henry, E. R. & Hochstrasser, R. M. (1987) Proc. Natl. Acad. Sci. USA 84, 6142–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantor, C. R. & Schimmel, P. R. (1980) Biophysical Chemistry (Freeman, San Francisco).
- 43.Deniz, A. A., Dahan, M., Grunwell, J. R., Ha, T. J., Faulhaber, A. E., Chemla, D. S., Weiss, S. & Schultz, P. G. (1999) Proc. Natl. Acad. Sci. USA 96, 3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kapanidis, A. N., Lee, N. K., Laurence, T. A., Doose, S., Margeat, E. & Weiss, S. (2004) Proc. Natl. Acad. Sci. USA 101, 8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hillisch, A., Lorenz, M. & Diekmann, S. (2001) Curr. Opin. Struct. Biol. 11, 201–207. [DOI] [PubMed] [Google Scholar]
- 46.Clegg, R. M., Murchie, A. I., Zechel, A. & Lilley, D. M. (1993) Proc. Natl. Acad. Sci. USA 90, 2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dale, R. E., Eisinger, J. & Blumberg, W. E. (1979) Biophys. J. 26, 161–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexiev, U., Rimke, I. & Pohlmann, T. (2003) J. Mol. Biol. 328, 705–719. [DOI] [PubMed] [Google Scholar]