

Abstract
Long non‐coding RNAs (lncRNAs) have been implicated in the regulation of chromatin conformation and epigenetic patterns. lncRNA expression levels are widely taken as an indicator for functional properties. However, the role of RNA processing in modulating distinct features of the same lncRNA is less understood. The establishment of heterochromatin at rRNA genes depends on the processing of IGS‐rRNA into pRNA, a reaction that is impaired in embryonic stem cells (ESCs) and activated only upon differentiation. The production of mature pRNA is essential since it guides the repressor TIP5 to rRNA genes, and IGS‐rRNA abolishes this process. Through screening for IGS‐rRNA‐binding proteins, we here identify the RNA helicase DHX9 as a regulator of pRNA processing. DHX9 binds to rRNA genes only upon ESC differentiation and its activity guides TIP5 to rRNA genes and establishes heterochromatin. Remarkably, ESCs depleted of DHX9 are unable to differentiate and this phenotype is reverted by the addition of pRNA, whereas providing IGS‐rRNA and pRNA mutants deficient for TIP5 binding are not sufficient. Our results reveal insights into lncRNA biogenesis during development and support a model in which the state of rRNA gene chromatin is part of the regulatory network that controls exit from pluripotency and initiation of differentiation pathways.
Keywords: DHX9, embryonic stem cells, heterochromatin, lncRNA
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology; Stem Cells
Introduction
The nucleolus is the compartment where ribosome biogenesis takes place, a process that is initiated by transcription of ribosomal RNA (rRNA) genes that synthesize ribosomal rRNA 1. In each somatic cell, a fraction of the 400 rRNA genes is transcriptionally silent and organized in heterochromatic structures, including CpG methylation and histone H3K9 methylation 2, 3. However, until now the role of silent heterochromatic rRNA repeats is not yet understood. Indeed, the formation of heterochromatin at rRNA genes appears to not be implicated in ribosome biogenesis since silent rRNA repeats do not become transcriptionally activated even under conditions of high metabolic activities of the cell 4.
The higher order organization of genomes is functionally important to ensure correct execution of gene expression programs. For instance, as cells differentiate into specialized cell types, chromosomes undergo diverse structural and organizational changes that affect gene expression and other cellular functions 5. The chromatin of embryonic stem cells (ESCs) is largely devoid of compact heterochromatin blocks when compared to lineage‐committed cells 6, 7. This structure well reflects the plasticity and transcriptional permissiveness of ESC genome that need to maintain the ability to enter any distinct transcriptional programs for lineage specification 8, 9. Upon differentiation, a large portion of the ESC genome remodels toward a highly condensed heterochromatic form 10. These changes are also accompanied by alterations of nuclear architecture such as the clustering of centromeres either at the nucleolus or at the nuclear envelope 11, 12. However, it still remains elusive how the switch from a lower to a higher order chromatin structure is achieved during ESC differentiation and whether this process plays a role in ESC differentiation.
Previous results have shown that also rRNA genes undergo chromatin changes during mouse ESC differentiation 13, 14. In ESCs, all 400 rRNA gene copies are active and only upon differentiation a fraction of these genes acquire heterochromatic marks such as CpG methylation and histone H3K9 methylation, as found in somatic cells 2, 13. In somatic mouse and human cells, establishment of heterochromatin at rRNA genes is mediated by TIP5 through its association with the long non‐coding (lnc)RNA pRNA, DNA methyltransferases, and histone modifier enzymes 3, 15, 16, 17. Although mouse and human pRNA sequences are lowly conserved in their sequences, they display a similar structure that serves for the association with TIP5 in vitro and in vivo 15, 16, 18. pRNA derives from the processing of the 2‐kb‐long IGS‐rRNA, a transcript originating from the spacer promoter that, in mouse, is located 2‐kb upstream the main rRNA gene promoter 16, 19. The production of mature pRNA is essential for the formation of heterochromatin at rRNA genes since only mature pRNA can guide TIP5 to nucleolus to establish silencing while the association of TIP5 with the unprocessed transcript abolishes this process 13. Mature pRNA promotes the association of TIP5 with TTF1, a nucleolar docking factor bound to the main promoter of rRNA genes, whereas IGS‐rRNA destroys the interaction with TTF1 and impairs TIP5 recruitment 13, 20. In ESCs, the maturation of IGS‐rRNA into pRNA is abolished and it is only upon ESC differentiation that the IGS‐rRNA processing is activated to produce pRNA, which leads to the establishment heterochromatin at a fraction of rRNA genes for the first time. Remarkably, addition of mature pRNA in ESCs was not only sufficient to establish nucleolar heterochromatin and to downregulate rRNA synthesis but also primed ESCs for differentiation. These results are consistent with recent studies proposing that elevated rRNA biosynthesis sustains pluripotency in mouse and human ESCs since rRNA downregulation through chemical inhibition of Pol I activity or deletion of fibrillarin induces differentiation 21, 22.
The critical role of pRNA biogenesis in the establishment of rRNA gene heterochromatin represents an important example of how the different features of the same lncRNA can be modulated to regulate chromatin conformation and epigenetic patterns during development. However, how this process is regulated is not understood. To this end, we performed a screening for IGS‐rRNA‐binding proteins and identified the RNA helicase DHX9 to be required for the production of mature pRNA. DHX9 binds to rRNA genes only upon ESC differentiation, and its activity to process IGS‐rRNA into pRNA is necessary to guide TIP5 to rRNA genes and establish rRNA gene heterochromatin. Remarkably, ESCs depleted of DHX9 are unable to differentiate and this phenotype can be reverted by the sole addition of mature pRNA. Taken together, these results provide molecular insights into lncRNA biogenesis that modulates different features of the same lncRNA and highlights the importance of lncRNA processing for the establishment of chromatin states during development.
Results
The RNA helicase DHX9 associates with IGS‐rRNA
pRNA is a 250–300 nucleotide transcript that derives from processing of the 2‐kb‐long IGS‐rRNA 16, 19. Production of pRNA is a key step for the formation of heterochromatin at rRNA genes since only mature pRNA can guide TIP5 to rRNA genes to establish repressive chromatin states 13. We sought to identify factors implicated in IGS‐rRNA processing by searching for proteins able to specifically bind to IGS‐rRNA. We performed GRNA chromatography, a method that relies on the interaction between a 19 nt RNA element called BoxB and a 22 amino acid long peptide λN22 23. We converted Glutathione Sepharose into an RNA affinity matrix by binding GST‐λN22 fusion protein to in vitro synthesized BoxB‐IGS‐rRNA or BoxB‐Control‐RNA and performed pull‐downs using nuclear extracts of NIH 3T3 or neural progenitors cells (NPCs), both proficient for IGS‐rRNA processing 13, 19 (Fig 1A). Proteins bound to IGS‐rRNA and control‐RNA sequences were eluted with RNase A and analyzed by mass spectrometry (Figs 1B and EV1A–C, Dataset EV1). To identify factors that specifically associate with IGS‐rRNA sequences, we considered only proteins that were present in all the experiments with a minimum of two peptides and that were associated with at least twofold higher peptide content to IGS‐rRNA compared to control‐RNA (Figs 1B and EV1B and C). Only six proteins fulfilled these criteria: DHX9, FUBP1, FUBP3, CSTF1, TIAR, and LSM6. As expected, all these factors are related to RNA pathways. The RNA helicase DHX9 (RNA Helicase A, RHA) caught our attention due to its reported localization in nucleoli of mouse and human cells, the compartment where processing of IGS‐rRNA takes place 24, 25, 26. Accordingly, immunofluorescence analysis revealed the presence of endogenous DHX9 or ectopic GFP‐DHX9 within nucleoli, as evidenced by the co‐localization with the nucleolar upstream binding factor UBF (Fig 1C). Moreover, in our effort to identify factors associated with TIP5 through immuno‐precipitation (IP) combined with mass‐spec analysis, DHX9 was consistently detected as a TIP5‐interacting protein (data not shown). Accordingly, co‐immunoprecipitation of TIP5 in HEK293T cells revealed the association of TIP5 with endogenous DHX9 and the N‐terminus domain of TIP5 was sufficient for this interaction (Fig 1D). The interaction between endogenous TIP5 and DHX9 was also detected in ESCs (Fig EV1D). Finally, the identification of DHX9‐interacting proteins in ESCs, ESCs after 4 days of differentiation, NIH 3T3 and HEK293 cells through anti‐FLAG immuno‐precipitation of ectopically expressed FLAG‐DHX9 followed by mass spectrometry revealed a strong enrichment in factors implicated in RNA processing (Datasets EV2 and EV3, and Fig EV2). In particular, components of the spliceosome (as determined by STRING database of interaction and KEGG pathway database) were remarkably abundant 27, 28. Consistent with these results, spliceosome was the major hit obtained through cellular component and pathway analysis in each of the GRNA chromatography experiments performed to identify IGS‐rRNA‐binding proteins (Dataset EV1). Interestingly, the absence of the spliceosome canonical DExH/RHA helicases (i.e., PRPF 2, 16, 22, and 43) 29 among DHX9 and IGS‐rRNA interacting factors suggests that DHX9 does not interact with the canonical spliceosome complex.
Figure 1. DHX9 is a nucleolar protein that associates with IGS‐rRNA and TIP5.
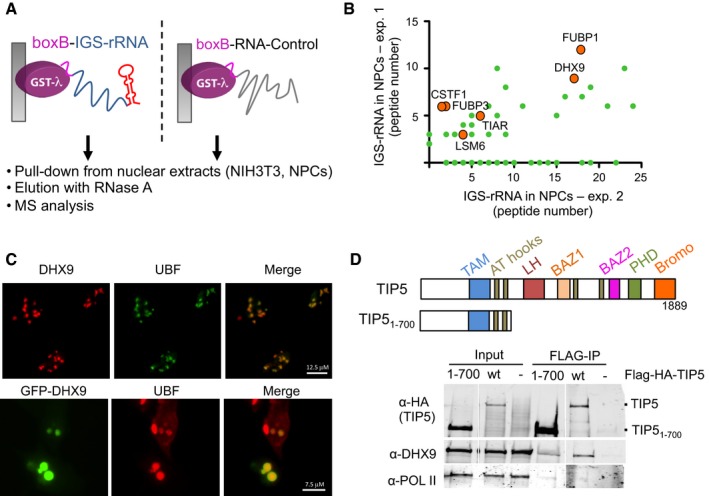
- Schema represents the GRNA chromatography employed for the identification of IGS‐rRNA‐binding proteins.
- Representative mass spectrometry analysis of proteins pulled down with IGS‐rRNA and found enriched in all three independent experiments (labeled with orange circles), using nuclear extracts from NIH 3T3 cells or NPCs. Values (green circles) refer to peptide numbers obtained in each experiment. For direct comparison with the other experiments, the same data are shown in Fig EV1B.
- DHX9 localizes in nucleoli. Immunofluorescence analysis with DHX9 antibodies in U2OS cells and with GFP antibodies in NIH 3T3 cells transfected with GFP‐DHX9 plasmids. Nucleoli are visualized with anti‐UBF.
- Endogenous DHX9 associates with TIP5. Schema represents the domain organization of TIP5. Anti‐FLAG immunoprecipitation from HEK293T cells expressing FLAG‐HA‐TIP5 or FLAG‐HA‐TIP51–700. Immunoblots show association of TIP5 with DHX9 but not with the RNA Pol II.
Source data are available online for this figure.
Figure EV1. DHX9 associates with IGS‐rRNA and TIP5.
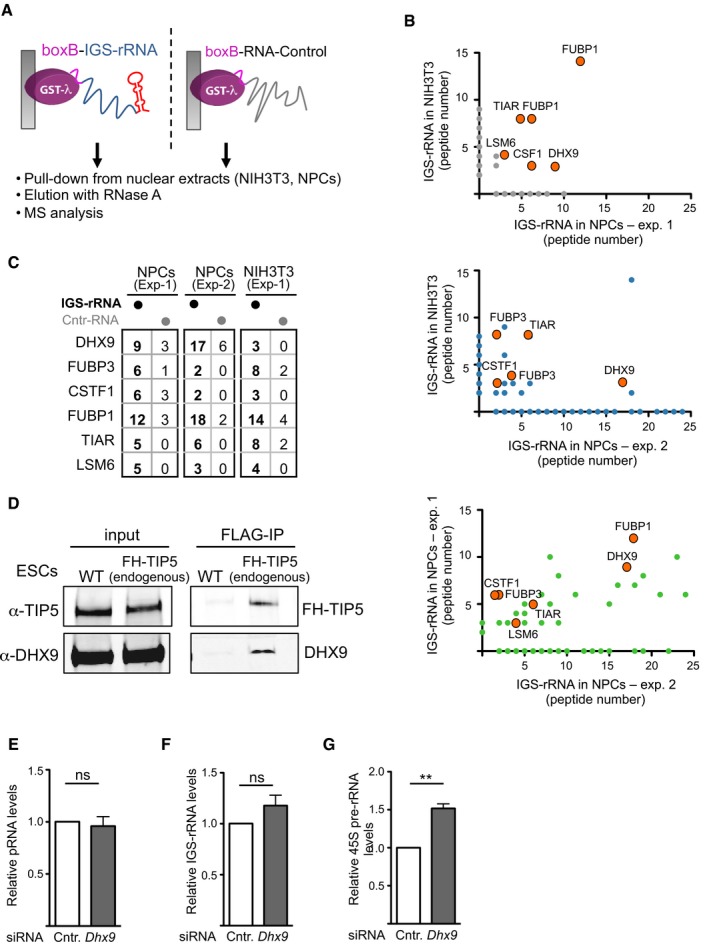
-
ASchema represents the GRNA chromatography employed for the identification of IGS‐rRNA‐binding proteins.
-
BMass spectrometry analysis of proteins pulled down with IGS‐rRNA and found enriched in all three independent experiments, which used nuclear extracts from NIH 3T3 cells or NPCs. Values refer to peptide numbers obtained in each experiment (gray, blue and green circles). Orange circles highlight the six IGS‐rRNA‐binding proteins found to associate with IGS‐rRNA in all experiments.
-
CComparison of peptides pulled down with IGS‐rRNA in three independent experiments using nuclear extracts from NIH 3T3 cells and NPCs. Values refer to peptide numbers obtained in each experiment.
-
DCo‐IP showing the interaction of endogenous TIP5 and DHX9 in embryonic stem cells (ESCs). Flag‐HA (FH)‐TIP5 ESCs were obtained by co‐transfection of the CRISPR/Cas9 vector expressing an sgRNA targeting the TIP5 genomic locus 3 nt upstream of the ATG start codon with a homology directed repair (HDR) template. Immunoblots of Flag‐IP and input show the association of endogenous FH‐TIP5 with DHX9.
-
E–GKnockdown of DHX9 upregulates 45S pre‐rRNA levels but does not affect the total amount of IGS‐rRNA and pRNA. qRT–PCR from NIH 3T3 cells transfected with siRNA‐control and siRNA‐DHX9. Values were obtained by amplification of cDNA generated in reverse transcription reactions using random hexamers. Values (mean ± SD) were from four independent experiments and calculated relative to GAPDH mRNA amounts and to control cells (siRNA‐control). Statistical significance (P‐values) for the experiments was calculated using the paired two‐tailed t‐test (**P < 0.01; ns, non‐significant).
Source data are available online for this figure.
Figure EV2. DHX9 associates with components of RNA processing and spliceosome.
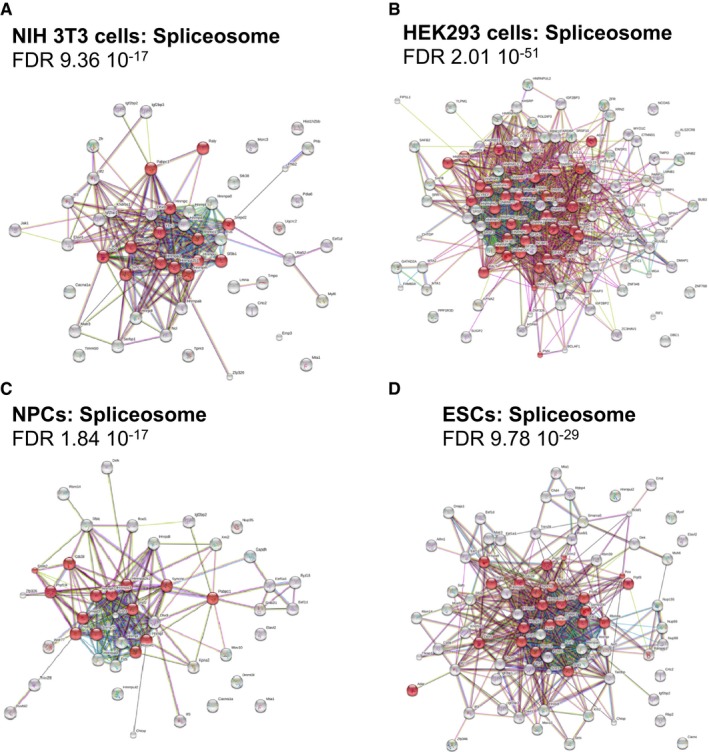
-
A–DSTRING analysis depicting functional protein association networks of DHX9 interacting proteins found in NIH 3T3 cells (A), HEK293T (B), ESCs after 3‐day differentiation (C), and ESCs (D). DHX9‐interacting proteins were identified by transfection of FLAG‐DHX9 plasmid, followed by anti‐FLAG immuno‐precipitation, FLAG peptide elution, and mass spectrometry analysis. Identified proteins are listed in Dataset EV1. Biological pathways and KEGG analysis are in Dataset EV2.
DHX9 belongs to the DExH/RHA family of helicase superfamily 2 and is characterized by two copies of a double‐stranded RNA‐binding domain (DSRM) at the amino terminus, a helicase core domain (HrpA) in the central region, and an RGG‐rich region at the carboxyl terminus, which confers both RNA and DNA helicase activities 30, 31, 32. DHX9 is the homolog of the maleless gene product MLE in Drosophila, where it is essential for dosage compensation between the two X chromosomes of females and the single one of males 33. In mammalian cells, DHX9 has been implicated in a variety of processes such as transcriptional activation 34, 35, 36, 37, 38 and genome stability 39, 40. However, the co‐activating function of DHX9 does not necessarily require ATP‐hydrolysis or DNA unwinding 36, 38.
The ability of DHX9 to bind to IGS‐rRNA, its nucleolar localization, the association with TIP5 and its link to factors implicated in spliceosome formation prompted us to investigate whether DHX9 plays a role in the processing of IGS‐rRNA. We initially employed a previously established assay based on the transfection of the IGS‐rRNA reporter plasmid in NIH 3T3 cells, proficient for IGS‐rRNA processing (Fig 2A) 13. Transcripts are measured by strand‐specific reverse transcription (RT) using a primer hybridizing vector sequences downstream the main rRNA gene promoter followed by amplification of pRNA and 5′‐IGS‐rRNA regions. Since IGS‐rRNA also includes pRNA sequences, values obtained through amplification with primers amplifying pRNA sequences represent the total amount of mature pRNA and unprocessed IGS‐rRNA transcripts whereas measurements of 5′‐IGS‐rRNA sequences correspond to the levels of unprocessed transcripts only. We made use of the IGS‐rRNA reporter assay in NIH 3T3 cells depleted of DHX9 by siRNA and found that processing of ectopic IGS‐rRNA was less efficient when compared to control cells (Fig 2A). Knockdown of DHX9 also induced a consistent accumulation of endogenous unprocessed IGS‐rRNA (Fig 2B). Depletion of DHX9 did not affect the total levels of IGS‐rRNA and pRNA sequences, indicating that DHX9 is not implicated in the synthesis of IGS‐rRNA itself but most likely acts on its processing (Fig EV1E and F). Remarkably, the defects in IGS‐rRNA processing upon DHX9 knockdown could be reversed upon ectopic expression of GFP‐DHX9, which is not targeted by the siRNA‐DHX9 (Fig 2C). Expression of GFP‐DHX9K417R that contains a mutation impairing the ATP‐helicase activity 36, 41 displayed a similar efficiency for restoring IGS‐rRNA processing (Fig 2C), suggesting that DHX9 RNA helicase activity is not required for IGS‐rRNA maturation. Together, these results indicate that DHX9 is implicated in the maturation of IGS‐rRNA into pRNA.
Figure 2. DHX9 is implicated in IGS‐rRNA processing.
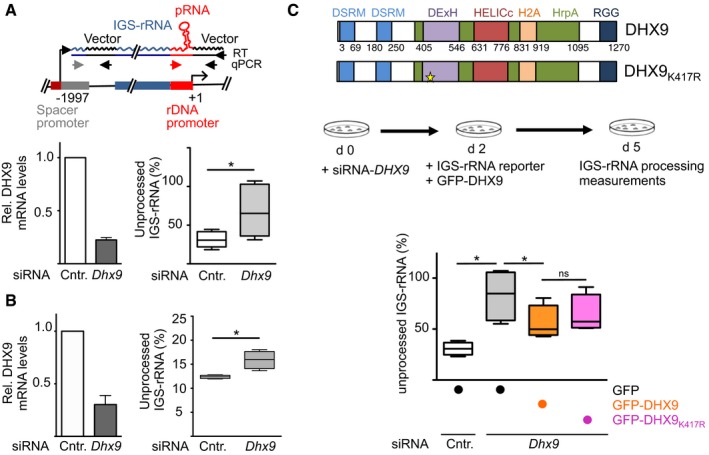
- Schema depicts the IGS‐rRNA reporter plasmid. Black arrows represent primers used to perform RT or to amplify plasmid sequences. Gray and red arrows indicate primers hybridizing to rRNA sequences. siRNA‐DHX9 or siRNA‐control‐treated NIH 3T3 cells were transfected with IGS‐rRNA reporter plasmid. Transcripts are measured by strand‐specific reverse transcription (RT) using a primer hybridizing vector sequences downstream the main rRNA gene promoter followed by amplification of pRNA (red and black arrows) and 5′‐IGS‐rRNA regions (gray and black arrows). Data from six experiments are represented as values of amplifications of the 5′‐region of IGS‐rRNA (unprocessed) normalized to amplifications of pRNA sequences (unprocessed + processed) of the reporter plasmid. Box plots depict the minimum and maximum values. The median is represented by a horizontal line within the boxes. DHX9 knockdown efficiency is shown on the left. Transcripts were normalized to GAPDH mRNA levels and siRNA‐control samples (mean ± SD).
- Measurements of endogenous IGS‐rRNA levels in NIH 3T3 cells depleted of DHX9 by siRNA. Data from four experiments have been measured for endogenous transcripts as described in (A).
- Schema depicts the domain organization of DHX9 and the strategy used to measure processing upon expression of plasmids expressing GFP‐DHX9 in NIH 3T3 cells depleted of DHX9 by siRNA. Values are from four independent experiments and were calculated as described in (A).
IGS‐rRNA is composed of three sequence elements, namely spacer region, enhancer repeats and pRNA, 16, 19 (Fig 3A). To determine whether DHX9 has a preferential association with one of these sequences, we performed electrophoretic mobility shift (EMSA) competition assays. Increasing amounts of in vitro transcripts corresponding to selected regions of murine rRNA were used to compete for binding of recombinant TIP5 or DHX9 to radiolabeled runoff transcripts from pBluescript (RNAMCS) 18. Consistent with previous results, TIP5 displayed a higher binding affinity for pRNA than a control‐RNA sequence since only pRNA can efficiently compete for the binding to TIP5 (Fig 3B) 13, 16, 18. DHX9 bound to RNA, forming high‐molecular‐weight complexes. However, the specificity of DHX9 for pRNA was lower than the one observed for TIP5. Indeed, control‐RNA could also compete for binding, although to a less extent since higher amounts of transcripts were required (Fig 3C). The interaction of DHX9 with nucleic acids is specific for RNA as shown by the higher affinity for pRNA than rDNA promoter sequences (Fig 3D). DHX9 displayed a slight preferential binding for spacer and enhancer RNA sequences as compared to pRNA (Fig 3E). These results are consistent with RNA immunoprecipitation (RIP) analyses of formaldehyde cross‐linked NIH 3T3 cells showing that DHX9 directly associates with IGS‐rRNA‐specific sequences but not with pRNA and that this interaction depends on its RNA‐binding domains DSRM and RGG (Fig 3F and G). Together these data support the GRNA chromatography results identifying DHX9 as an IGS‐rRNA‐specific binding factor. Finally, the preferential association of DHX9 for IGS‐rRNA sequences upstream of the pRNA element is further supported by the lack of DHX9 in GRNA chromatography experiments using pRNA as bait (data not shown).
Figure 3. DHX9 associates with RNA .
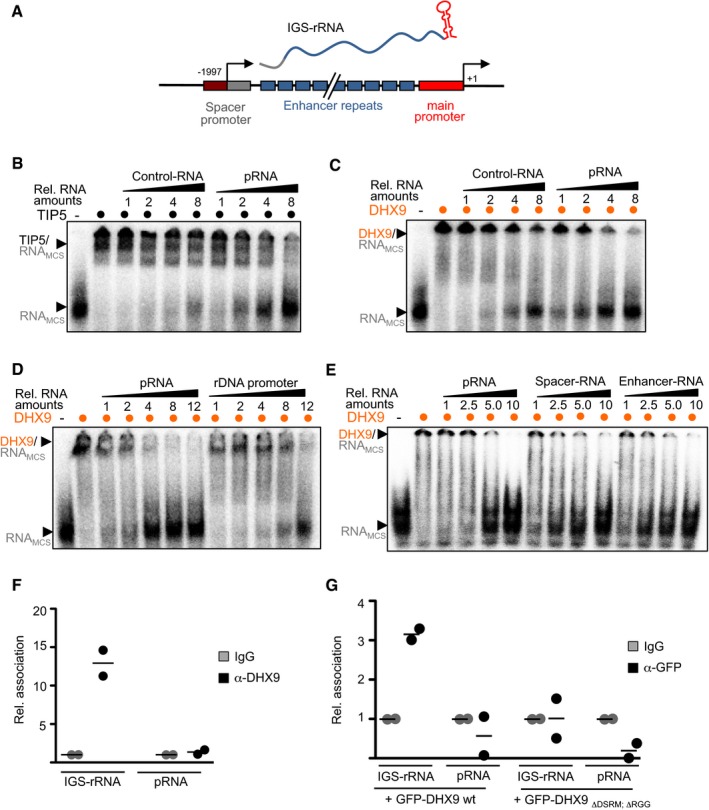
- Schema depicts the mouse 5′‐rRNA gene organization and IGS‐rRNA sequence composition: Spacer promoter (gray), enhancer repeats (blue), main promoter (red, pRNA), and transcription start sites of IGS‐rRNA (−1,997) and 45S pre‐rRNA (+1).
- Increasing equal moles of in vitro transcripts corresponding to control‐RNA or pRNA (starting from 0.32 pmoles) were used to compete for binding of 50 ng recombinant TIP51–600 to radiolabeled runoff transcripts from pBluescript (RNAMCS). RNA/protein complexes were analyzed by EMSA.
- Binding 75 ng of recombinant DHX9 to RNA was analyzed as described in (B). Competition was performed using increasing equal moles of in vitro transcripts corresponding to control‐RNA or pRNA (starting from 0.08 pmoles).
- DHX9 preferentially binds to RNA than DNA. The same nucleotide sequences as RNA (pRNA) or double‐stranded DNA (rDNA; starting from 0.08 pmoles) were used to compete for binding of 75 ng DHX9 to radiolabeled MCS‐RNA.
- The binding of 75 ng DHX9 was analyzed with equal moles of pRNA, spacer, and enhancer repeat transcripts (starting from 0.08 pmoles).
- DHX9 associates in vivo with IGS‐rRNA. RNA immunoprecipitation (RIP) analysis from formaldehyde cross‐linked NIH 3T3 cells. Scatter plot represents the values and the mean of two independent experiments.
- DHX9 RNA‐binding domains are required for the association with IGS‐rRNA. RIP analysis from NIH 3T3 cells transfected with GFP‐DHX9 and GFP‐DHX9ΔDSRM‐ΔRGG expression plasmids. RNA was measured by qPCR through amplification of IGS‐rRNA (−1,997/−1,905) and pRNA (−165/−1) sequences. Values are normalized to input and IgG values from two independent experiments. Scatter plot represents the values and the mean of two independent experiments.
DHX9 associates with TIP5 independently of RNA
The ability of DHX9 to bind IGS‐rRNA prompted us to investigate whether RNA mediates the association of DHX9 with TIP5. We performed co‐immunoprecipitations from HEK293T cells transfected with plasmids expressing Flag‐HA‐TIP5 and GFP‐DHX9 or GFP‐DHX9 mutants (DHX9K417R, DHX9ΔDSRM, DHX9ΔRGG; Fig 4A). As shown in Fig 4B, DHX9 mutations inactivating the ATPase activity (GFP‐DHX9K417R) did not affect the association with TIP5. Remarkably, RNase A treatment did not alter DHX9‐TIP5 interaction. Consistent with these results, the RNA‐binding domains of DHX9 (DSRM and RGG) were not required for TIP5‐DHX9 interaction (Fig 4C). Although co‐expression of DHX9ΔDSRM and DHX9ΔRGG mutants negatively affects expression of ectopic Flag‐HA‐TIP5 (but not of the endogenous protein, Fig EV3), their association with TIP5 was similar to the one observed with wild‐type DHX9. Similarly, TIP5W531G,Y532A, a mutant with impaired RNA‐binding ability 16, efficiently interacted with DHX9 (Fig 4D). Taken together, these results indicate that the association of DHX9 with TIP5 does not require RNA. Considering the preferential binding of TIP5 to pRNA sequences 13 and the association of DHX9 with IGS‐rRNA, it may be possible the TIP5‐DHX9 complex can simultaneously associate with IGS‐rRNA through the interaction of TIP5 with pRNA sequences and the binding of DHX9 to the rest of the unprocessed transcript.
Figure 4. TIP5‐DHX9 interaction is not mediated by RNA .
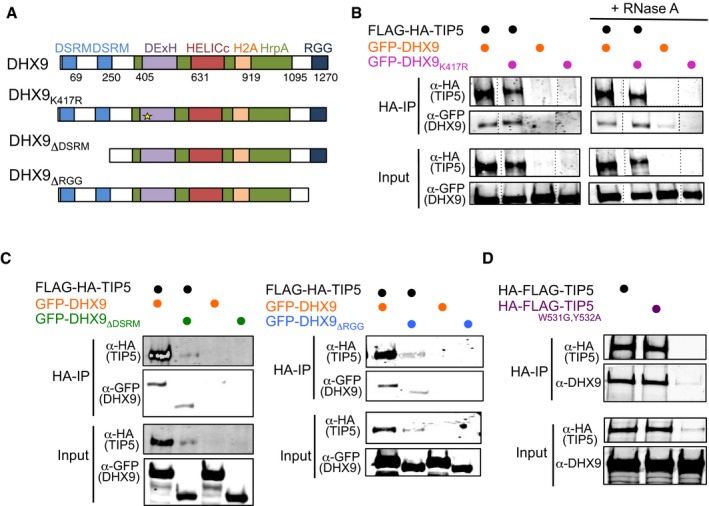
- Schema represents the domain organization of DHX9 and the mutants used for the interaction study.
- Anti‐FLAG immunoprecipitation from HEK293T cells expressing FLAG‐HA‐TIP5 and GFP‐DHX9 or GFP DHX9K4173. Bead‐bound FLAG‐TIP5 immunoprecipitates were incubated with RNase A. After washing, co‐precipitated proteins were visualized with anti‐HA and ‐GFP antibodies. The data show one representative experiment out of two independent experiments.
- Anti‐FLAG immunoprecipitation from HEK293T cells expressing FLAG‐HA‐TIP5, GFP‐DHX9, and GFP‐DHX9 mutants ΔDSRM (left panel) or ΔRGG (right panel).
- Anti‐FLAG immunoprecipitation from HEK293T cells expressing FLAG‐HA‐TIP5 or the RNA‐binding deficient mutant FLAG‐HA‐TIP5W513G,Y532A.
Source data are available online for this figure.
Figure EV3. Ectopic expression of DHX9 wild type and mutants does not affect TIP5 endogenous levels.
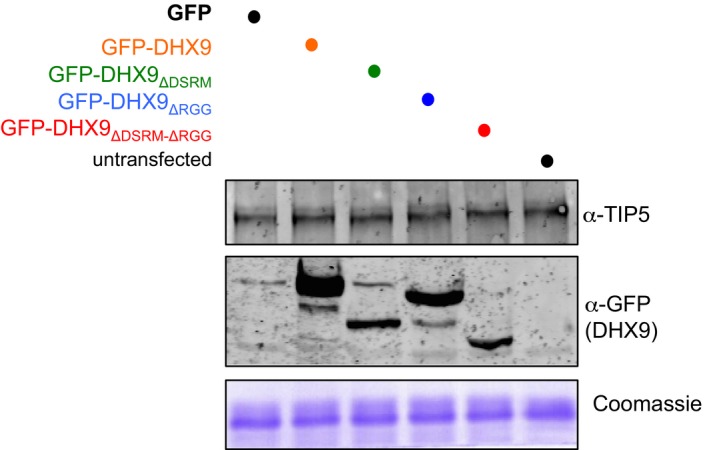
Western blot of lysates of HEK293 cells transfected with the indicated DHX9 expression vectors. Coomassie staining serves as control for equal loading between samples. Source data are available online for this figure.
DHX9 is required for the formation of heterochromatin at rRNA genes through processing of IGS‐rRNA into pRNA
The results above indicated that DHX9 is required for processing of IGS‐rRNA into pRNA. Previous findings showed that only mature pRNA could guide TIP5 to rRNA genes by promoting the association of TIP5 with TTF1, a nucleolar docking factor bound to the main promoter of rRNA genes 13. The unprocessed IGS‐rRNA alone, on the other hand, destroys the interaction with TTF1 and impairs TIP5 recruitment to rRNA genes. We therefore investigated whether depletion of DHX9 affects the association of TIP5 with rRNA genes, TIP5 nucleolar localization, and rRNA gene silencing. We analyzed NIH 3T3 cells, in which the function of TIP5 and pRNA in establishing silencing at 40–50% of rRNA genes has been well characterized 3, 16, 19. While knockdown of DHX9 did not alter TIP5 levels (Fig 5A), TIP5 occupancy at the rRNA genes was impaired in the absence of DHX9 (Fig 5B). Consistent with these results, the nucleolar localization of TIP5 was lost upon depletion of DHX9 (Fig 5C). Remarkably, upon transfection of pRNA in cells depleted of DHX9 the localization of TIP5 was retained in nucleoli (Fig 5D). Taken together, these results indicate that DHX9 is required for the association of TIP5 with rRNA genes through its activity in processing IGS‐rRNA into pRNA. We then analyzed whether the nucleolar localization of DHX9 depends on TIP5. Since all DHX9 antibodies tested so far failed to detect a specific signal of DHX9 in mouse cells by immunofluorescence analyses (data not shown), we measured the cellular localization of DHX9 upon TIP5 knockdown in U2OS cells (human osteosarcoma cells). U2OS cells showed a strong enrichment and specific signal of DHX9 in nucleoli (Figs 1C and 5E). Knockdown of TIP5 did not affect DHX9 levels (Fig EV4); however, the enrichment of DHX9 in nucleoli was lost and DHX9 was mostly localized in nucleoplasm (Fig 5E). Consistent with these results, the association of DHX9 with rRNA genes in NIH 3T3 cells decreased upon depletion of TIP5 (Fig 5F), suggesting that DHX9 requires the association of TIP5 with mature pRNA to bind to rRNA genes. Finally, NIH 3T3 cells depleted of DHX9 showed a reduction in rRNA gene silencing. Upon DHX9 knockdown, rRNA genes displayed decreased levels of repressive epigenetic marks such as CpG methylation and H3K9me2 and increased 45S pre‐rRNA transcription to levels similar to what reported upon TIP5 knockdown in NIH 3T3 cells 42 (Figs 5G–I and EV1G). Taken together, these results support a role of DHX9 in guiding TIP5 to rRNA genes and subsequent establishment of nucleolar heterochromatin in a manner dependent on the production of mature pRNA.
Figure 5. DHX9 mediates recruitment of TIP5 and establishment of rRNA gene heterochromatin through processing of IGS‐rRNA into pRNA .
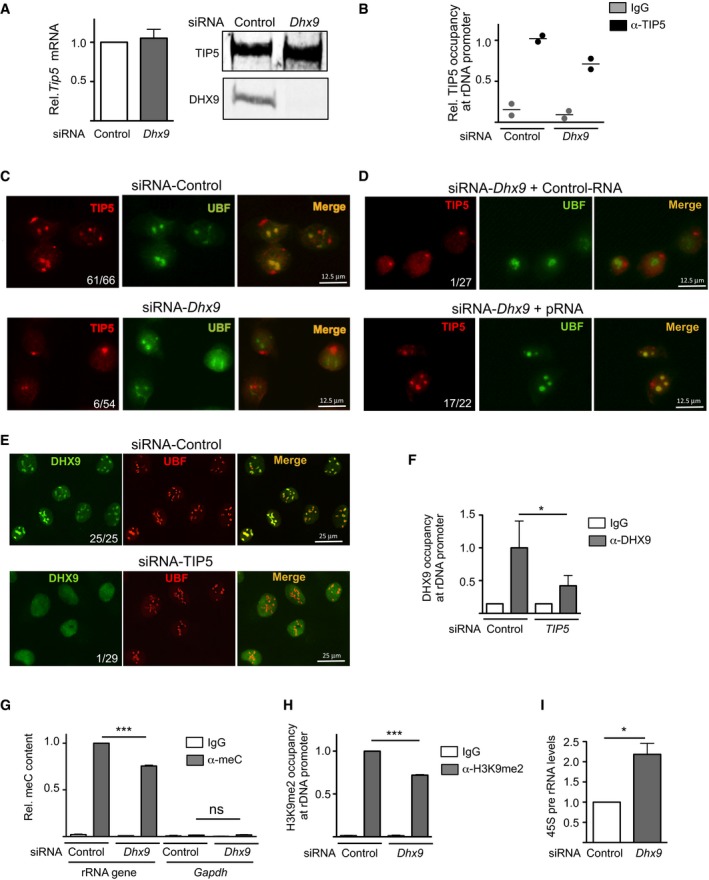
- DHX9 knockdown does not affect TIP5 levels. mRNA and protein levels of TIP5 in NIH 3T3 cells depleted of DHX9 by siRNA. TIP5 mRNA values were normalized to GAPDH mRNA and to siRNA‐control cells. Values (mean ± SD) are from three independent experiments.
- DHX9 is required for the association of TIP5 with rRNA genes. ChIP analysis of TIP5 occupancy at rRNA genes in NIH 3T3 cells depleted of DHX9 by siRNA. Data from two independent experiments are represented as bound over input and normalized to siRNA‐Control cells. Scatter plot represents the values and the mean of two independent experiments.
- DHX9 is required for the localization of TIP5 in nucleoli. Immunofluorescence with anti‐TIP5 and anti‐UBF of NIH 3T3 cells treated with siRNA‐Control or‐DHX9. Numbers refer to cells showing TIP5 nucleolar localization relative to the number of analyzed cells.
- Retention of TIP5 in nucleoli depends on DHX9‐mediated production of mature pRNA. Immunofluorescence with anti‐TIP5 and anti‐UBF of DHX9‐depleted NIH 3T3 cells transfected with Control‐RNA or pRNA.
- Nucleolar localization of DHX9 depends on TIP5. Immunofluorescence with anti‐DHX9 and anti‐UBF of U2OS cells treated with siRNA‐Control or siRNA‐TIP5. Numbers refer to cells showing DHX9 nucleolar localization relative to the number of analyzed cells.
- TIP5 is required for the association of DHX9 with rRNA genes. ChIP analysis of DHX9 occupancy at rRNA genes in U2OS cells depleted of TIP5 by siRNA. Values (mean ± SD) from three independent experiments are represented as bound over input and normalized to siRNA‐Control cells.
- DHX9 is required for rRNA gene silencing. Methylated DNA immunoprecipitation (MeDIP) analysis of rRNA gene promoter using anti‐5mC antibodies in NIH 3T3 cells upon DHX9 knockdown. Enrichments were calculated relative to input and normalized to rRNA genes in control cells. Low enrichment of GAPDH sequences (free of CpG methylation) ensures for the specificity of the measurement. Values (mean ± SD) are from three independent experiments.
- ChIP analysis of H3K9me2 at rRNA gene promoter in NIH 3T3 cells upon DHX9 knockdown. Enrichments were calculated relative to input and normalized to control cells. Values (mean ± SD) are from three independent experiments.
- Knockdown of DHX9 upregulates rRNA transcription. RT–qPCR of 45S pre‐rRNA levels in NIH 3T3 cells upon DHX9 knockdown. Values (mean ± SD) from three independent experiments were normalized to GAPDH mRNA and to control cells.
Figure EV4. TIP5 knockdown does not affect DHX9 levels.
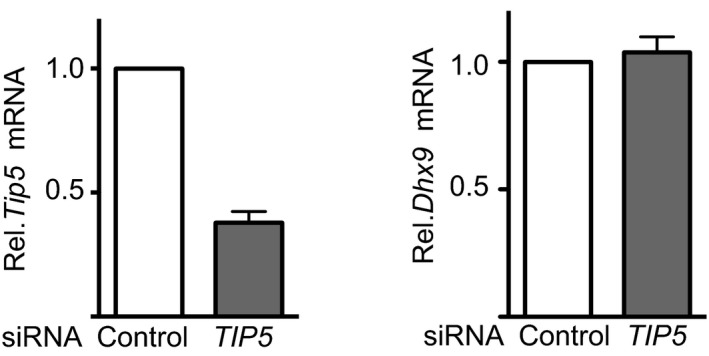
mRNA of TIP5 and DHX9 in NIH 3T3 cells depleted of TIP5 by siRNA. TIP5 mRNA values were normalized to GAPDH mRNA and to siRNA‐control cells. Average and standard deviation (n = 3) are shown for each point.
DHX9 localizes at nucleoli only upon ESC differentiation
Processing of IGS‐rRNA into pRNA is impaired in ESCs and activated only upon ESC differentiation, which coincides with the timing of de novo heterochromatin formation at rRNA genes 13. To determine whether the inhibition of IGS‐rRNA processing in ESCs is dependent on alterations in DHX9 expression or cellular localization, we analyzed and compared ESCs before and after 4 days of differentiation achieved upon leukemia inhibitory factor (LIF) withdrawal. DHX9 is expressed in both ESCs and differentiated cells, the latter showing a slight increase at mRNA and protein levels (Fig EV5A and B). Remarkably, ChIP analysis revealed that the association of DHX9 with rRNA genes was higher in differentiated cells than in ESCs (Fig 6A). Therefore, although DHX9 and TIP5 associate in ESCs (Fig EV1D), in the absence of mature pRNA this interaction is not sufficient to recruit DHX9 or TIP5 to rRNA genes. Consistent with these results, analysis of GFP‐DHX9 in ESCs revealed its localization in nuclei without any evident enrichment in nucleoli (Fig 6B–D). In contrast, upon differentiation GFP‐DHX9 drastically changed its position within the nucleus and all cells showed DHX9 with an exclusive localization within nucleoli and a weak signal in the rest of nucleoplasm (Fig 6C). The accumulation of DHX9 in nucleoli and its association with rRNA genes only upon ESC differentiation correlates well with the activation of IGS‐rRNA processing into pRNA, which takes place in nucleoli 19. However, how DHX9 is excluded from nucleoli of ESCs still remains unknown.
Figure EV5. Expression levels of DHX9 in ESCs and NPCs.
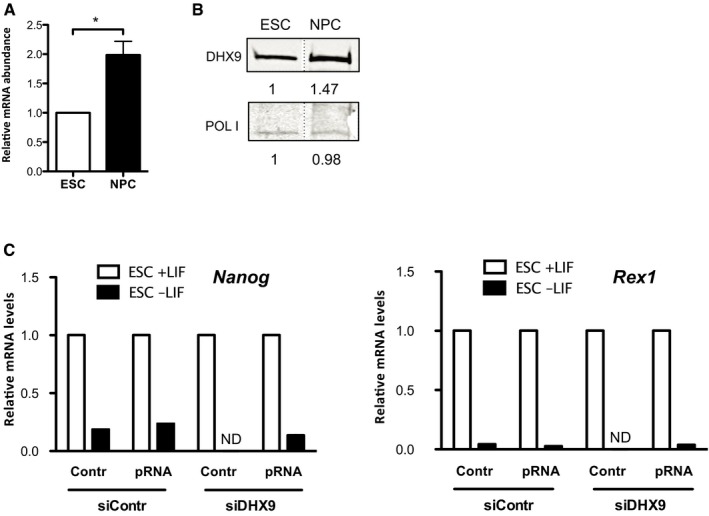
-
A, BDHX9 transcript (A) and protein (B) levels in ESCs and NPCs. RT–qPCR values (mean ± SD) from three independent experiments were normalized to Rps12 mRNA levels and to ESCs. Statistical significance (P‐values) for the experiments was calculated using the paired two‐tailed t‐test (*P < 0.05).
-
CqRT–PCR showing expression levels of pluripotency markers Nanog and Rex1 in ESCs + LIF and differentiated cells obtained upon withdrawal of LIF (ESCs−LIF). Values were normalized to rsp12 mRNA and the corresponding ESCs. Because of the few amounts of differentiated cells obtained upon DHX9 knockdown, mRNA levels in these samples could not be determined (ND). Data represent the mean of two independent experiments.
Source data are available online for this figure.
Figure 6. DHX9‐mediated production of mature pRNA is required for ESC differentiation.
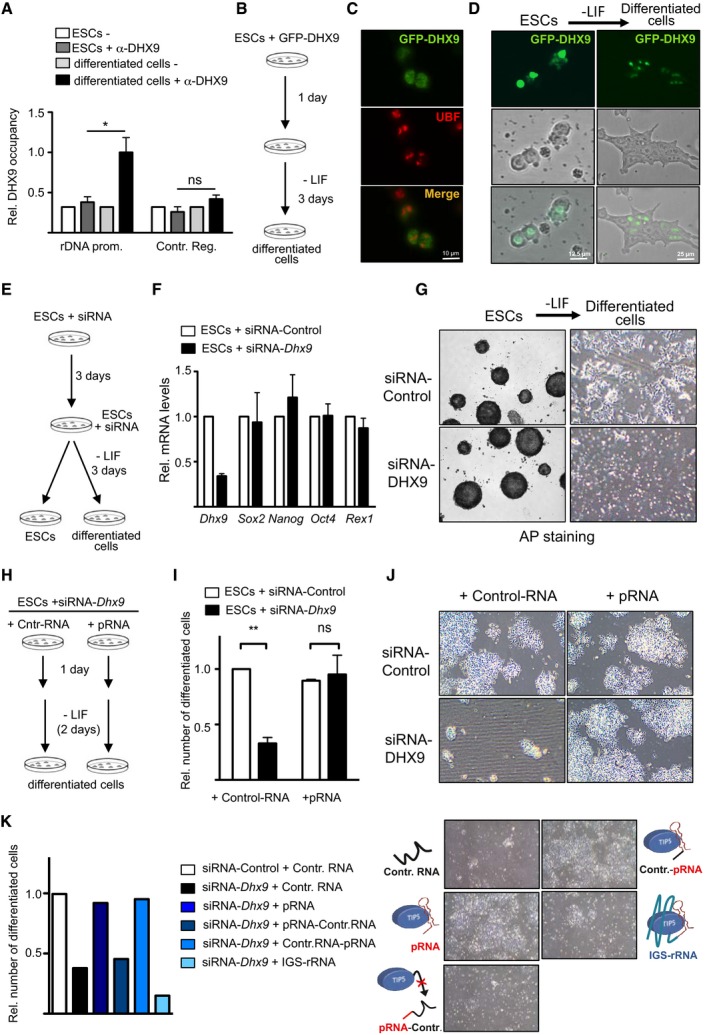
-
ADHX9 associates with rRNA genes only upon ESC differentiation. ChIP analysis using DHX9 antibodies in ESCs and cells after 4 days of differentiation. Values (mean ± SD) are from three independent experiments and calculated relative to input and to values of rRNA sequences in ESCs.
-
B–DDHX9 is not enriched in nucleoli of ESCs. (B) Schema depicts the experimental strategy to measure DHX9 localization in ESCs and after 3 days of differentiation. (C) Immunofluorescence analysis with GFP and UBF antibodies of ESCs transfected with GFP‐DHX9. (D) Life cell imaging of ESCs transfected with GFP‐DHX9 (right). Nucleoli can be visualized by phase contrast.
-
E–GDHX9 is required for ESC differentiation. (E) Schema depicts the experimental strategy to analyze differentiation of ESCs depleted of DHX9 by siRNA. (F) RT–qPCR analysis of DHX9 and pluripotency factors in ESCs upon DHX9 knockdown. Expression levels were normalized to Rps12 mRNA amounts and control ESCs. Average and standard deviation (n = 3) are shown for each point. (G) Representative images of alkaline phosphatase staining of ESCs and cells after 3 days of differentiation.
-
H–JDHX9‐mediated production of mature pRNA is required for ESC differentiation. (H) Schema depicts the experimental strategy to analyze differentiation of ESCs depleted of DHX9 by siRNA in the presence of mature pRNA. (I, J) Quantification and representative images of ESCs after 3 days of differentiation. Values (mean ± SD) represent number of differentiated cells from three independent experiments relative to control cells (siRNA‐control + Control‐RNA).
-
KThe association of TIP5 with mature pRNA is required for ESC differentiation. Quantification and representative images of ESCs treated with siRNA‐DHX9 and transfected with pRNA mutants and IGS‐rRNA after 3 days of differentiation. Values represent number of differentiated cells to control differentiated cells (siRNA‐control + Control‐RNA). Data represent the mean of two independent experiments.
DHX9‐mediated processing of IGS‐rRNA is required for ESC differentiation
Previous work has shown that during differentiation, the euchromatic and transcriptional permissive ESC genome remodels into a more repressive and heterochromatic structure 10, 43. Similarly, rRNA genes in ESCs are euchromatic and only upon ESC differentiation heterochromatin in the nucleolus is established 13. Determinant for the lack of heterochromatin in nucleoli is the impairment of IGS‐rRNA processing since addition of mature pRNA into ESCs was sufficient to induce heterochromatin formation at rRNA genes. Remarkably, the presence of ectopic pRNA in ESCs also initiated the establishment of highly condensed chromatin structures outside of the nucleolus, which was similar to what observed in differentiated cells. Moreover, such ESCs were primed for differentiation as indicated by the increased expression of genes implicated in developmental and differentiation processes. Therefore, we asked whether the production of mature pRNA mediated by DHX9 is necessary for ESC differentiation. Upon DHX9 knockdown, ESCs did not show alterations in important molecular features of the undifferentiated state such as cell proliferation, expression of the pluripotency genes, cell morphology, and alkaline phosphatase (AP) staining (Fig 6E–G). To determine whether DHX9 depletion affects ESC differentiation, we induced monolayer differentiation of ESCs treated with siRNA‐control or siRNA‐Dhx9. After 3 days, control cells displayed morphological structures typical of differentiated cells were negative for AP staining and had downregulation of pluripotency markers (Figs 6G and EV5C). In contrast, after induction of differentiation, cells depleted of DHX9 did not attach to the plate and underwent cell death (Fig 6G). To assess whether the requirement of DHX9 for ESC differentiation depends on its role in IGS‐rRNA processing, we transfected mature pRNA into ESCs treated with siRNA‐control or siRNA‐Dhx9 and subsequently induced differentiation 1 day later (Fig 6H). Both ESCs transfected with siRNA‐control and with control‐RNA or pRNA efficiently differentiated whereas ESCs depleted of DHX9 and transfected with control‐RNA underwent cell death (Fig 6I and J). Remarkably, transfection of mature pRNA in ESCs knocked down for DHX9 was sufficient to entirely restore differentiation capability to same efficiencies as in control cells (Fig 6I and J). Consistent with the results described above, DHX9‐depleted ESCs transfected with IGS‐rRNA were unable to differentiate (Fig 6K), supporting the role of mature pRNA to drive cells into differentiation. Since the association of TIP5 with pRNA is key to establish heterochromatin at rRNA genes, we analyzed the ability of DHX9‐depleted ESCs to differentiate using a pRNA mutant whose 3′‐region necessary for TIP5 association was replaced with a control‐RNA sequence (pRNA‐Control) 13, 15, 16. As shown in Fig 6K, DHX9‐depleted ESCs transfected with pRNA‐Control failed to differentiate. In contrast, replacement of the 5′‐pRNA region (Control‐pRNA), which is not required for the association of TIP5 and recruitment to rRNA genes 13, was sufficient to entirely restore ESC differentiation capacity, underscoring the importance of TIP5/pRNA‐mediated heterochromatin formation at rRNA genes during ESC differentiation. Together, these results indicate that processing of IGS‐rRNA into pRNA mediated by DHX9 is an event required for early differentiation and highlight the role of the chromatin state of rRNA genes in controlling this process.
Discussion
In this work, we have shown that the RNA helicase DHX9 is required to process IGS‐rRNA into pRNA and this activity is required for ESC differentiation. Because of the role of pRNA to establish heterochromatin at rRNA genes and the fact that mature pRNA is produced only upon ESC differentiation, these results strongly support a role of the chromatin state of rRNA genes for the ability of ESCs to undergo differentiation.
The remodeling of the open and euchromatic genome structure of ESCs toward the formation of highly condensed heterochromatic structures marks the exit of ESCs from pluripotency and the entry into differentiation program 8. This change in chromatin state is also characteristic of rRNA genes, a fraction of which only acquires epigenetic silent marks upon differentiation 13. The formation of heterochromatin in the nucleolus during differentiation is also timely coincident with the clustering of highly condensed heterochromatin at nucleoli or at the nuclear periphery 11, 12. The link between the nucleolus and heterochromatin is also true for the inactive X‐chromosome that contacts the nucleolus to duplicate its silent chromatin structures during mid‐to‐late S phase 44. Similarly, lamina‐associated domains (LADs) that are relatively gene poor and have a repressive chromatin signature were also found relocated next to the nucleolus 45, 46. Thus, by analogy with the nuclear periphery 47, 48 also the clustering of heterochromatin at the nucleolus might play a role for the establishment of mammalian heterochromatin.
Our previous work has started to highlight the nucleolus as an important regulator that orchestrates the formation of heterochromatin. Indeed, inducing heterochromatin at rRNA genes in ESCs through addition of mature pRNA resulted in a drastic change of genome architecture with the appearance of highly condensed heterochromatic blocks outside the nucleolus 13. These changes were also accompanied by a global increase in H3K9me2, maturation of heterochromatin at repetitive sequences—such as major and minor satellites—and their transcriptional repression as found in differentiated cells. Although the mechanisms through which the nucleolus acts in this global restructuring of genome architecture is yet unknown, what is clear is that it depends on the chromatin state of rRNA genes since the gain of heterochromatin at rRNA genes induces the rest of the genome to remodel into highly condensed structures.
The finding that DHX9 is required for processing of IGS‐rRNA into pRNA, a reaction that is activated only upon ESC differentiation, allowed the determination that inhibition of this process impairs ESC differentiation. Indeed, the defects of differentiation observed upon DHX9 knockdown could be reverted solely by the addition of mature pRNA, underscoring the importance of DHX9 in the processing of IGS‐rRNA into pRNA and subsequent establishment of nucleolar heterochromatin in early development. Remarkably, the requirement of DHX9 in early development is also supported by previous data indicating that DHX9 is essential for gastrulation and Dhx9 −/− embryos do not progress further than E7.5 in development 49. Moreover, the switch in the cellular localization of DHX9, which is lowly enriched in nucleoli of ESCs but exclusively present in nucleoli of differentiated cells, correlates well with the activation of IGS‐rRNA processing that takes place in nucleoli only upon exit of pluripotency. However, how the cellular localization of DHX9 is regulated in early development is yet unclear and will be an aim of our future studies.
The contribution of DHX9‐mediate production of pRNA in ESCs differentiation supports previous observations showing defects in ESC differentiation upon depletion of TIP5 13. Thus, impairment of rRNA gene heterochromatin achieved by alteration of pathways upstream TIP5 silencing activity without altering TIP5 expression levels brought to similar conclusion, that is the chromatin state of rRNA genes plays a role for the exit from pluripotency. This is further supported by the fact that pRNA mutants with impaired ability to associate with TIP5 failed to rescue the differentiation defects observed in ESCs upon depletion of DHX9. Recent studies have proposed that elevated rRNA biosynthesis sustains pluripotency in mouse and human ESCs since rRNA downregulation through chemical inhibition of Pol I activity or deletion of fibrillarin induces differentiation 21, 22. Although the set‐up of our experiments does not allow us to determine whether the impairment of ESC differentiation is due to the maintenance of high rRNA transcription or to the lack of heterochromatin in nucleolus, we favor the latter case. Indeed, it is unlikely that cells with high rRNA level undergo cell death since ribosome biogenesis is well known to be positively correlated with cell viability and proliferation 1. Thus, our data favor a model in which the nucleolus is not only the cellular compartment where ribosomes are produced but it is also a central component of nuclear architecture that coordinates the balance between euchromatin and heterochromatin according to developmental stages. Likewise, the function of rRNA genes might not only be limited to the synthesis of rRNA, a model which is in agreement with early studies showing that the fraction of silent rRNA genes present in each differentiated cell does not change its transcriptional state even under conditions of high metabolic activities 4. The link between rRNA genes and the chromatin architecture of the rest of the genome is also supported by previous results in Drosophila showing that deletion of rRNA repeats reduced heterochromatin content elsewhere in the genome 50. A similar observation was also found upon knockdown of TIP5 in NIH 3T3 cells, which induced not only a decrease of rRNA gene silencing but also the loss of perinucleolar heterochromatic blocks and the reduction of silent histone marks at pericentric heterochromatin 42.
Our data suggested that the splicing pathway might be implicated in the processing of IGS‐rRNA into pRNA. The spliceosome was indeed the major hit obtained through cellular component and pathway analysis of both IGS‐rRNA pulled‐down proteins and DHX9‐associated factors from ESCs, differentiated ESCs, NIH 3T3, and HEK293 cells. Remarkably, spliceosome core components (i.e., U2 Small Nuclear RNA Auxiliary Factor 2 U2AF2, Splicing factor 3B subunit 3 SF3B3, pre‐mRNA Processing Factor 3 and 8, PRP3 and 8) have been found in the analysis of the proteome of nucleoli 51. The possibility of a role of the spliceosome in the production of mature pRNA is also supported by the absence of ribonucleases among the identified IGS‐rRNA interacting proteins. Xrn2 (5′–3′ exoribonuclease 2) was the unique ribonuclease we identified as DHX9‐associated proteins in three out of four experiments. However, analyses of IGS‐rRNA processing in cells depleted of Xrn2 by siRNA did not reveal any substantial defect as the ones found upon DHX9 knockdown (data not shown). Thus, the strong interaction of DHX9 with spliceosome components and the lack of association with ribonucleases with function in IGS‐rRNA processing suggest a mechanism where DHX9 through binding to TIP5 and/or IGS‐rRNA recruits the spliceosome complex for IGS‐rRNA processing. The mechanisms of this reaction are currently under investigation.
Taken together, the results provided molecular insights into the biogenesis of lncRNAs that modulates different features of the same lncRNA according to developmental stage. Moreover, the data supported a model in which the state of rRNA gene chromatin is part of the regulatory network that controls the exit from pluripotency and the initiation of differentiation pathways.
Materials and Methods
Cell culture
NIH 3T3, U2OS, and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Gibco).
One hundred and twenty‐nine mouse embryonic stem cells (E14 line) were cultured in DMEM‐F12 and Neurobasal medium (1:1, Gibco), supplemented with 1× N2/B27 (Gibco), 1× penicillin/streptomycin/l‐glutamine (Gibco), 50 μM β‐mercaptoethanol (Gibco), recombinant leukemia inhibitory factor, LIF (ESGRO, 1,000 U/ml) and MEK and GSK3β inhibitors, 2i (Stemolecule CHIR99021 and PD0325901, 3 and 1 μM, respectively). ESCs were seeded at a density of 50,000 cells/cm2 in culture dishes (Corning® CellBIND® surface) treated with 0.1% gelatine without feeder layer. Propagation of cells was carried out every 2 days using trypsin 0.5× for enzymatic cell dissociation. ESCs were differentiated by culturing for 48–72 h in complete medium: DMEM, 10% FCS, 1 mM sodium piruvate (Sigma), 1× NEAA (Gibco), 1× penicillin/streptomycin/l‐glutamine, 100 μM β‐mercaptoethanol.
The differentiation toward neural progenitor cells (NPC) was obtained according to previously established protocol 52. In brief, differentiation employed a suspension‐based embryoid bodies formation (Bacteriological Petri Dishes, Bio‐one with vents, Greiner®) in neural differentiation media (DMEM, 10% FCS, 1× MEM NEAA, 1× penicillin/streptomycin/l‐glutamine, 100 μM β‐mercaptoethanol). During the 8‐day differentiation procedure, media were exchanged every 2 days. In the last 4 days of differentiation, the media were supplemented with 2 μM retinoic acid (RA) to generate neuronal precursors (i.e., Pax‐6‐positive radial glial cells).
Transfections
Plasmids and siRNAs were transfected in HEK293T cells using calcium phosphate protocol. NIH 3T3 mouse fibroblasts, U2OS and ESCs were transfected with the indicated siRNAs (50 nM siRNA) using Lipofectamine® RNAiMAX (Life Technologies) in Opti‐MEM® GlutaMAX™ (GIBCO) reduced‐serum medium. Analysis of differentiated transfected ESCs was performed using consecutive transfections. To test ESC ability to differentiate upon DHX9 knockdown, 3 days after the first transfection, equal amounts of ESCs (e.g., siRNA‐control and siRNA‐DHX9 treated cells) were again transfected and induced to differentiate in complete media (G‐MEM, 10% FCS, sodium pyruvate 100 mM, 1× MEM NEAA, l‐glutamine) by withdrawal of LIF and 2i. Transfections of siRNA together with synthetic RNAs (1 mg/ml) were performed using Transit‐X2® transfection reagent (Mirus), and the differentiation was induced 1 day after transfection by withdrawal of LIF and 2i.
Efficiencies of siRNA‐mediated depletions and synthetic RNA levels were monitored by qRT–PCR 3–4 days post‐transfection.
RNA extraction, reverse transcription, and quantitative PCR (RT–qPCR)
RNA was purified with TRIzol reagent (Life Technologies). Residual contaminating genomic DNA was removed with Ambion® TURBO™ DNase according to manufacturer's instructions. RNA was primed with random hexamers and reverse‐transcribed to cDNA. Amplification of samples without reverse transcriptase assured absence of genomic or plasmid DNA (data not shown). The relative transcription levels were determined by normalizing to Rps12 or GAPDH mRNA levels, as indicated. Measurements of IGS‐rRNA processing were performed as previously described 53. Reverse transcription was obtained using primers −20/−1 Rev (endogenous transcripts) or Vector RT primer hybridizing vector sequences (reporter assay). Processing efficiency was calculated by normalizing amplifications of the 5′‐IGS‐rRNA region (endogenous: primers −1,994/−1,975 For and −1,922/−1,905 Rev; reporter: primers −1,994/−1,975 For and Vector 1 Rev) to amplification of pRNA sequences (endogenous: primers −165/−145 For and −39/−20 Rev; reporter: primers −165/−145 For and Vector 2 Rev). qRT–PCR was performed with SensiMix SYBR Hi‐ROX Mix (Bioline) on a Rotor‐Gene Q (Qiagen). Statistical significance (P‐values) of the difference in expression levels between genes was calculated using the two‐sample paired t‐test. Primer sequences are listed in Table EV1.
Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed as previously described 54. Briefly, formaldehyde 1% was added to cultured cells to cross‐link proteins to DNA. Isolated nuclei were then lysed and sonicated using a Bioruptor ultrasonic cell disruptor to shear genomic DNA to an average fragment size of 200 bp. 20 μg of chromatin was diluted to a total volume of 500 μl with IP buffer (16.7 mM Tris–HCl, pH 8.1, 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton X‐100) and pre‐cleared with 10 μl packed Sepharose beads for 2 h at 4°C. Pre‐cleared chromatin was incubated overnight with the indicated antibodies. The next day, Dynabeads protein‐A (or ‐G, Millipore) were added and incubated for 4 h at 4°C. After washing, bound chromatin was eluted with the elution buffer (1% SDS, 100 mM NaHCO3). Upon proteinase K digestion (50°C for 1 h) and reversion of cross‐linking (65°C, overnight), DNA was purified with phenol/chloroform, ethanol precipitated and quantified by qPCR using the primers listed in Table EV1.
Methylated DNA immunoprecipitation (MeDIP)
200‐bp length DNA fragments were denatured and incubated overnight with 5mC‐antibody (Diagenode) in 200 μl IP buffer (10 mM Na‐phosphate buffer, pH 7.0, 140 mM NaCl, 0.05% Triton X‐100). The day after, 20 μl of protein‐G Dynabeads (Millipore) was added and incubated for 2 h at 4°C. Beads were then washed three times at RT for 10 min with IP buffer and subsequently incubated with 250 μl proteinase K digestion mix (50 mM Tris–HCl, pH 8, 0.5% SDS, 10 mM EDTA, 2 μg proteinase K) overnight at 42°C. DNA was purified with phenol/chloroform, ethanol precipitated and quantified by qPCR.
AP staining
Cells were fixed in 4% paraformaldehyde for 10 min, washed with AP buffer (100 mM Tris‐Cl pH 9.5, 100 mM NaCl, 50 mM MgCl2), and then incubated for 5–10 min in BCIP®/NBT liquid substrate system (Sigma). The staining was blocked with 10 mM Tris and 1 mM EDTA for 10 min.
GRNA chromatography
GRNA chromatography was performed as previously described 23. Briefly, Glutathione Sepharose matrix was incubated for 1 h rotating at 4°C with GST‐λ phage N antiterminator fusion protein and equimolar amounts of in vitro synthesized BoxB‐RNAs (i.e., BoxB‐Control‐RNA and BoxB‐IGS‐rRNA) to obtain the RNA affinity matrix (GRNA resins). The GRNA resins were incubated overnight rotating at 4°C with nuclear extracts from NIH 3T3 cells or NPC in BB buffer (50 mM Tris–HCl pH 7.5, 100 mM KCl, 2 mM MgSO2, 0.1% NP‐40, 0.1 mg/ml yeast tRNA, 0.01 mg/ml heparin, 1× protease inhibitor cOmplete EDTA‐free Roche), followed by extensive washing and elution of bound proteins using 20 μg/ml of RNase A (Thermo Scientific). Eluted proteins were precipitated in 20% trichloroacetic acid, washed extensively with pure acetone, and dried at 95°C for 5–10 min. The samples were solubilized and incubated overnight at 37°C in 30 μl of buffer (10 mM Tris, pH 8.2, 2 mM CaCl2), 10 μl acetonitrile, 5 μl trypsin (100 ng/μl in 10 mM HCl), and 5 μl of RapiGest™ (1% in water). After centrifugation, the supernatants were dried and resuspended in 0.1% formic acid and analyzed by LC‐MS/MS.
RNA immunoprecipitation
RNA immunoprecipitation was performed as previously described 55. Briefly, 1% final concentration formaldehyde was added to the cell medium to cross‐link proteins to RNA. Nuclei were then isolated in swelling buffer (5 mM HEPES pH 8.0, 85 mM KCl, 0.5% Nonidet P‐40, 1× protease inhibitor cOmplete EDTA‐free Roche) and lysed in nuclei lysis buffer (50 mM Tris–HCl pH 8.0, 10 mM EDTA pH 8.0, 1% w/v SDS, 1× protease inhibitor, 40 U/ml RNase inhibitor). The extracts were diluted tenfold with FA lysis buffer (1 mM EDTA pH 8.0, 50 mM HEPES pH 7.5, 140 mM NaCl, 0.1% w/v sodium deoxycholate, 1% v/v Triton X‐100, 1× protease inhibitor, 40 U/ml RNase inhibitor) and sonicated for 5 min with 30 s on/off cycles in a Bioruptor sonicator (Diagenode). The extracts were pre‐cleared with 20 μl packed Sepharose beads for 1 h rotating at 4°C. Pre‐cleared extracts were then adjusted to 25 mM MgCl2, and 5 mM CaCl2 and incubated at 37°C for 30 min. with 120 μg/ml of DNase I (Fermentas). 5% of extracts was retained as input. Pre‐cleared extracts were incubated overnight with the DHX9 antibody (Abcam ab26271) or GFP Trap® beads (Chromotek) to IP the endogenous or GFP‐tagged overexpressed protein, respectively. The next day, in case of endogenous protein, Dynabeads protein‐A (Millipore) were added and incubated for 4 h at 4°C. After stringent washing, bound RNA–protein complexes were eluted with RIP elution buffer (10 mM EDTA, 100 mM Tris–HCl pH 8.0, 1% SDS, 40 U/ml RNase inhibitor). Upon proteinase K digestion (42°C for 1 h) and reversion of cross‐linking (65°C, 1 h), RNA was purified with an equal volume of acid‐equilibrated phenol/chloroform and isopropanol precipitated. Residual contaminating genomic DNA was removed with Ambion® TURBO™ DNase according to manufacturer's instructions. After isopropanol precipitation, RNA was primed with random hexamers and reverse‐transcribed to cDNA (as described above) and quantified by qPCR using the primers listed in Table EV1.
Immunoprecipitation
Nuclei were obtained by resuspending cells in hypotonic buffer (0.5% NP‐40, 85 mM KCl, 5 mM HEPES, pH 7.4) and subsequent centrifugation at 6,000 g for 10 min at 4°C. Nuclei were then resuspended in nuclear extraction buffer (50 mM Tris–HCl pH 7.5, 0.15 M KCl, 5 mM MgCl2, 0.2 mM EDTA, 20% glycerol, 0.5 mM DTT, 0.5% NP‐40 and 1× protease inhibitor cOmplete EDTA‐free Roche) and sonicated two times for 30 s with a Bioruptor ultrasonic cell disruptor. After DNase I treatment for 1 h at 4°C, extracts were sonicated again for 30 s and centrifuged for 10 min at 4°C with 3,400 g. 0.5 mg of proteins from the resulting supernatant was immunoprecipitated overnight at 4°C using anti‐HA‐Agarose (Sigma) or M2 beads (Sigma). 0.05 mg (10%) of the extracts was later used to check for equal input material. Immunoprecipitates were washed three times at 4°C for 5 min with wash buffer (20 mM Tris–HCl, pH 7.8, 150 mM KCl, 5 mM MgCl2, 0.2 mM EDTA, 10% glycerol, 0.1% Tween and 1× protease inhibitor cOmplete EDTA‐free Roche). Beads were collected by centrifuging for 5 min at 4°C with 500 rcf. Proteins were denatured with 1× Lämmli buffer, separated on SDS–polyacrylamide gel, and analyzed by immunoblotting.
For the analysis of DHX9 interactome, nuclei were obtained by resuspending FLAG‐DHX9 expressing cells in nuclei preparation buffer (10 mM HEPES pH7.6; 1.5 mM MgCl2; 10 mM KCl; 0.5 mM DTT and 1× protease inhibitor cOmplete EDTA‐free Roche). After 10‐min (HEK293T, ESC and ESC differentiated cells) or 30‐min (NIH 3T3) incubation, cells were centrifuged at 3,800 g for 5 min, resuspended in nuclei preparation buffer, and subjected to 20–40 strokes in a dounce homogenizer. Nuclei were collected by centrifugation at 6,000 rcf, resuspended in nuclear extraction buffer, and immunoprecipitation was performed as described above. Elution of DHX9‐interacting proteins was obtained by competition with 100 μg/ml of free FLAG peptide in wash buffer. Proteins of each elution were precipitated with trichloroacetic acid as described above for GRNA chromatography and analyzed by LC‐MS/MS.
Immunofluorescence
TIP5 immunofluorescence was performed as described in ref. 13. Briefly cells were seeded on glass coverslips 72–96 h before analysis and in the case of knockdown experiments transfected with siRNAs as described above. To perform the immunofluorescence, coverslips were washed with 1× PBS and cells were incubated with permeabilization buffer (20 mM Tris, pH 8, 5 mM MgCl2, 0.5 mM EDTA, 25% glycerol, 0.05% Triton X‐100) for 4 min at RT. After three washes with 1× PBS, cells were fixed with ice‐cold pure methanol for 7 min at −20°C, washed again with 1× PBS, and incubated overnight with anti‐TIP5 and anti‐UBF. After washing three times with 1× PBS, cells were incubated with fluorescently labeled secondary antibodies for 1 h at RT, washed again, and stained with DAPI. Immunofluorescent images were digitally recorded.
DHX9 immunofluorescence was performed according to the protocol indicated by the DHX9 antibody manufacturer (Abgent). Briefly cells were seeded on glass coverslips 72–96 h before analysis. To perform the immunofluorescence, the coverslips were washed with 1× PBS and cells were fixed and permeabilized in 1× PBS containing 4% paraformaldehyde and 0.1% Triton X‐100 for 20 min at RT followed by an incubation in blocking solution (1% BSA in PBS) for 30–60 min. After washing three times with 1× PBS, cells were incubated with anti‐DHX9 for 1–3 h at RT, washed three times with 1× PBS, and incubated with fluorescently labeled secondary antibodies for 1 h at RT, washed again, and stained with DAPI. Immunofluorescent images were digitally recorded.
In vitro RNA synthesis
The following RNA and control sequences were cloned by PCR into pJET1.2 plasmids: Control‐RNA (pJET1.2 backbone); pRNA (mrDNA from −232 to −1); Spacer‐rRNA (mrDNA from −1,994 to −1,905); Enhancer‐rRNA (mrDNA from −554 to −447); IGS‐rRNA (mrDNA from −1,997 to +1). pRNA mutants (pRNA‐Control and Control‐pRNA were described in ref. 13. BoxB‐Control (BoxB sequence fused to pJET1.2 backbone from 345 to 2,134) and BoxB‐IGS‐rRNA (BoxB sequence fused to mrDNA from −1,994 to −1) were cloned by PCR into pTOPO 2.1 plasmid. Synthetic RNAs were synthesized using T7 polymerase and as substrate XbaI (pJET1.2), BamHI (pTOPO 2.1), or AvaII (pJET1.2 BoxB‐Control) linearized vectors containing the indicated sequences. IGS‐rRNA used for ESCs transfection was synthesized using the HiScribe™ T7 ARCA Kit (Neb E2065S) according manufacturer's instructions, in order to increase its stability upon transfection. After treatment with DNase I, transcripts were double purified using TRIzol reagent (Invitrogen) according to the manufacture's protocol, quantified and analyzed by agarose gel electrophoresis.
EMSA competition assay
Radiolabeled RNAMCS was synthesized by T7 RNA polymerase using EcoRI linearized pBluescript‐KS(+) plasmid as template. After treatment with DNase I, transcripts were purified and 50,000 cpm of MCS‐RNA was incubated for 15 min on ice with 75 ng recombinant TIP5 or 50 ng DHX9 in EMSA buffer (20 mM Tris–HCl pH 8.0, 5 mM MgCl2, 100 mM KCl, and 0.2 mM EDTA). The amounts of TIP5 and DHX9 moieties to be used in EMSA competition assay were determined by pilot titration experiments as the minimal amount of proteins necessary to obtain a complete shift of radiolabeled RNAMCS in the absence of competitor RNA (shown in the second lane of each EMSA competition experiment). Cold competitor RNA was added, and incubation was continued for 30 min. RNA–protein complexes were analyzed by electrophoresis on 6% (w/v) native polyacrylamide gels and visualized by autoradiography. To produce recombinant DHX9, HEK 293T cells overexpressing FLAG‐DHX9‐His were resuspended in lysis buffer (10 mM Tris–HCl pH 7.5, 500 mM NaCl, 5 mM MgCl2, 15 mM imidazole, 10% glycerol, 2 mM β‐mercaptoethanol, 1× protease inhibitor cOmplete EDTA‐free Roche), incubated for 10 min at 4°C, and sonicated two times for 30 s with a Bioruptor ultrasonic cell disruptor. Extracts were then treated with DNase I (10 U/ml, Thermo Scientific) and RNase A (40 μg/ml, Thermo Scientific) for 1 h at 4°C and centrifuged at 20,000 g for 30 min. Supernatant was incubated with ProBond™ resin (Invitrogen) at 4°C for 3 h to capture His‐tagged proteins. After washing using lysis buffer, DHX9‐bound bead were resuspended in lysis buffer and treated again with DNase I and RNase A as described above. After extensive washing using lysis buffer containing 500 mM NaCl, DHX9 was eluted using lysis buffer containing 300 mM NaCl and 300 mM imidazole. To produce recombinant TIP5, the N‐terminal region of TIP5 fused to His tag (TIP51–600‐His), which contains the RNA‐binding domain TAM, was expressed in Escherichia coli BL21. Cell pellets were resuspended in lysis buffer (20 mM Tris–HCl, pH 8, 300 mM KCl, 5 mM imidazole, 1 mM 2‐mercaptoethanol, and 1× protease inhibitor cOmplete EDTA‐free Roche), treated with 50 U DNaseI for 30 min, and centrifuged for 30 min at 20,000 g. Supernatants were incubated with ProBond™ resin for 4 h, washed with wash buffer (50 mM Tris–HCl, pH 8, 500 mM KCl, 20 mM imidazole, 1 mM 2‐mercaptoethanol and protease inhibitors), treated with RNase A, followed by further washing steps. TIP51–600 was eluted using wash buffer containing 250 mM imidazole. The quality and identity of purified proteins were assessed by Coomassie Brilliant Blue staining and Western blot analysis.
Antibodies
The following antibodies were used: anti‐DHX9 (AW5241) from Abgene; anti‐5mc (MAb‐081‐100) and anti‐TIP5 (CS‐090‐100) from Diagenode; anti‐UBF (sc‐13125) and anti‐DHX9 (sc‐66997) from Santa Cruz; anti‐GFP (11814460001) from Roche; anti‐H3K9me2 (17‐648) from Millipore; anti‐HA (MMS‐101P) from Covance.
Author contributions
SL performed screening for IGS‐rRNA interacting proteins, IGS‐rRNA processing analysis, biochemical assays, immunofluorescence experiments, ESC differentiation experiments, contributed to experimental design and data analysis, and wrote the manuscript; DB performed EMSA assays; CFS performed FLAG‐DHX9 IP and contributed to the analysis of the interactome; DD performed the co‐IP for endogenous TIP5 and DHX9; RS conceived and supervised the project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Acknowledgements
This work was supported by the Swiss National Science Foundation (31003A_173056 and 310003A‐152854), Krebsliga Zurich, Julius Müller Stiftung, Olga Mayenfisch Stifung, Stiftung für wissenschaftliche Forschung an der Universität Zürich, and Forschungskredit of the University of Zurich. We thank Kevin Czaplinski for plasmids and protocols for GRNA chromatography, Li Xing for DHX9 expressing plasmids, and Peter Hunziker and the Functional Genomic Center Zurich for the assistance in proteomic analysis. We also thank Ulrike Kutay, Michael O. Hottiger, Christian Lehner, and Davide Gabellini for helpful discussions during the progression of this work.
EMBO Reports (2017) 18: 1248–1262
References
- 1. Moss T, Stefanovsky VY (2002) At the center of eukaryotic life. Cell 109: 545–548 [DOI] [PubMed] [Google Scholar]
- 2. Santoro R, Grummt I (2001) Molecular mechanisms mediating methylation‐dependent silencing of ribosomal gene transcription. Mol Cell 8: 719–725 [DOI] [PubMed] [Google Scholar]
- 3. Santoro R, Li J, Grummt I (2002) The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 32: 393–396 [DOI] [PubMed] [Google Scholar]
- 4. Conconi A, Widmer RM, Koller T, Sogo JM (1989) Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell 57: 753–761 [DOI] [PubMed] [Google Scholar]
- 5. Pombo A, Dillon N (2015) Three‐dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol 16: 245–257 [DOI] [PubMed] [Google Scholar]
- 6. Fussner E, Ahmed K, Dehghani H, Strauss M, Bazett‐Jones DP (2010) Changes in chromatin fiber density as a marker for pluripotency. Cold Spring Harb Symp Quant Biol 75: 245–249 [DOI] [PubMed] [Google Scholar]
- 7. Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T (2006) Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell 10: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaspar‐Maia A, Alajem A, Meshorer E, Ramalho‐Santos M (2011) Open chromatin in pluripotency and reprogramming. Nat Rev Mol Cell Biol 12: 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gorkin DU, Leung D, Ren B (2014) The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 14: 762–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meshorer E, Misteli T (2006) Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol 7: 540–546 [DOI] [PubMed] [Google Scholar]
- 11. Bartova E, Galiova G, Krejci J, Harnicarova A, Strasak L, Kozubek S (2008) Epigenome and chromatin structure in human embryonic stem cells undergoing differentiation. Dev Dyn 237: 3690–3702 [DOI] [PubMed] [Google Scholar]
- 12. Wiblin AE, Cui W, Clark AJ, Bickmore WA (2005) Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. J Cell Sci 118: 3861–3868 [DOI] [PubMed] [Google Scholar]
- 13. Savic N, Bar D, Leone S, Frommel SC, Weber FA, Vollenweider E, Ferrari E, Ziegler U, Kaech A, Shakhova O et al (2014) lncRNA maturation to initiate heterochromatin formation in the nucleolus is required for exit from pluripotency in ESCs. Cell Stem Cell 15: 720–734 [DOI] [PubMed] [Google Scholar]
- 14. Schlesinger S, Selig S, Bergman Y, Cedar H (2009) Allelic inactivation of rDNA loci. Genes Dev 23: 2437–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guetg C, Scheifele F, Rosenthal F, Hottiger MO, Santoro R (2012) Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol Cell 45: 790–800 [DOI] [PubMed] [Google Scholar]
- 16. Mayer C, Schmitz KM, Li J, Grummt I, Santoro R (2006) Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol Cell 22: 351–361 [DOI] [PubMed] [Google Scholar]
- 17. Zhou Y, Santoro R, Grummt I (2002) The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J 21: 4632–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mayer C, Neubert M, Grummt I (2008) The structure of NoRC‐associated RNA is crucial for targeting the chromatin remodelling complex NoRC to the nucleolus. EMBO Rep 9: 774–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santoro R, Schmitz KM, Sandoval J, Grummt I (2010) Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep 11: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grummt I, Kuhn A, Bartsch I, Rosenbauer H (1986) A transcription terminator located upstream of the mouse rDNA initiation site affects rRNA synthesis. Cell 47: 901–911 [DOI] [PubMed] [Google Scholar]
- 21. Watanabe‐Susaki K, Takada H, Enomoto K, Miwata K, Ishimine H, Intoh A, Ohtaka M, Nakanishi M, Sugino H, Asashima M et al (2014) Biosynthesis of ribosomal RNA in nucleoli regulates pluripotency and differentiation ability of pluripotent stem cells. Stem Cells 32: 3099–3111 [DOI] [PubMed] [Google Scholar]
- 22. Woolnough JL, Atwood BL, Liu Z, Zhao R, Giles KE (2016) The regulation of rRNA gene transcription during directed differentiation of human embryonic stem cells. PLoS One 11: e0157276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Czaplinski K, Kocher T, Schelder M, Segref A, Wilm M, Mattaj IW (2005) Identification of 40LoVe, a Xenopus hnRNP D family protein involved in localizing a TGF‐beta‐related mRNA during oogenesis. Dev Cell 8: 505–515 [DOI] [PubMed] [Google Scholar]
- 24. Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI (2002) Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1–11 [DOI] [PubMed] [Google Scholar]
- 25. Fuchsova B, Hozak P (2002) The localization of nuclear DNA helicase II in different nuclear compartments is linked to transcription. Exp Cell Res 279: 260–270 [DOI] [PubMed] [Google Scholar]
- 26. Zhang S, Kohler C, Hemmerich P, Grosse F (2004) Nuclear DNA helicase II (RNA helicase A) binds to an F‐actin containing shell that surrounds the nucleolus. Exp Cell Res 293: 248–258 [DOI] [PubMed] [Google Scholar]
- 27. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al (2015) STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44: D457–D462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jarmoskaite I, Russell R (2014) RNA helicase proteins as chaperones and remodelers. Annu Rev Biochem 83: 697–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang S, Grosse F (1994) Nuclear DNA helicase II unwinds both DNA and RNA. Biochemistry 33: 3906–3912 [DOI] [PubMed] [Google Scholar]
- 31. Zhang S, Grosse F (1997) Domain structure of human nuclear DNA helicase II (RNA helicase A). J Biol Chem 272: 11487–11494 [DOI] [PubMed] [Google Scholar]
- 32. Lee CG, Hurwitz J (1992) A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3′ to 5′ direction. J Biol Chem 267: 4398–4407 [PubMed] [Google Scholar]
- 33. Kuroda MI, Kernan MJ, Kreber R, Ganetzky B, Baker BS (1991) The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila . Cell 66: 935–947 [DOI] [PubMed] [Google Scholar]
- 34. Myohanen S, Baylin SB (2001) Sequence‐specific DNA binding activity of RNA helicase A to the p16INK4a promoter. J Biol Chem 276: 1634–1642 [DOI] [PubMed] [Google Scholar]
- 35. Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD (1998) BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet 19: 254–256 [DOI] [PubMed] [Google Scholar]
- 36. Nakajima T, Uchida C, Anderson SF, Lee CG, Hurwitz J, Parvin JD, Montminy M (1997) RNA helicase A mediates association of CBP with RNA polymerase II. Cell 90: 1107–1112 [DOI] [PubMed] [Google Scholar]
- 37. Zhong X, Safa AR (2004) RNA helicase A in the MEF1 transcription factor complex up‐regulates the MDR1 gene in multidrug‐resistant cancer cells. J Biol Chem 279: 17134–17141 [DOI] [PubMed] [Google Scholar]
- 38. Aratani S, Fujii R, Oishi T, Fujita H, Amano T, Ohshima T, Hagiwara M, Fukamizu A, Nakajima T (2001) Dual roles of RNA helicase A in CREB‐dependent transcription. Mol Cell Biol 21: 4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jain A, Bacolla A, Del Mundo IM, Zhao J, Wang G, Vasquez KM (2013) DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res 41: 10345–10357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chakraborty P, Grosse F (2011) Human DHX9 helicase preferentially unwinds RNA‐containing displacement loops (R‐loops) and G‐quadruplexes. DNA Repair (Amst) 10: 654–665 [DOI] [PubMed] [Google Scholar]
- 41. Xing L, Liang C, Kleiman L (2011) Coordinate roles of Gag and RNA helicase A in promoting the annealing of formula to HIV‐1 RNA. J Virol 85: 1847–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guetg C, Lienemann P, Sirri V, Grummt I, Hernandez‐Verdun D, Hottiger MO, Fussenegger M, Santoro R (2010) The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J 29: 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhattacharya D, Talwar S, Mazumder A, Shivashankar GV (2009) Spatio‐temporal plasticity in chromatin organization in mouse cell differentiation and during Drosophila embryogenesis. Biophys J 96: 3832–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang LF, Huynh KD, Lee JT (2007) Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 129: 693–706 [DOI] [PubMed] [Google Scholar]
- 45. Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B (2013) Single‐cell dynamics of genome‐nuclear lamina interactions. Cell 153: 178–192 [DOI] [PubMed] [Google Scholar]
- 46. Kind J, van Steensel B (2010) Genome‐nuclear lamina interactions and gene regulation. Curr Opin Cell Biol 22: 320–325 [DOI] [PubMed] [Google Scholar]
- 47. Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M et al (2012) Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150: 948–960 [DOI] [PubMed] [Google Scholar]
- 48. Towbin BD, Gonzalez‐Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM (2012) Step‐wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150: 934–947 [DOI] [PubMed] [Google Scholar]
- 49. Lee CG, da Costa Soares V, Newberger C, Manova K, Lacy E, Hurwitz J (1998) RNA helicase A is essential for normal gastrulation. Proc Natl Acad Sci USA 95: 13709–13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paredes S, Maggert KA (2009) Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci USA 106: 17829–17834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M (2005) Nucleolar proteome dynamics. Nature 433: 77–83 [DOI] [PubMed] [Google Scholar]
- 52. Bibel M, Richter J, Schrenk K, Tucker KL, Staiger V, Korte M, Goetz M, Barde YA (2004) Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat Neurosci 7: 1003–1009 [DOI] [PubMed] [Google Scholar]
- 53. Savić N, Bär D, Leone S, Frommel SC, Weber FA, Vollenweider E, Ferrari E, Ziegler U, Kaech A, Shakhova O et al (2014) lncRNA maturation to initiate heterochromatin formation in the nucleolus is required for exit from pluripotency in ESCs. Cell Stem Cell 720–734 [DOI] [PubMed] [Google Scholar]
- 54. Santoro R (2014) Analysis of chromatin composition of repetitive sequences: the ChIP‐Chop assay. Methods Mol Biol 1094: 319–328 [DOI] [PubMed] [Google Scholar]
- 55. Selth LA, Close P, Svejstrup JQ (2011) Studying RNA‐protein interactions in vivo by RNA immunoprecipitation. Methods Mol Biol 791: 253–264 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 4