

ABSTRACT
Caldicellulosiruptor bescii is the most thermophilic cellulose degrader known and is of great interest because of its ability to degrade nonpretreated plant biomass. For biotechnological applications, an efficient genetic system is required to engineer it to convert plant biomass into desired products. To date, two different genetically tractable lineages of C. bescii strains have been generated. The first (JWCB005) is based on a random deletion within the pyrimidine biosynthesis genes pyrFA, and the second (MACB1018) is based on the targeted deletion of pyrE, making use of a kanamycin resistance marker. Importantly, an active insertion element, ISCbe4, was discovered in C. bescii when it disrupted the gene for lactate dehydrogenase (ldh) in strain JWCB018, constructed in the JWCB005 background. Additional instances of ISCbe4 movement in other strains of this lineage are presented herein. These observations raise concerns about the genetic stability of such strains and their use as metabolic engineering platforms. In order to investigate genome stability in engineered strains of C. bescii from the two lineages, genome sequencing and Southern blot analyses were performed. The evidence presented shows a dramatic increase in the number of single nucleotide polymorphisms, insertions/deletions, and ISCbe4 elements within the genome of JWCB005, leading to massive genome rearrangements in its daughter strain, JWCB018. Such dramatic effects were not evident in the newer MACB1018 lineage, indicating that JWCB005 and its daughter strains are not suitable for metabolic engineering purposes in C. bescii. Furthermore, a facile approach for assessing genomic stability in C. bescii has been established.
IMPORTANCE Caldicellulosiruptor bescii is a cellulolytic extremely thermophilic bacterium of great interest for metabolic engineering efforts geared toward lignocellulosic biofuel and bio-based chemical production. Genetic technology in C. bescii has led to the development of two uracil auxotrophic genetic background strains for metabolic engineering. We show that strains derived from the genetic background containing a random deletion in uracil biosynthesis genes (pyrFA) have a dramatic increase in the number of single nucleotide polymorphisms, insertions/deletions, and ISCbe4 insertion elements in their genomes compared to the wild type. At least one daughter strain of this lineage also contains large-scale genome rearrangements that are flanked by these ISCbe4 elements. In contrast, strains developed from the second background strain developed using a targeted deletion strategy of the uracil biosynthetic gene pyrE have a stable genome structure, making them preferable for future metabolic engineering studies.
KEYWORDS: Southern blotting, genome sequencing, insertion elements, kanamycin resistance
INTRODUCTION
Caldicellulosiruptor bescii is a strict anaerobe that grows optimally at 78°C and is the most thermophilic cellulose degrader known. It produces acetate, lactate, and hydrogen from the fermentation of a variety of sugars, as well as from nonpretreated plant biomass (1). Its ability to deconstruct lignocellulosic biomass, combined with its high optimal growth temperature, makes C. bescii of great biotechnological interest for metabolic engineering efforts toward lignocellulosic bio-based fuel and chemical production. To this end, a genetic system was developed for C. bescii utilizing a uracil auxotrophic mutant background strain and the counterselectable marker pyrF, a gene required for biosynthesis of uracil that also confers sensitivity to 5-fluoroorotic acid (5-FOA) (2, 3). Because there was no method for direct selection of a targeted deletion of pyrF in wild-type C. bescii, the initial development of a genetic background strain relied on the selection of random mutants containing deletions in uracil biosynthesis pathway genes (4). This method resulted in strain JWCB005, which has a partial deletion in both the pyrF and pyrA genes (4). A more recent development leading to the improvement of C. bescii genetic methodologies was the use of a high-temperature kanamycin resistance gene (htk codon optimized for C. bescii [Cbhtk]) that enables antibiotic resistance to be utilized for gene insertion or deletion (5). This strategy allowed for the clean deletion of the pyrE gene from wild-type C. bescii, generating the uracil-auxotrophic 5-FOA-resistant strain MACB1018. This strain has also been used as a genetic background via the utilization of Cbhtk and kanamycin to select for transformants and 5-FOA resistance to select for loss of the pyrE marker (5).
To date, the JWCB005 genetic background strain has been the basis for the majority of the genetically engineered strains of C. bescii (Fig. 1 and Table 1). Efforts to improve the genetic system included the development of a replicating shuttle vector, as well as deletion of the cbeI gene to generate strain JWCB018, allowing for transformation without prior methylation of the DNA (3). Other work has led to a better understanding of plant biomass degradation through the deletion of the major cellulose-degrading enzymes CelA and pectate-lyase (6, 7). Additional studies included engineering-improved biomass utilization via heterologous expression of cellulose-degrading enzymes from other members of the Caldicellulosiruptor genus or other thermophilic cellulolytic organisms (8–10). Detoxification of furan aldehydes found in pretreated plant materials has also been addressed (11). Utilizing this genetic system also led to the discovery of the unexpected ability of C. bescii to utilize tungsten, a metal seldom used in biology (12).
FIG 1.

Tree of genetically modified strains of C. bescii originating from two genetic background lineages. Strains included in the Southern blot analyses are shown in bold text. Strains sequenced using PacBio technology are shown in red. The strain genotypes and the publications in which they are described can be found in Table 1.
TABLE 1.
Genetically modified strains of C. bescii to date, including genotype, parent strain, and reference
| Strain | Genotype or description | Parent strain | Reference |
|---|---|---|---|
| C. bescii DSM 6725 | None | 1 | |
| JWCB002 | ΔpyrBCF | C. bescii DSM 6725 | 2 |
| JWCB003 | pyrBCF restored by marker replacement | JWCB002 | 2 |
| JWCB005 | ΔpyrFA | C. bescii DSM 6725 | 4 |
| JWCB010 | ΔpyrFA ΔpecABCR | JWCB005 | 6 |
| JWCB011 | Transformed with pDCW89 | JWCB005 | 4 |
| JWCB014 | Transformed with pDCW129 | JWCB005 | 4 |
| JWCB017 | ΔpyrFA Δldh | JWCB005 | 13 |
| JWCB018 | ΔpyrFA ΔcbeI ldh::ISCbe4 | JWCB005 | 3 |
| JWCB021 | Transformed with pDCW89 | JWCB018 | 4 |
| JWCB029 | ΔpyrFA ΔcbeI ldh::ISCbe4 ΔcelA | JWCB018 | 7 |
| JWCB032 | ΔpyrFA ldh::ISCbe4 Δcbe1 PS-layer Cthe-adhE | JWCB018 | 15 |
| JWCB033 | ΔpyrFA ldh::ISCbe4 Δcbe1 PS-layer Cthe-adhE*(EA) | JWCB018 | 15 |
| JWCB036 | ΔpyrFA Δldh CIS1::PS-layer Cthe-adhE | JWCB017 | 14 |
| JWCB038 | ΔpyrFA Δldh CIS1::PS-layer Cthe-adhE ΔhypADFCDE | JWCB017 | 14 |
| JWCB044 | ΔpyrFA ldh::ISCbe4 ΔcbeI PS-layer bdhA | JWCB018 | 11 |
| JWCB046 | Transformed with pDCW173 | JWCB029 | 45 |
| JWCB049 | ΔpyrFA Δldh CIS1::PS-layer adhE(Teth39_0206) | JWCB017 | 16 |
| JWCB052 | ΔpyrFA ldh::ISCbe4 Δcbe1 PS-layer E1 | JWCB018 | 8 |
| JWCB054 | ΔpyrFA Δldh CIS1::PS-layer adhB(Teth39_0218) | JWCB017 | 16 |
| JWCB069 | Transformed with pSKW06 | JWCB029 | 46 |
| JWCB070 | Transformed with pSKW07 | JWCB029 | 46 |
| JWCB071 | Transformed with pSKW09 | JWCB029 | 46 |
| JWCB073 | Transformed with pJGW07 containing C. thermocellum pyrF | JWCB052 | 10 |
| JWCB074 | Transformed with pSKW10 containing PS-layer Acel_0180 | JWCB052 | 10 |
| JWCB075 | Transformed with pSKW11 containing PS-layer Acel_0372 | JWCB052 | 10 |
| JWCB079 | Transformed with pSKW14 | JWCB029 | 46 |
| JWCB080 | Transformed with pSKW15 | JWCB029 | 46 |
| JWCB081 | Transformed with pSKW16 | JWCB029 | 46 |
| RKCB103 | ΔpyrFA ΔcbeI ldh::ISCbe4 pJMC009 (PS-layer Calkro_0402) | JWCB018 | 9 |
| RKCB106 | ΔpyrFA ΔcbeI ldh::ISCbe4 PS-layer Cbhtk | JWCB018 | 5 |
| MACB1002 | ΔpyrFA ΔcbeI ldh::ISCbe4 pIMSPfAOR (PS-layer-His6-PF0346) | JWCB018 | 12 |
| MACB1013 | ΔpyrFA ΔcbeI PS-layer aor adhA (PF0346, Teth514_0564) | JWCB018 | This study |
| MACB1015 | Transformed with pSBS4 | C. bescii DSM 6725 | 5 |
| MACB1017 | ΔpyrE | C. bescii DSM 6725 | 5 |
| MACB1018 | ΔpyrE | C. bescii DSM 6725 | 5 |
| MACB1019 | ΔpyrE | C. bescii DSM 6725 | 5 |
| MACB1020 | ΔpyrE | C. bescii DSM 6725 | 5 |
| MACB1021 | ΔpyrE | C. bescii DSM 6725 | 5 |
| MACB1032 | ΔpyrE ΔcbeI | MACB1018 | 5 |
| MACB1034 | ΔpyrE Δldh | MACB1018 | 5 |
Of particular interest for industrial applications are strains where metabolism has been altered to engineer C. bescii for ethanol production. Deletion of the lactate dehydrogenase gene (ldh) in C. bescii eliminated lactate production and increased acetate and hydrogen production by 21% and 34%, respectively, compared to the parent strain (13). The deletion of the maturation genes required for the nickel-iron hydrogenase showed that this enzyme was not responsible for the majority of the hydrogen production by C. bescii (14). The addition of a bifunctional alcohol dehydrogenase gene (adhE) from Clostridium thermocellum, resulting in strain JWCB032, allowed for the production of ethanol from plant biomass at 65°C (15), and production at 75°C was obtained by expressing the genes encoding AdhE and AdhB from Thermoanaerobacter pseudethanolicus 39E, although the ethanol yield was much lower (16). JWCB032 is the best ethanol-producing strain of C. bescii to date, making it thus far the most promising strain for future industrial development.
Genetic stability of microbial strains is paramount for their industrial application. This is especially important as strains of relatively unstudied nonmodel microorganisms are developed for biotechnological applications. Insertion sequence (IS) elements can contribute to instability via transposition into other parts of the chromosome, thereby potentially modifying or eliminating gene function or expression. However, little is known currently about IS elements and genome stability in Clostridiales and Firmicutes. In C. bescii, there are 45 annotated IS elements from 7 classes of transposon families, 25 of which are full length and 20 of which are truncated and presumably inactive. One of these classes of IS elements, ISCbe4, was shown to be active in C. bescii. It was observed in the generation of strain JWCB018, where its transposition into the ldh gene eliminated lactate production (17). Herein, we have investigated the mutations and IS element movement in strains within the two genetic background lineages, JWCB005 (ΔpyrFA) and MACB1018 (ΔpyrE), by genome sequencing and Southern blot analyses to assess their genomic stability for future studies. These analyses show that JWCB005 and its daughter strains have a significantly higher number of mutations and active ISCbe4 elements than the wild type, and this contributes to genome instability that can result in large genome rearrangements. In contrast, the genetic background lineage with a clean deletion of pyrE (MACB1018) had no significant genome rearrangements and significantly fewer mutations.
RESULTS AND DISCUSSION
Phenotypic abnormalities and IS element movements in JWCB005 lineage strains.
In 2012, the first genetically tractable strain of C. bescii, JWCB005, was reported, obtained via selection for a random mutation in the uracil biosynthetic genes (4). JWCB005 contained a deletion in the pyrFA genes, and growth without uracil was possible with only the addition of the pyrF gene (4). Strain JWCB005 and its ΔcbeI daughter strain, JWCB018, have since been the basis for 30 of the 40 strains of C. bescii that have been developed (Fig. 1 and Table 1) (3). A second genetic background lineage of C. bescii was recently developed, based on strain MACB1018, which contains a targeted deletion of pyrE (2, 5) (Fig. 1 and Table 1). So far, MACB1018 has been used to develop only two strains, MACB1032 and MACB1034, which are described below. However, several phenotypic abnormalities in strains in the JWCB005 lineage have become apparent, including decreased swimming motility, clumping when grown without shaking, and the spontaneous production of lactate, despite the fact that the gene for ldh was assumed to be inactivated.
To further investigate the clumping phenotype in the JWCB005 lineage, a swimming motility assay was used to compare various strains (Fig. 2). We utilized a wild-type stock of the originally published C. bescii DSM 6725 strain (wild-type reference [WT-Ref]), JWCB005 (ΔpyrFA), JWCB018 (ΔpyrFA ΔcbeI), MACB1018 (ΔpyrE), and the wild-type parent to MACB1018 (WT-Parent). JWCB005 and WT-Parent both generated discrete colonies, indicative of a swimming motility defect, while all other strains gave diffuse colonies (Fig. 2). Of these five strains, only JWCB018 showed evidence of clumping and settling in liquid growth medium (see Fig. S2 in the supplemental material). These results indicate that the motility and clumping phenotypes are likely not related, and, since they were not previously documented, no genetic basis for their existence is known. However, JWCB018 was reported to be unable to produce lactate, and this phenotype was shown to be the result of an insertion sequence element (ISCbe4) transposition into the lactate dehydrogenase gene (ldh) (17).
FIG 2.
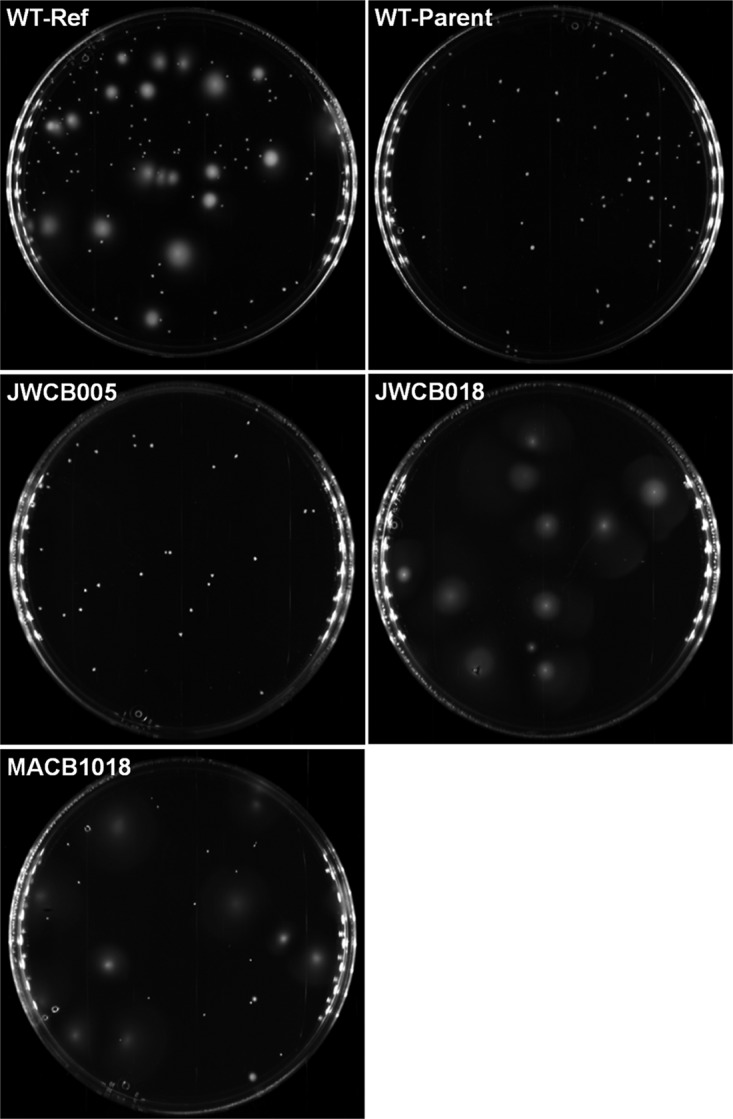
Motility test using 0.03% Bacto agar plates. Mobile cells generate diffuse colonies, and nonmotile cells generate discrete colonies. WT-Ref, JWCB018, and MACB1018 are motile, while WT-Parent and JWCB005 lose their motility.
We have also observed two instances of ISCbe4 transposition during the construction of strains using the JWCB005 lineage. The first occurred when a strain, MACB1002, was constructed to heterologously express the gene (aor) encoding the aldehyde oxidoreductase of Pyrococcus furiosus, driven by the slp promoter on a replicating shuttle vector (12). The replicating plasmid was isolated from the recombinant strain, but the plasmid size differed from that of the original transformation plasmid by about 1.5 kb. PCR and sequence analysis confirmed the presence of an ISCbe4 element located 73 bases inside the 5′ end of the 200-bp slp promoter sequence (Fig. 3; see also Fig. S3). A 10-base sequence of the promoter was repeated on each side of the IS element. Expression of the aor gene from P. furiosus (Pfaor) did not appear to be negatively affected, as 127 bp of the 3′ end of the promoter were still intact, and the expression level of Pfaor was higher than that of the native slp gene (12).
FIG 3.
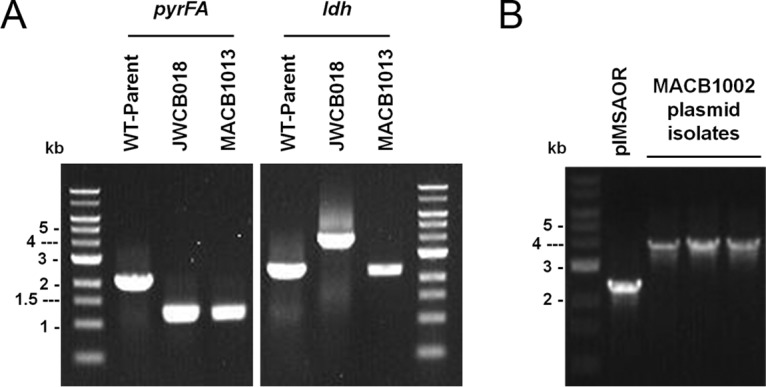
Movement of ISCbe4 elements observed in strains derived from JWCB005. (A) PCR verification of pyrFA and ldh loci of MACB1013. MACB1013 has the ΔpyrFA mutation matching its parent strain JWCB018 (1.1 kb) and a wild-type ldh gene (2.3 kb). (B) PCR products showing ISCbe4 element insertion in plasmids originating from strain MACB1002 (4.1 kb) compared to the control plasmid used for constructing the strain carrying pIMSAOR (2.4 kb).
The second IS element movement that we observed in a JWCB005-derived strain was in strain MACB1013. This strain was constructed to express Pfaor and the adhA gene encoding the primary alcohol dehydrogenase of Thermoanaerobacter sp. strain X514 to investigate ethanol production via the novel AOR-AdhA pathway (18). JWCB018 was used as the parent strain, which contains an ISCbe4 insertion in the ldh locus, thereby eliminating lactate production (17). JWCB018 was transformed with pGR002 to insert the aor-adhA expression construct at the cbeI locus to generate strain MACB1013 (Fig. S1). However, MACB1013 was unexpectedly found to produce lactate. Sequencing of the ldh locus showed that the ISCbe4 element was no longer present in the ldh gene, which instead had the wild-type sequence (Fig. 3). The mechanism of the transposase encoded within the ISCbe4 element has not been determined, and it is unknown whether the loss of ISCbe4 in this instance was the result of a rare recombination event or if it was transposase mediated (19, 20). All of these observations established the need to investigate IS element movements and genome stability within the JWCB005 lineage, as well as the newer MACB1018 (ΔpyrE-derived) lineage (Fig. 1).
Southern blot analyses of C. bescii strains.
Given the phenotypic abnormalities and observed IS element movements in the ΔpyrFA mutant (JWCB005) lineage strains described above, several strains were selected to probe for IS element movement via Southern blot analyses: JWCB005, JWCB018, JWCB032, and MACB1013 from the ΔpyrFA mutant lineage, MACB1018, MACB1032, and MACB1034 from the ΔpyrE mutant lineage, and WT-Ref as a control. As shown in Fig. 1, JWCB032 and MACB1013 are daughters of JWCB018 that were engineered for ethanol production using two different pathways, with JWCB032 having the highest ethanol yield of any strain of C. bescii to date (15). MACB1018 is a recently developed alternative genetic background strain that was used for the construction of MACB1032 and MACB1034, containing deletions of ldh and cbeI, respectively (5).
Southern blot analysis for ISCbe4 was performed on NsiI-digested genomic DNA from the various strains of C. bescii, and the results are shown in Fig. 4. The probe was designed to hybridize to a 444-bp region of the transposase gene and contains homology to all intact copies of ISCbe4. Wild-type C. bescii and strains MACB1018, MACB1032, and MACB1034 all have the expected pattern of digestion and hybridization with the ISCbe4 probe, with one additional band present for MACB1018 and its daughter strains. However, all strains from the JWCB005 lineage, including JWCB018, JWCB032, and MACB1013, have a clear increase in the instances of ISCbe4 in their genomes, although resolution of the exact number from the Southern blot analysis alone is difficult, and more than one transposon may be present on a given restriction fragment. Furthermore, resolution of bands of similar size may not have occurred under the conditions used for this Southern blot analysis, which would result in a potential underestimation of the movements that have occurred. The same membrane used in Fig. 4 was stripped and hybridized repeatedly with probes designed for four of the other IS elements in C. bescii, ISCbe1, ISCbe2, ISCbe3, and ISCbe5 (Fig. S4). For these additional four probes, either limited or no movement was observed for each insertion element.
FIG 4.
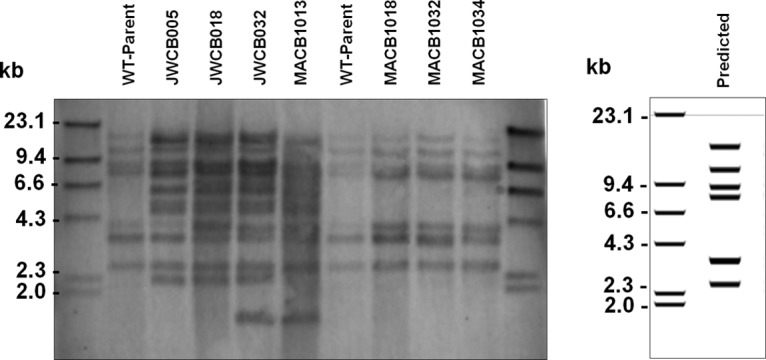
Southern blot probed for ISCbe4 element. The predicted banding pattern is shown at the right, with a digital blot generated using the published WT-Ref sequence. WT-Parent gives the expected banding pattern based on sequencing results. There is only one additional band observable for MACB1018, MACB1032, and MACB1034, the strains from the targeted deletion of pyrE lineage. JWCB005, JWCB018, JWCB032, and MACB1013 (which was derived from JWCB018) all have a dramatic increase in ISCbe4 elements, the exact number of which is difficult to determine from the Southern blot analysis alone.
Sequencing of C. bescii strains.
To better understand genome stability and IS element movement within the two C. bescii lineages, PacBio sequencing was performed on the following strains: JWCB005 (ΔpyrFA mutant) and JWCB018 (daughter of JWCB005), as well as MACB1018 (ΔpyrE mutant), its wild-type C. bescii DSM 6725 parent (WT-Parent), and four of its sister strains generated concurrently with MACB1018: MACB1017, MACB1019, MACB1020, and MACB1021. The sister strains of MACB1018 were included to gain insight into the variation that can occur among isolates within one round of genetic manipulation. PacBio sequencing was used in order to observe the transpositions of ISCbe4, since the longer read length allows for greater accuracy in identification of IS element localization. The sequencing and assembly resulted in a single draft-quality contig for each the strains (see Materials and Methods for processing details). These eight draft genomes were compared to the previously published level 6 finished Sanger/454-based reference genome of wild-type C. bescii DSM 6725 (WT-Ref; accession no. CP001393.1) (21).
IS element analyses.
The Web-based ISsaga2 analysis tool (22) was used to predict the location and type of all known IS elements in all nine genomes (eight strains plus the WT parent). Table 2 shows the number of all known complete and partial IS elements by family in the wild-type reference sequence and in the strains sequenced in this study. IS elements defined as complete by ISsaga are those that meet or exceed both global and local alignment thresholds for IS ends, inverted repeats, direct repeats, and associated open reading frames (ORFs). Partial IS elements score below the threshold and/or are missing components (e.g., inverted repeats). The numbers of complete and partial IS elements remain the same in each of the genomes, with the notable exception of ISCbe4, as well as one instance of ISCbe2 (Table 2). The only apparently active IS element is ISCbe4, and the one ISCbe2 element that is lost from JWCB018 can be explained by a recombination event between two ISCbe4 elements that flank the lost ISCbe2 element in the parent strain (Fig. 5).
TABLE 2.
Number of all known insertion elements in each of the resequenced strains of C. bescii
| IS type | No. of insertion elements (no. of partial elements) by strain |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| WT-Ref | WT-Parent | JWCB005 | JWCB018 | MACB1017 | MACB1018 | MACB1019 | MACB1020 | MACB1021 | |
| ISCbe1 | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) |
| ISCbe2 | 5 (2) | 5 (2) | 5 (2) | 4 (2) | 5 (2) | 5 (2) | 5 (2) | 5 (2) | 5 (2) |
| ISCbe3 | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) | 4 (2) |
| ISCbe4 | 7 (5) | 7 (5) | 19 (5) | 23 (5) | 8 (5) | 9 (5) | 10 (5) | 9 (5) | 8 (5) |
| ISCbe6 | 4 (3) | 4 (3) | 4 (3) | 4 (3) | 4 (3) | 4 (3) | 4 (3) | 4 (3) | 4 (3) |
| ISCsa1 | 0 (2) | 0 (2) | 0 (2) | 0 (2) | 0 (2) | 0 (2) | 0 (2) | 0 (2) | 0 (2) |
| ISCsa9 | 1 (4) | 1 (4) | 1 (4) | 1 (4) | 1 (4) | 1 (4) | 1 (4) | 1 (4) | 1 (4) |
FIG 5.

Overall genome arrangements for the WT-Parent, JWCB005, MACB1018, and JWCB018. ISCbe4 elements are shown in black lines along the outside of the circular genome diagrams. There are no large-scale rearrangements in JWCB005 and MACB1018 compared to wild type. There are two large rearrangements and inversions in JWCB018 (green and red regions) compared to the wild type and its parent, JWCB005.
The genome positions of each IS element were identified using the genome sequences upstream and downstream from them in order to examine the stability of individual elements from each parent strain to its daughter(s). With the exception of ISCbe4, the positions of all the IS elements remain the same; however, as expected from the IS element counts in Table 2, the genome positions of ISCbe4 elements in the two lineages shown in Fig. 1 are very different. There was one instance that appeared to be an increase in one ISCbe3 element in MACB1018; however, further analysis indicated an assembly error, as regions upstream and downstream of the element were also duplicated. PCR analysis confirmed that this region in MACB1018 was identical to the WT-Parent (Fig. S5). This finding highlights the occasional errors that are present in the draft sequences and the need to verify results when questions arise.
In most cases where strains gained ISCbe4 elements, the existing ISCbe4 elements from the parent strains were retained at the same genome location in the daughter strains. As shown in Table 2, the sister strains MACB1017 to MACB1021 have different numbers of ISCbe4 elements. However, many of these insertion sites are the same, and there are only three new locations for ISCbe4 compared to the parent (WT-Parent). The new ISCbe4 in MACB1017 is shared by all five sister strains. The second new ISCbe4 in MACB1018 is shared by MACB1019 and MACB1020. The final ISCbe4 is unique to MACB1019. An examination of the ISCbe4 elements in the JWCB005 lineage shows that most of the ISCbe4 elements are shared between JWCB005 and JWCB018, except for those new to JWCB018. The individual elements were slightly more difficult to identify, as a number of elements had swapped flanking regions, indicating that recombination had occurred at these ISCbe4 elements. There was one notable instance where one ISCbe4 element was lost between WT-Parent and JWCB005. This loss did not result in a duplication of the direct repeats flanking ISCbe4; instead, only one copy of the direct repeat remained. This observation is particularly important, as it is the second instance of the loss of ISCbe4 without the duplication of the direct repeats. Interestingly, one ISCbe4 element in JWCB018 disrupted a gene previously identified as a cellulose-binding protein termed a tāpirin (Athe_1870) (23, 24), and this could potentially give rise to changes in attachment to and degradation of biomass substrates (Table S5). However, the cause of the swimming motility defect seen in strain WT-Parent and JWCB005 or the clumping phenotype in JWCB018 could not be attributed to movement of the known insertion elements.
While the mechanism of transposition of ISCbe4 has not been biochemically characterized, the results presented here have implications for its mode of action. The increase in its copy number within the genomes of all strains that have undergone genetic manipulation without loss of the transposon at the wild-type or parent position suggests that this IS element may use a replicative method of transposition (20). A “copy-out paste-in” mechanism, as described by Curcio and Derbyshire, seems to be the most likely mode of replication (20). Additionally, Guérillot et al. reported circular forms of other ISLre2 family transposons, of which ISCbe4 is a member (25). This mechanism would also explain the loss of ISCbe4 without duplication of the direct repeats, as this would be a rare event occurring during the repair of the single-strand break caused during movement of the IS element, resulting in the clean loss of the element rather than duplication.
Comparison of published and resequenced wild-type strains.
To evaluate the quality of the draft contigs initially, a whole-genome alignment of the published wild-type reference genome (WT-Ref) to the PacBio resequenced wild-type contig (WT-Parent) was performed. This showed that there are four single nucleotide polymorphisms (SNPs) (three occurring within genes) between these two genome sequences, and seven deletions (three within genes) and 14 insertions (nine within genes) of two or more nucleotides in the reference genome (Table S2). Upon closer inspection of the insertions and deletions in the alignment, most were located in homopolymer runs of four or more nucleotides and were on average less than one nucleotide long, so these may be attributed to sequencing or assembly artifacts. Overall, the two genome sequences generally agree but with only one notable exception, and this is at the so-called glucan degradation locus (GDL) (23). As shown in Fig. 6, these two genome assemblies have different arrangements within this locus, accounting for two of nine insertions into genes and two of three deletions within genes. Determining which orientation is correct is of biological relevance, since the GDL is critically important for biomass degradation in C. bescii. To confirm the correct orientation, we designed primers to bridge these regions and performed PCR (Fig. 6). Genomic DNA from DSM 6725 was used as the template for the six reactions, and the primers were paired to give products for either WT-Parent or WT-Ref (Fig. 6). All expected products were generated for the orientation presented by the PacBio resequencing, while bands of the incorrect size were observed for the PCR products predicted by the orientation in the original Sanger/454-sequenced C. bescii genome. Hence, the PacBio assembly is the correct and biologically relevant sequence (Fig. 6). This region of the C. bescii genome has stretches of identical nucleotides up to 2.5 kb in length, and the longer read length of PacBio sequencing is advantageous for assembling this highly repetitive region. Ultimately, there is no change to the sequence of any particular gene in this region; however, the gene order is affected by these new results.
FIG 6.
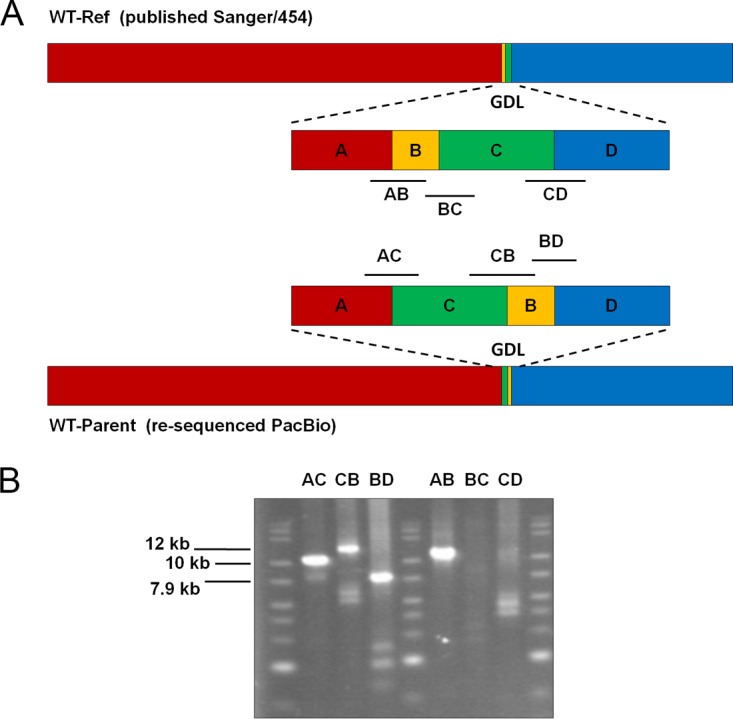
Differences in genome arrangement between previously published genome sequence (WT-Ref) and resequencing results (WT-Parent). (A) The overall arrangement of the wild-type genome sequenced by the two methods. There is only one major disagreement at the glucan degradation locus (GDL). The black lines at each region show the locations of the PCR products used to validate the biologically relevant genome region. (B) PCR validation of the arrangement at the GDL. Expected product sizes for the resequenced genome are AC, 10.0 kb; CB, 12.0 kb; and BD, 7.9 kb. Product sizes for the published genome are AB, 10.5 kb; BC, 9.0 kb; and CD, 10.6 kb. The expected products were attained for the orientation reported by the PacBio resequencing. For reactions targeting the orientation in the published genome, one larger than expected product was observed for reaction AB, and all other bands were faint or smaller than expected, indicative of nonspecific PCR products.
Genome rearrangements in C. bescii strains.
To examine genome rearrangements in the two lineages, the JWCB005, JWCB018, and MACB1018 sequences were aligned to the WT-Parent sequence. The WT-Parent sequence was selected as the reference for these alignments because of the corrected GDL orientation. The alignment results show that dramatic rearrangements have occurred in JWCB018 (Fig. 5). The overall organization of the WT-Parent, MACB1018, and JWCB005 genomes is the same, although JWCB005 has a notable increase in the number of ISCbe4 elements. JWCB018, however, has two large genome rearrangements and inversions (Fig. 5). Intriguingly, each of these rearrangements is flanked by an ISCbe4 element, suggesting that the increased number of ISCbe4 elements is responsible for the rearrangements observed. There is also evidence for the loss of 10 kb of DNA coding for 11 genes (Athe_0717 to Athe_0727) from JWCB018, which is located between two ISCbe4 elements in JWCB005. These genes include those encoding five Leptospira repeat proteins, two hypothetical proteins, a transglutaminase, a transpeptidase, and one instance of ISCbe2. These genes have not been studied in detail in C. bescii and clearly are not essential for growth under laboratory conditions. However, these dramatic changes to the genome, combined with the dramatic increase in the number of ISCbe4 elements, demonstrate the instability of JWCB005 and of its daughters. In contrast, the alignment of WT-Parent to MACB1018 or any of its sister strains (MACB1017, MACB1019, MACB1020, and MACB1021) shows no genome rearrangements (Fig. S6).
Mutations in C. bescii strains.
While rearrangements can have major impacts on a genome's stability, so too can smaller variations. Thus, whole-genome alignments of the eight non-wild-type genomes were performed against WT-Ref to investigate SNPs and indels. JWCB005 and JWCB018 had 50 and 56 SNPs in 13 and 16 unique genes, respectively, compared to MACB1018 and its sisters (MACB1017 and MACB1019 to MACB1021), which ranged from six to nine SNPs in five to six unique genes (Table S3). As expected, each lineage contained SNPs that were highly conserved among related strains, indicating they are true mutations and are not sequencing or assembly errors. However, it is not clear that any of these SNPs can be attributed to the movements of ISCbe4, as none were in close proximity to the insertion elements.
Several SNPs are found in the 23S rRNA gene (Athe_R0035) of strains JWCB005 (3 SNPs), JWCB018 (3 SNPs), and MACB1017 (1 SNPs). However, a disproportionately high number of the SNPs from the ΔpyrFA mutant lineage strains (JWCB005 and JWCB018) were in an annotated pseudogene, Athe_2202. Yet, no SNPs were found in this pseudogene in the ΔpyrE mutant lineage strains (MACB1017 to MACB1021), suggesting it is not a sequencing or assembly error. Unfortunately, the swimming motility defect in WT-Parent and JWCB005 could not be easily attributed to any SNP. The clumping phenotype in JWCB018 could be related to an SNP in a gene that encodes a transketolase domain protein (Athe_2060). Athe_2060 is homologous to a gene in Mycoplasma genitalium previously reported to cause cells to clump when disrupted (26). However, in C. bescii, this SNP cannot be the sole cause for the clumping phenotype, because it is also found in JWCB005, which does not exhibit this phenotype.
JWCB005 and JWCB018 had 34 and 79 deletions (16 and 25 in unique genes) and 39 and 46 insertions (12 and 16 in unique genes) compared to the reference genome, respectively (Table S4). In contrast, the ΔpyrE mutant lineage strains MACB1017 to MACB1021 had 15 to 19 deletions (10 to 12 in unique genes) and 9 to 14 insertions (5 to 7 in unique genes) compared to the WT-Ref genome (Table S4). It should be noted that the error in the WT-Ref GDL, as described above, does account for two of the gene insertions and two of the gene deletions in all eight strains. One of the deletions in JWCB005 caused a frameshift in the flagellar M-ring protein, Flif (Athe_2173), which is likely the source of the swimming motility defect based on previous studies in a different organism (27). Interestingly, the WT-Parent, but not the ΔpyrE mutant daughter strains, also shares this mutation in a thymine-rich region. Therefore, it was further confirmed by PCR amplification and sequencing. The results suggest that this region of the fliF gene seems to be highly unstable, as the phenotype switches readily between generations of C. bescii strains.
Establishing a screen for C. bescii strains.
Although genome sequencing analysis gives a much more precise picture of the movements of ISCbe4 and mutations, the approach is not high throughput and is not feasible for every new C. bescii strain that is generated. To that aim, we sought to develop and validate a faster method to determine the genome stability of C. bescii strains. For this analysis, we compared the Southern blot results shown in Fig. 4 with digital Southern blot analysis generated from the new sequencing results for those strains that were sequenced. This comparison shows that the Southern blot and sequencing results are very closely related, allowing for the potential use of Southern blot analysis as a preliminary screen to estimate changes in the genomes of strains (Fig. 4; see also Fig. S7). Since a goal of this study was to find a method to quickly evaluate the genomic stability of strains over time, it is noteworthy that ISCbe4 was the only IS element in C. bescii that changed significantly on the Southern blots, making it an ideal single marker for genome stability in these lineages. Since the Southern blots serve as a proxy for IS movements, we set out to calculate the median number of mutations per ISCbe4 element so that we could estimate the total number of mutations in a new strain using only Southern blotting. As a relative control for sequencing and assembly errors, we subtracted the WT-Refseq total IS and total mutations from the total IS and total mutations for each of the newly sequenced strains. While not all IS elements and mutations are phenotypically equal, we assumed they were for ease of calculation. Next, we divided the number of mutations by the number of ISCbe4 elements per strain to get the ratio of mutations per ISCbe4. This yielded a median of 8.2 mutations per ISCbe4 over the eight strains, with a range of 3 to 10 mutations per IS. This means that for every new band on a Southern blot, there are approximately 8 or 9 additional mutations accompanying it. While ISCbe4 elements provide a good estimation of the mutation rate, this method is still susceptible to the limitations of Southern blotting, so that not all elements will be counted as separate bands when the fragment sizes overlap. However, since the IS elements predicted from the sequencing data are almost identical to those on the blot, this is not a major concern.
Conclusions.
Strains constructed using the ΔpyrFA mutant lineage (JWCB005) generated by random mutagenesis have improved the understanding of C. bescii metabolism and biomass deconstruction, as well as expanded the possibilities for engineering biofuel production in this organism. However, moving forward, it will be important to utilize the most stable strains for metabolic engineering purposes. The phenotypic abnormalities, chromosomal rearrangement, and genetic variations that we have documented in the ΔpyrFA mutant lineage (JWCB005) throughout this study are significant. In contrast, our analyses indicate that the newer ΔpyrE mutant lineage (MACB1018), constructed via the targeted deletion of pyrE, shows no major rearrangements, fewer IS element movements, and significantly fewer mutations. Thus, as the ΔpyrE-derived lineage is more similar to the wild type and appears to be significantly more stable, it will be a better choice for use in future metabolic engineering efforts. Additionally, with the aid of the long reads generated from PacBio sequencing technology, we have shown the glucan degradation locus (GDL) in the originally published C. bescii wild-type genome is in the incorrect orientation. In the future, Southern blot analysis can be used to initially examine the movement of IS elements in new strains, with particular attention given to ISCbe4, and to provide an estimate of the number of genetic mutations. Genome sequencing will then be used to verify these results, where the time and cost deem it necessary.
MATERIALS AND METHODS
Growth of C. bescii.
C. bescii DSM 6725 was obtained from the DSMZ German Collection of Microorganisms and Cell Cultures, and a glycerol stock of it was used to represent wild-type reference (WT-Ref) for assays. Another stock from 2015 that was used as the parent strain for MACB1017 to MACB1021 was used for resequencing (WT-Parent) and assays. C. bescii strains JWCB005, JWCB018, and JWCB032 were obtained from J. Westpheling (University of Georgia). C. bescii strains MACB1013, MACB1017 to MACB1021, MACB1032, and MACB1034 were generated as described previously (5). Strains of C. bescii were grown on the glucose-containing modified DSM 516 (CG516) medium containing 20 μM uracil, as previously described (5). For sequencing analysis, cultures were grown in 500 ml of cellobiose-containing low-osmolarity complex (LOC) medium, as described previously (28). The 500-ml cultures for sequencing analysis were grown overnight statically at 70°C under anaerobic conditions. All other cultures were grown overnight at 75°C with shaking at 150 rpm under anaerobic conditions. All strains were revived from glycerol stocks and transferred to fresh medium before genomic DNA was extracted for Southern blot and/or DNA sequencing analyses.
Swimming motility assay.
The swimming motility assay is based on previous studies with mesophilic microorganisms (27). Cultures of each strain were grown overnight and then serially diluted in 1× low-osmolarity defined (LOD) salts. In an anaerobic chamber containing N2/H2 (98%/2% [vol/vol]), cells were distributed onto plates and grown while embedded in LOC medium containing 0.3% Bacto agar. Plates were incubated at 65°C for 2 days.
Clumping assay.
Glycerol stocks of WT-Parent, JWCB005, JWCB018, and MACB1018 were revived overnight in CG516 medium supplemented with uracil. Cell density was determined using a Petroff-Hauser counting chamber. Cultures were transferred to 50-ml glass serum bottles containing 20 ml CG516 medium with uracil at a starting cell density of 2 × 106 cells · ml−1 and grown at 78°C for 36 h with no shaking or disturbance. Cultures were then swirled gently just prior to being imaged.
Genomic DNA extraction.
Cells from 50-ml and 500-ml cultures were harvested at 6,000 × g for Southern blot and DNA sequencing analyses, respectively. A phenol-chloroform-isoamyl alcohol extraction was used, as described previously (23). Ethanol precipitation was performed to obtain highly pure DNA. The DNA concentration was determined using a Thermo Scientific NanoDrop 2000c spectrophotometer.
Strain construction.
pGR002 for transformation into strain JWCB018 was generated using Gibson Assembly from New England BioLabs (29). The structure of the plasmid is shown in Fig. S1 and was sequence verified. Competent cells were prepared by growing 500-ml cultures in LOD medium with amino acids (28) and washed with 10% (wt/vol) sucrose, as described previously (5). Cells were mixed with 0.5 to 1 μg of plasmid DNA and transferred to a 1-mm-gap electroporation cuvette, and electroporation was carried out as described previously (5). Recovery was in 20 ml of LOC medium at various intervals, 1-ml samples of recovery cultures were centrifuged for 1 min at 14,000 rpm, and the supernatant was removed to minimize uracil carryover to selective medium. Cell pellets were resuspended in 0.2 ml of selective LOD medium without uracil supplementation and transferred to the selective medium (28). Transformation isolates were purified once on solid LOD medium without uracil and screened for complete plasmid insertion at the ΔcbeI locus. Counterselection was then performed on solid LOD medium with 4 mM 5-FOA and 40 μM uracil for loss of the plasmid backbone or reversion to the parent strain. Colony isolates were once again screened and subjected to a final round of purification on solid LOD medium with 40 μM uracil, and the insertion of Pfaor (the Pyrococcus furiosus aldehyde ferredoxin oxidoreductase gene) and adhA was confirmed by sequencing. This strain was designated MACB1013.
Genome sequencing and assembly.
All aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov. The raw reads from each genome were de novo assembled using HGAP (version 2.3.0) (32) into a single chromosomal contig. However, the parameters were not optimal for the correct assembly of the two native plasmids of C. bescii, and thus, these data were not used for further analyses. Read coverage ranged from 72.6× to 499.1×. No genome polishing was performed beyond the PCR verifications listed in this study.
Genome annotation.
Genome features and annotations were predicted using JGI's standard pipeline, but that information was not utilized in this study. Instead, the features and annotations from the reference genome sequence were transferred to the draft genome sequences using the Rapid Annotation Transfer Tool (RATT; version 1.0 using the “strain” settings profile) (33) for comparison with other analyses in this study. In the publicly available genome drafts, the genes were identified using Prodigal (34), followed by a round of manual curation using GenePRIMP (35). The predicted coding sequences (CDSs) were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant, UniProt, TIGRFam, Pfam, KEGG, COG, and InterPro databases. The tRNAscan-SE tool (36) was used to find tRNA genes, whereas rRNA genes were found by searches against models of the rRNA genes built from SILVA (37). Other noncoding RNAs, such as the RNA components of the protein secretion complex and RNase P, were identified by searching the genome for the corresponding Rfam profiles using INFERNAL (38). Additional gene prediction analysis and manual functional annotation were performed within the Integrated Microbial Genomes (IMG) platform developed by the Joint Genome Institute, Walnut Creek, CA, USA (39).
Genome alignments and variations.
The assembled draft genomes were downloaded from the JGI genome portal (30), and the plasmid contigs were removed due to assembly errors (data not shown). The reference genome was that of wild-type C. bescii DSM 6725 (WT-Ref; accession no. CP001393.1) from NCBI's GenBank repository, and the plasmids were removed for consistency in analyses (21). Whole-genome alignments to this reference were created using progressiveMauve (version 20150226 build 10) (40), with the following nondefault settings: default seed weight, false; use seed families, true; match seed weight, 15; and minimum LCB weight, 3,000. Based on an initial alignment of each draft genome sequence to the reference, the first base pair of each chromosomal contig was shifted, and the reverse complement was generated, if required, using Geneious version 8.1.8 (41) (see Table S1 for specific changes). The final alignments and all other analyses were based on these newly generated draft genome sequences. The locally colinear block (LCB) coordinates from the final progressiveMauve (version 20150226 build 10) alignments were extracted and added to the genomes as GFF3 tracks. In addition, the final alignments produced the coordinates for SNPs and indels (or gaps) identified by progressiveMauve. Those coordinates were mapped on the reference sequence features (e.g., genes, CDSs, etc.) using a custom BioPerl script (42) and tabulated (Tables S2 to S4).
Southern blot analysis probe generation.
Primers for insertion sequence elements ISCbe1, ISCbe2, ISCbe3, ISCbe4, and ISCbe5, as identified by the ISfinder website (43), were designed using the wild-type strain C. bescii DSM 6725 (accession no. CP001393.1) from NCBI's GenBank repository (21). Probes were designed to generate 400- to 600-bp PCR products from C. bescii DSM 6725 genomic DNA using the primer pairs shown in Table 3. PrimeSTAR Max polymerase (TaKaRa Bio) was used according to the manufacturer's recommendations, and PCR products were then concentrated and purified using a DNA Clean & Concentrator kit (Zymo Research). Probes were labeled with digoxigenin (DIG) using the DIG-High Prime DNA labeling and detection starter kit I.
TABLE 3.
Primers used in this study, including those used to amplify IS element probes for Southern blot analysis, GDL arrangement confirmation, and ISCbe3 duplication confirmation
| Target | Primer name | Sequence (5′ to 3′) |
|---|---|---|
| ISCbe4 | JD001 | GGAGCGATAATAGAGAGAGTAGTAGAC |
| JD002 | CTTTATCCAATTAGCTCCATCTCC | |
| ISCbe1 | JD003 | GCATACTTCAACATCCCTATCACAG |
| JD004 | GTACGTATATGCGACGATACAAGC | |
| ISCbe2 | JD005 | CTGGCTTCAGATACAGACG |
| JD006 | GCTCTTGGTGGAATAGGATTTAGTTC | |
| ISCbe3 | JD007 | CAGTCAAGGGTGTTTATGAG |
| JD008 | CACCCTGATATGGCAGTATC | |
| ISCbe6 | JD009 | CTCAACTGGTGGATTATACATC |
| JD010 | CCAAGAGACAGAGAAGGTG | |
| GDL A | AR073 | CCACTTGGTGCCACATAAATAGC |
| GDL B | AR075 | GCAAGAAGGTTAGGTGGAAACAG |
| AR076 | CTGTTTCCACCTAACCTTCTTGC | |
| GDL C | AR077 | AAGAAGAAATTCAATCAAAGTTGATG |
| AR074 | TCACGTATGACGATTGAAGC | |
| GDL D | AR078 | GAGGTTAGAGATTTATGAAGCGTTACAG |
| ISCbe3 region A | AR082 | TAAAAGCTGTATCGCACCACC |
| AR084 | AATTGAAGCAGAGTGTGGAGC | |
| ISCbe3 region B | AR083 | CTCAGCTTATTCAAGGACGAC |
| AR081 | CACTTCTCAGTGGAGTAGAGTC |
Southern blot analyses.
Southern blots were generated with 3 μg of genomic DNA from each strain digested with the NsiI–high-fidelity (HF) restriction enzyme (New England BioLabs). The restriction fragments were separated by electrophoresis at 100 V for 2 h in a 0.7% (wt/vol) agarose gel containing 0.5 μg · ml−1 ethidium bromide. The gels were subjected to depurination and denaturation, followed by neutralization. Gels were then equilibrated in 1× Tris-borate-EDTA (TBE) for 10 min. Restriction fragments from the gel were transferred onto positively charged membranes (Roche) using the Thermo Scientific semidry electroblotter at 120 mA for 45 min. The DNA fragments were fixed to the membrane by UV cross-linking in a Stratagene UV Stratalinker 2400 using the auto-cross-link setting. After prehybridization, the nylon membrane with the restriction fragments was incubated with the probe with shaking at 45°C overnight, in accordance with the DIG-High Prime DNA labeling and detection starter kit I (Roche) protocol. Stringency washes were performed using 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and 0.1% SDS at 65°C for two 30-min washes. Immunological detection was performed according to the manufacturer's instructions, and membranes were stripped and reprobed according to the DIG protocol.
In silico Southern blot analyses.
The reference and draft genome sequences were digitally digested with the NsiI restriction enzyme using Geneious version 8.1.8, and the individual sequence fragments were assigned unique identification (ID) numbers. Theses fragments were added to a custom BLAST database queried with the ISCbe1, ISCbe2, ISCbe3, ISCbe4, and ISCbe5 probes (as described above) using blastn (version 2.2.29 with default settings, except an E value cutoff of 1e−10, yet no hit was greater than 1e−100) (44). Then, only the probe hits were temporarily reassembled into a single pseudocontig for each genome using a custom BioPerl script. This pseudocontig was digitally redigested with NsiI using Geneious (version 8.1.8), yielding the digital Southern blot images.
Insertion sequence analyses.
The insertion sequence elements were predicted for the reference and draft genome sequences using the semiautomatic annotation engine in the Web-based ISsaga 2 tool (http://issaga.biotoul.fr/). Predicted complete and partial IS elements were extracted and added to the genomes as GFF3 tracks.
Accession number(s).
The assembled level 3 improved high-quality draft genome sequences were generated for wild-type C. bescii DSM 6725 (WT-Parent; European Nucleotide Archive [ENA] accession no. FXXF01000001), JWCB005 (ENA accession no. FUZN01000002), JWCB018 (ENA accession no. FXXD01000001), MAC1017 (ENA accession no. FXXE01000001), MAC1018 (ENA accession no. FUZJ01000001), MAC1019 (ENA accession no. FUZL01000001), MAC1020 (ENA accession no. FXXC01000001), and MAC1021 (ENA accession no. FWDH01000001) by the U.S. Department of Energy's Joint Genome Institute (JGI) (30) using Pacific Biosciences (PacBio) SMRTbell libraries, with sequencing on the PacBio RS/RS II platform (31).
Supplementary Material
ACKNOWLEDGMENTS
We thank Daehwan Chung and Janet Westpheling for providing C. bescii strains JWCB005, JWCB018, and JWCB032.
This research was supported by a grant (DE-PS02-06ER64304) from the Bioenergy Science Center (BESC), Oak Ridge National Laboratory, a U.S. Department of Energy (DOE) Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00444-17.
REFERENCES
- 1.Yang SJ, Kataeva I, Wiegel J, Yin Y, Dam P, Xu Y, Westpheling J, Adams MW. 2010. Classification of 'Anaerocellum thermophilum' strain DSM 6725 as Caldicellulosiruptor bescii sp. nov. Int J Syst Evol Microbiol 60:2011–2015. doi: 10.1099/ijs.0.017731-0. [DOI] [PubMed] [Google Scholar]
- 2.Chung D, Farkas J, Huddleston JR, Olivar E, Westpheling J. 2012. Methylation by a unique α-class N4-cytosine methyltransferase is required for DNA transformation of Caldicellulosiruptor bescii DSM6725. PLoS One 7:e43844. doi: 10.1371/journal.pone.0043844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung D, Farkas J, Westpheling J. 2013. Overcoming restriction as a barrier to DNA transformation in Caldicellulosiruptor species results in efficient marker replacement. Biotechnol Biofuels 6:82. doi: 10.1186/1754-6834-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung D, Cha M, Farkas J, Westpheling J. 2013. Construction of a stable replicating shuttle vector for Caldicellulosiruptor species: use for extending genetic methodologies to other members of this genus. PLoS One 8:e62881. doi: 10.1371/journal.pone.0062881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipscomb GL, Conway JM, Blumer-Schuette SE, Kelly RM, Adams MWW. 2016. Highly thermostable kanamycin resistance marker expands the toolkit for genetic manipulation of Caldicellulosiruptor bescii. Appl Environ Microbiol 82:4421–4428. doi: 10.1128/AEM.00570-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung D, Pattathil S, Biswal AK, Hahn MG, Mohnen D, Westpheling J. 2014. Deletion of a gene cluster encoding pectin degrading enzymes in Caldicellulosiruptor bescii reveals an important role for pectin in plant biomass recalcitrance. Biotechnol Biofuels 7:1–12. doi: 10.1186/1754-6834-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young J, Chung D, Bomble YJ, Himmel ME, Westpheling J. 2014. Deletion of Caldicellulosiruptor bescii CelA reveals its crucial role in the deconstruction of lignocellulosic biomass. Biotechnol Biofuels 7:1–8. doi: 10.1186/1754-6834-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung D, Young J, Cha M, Brunecky R, Bomble YJ, Himmel ME, Westpheling J. 2015. Expression of the Acidothermus cellulolyticus E1 endoglucanase in Caldicellulosiruptor bescii enhances its ability to deconstruct crystalline cellulose. Biotechnol Biofuels 8:113. doi: 10.1186/s13068-015-0296-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conway JM, Pierce WS, Le JH, Harper GW, Wright JH, Tucker AL, Zurawski JV, Lee LL, Blumer-Schuette SE, Kelly RM. 2016. Multidomain, surface layer associated glycoside hydrolases contribute to plant polysaccharide degradation by Caldicellulosiruptor species. J Biol Chem 291:6732–6747. doi: 10.1074/jbc.M115.707810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SK, Chung D, Himmel ME, Bomble YJ, Westpheling J. 2016. Heterologous expression of family 10 xylanases from Acidothermus cellulolyticus enhances the exoproteome of Caldicellulosiruptor bescii and growth on xylan substrates. Biotechnol Biofuels 9:176. doi: 10.1186/s13068-016-0588-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung D, Verbeke TJ, Cross KL, Westpheling J, Elkins JG. 2015. Expression of a heat-stable NADPH-dependent alcohol dehydrogenase in Caldicellulosiruptor bescii results in furan aldehyde detoxification. Biotechnol Biofuels 8:102. doi: 10.1186/s13068-015-0287-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott IM, Rubinstein GM, Lipscomb GL, Basen M, Schut GJ, Rhaesa AM, Lancaster WA, Poole FL Jr, Kelly RM, Adams MWW. 2015. A new class of tungsten-containing oxidoreductase in the genus of the plant biomass-degrading, thermophilic bacteria Caldicellulosiruptor. Appl Environ Microbiol 81:7339–7347. doi: 10.1128/AEM.01634-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cha M, Chung D, Elkins JG, Guss AM, Westpheling J. 2013. Metabolic engineering of Caldicellulosiruptor bescii yields increased hydrogen production from lignocellulosic biomass. Biotechnol Biofuels 6:1–8. doi: 10.1186/1754-6834-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cha M, Chung D, Westpheling J. 2016. Deletion of a gene cluster for [Ni-Fe] hydrogenase maturation in the anaerobic hyperthermophilic bacterium Caldicellulosiruptor bescii identifies its role in hydrogen metabolism. Appl Microbiol Biotechnol 100:1823–1831. doi: 10.1007/s00253-015-7025-z. [DOI] [PubMed] [Google Scholar]
- 15.Chung D, Cha M, Guss AM, Westpheling J. 2014. Direct conversion of plant biomass to ethanol by engineered Caldicellulosiruptor bescii. Proc Natl Acad Sci U S A 111:8931–8936. doi: 10.1073/pnas.1402210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung D, Cha M, Snyder EN, Elkins JG, Guss AM, Westpheling J. 2015. Cellulosic ethanol production via consolidated bioprocessing at 75°C by engineered Caldicellulosiruptor bescii. Biotechnol Biofuels 8:1–13. doi: 10.1186/s13068-014-0179-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cha M, Wang H, Chung D, Bennetzen JL, Westpheling J. 2013. Isolation and bioinformatic analysis of a novel transposable element, ISCbe4, from the hyperthermophilic bacterium, Caldicellulosiruptor bescii. J Ind Microbiol Biotechnol 40:1443–1448. doi: 10.1007/s10295-013-1345-8. [DOI] [PubMed] [Google Scholar]
- 18.Basen M, Schut GJ, Nguyen DM, Lipscomb GL, Benn RA, Prybol CJ, Vaccaro BJ, Poole FL, Kelly RM, Adams MWW. 2014. Single gene insertion drives bioalcohol production by a thermophilic archaeon. Proc Natl Acad Sci U S A 111:17618–17623. doi: 10.1073/pnas.1413789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hennig S, Ziebuhr W. 2008. A transposase-independent mechanism gives rise to precise excision of IS256 from insertion sites in Staphylococcus epidermidis. J Bacteriol 190:1488–1490. doi: 10.1128/JB.01290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curcio MJ, Derbyshire KM. 2003. The outs and ins of transposition: from Mu to kangaroo. Nat Rev Mol Cell Biol 4:865–877. doi: 10.1038/nrm1241. [DOI] [PubMed] [Google Scholar]
- 21.Kataeva IA, Yang SJ, Dam P, Poole FL Jr, Yin Y, Zhou F, Chou WC, Xu Y, Goodwin L, Sims DR, Detter JC, Hauser LJ, Westpheling J, Adams MWW. 2009. Genome sequence of the anaerobic, thermophilic, and cellulolytic bacterium “Anaerocellum thermophilum” DSM 6725. J Bacteriol 191:3760–3761. doi: 10.1128/JB.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. 2011. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol 12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blumer-Schuette SE, Giannone RJ, Zurawski JV, Ozdemir I, Ma Q, Yin Y, Xu Y, Kataeva I, Poole FL Jr, Adams MWW, Hamilton-Brehm SD, Elkins JG, Larimer FW, Land ML, Hauser LJ, Cottingham RW, Hettich RL, Kelly RM. 2012. Caldicellulosiruptor core and pangenomes reveal determinants for noncellulosomal thermophilic deconstruction of plant biomass. J Bacteriol 194:4015–4028. doi: 10.1128/JB.00266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blumer-Schuette SE, Alahuhta M, Conway JM, Lee LL, Zurawski JV, Giannone RJ, Hettich RL, Lunin VV, Himmel ME, Kelly RM. 2015. Discrete and structurally unique proteins (tāpirins) mediate attachment of extremely thermophilic Caldicellulosiruptor species to cellulose. J Biol Chem 290:10645–10656. doi: 10.1074/jbc.M115.641480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guérillot R, Siguier P, Gourbeyre E, Chandler M, Glaser P. 2014. The diversity of prokaryotic DDE transposases of the mutator superfamily, insertion specificity, and association with conjugation machineries. Genome Biol Evol 6:260–272. doi: 10.1093/gbe/evu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA III, Smith HO, Venter JC. 2006. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A 103:425–430. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attmannspacher U, Scharf BE, Harshey RM. 2008. FliL is essential for swarming: motor rotation in absence of FliL fractures the flagellar rod in swarmer cells of Salmonella enterica. Mol Microbiol 68:328–341. doi: 10.1111/j.1365-2958.2008.06170.x. [DOI] [PubMed] [Google Scholar]
- 28.Farkas J, Chung D, Cha M, Copeland J, Grayeski P, Westpheling J. 2013. Improved growth media and culture techniques for genetic analysis and assessment of biomass utilization by Caldicellulosiruptor bescii. J Ind Microbiol Biotechnol 40:41–49. doi: 10.1007/s10295-012-1202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, Smirnova T, Grigoriev IV, Dubchak I. 2014. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res 42:D26–D31. doi: 10.1093/nar/gkt1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 32.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 33.Otto TD, Dillon GP, Degrave WS, Berriman M. 2011. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res 39:e57. doi: 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. 2010. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 7:455–457. doi: 10.1038/nmeth.1457. [DOI] [PubMed] [Google Scholar]
- 36.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. 2009. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 40.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, Lehvaslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res 12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung D, Young J, Bomble YJ, Vander Wall TA, Groom J, Himmel ME, Westpheling J. 2015. Homologous expression of the Caldicellulosiruptor bescii CelA reveals that the extracellular protein is glycosylated. PLoS One 10:e0119508. doi: 10.1371/journal.pone.0119508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SK, Chung D, Himmel ME, Bomble YJ, Westpheling J. 2016. Engineering the N-terminal End of CelA results in improved performance and growth of Caldicellulosiruptor bescii on crystalline cellulose. Biotechnol Bioeng 114:945–950. doi: 10.1002/bit.26242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.