

ABSTRACT
Cellular pigmentation is an important virulence factor of the oral pathogen Porphyromonas gingivalis. Pigmentation has been associated with many bacterial functions, including but not limited to colonization, maintaining a local anaerobic environment by binding oxygen molecules, and defense against reactive oxygen species (ROS) produced by immune cells. Pigmentation-associated loci identified to date have involved lipopolysaccharide, fimbriae, and heme acquisition and processing. We utilized a transposon mutant library of P. gingivalis strain ATCC 33277 and screened for pigmentation-defective colonies using massively parallel sequencing of the transposon junctions (Tn-seq) to identify genes involved in pigmentation. Transposon insertions at 235 separate sites, located in 67 genes and 15 intergenic regions, resulted in altered pigmentation: 7 of the genes had previously been shown to be involved in pigmentation, while 75 genes and intergenic regions had not. To further confirm identification, we generated a smaller transposon mutant library in P. gingivalis strain W83 and identified pigment mutations in several of the same loci as those identified in the screen in ATCC 33277 but also eight that were not identified in the ATCC 33277 screen. PGN_0361/PG_0264, a putative glycosyltransferase gene located between two tRNA synthetase genes and adjacent to a miniature inverted-repeat transposable element, was identified in the Tn-seq screen and then verified through targeted deletion and complementation. Deletion mutations in PGN_0361/PG_0264 glycosyltransferase abolish pigmentation, modulate gingipain protease activity, and alter lipopolysaccharide. The mechanisms of involvement in pigmentation for other loci identified in this study remain to be determined, but our screen provides the most complete survey of genes involved in pigmentation to date.
IMPORTANCE P. gingivalis has been implicated in the onset and progression of periodontal disease. One important virulence factor is the bacterium's ability to produce pigment. Using a transposon library, we were able to identify both known and novel genes involved in pigmentation of P. gingivalis. We identified a glycosyltransferase, previously not associated with pigmentation, that is required for pigmentation and determined its mechanism of involvement. A better understanding of the genes involved in pigmentation may lead to new insights into the complex mechanisms involved in this important virulence characteristic and could facilitate development of novel therapeutics.
KEYWORD: Porphyromonas gingivalis
INTRODUCTION
Microbial pigments serve diverse functional roles in the life cycle and physiology of their producers and may affect interactions within the microbial community and with host cells (1). In bacterial pathogens, pigmentation is associated with virulence and protection against antimicrobial compounds, and microbial pigments can be components of toxins (1). Porphyromonas gingivalis, a bacterium found in diseased human oral gingiva, produces a characteristic black pigment that consists of μ-oxo bis-heme dimers tethered to the outer surface of the cell (2). Pigmentation is considered a major virulence factor of the species, involved in sequestering iron to fulfill nutritional requirements as well as serving as a defense against oxidative stress (1, 3, 4).
Components of the colony pigmentation biosynthetic and regulatory interaction network have been identified through targeted deletions and Tn4400- or Tn4351-based transposon mutagenesis (5). Among the pigmentation-associated loci identified were genes involved in lysine gingipain production, lipopolysaccharide (LPS) biosynthesis and modification, fimbriae, heme acquisition, and transport and processing functions. P. gingivalis LPS can be extensively modified. Four different lipid A moieties and at least two distinct repeating oligosaccharides attached to the core oligosaccharide have been identified (6, 7). P. gingivalis LPS can be an agonist or antagonist for Toll-like receptor 2 (TLR2) and TLR4, causes alveolar bone resorption, and is an important target for antimicrobial compounds (8, 9). Alterations or loss of LPS structure may lead to changes on the outer surface of the bacterial cell, which in turn may result in loss of heme binding.
Changes in other cell surface structures such as fimbriae may also alter heme binding sites. The fimbriae of P. gingivalis, both the major (fimA) and minor (mfa1) types, serve as attachment points to bridge bacterium-bacterium interactions as well as bacterium-host interactions (10).
It is thought that P. gingivalis growth is dependent on exogenous sources of heme, and several heme acquisition, uptake, and processing loci have been found to be necessary for growth and virulence (11). Limited heme acquisition or processing may lead to lack of sufficient heme to display on the cell surface. Additionally, modifications to heme or regulatory signals may be necessary for P. gingivalis to display heme on the cell surface, as the absence of red blood cells (RBCs) in growth medium essentially abolishes colony pigmentation regardless of heme or protoporphyrin IX concentration (our unpublished data).
In this study, we utilized transposon mutant libraries of P. gingivalis to further characterize the colony pigmentation phenotype (12). These libraries were constructed using a Mariner transposon system that results in a greater number of insertions into different loci than those obtained with other methods. Our studies identify 75 genes and sites that have not previously been associated with pigmentation. We selected one gene, coding for a putative glycosyltransferase, confirmed its role in pigmentation, and characterized the mechanism of its involvement.
RESULTS
Transposon mutant library colony pigmentation screen and Tn-seq.
Preliminary colony pigmentation screens were performed with strain ATCC 33277 and W83 mutant libraries to optimize the selection conditions. Clones with altered pigmentation selected in the preliminary screens had transposon insertions at multiple sites as determined by nested semirandom sequencing as previously described (12, 13). Several genes known to be involved in pigmentation were identified in this manner, and few repetitive insertion sites between clones were found (data not shown), suggesting that a larger screen was appropriate. Based on the growth and pigmentation rates of mutants as colonies on blood agar plates containing Trypticase soy agar supplemented with defibrinated sheep’s blood (5% vol/vol), hemin (5 µg/ml), and menadione (0.5 µg/ml) (BAPHK), we chose a range of 7 to 21 days for screening based on colony size, pigmentation, and hemolysis patterns.
Four separate screens were performed, each using a single aliquot of a pooled P. gingivalis ATCC 33277 mutant library that has been previously characterized (12). Clones were isolated between 7 and 14 days of growth. Each clone was replated to confirm the pigmentation defect and then prepared for massively parallel sequencing of the transposon junctions (Tn-seq). Equal amounts of each isolated pigmentation mutant were combined into a single sample for sequencing. Sequencing of transposon junctions identified a total of 235 unique transposon insertions from colonies with pigmentation defects (Table 1; see also Table S1 in the supplemental material). Of the total of 235 insertions, 121 are within kgp, hagA, and rgpA, at 107 unique sites.
TABLE 1.
P. gingivalis colony pigmentation-associated screen Tn-seq (limited detail)a
| Locus | Gene | Product or description |
|---|---|---|
| PGN_0026 | Cytidine deaminase-like protein (IPR016193) | |
| PGN_0048 | PcfK-like protein | |
| PGN_0100 | Diaminopimelate decarboxylase LysA | |
| PGN_0103 | tonB | TonB |
| PGN_0123 | PorSS C-terminal domain protein | |
| PGN_0184 | fimD | Minor component FimD |
| PGN_0215 | Hypothetical protein | |
| PGN_0287 | mfa1 | Mfa1 fimbrillin |
| PGN_0289 | mfa2 | Fimbrillin A-associated anchor protein Mfa2 |
| PGN_0361 | Glycosyltransferase family 2 | |
| PGN_0380 | nagC | Transcriptional regulator/sugar kinase |
| PGN_0413 | gyrB | DNA gyrase B subunit |
| PGN_0438 | Hypothetical protein | |
| PGN_0465 | relA | GTP pyrophosphokinase |
| PGN_0504 | scpB | Methylmalonyl coenzyme A decarboxylase beta subunit |
| PGN_0533 | nadA | Quinolinate synthetase A (IPR003473) |
| PGN_0637 | htrA | Heat shock-related protease HtrA protein |
| PGN_0715 | Outer membrane efflux protein (IPR003423) | |
| PGN_0778 | porT | PorT |
| PGN_0789 | TPR domain protein | |
| PGN_0862 | Type III restriction enzyme, Res subunit | |
| PGN_0903 | fimR | Two-component system response regulator FimR |
| PGN_0946 | Hypothetical protein | |
| PGN_0949 | ABC transporter ATP-binding protein | |
| PGN_0950 | ABC transporter ATP-binding protein | |
| PGN_0952 | Carboxyl-terminal processing protease | |
| PGN_1105 | FKBP-type peptidyl-prolyl cis-trans isomerase | |
| PGN_1116 | Aminotransferase, class I/class II (IPR004839) | |
| PGN_1120 | NADPH-NAD transhydrogenase/alanine dehydrogenase/PNT | |
| PGN_1234 | Hypothetical protein | |
| PGN_1272 | Diaminopimelate decarboxylase LysA | |
| PGN_1275 | tonB | TonB |
| PGN_1282 | traN | Conjugate transposon protein TraN |
| PGN_1284 | traP | DNA primase involved in conjugation TraP |
| PGN_1302 | waaL | O-antigen ligase |
| PGN_1303 | Lipoprotein | |
| PGN_1313 | Hypothetical protein | |
| PGN_1591 | Hypothetical protein | |
| PGN_1618 | Methionine gamma-lyase | |
| PGN_1643 | PF07610 family protein/PapD-like (IPR008962) | |
| PGN_1666 | purL | Phosphoribosylformylglycinamidine synthase |
| PGN_1675 | porL | Por secretion system protein PorL/GldL |
| PGN_1690 | Alpha-l-fucosidase | |
| PGN_1722 | Phosphoribulokinase/uridine kinase (IPR006083) | |
| PGN_1728 | kgp | Lysine-specific cysteine proteinase Kgp |
| PGN_1733 | hagA | Hemagglutinin protein HagA |
| PGN_1770 | PorSS C-terminal domain protein | |
| PGN_1777 | Peptidase C1B, bleomycin hydrolase (IPR004134) | |
| PGN_1812 | ppk | Polyphosphate kinase |
| PGN_1914 | Carboxyl-terminal processing protease | |
| PGN_1916 | ABC transporter ATP-binding protein | |
| PGN_1917 | ABC transporter ATP-binding protein | |
| PGN_1920 | Membrane transport protein, MMPL domain (IPR004869) | |
| PGN_1970 | rgpA | Arginine-specific cysteine proteinase RgpA |
| PGN_1998 | Outer membrane protein, OmpA/MotB, C terminal (IPR006665) | |
| PGN_2010 | Secreted protein, YngK-like | |
| PGN_2017 | YjeF family protein | |
| PGN_2047 | Hypothetical protein | |
| PGN_2080 | Pectin lyase fold/virulence factor (IPR011050)/PorSS C-terminal domain | |
| PGN_r0001 | 16S rRNA | |
| PGN_r0002 | 23S rRNA | |
| PGN_r0002 | 23S rRNA | |
| PGN_r0004 | 16S rRNA | |
| PGN_r0005 | 23S rRNA | |
| PGN_r0008 | 23S rRNA | |
| PGN_r0009 | 16S rRNA | |
| PGN_r0010 | 16S rRNA | |
| PGN_r0011 | 23S rRNA | |
| Intergenic region | 3′ to hagB | |
| Intergenic region | 3′ to PGN_0136 | |
| Intergenic region | 3′ to murQ and 5′ to PGN_1195 | |
| Intergenic region | 3′ to PGN_0840 (ispD) | |
| Intergenic region | 3′ to PGN_0326 | |
| Intergenic region | 3′ to PGN_1770 | |
| Intergenic region | 5′ to PGN_0329 (transglutaminase gene) | |
| Intergenic region | 3′ to PGN_1523 (polysaccharide export protein gene) | |
| Intergenic region | 3′ to PGN_1791 (flavodoxin gene fldA) | |
| Intergenic region | 5′ to PGN_0904 (fimS) | |
| Intergenic region | 3′ to PGN_0174 (AraC transcriptional regulator) | |
| Intergenic region | ISPg1 | |
| Intergenic region | 5′ to rRNA-16S rRNA (PGN_r0009) | |
| Intergenic region | 3′ to PGN_0170 (l-asparaginase gene) | |
| Intergenic region | 5′ to PGN_0889 (tkrA) | |
| Intergenic region | CRISPR 30–36 | |
| Intergenic region | ISPg3 TIR |
Genes and intergenic regions identified through Tn-seq analysis. The ATCC 33277 strain locus name, gene designation, and protein functional characterization are provided. Protein functional characterizations were either found through NCBI or determined using Pfam. Gene designations are noted if they are NCBI listed or if a previous article had characterized the gene in P. gingivalis.
We also screened a nonsaturated transposon mutant library in P. gingivalis strain W83, which contains approximately 12,000 transposon insertions, to confirm findings obtained using the 33277 transposon library. Because this library is missing insertions into many nonessential genes, we would not expect the screen of this library to yield complete results. Approximately 50 pigmentation-deficient colonies were selected, and the transposon insertion sites were identified by direct sequencing of PCR amplicons of the transposon insertional junction as previously described (12, 13) (see Table S2 in the supplemental material). A total of 19 genes were identified in this screen, of which 8 overlapped with those identified in the screen using the 33277 transposon library, porU, porL, PG_0264 (glycosyltransferase gene), rgp, traP, waaL, kgp, and hagE, while 11 genes were identified only in this screen of the W83 transposon library.
In silico analyses of PGN_0361/PG_0264.
Two nonpigmenting mutants with insertions in PGN_0361 were isolated from the ATCC 33277 library, and one mutant with an insertion in the corresponding gene (PG_0264) was isolated from the W83 library. The insertion site within the W83 isolate is identical to that in one of the ATCC 33277 isolates, and these strains were used for all subsequent assays. The transposon insertion is 174 nucleotides (corresponding to 58 amino acids) into the gene, which is a total of 1,068 nucleotides.
We performed in silico analyses to characterize PGN_0361/PG_0264. Pfam analysis identified a glycosyltransferase 2_3 domain that encompasses the N-terminal half of the gene product (E value, 3.6 × 10−23), a motif that is widely distributed among prokaryotic and eukaryotic species (see Fig. S1A in the supplemental material). Interproscan analysis confirmed the glycosyltransferase 2_3 domain, which is also recognized in GENE3D and PANTHER. The C-terminal third of the protein contains no predicted domains.
The PGN_0361/PG_0264 gene is located immediately downstream of the tyrosyl-tRNA synthetase gene (tyrS). tyrS is essential and presumably supplies a promoter for transcription of PGN_0361/PG_0264, given the lack of space or a predictable promoter upstream of PGN_0361/PG_0264 (12). The first true protein-encoding sequence downstream of PGN_0361/PG_0264 is that for arginyl-tRNA synthetase, encoded by argS, another essential gene. However, between the 3′ end of PGN_0361/PG_0264 and the 5′ end of argS is a nonautonomous transposable element (14). Thus, the PGN_0361/PG_0264 gene is confined between two essential tRNA synthetase genes and adjacent to a repetitive nonautonomous transposable element (see Fig. S2 in the supplemental material).
The NCBI program BLAST and Conserved Domain Database (CDD) were queried to find homologues of PGN_0361/PG_0264, the genomic landscape of PGN_0361/PG_0264 homologues, and potential active sites of the glycosyltransferase domain. Following BLASTn and BLASTp for PGN_0361/PG_0264 (using default BLAST settings), all the matches with E values greater than 1 × 10−100 were within the phylum Bacteroidetes. Porphyromonas gingivicanis encodes the tyrosyl-tRNA synthetase upstream of the PGN_0361/PG_0264 homologue, similar to P. gingivalis, but there has been downstream rearrangement resulting in truncation of the protein. Recombination and repair factor recR is the upstream gene in the majority of other Porphyromonas, Parabacteroides, Haliscomenobacter, Sporocytophaga, Owenweeksia, and Tannerella species. The Fluviicola taffensis homologue contains the endoribonuclease L-PSP gene directly upstream; this gene has a repetitive nonautonomous transposable element associated with it in P. gingivalis. Of note, the C-terminal regions of the proteins have less homology between strains and species than the N-terminal regions, which contain the glycosyltransferase 2_3 domain. The CDD queries revealed nonoverlapping predicted metal binding and transferase active sites, which were conserved throughout PGN_0361/PG_0264 homologues (Fig. S1B). An “unextendable partial coding region” is present in the Barnesiella viscericola DSM 18177 PGN_0361/PG_0264 homologue but not in Barnesiella intestinihominis YIT 11860. An “unextendable partial coding region” is present in the Porphyromonas crevioricanis strain COT-253_OH1447 homologue and is followed by a pseudogene involved in cell wall biosynthesis. In the instances of shortened sequences, the stop codon always occurs in the C-terminal region after the glycosyltransferase domain.
Protein structure predictions were determined using the PHYRE2 platform. The best model template found was n-acetylgalactosaminyltransferase 2 from humans, which is involved in mucin type O-glycan biosynthesis. O-glycan biosynthesis involves the addition of carbohydrate moieties in a repeating fashion, similar to LPS biosynthesis in bacteria, and analogous O-glycosylation systems have been described in Neisseria and Pseudomonas species (15). The carbohydrate active enzyme database (CAZY) glycosyltransferase (GT) type of human n-acetylgalactosaminyltransferase 2 is GT27; however, GT27 and GT2 are noted as having high similarities (compared to all 97 GT types) (see Table S3 in the supplemental material).
PGN_0361/PG_0264 is required for pigmentation in P. gingivalis.
To confirm that inactivation of PGN_0361/PG_0264 is responsible for the nonpigmenting phenotype, we constructed targeted gene deletions and then complemented the deletion in trans. Deletions were constructed via insertion of the erythromycin resistance-encoding ermG gene into the PGN_0361 and PG_0264 genes by homologous recombination that removed the entire target gene. All deletion clones isolated recapitulated the nonpigmenting phenotype (Fig. 1). Complementation of the transposon insertion and the targeted deletion in the strain W83 background was carried out by cloning full-length PGN_0361 into the replicating vector pT-COW and conjugating the resultant pT-COW::PGN_0361 back into P. gingivalis strains. All complemented strains showed wild-type (WT) pigmentation when cultured on medium containing tetracycline selective for the complement vector (Fig. 1). We also quantified the amounts of heme bound to W83, the PG_0264 mutant, and the complemented strain grown on solid medium. The PG_0264 mutant strain bound significantly less heme than the wild-type strain (P < 0.05), and complementation resulted in a significant increase in heme binding (P < 0.01) (Fig. 1E).
FIG 1.
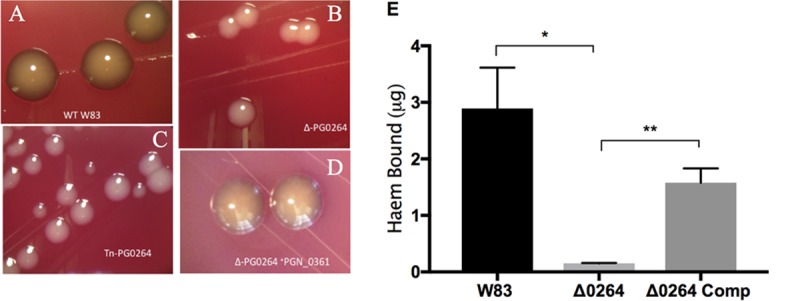
Pigmentation of wild-type and putative glycosyltransferase mutant strains on blood agar. (A to D) Close-up images of individual colonies of W83 (wild type) and the putative glycosyltransferase deletion mutant (Δ-PG0264), the putative glycosyltransferase transposon mutant (Tn-PG0264), and the complemented deletion mutant (Δ-PG0264 +PGN_0361) in the W83 strain background are shown. (E) Heme bound to W83, ΔPG_0264, and ΔPG_0264 complemented (Δ0264 Comp) strains was quantified from colonies grown on blood agar plates for 6 days. Cells were resuspended in PBS and adjusted to equivalent optical densities. Heme was quantified from 50 μl of these cell suspensions. Shown are the means ± standard errors from three independent experiments, each performed in triplicate. Statistical significance was determined by two-tailed t test (*, P < 0.05; **, P < 0.01).
PG_0264 mutant growth characteristics.
To determine the effects and mechanism of action of the PGN_0361/PG_0264 gene, we employed assays to assess heme metabolism, gingipain protease activity and expression, and lipopolysaccharide modifications, as these have been identified as critical to pigmentation.
Three different types of broth growth experiments were carried out. First, wild-type and mutant strains were grown anaerobically at 37°C for 48 h to observe growth rates. Growth of the glycosyltransferase mutant did not substantially differ from that of the wild type in standard broth medium supplementation (Fig. 2). However, both the complemented strains have growth curves that are significantly different from that of W83 at time points between 16 and 35 h, before reaching an endpoint similar to that of the wild-type and mutant strains (P < 0.05). In addition, the growth curve of a strain with a transposon insertion in the O-antigen ligase gene (the Tn-waaL mutant) significantly deviates from that of W83 between 18 and 29 h (P < 0.05).
FIG 2.
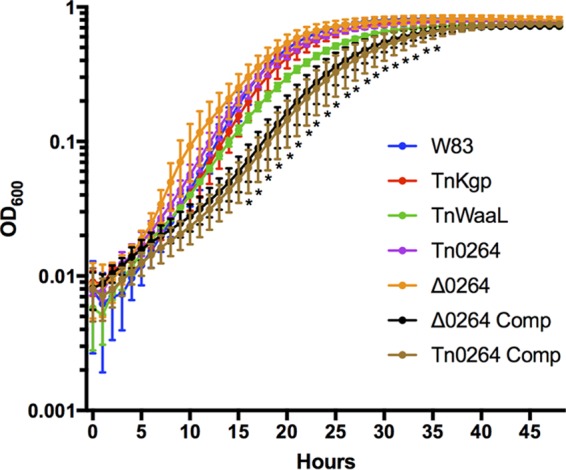
Growth curves of wild-type and pigmentation-defective mutant strains. Forty-eight-hour cultures of each strain were diluted into fresh BHIHKSbcStgC medium to an OD600 of 0.02. Growth was monitored under anaerobic conditions for 48 h. Strains include W83 (wild type), a lysine gingipain transposon mutant (TnKgp), an O-antigen ligase transposon mutant (TnWaaL), a PG_0264 glycosyltransferase transposon mutant (Tn0264), a PG_0264 glycosyltransferase deletion mutant (Δ0264), a complemented PG_0264 glycosyltransferase deletion mutant (Δ0264 Comp), and a complemented PG_0264 transposon mutant (Tn0264 Comp). The growth curves represent the averages from three independent experiments. Error bars represent the standard errors. Statistical significance was determined by two-way analysis of variance (ANOVA) and corrected for multiple comparisons (P < 0.05).
Wild-type and mutant strains were grown in BHIHKSbcStgC medium (see Materials and Methods) anaerobically at 37°C for 72 h. Cells from wild-type and PG_0264 mutants were visually identical at endpoints after static growth. In contrast, the control, O-antigen ligase (waaL) mutant culture resulted in a significant pelleting phenotype (see Fig. S3 in the supplemental material). Wild-type W83, the PG_0264 deletion strain, and the O-antigen ligase waaL transposon insertion mutant were also grown on blood agar either with or without hemin supplementation. No colony morphology differences were apparent for any of the three strains due to exogenous hemin supplementation of blood agar (see Fig. S4 in the supplemental material).
We also examined the PG_0264 mutant's susceptibility to hydrogen peroxide stress since pigmentation has been implicated in protection from oxidative stress. Indeed, we found that the nonpigmented PG_0264 mutant is more susceptible to hydrogen peroxide stress than the wild type and that complementation restores susceptibility to wild-type levels (P < 0.05) (see Fig. S5 in the supplemental material).
The PG_0264 mutant lacks arginine and lysine gingipain activity.
The PG_0264 deletion mutant was completely deficient in Rgp and Kgp gingipain activity, in contrast to the wild-type strain. Complementation of the deletion mutant restored Rgp and Kgp gingipain activity. The O-antigen ligase transposon mutant (Tn-waaL), identified as deficient in pigmentation in this screen, was also deficient in both arginine gingipain and lysine gingipain activities. Similarly, as expected, the control lysine gingipain transposon mutant, which also was deficient in pigmentation, lacked lysine gingipain activity but retained arginine gingipain activity (Fig. 3).
FIG 3.

Lysine and arginine gingipain activities of pigmentation-defective mutants. Wild-type and mutant lines were grown either on solid medium (TSB blood agar) or in liquid culture (BHI) and assayed for gingipain activity using fluorogenic substrates. Whole, live cells grown on solid medium were removed from agar and assayed directly. Liquid cultures were separated by centrifugation, and washed cells and supernatant media were assayed separately. The activities of cells grown on solid medium (S), cells grown in liquid medium (L), and supernatant media of liquid cultures are compared. Values indicate lysine gingipain and arginine gingipain activities of mutant lines as a ratio to the activities of the wild-type control (parent strain W83). Samples differing from the WT with statistical significance (P ≤ 0.05) are indicated with an asterisk.
rgpA expression is downregulated in the PG_0264 mutant.
To determine whether the lack of gingipain activity observed was due to altered gingipain expression patterns in the PG_0264 mutant, we measured the expression of rgpA and kgp. There was significantly less rgpA expression in the PG_0264 mutant than in the wild-type control (P < 0.001), and the complemented strain significantly increased the expression of rgpA compared to that of the PG_0264 mutant (P < 0.001) (Fig. 4). kgp expression was not statistically significantly different between the strains. We also examined the expression level of hagA, a hemagglutinin protein important in pigmentation, and found there to be no difference in expression between the PG_0264 mutant and the wild-type control (Fig. 4) (16). Of note, the Kgp precursor is known to be cleaved by Rgp to form the mature Kgp enzyme (17). Therefore, Kgp activity can be substantially reduced if expression of rgpA is low.
FIG 4.

Expression of rgpA, kgp, and hagA in the PG_0264 mutant. Shown are the results of qRT-PCR experiments performed with the wild-type strain W83, its derivative PG_0264 mutant strain, and the complemented strain. Transcript levels of rgpA, kgp, and hagA were measured from mid-log cultures of the indicated strains and normalized to 16S transcript levels. Shown are the means ± standard errors from six independent experiments, each performed in triplicate. Statistical significance was determined by a one-way ANOVA corrected for multiple comparisons (***, P < 0.001; ns, not significant).
PG_0264 deletion results in altered LPS structure.
Since pigmentation and gingipain activity were affected in the PG_0264 deletion mutant, we hypothesized that the putative glycosyltransferase may modify LPS structure and thereby affect pigmentation and gingipain activity (18, 19). We employed three different methods to analyze lipopolysaccharide. First, LPS preparations were examined via silver staining following SDS-PAGE. Variations in the banding patterns between wild-type and deletion mutant strains are apparent (Fig. 5A). The typical “laddering” that is attributed to repeating O-antigen units is absent in the deletion strains. Notably, using SDS-PAGE we observed the reverse-laddering phenotype when examining the LPS via glycan staining to visualize sugar moieties. The PG_0264 transposon insertion and deletion strains have laddering, while it is absent in the wild-type strains (Fig. 5B). The control O-antigen ligase mutant lacks all ladder-type staining, which is expected because without the activity of O-antigen ligase, only the lipid A portion of LPS is present on the cell surface (15). Complementation of both the PG_0264 transposon mutant and the PG_0264 deletion mutant with the pT-COW plasmid expressing PGN_0361 restores the wild-type LPS staining pattern, confirming that the change is due to the function of PGN_0361 (Fig. 5B). Western blotting performed with antibody 1B5, which is specific to the anionic lipopolysaccharide (APS) repeating unit of P. gingivalis LPS, reveals a complete lack of APS in both the PG_0264 transposon mutant and the PG_0264 deletion mutant; APS is restored in the complemented mutant (Fig. 5C). The waaL transposon mutant lacks APS, which has been previously reported and serves as a control for the 1B5 antibody (20).
FIG 5.

Lipopolysaccharide structural staining. (A) Silver staining of lipopolysaccharide preparations from the wild-type strain W83 and the PG_0264 deletion strain. (B) Glycan stain of the lipopolysaccharide preparations from WT W83, the PG_0264 transposon insertion mutant (TnPG0264), the PG_0264 deletion strain, the O-antigen ligase transposon insertion mutant (TnWaaL), the complemented PG_0264 transposon insertion mutant (Tn-1 Comp), and the complemented PG_0264 deletion strain (Δ-1 Comp). (C) Western blotting with monoclonal antibody 1B5 specific to APS of lipopolysaccharide preparations from WT W83, the PG_0264 transposon insertion mutant, the PG_0264 deletion strain, the O-antigen ligase transposon insertion mutant, the complemented PG_0264 transposon insertion mutant, and the complemented PG_0264 deletion strain.
DISCUSSION
Using our transposon mutant library and sequencing strategy, we sought to identify new genes that are involved in pigmentation of P. gingivalis. The majority of mutants identified from the Tn-seq screen had insertions into genes already known to be involved in colony pigmentation: kgp, rgpA, and hagA. These serve to validate the screen. The kgp, hagA, and rgpA genes have highly repetitive hemagglutinin domains and were previously shown to be involved in colony pigmentation (16, 21). The kgp, hagA, and rgpA genes as well as the other gingipain and hemagglutinin genes constitute the largest protein coding sequences in the genome and are the most highly saturated with transposon insertions in the mutant library. Therefore, insertions in these genes would be expected to be present in the majority of mutants selected in the screen.
Other genes known or likely to be involved in pigmentation identified in our screen include tonB and lysA, each of which is found twice within the genome. Pigmentation defects were identified in both copies of tonB and lysA. TonB facilitates active transport of substrates across the outer membrane (22). Importantly, in the absence of TonB, outer membrane transporters bind their cognate substrate but cannot transport the substrate into the periplasm. In P. gingivalis, TonB is involved in the uptake of iron and heme by acting with each of the four known heme uptake systems: those encoded by hmu, iht, tlr, and rag (23, 24). LysA, the meso-diaminopimelate decarboxylase, is involved in peptidoglycan biosynthesis, thus potentially affecting the outer membrane components that bind and complex heme to the outer surface of the colony.
We identified several rRNA genes in which transposon insertion disrupted pigmentation. Multiple insertions were seen within 16S rRNA and 23S rRNA loci and in ISPg1 regions. Mutations in these ribosomal genes can affect translation and growth rates in general, which may delay colony pigmentation rather than completely abolish pigmentation.
Candidate genes lacking a currently recognized functional link to pigmentation include PGN_0361, PGN_0637 (heat shock-related protease-encoding gene htrA), PGN_1770/PGN_0123/PGN_2080 (encoding the Por secretion system [PorSS] C-terminal domain), and PGN_1116 and PGN_1234 (encoding aminotransferase class I/classII). The heat shock-related protease-encoding gene htrA (PGN_0637) was identified three independent times in the screen. htrA and its homologues have been shown to affect protein maturation as well as outer membrane morphology (25). Two of the three PorSS C-terminal domain-encoding genes (PGN_1770, PGN_0123, and PGN_2080) identified in the screen are adjacent to known pigment- or heme-affecting loci. The Por secretion system is a Bacteroides-specific secretion system that in P. gingivalis is responsible for gingipain protease secretion, and interruption of most PorSS genes leads to pigmentation defects (26). Since these PorSS C-terminal domain genes are located downstream of the previously identified pigment-affecting genes (omp28 and gppX), it is tempting to speculate that the effects on pigmentation are due to alterations in protein transport or localization. The two aminotransferase genes (PGN_1116 and PGN_1234) are each located adjacent to known pigment-affecting loci, genes encoding a PorSS C-terminal domain protein and the PorR/PorS two-component system, respectively. Given the gene orientations, polarity of insertions should not be a factor in the phenotype. Of note, aminotransferase domains are found within proteins that are part of or feed into tetrapyrrole biosynthesis pathways (27).
A third group of candidates identified in our screen involve mobile elements. Each of the eight genes from TnPg17-A and TnPg17-B were identified once, and none of the sequences were repetitive. The two mobile elements are distinct four-gene clusters located in separate parts of the genome and comprise two ABC transporter protein genes, a carboxyl-terminal processing protein gene, and a membrane protein gene. There are 31 full (intact) versions of ISPg1 in strain ATCC 33277 (28). The IS elements are transcribed and may have outward-reading promoters that affect upstream or downstream transcription. Such mutations within these regions are likely to cause pigmentation defects when associated with other protein-encoding sequences in the pigmentation network. The insertions into the region of clustered regularly interspaced short palindromic repeats (CRISPR) 30 to 36 are located within the promoter region of an ISPg1 element. As a result, their effect on pigmentation may be indirect. Downstream of the ISPg1 element is a miniature inverted-repeat transposable element (MITE) and a phosphoglucomutase (Pgm)-encoding gene (pgm) (14). This MITE, named BrickBuilt, is comprised of 23-nucleotide tandem repeats flanked by small inverted repeats. Interestingly, BrickBuilt can drive transcription bidirectionally from promoter elements in the MITE (14). Phosphoglucomutase is involved in carbohydrate metabolism, and mutations in pgm are pleiotropic. Additionally, in Yersinia pestis, phosphoglucomutase activity modulates surface components that dramatically change polymyxin B susceptibility, a connection that could potentially result in pigmentation changes in P. gingivalis (29).
We also performed the same screen using a less complex transposon library in P. gingivalis strain W83. Of the 19 genes identified, 8 were also identified in the screen of the P. gingivalis 33277 transposon library while 11 genes were identified in the W83 screen only. Three of these 11 genes do not have transposon insertions in the 33277 library, which explains their absence from the genes identified in the 33277 transposon library screen. The remaining eight genes found only in the W83 screen do have transposon insertions in the 33277 transposon library, suggesting that the gene list derived from the Tn-seq screen using the 33277 library may not be a fully comprehensive list of pigmentation-associated genes.
We chose to focus our studies on the putative glycosyltransferase gene PGN_0361/PG_0264 because insertions were identified during the screen in both wild-type backgrounds. Deletion of PGN_0361/PG_0264 leads to pigmentation defects and altered LPS structure. Furthermore, we show that rgpA expression in the mutant is significantly less than that in the wild type. Rgp is known to process the precursor form of Kgp to yield the mature, catalytically active form of the enzyme (17). In addition, Rgp converts oxyhemoglobin to methemoglobin, which is more readily degraded by Kgp to release free heme (30). Therefore, we propose that the PG_0264 mutant is nonpigmented at least in part due to its decreased expression of rgpA and therefore a smaller population of catalytically active Kgp proteins and lower levels of the methemoglobin substrate for Kgp. As both Rgp and Kgp are involved in pigmentation, the rgpA expression pattern observed in the PG_0264 mutant could very well impair pigmentation. The altered LPS structure, most notably the lack of APS, also likely contributes to the decreased heme binding and lack of pigmentation observed in the PG_0264 mutant. In fact, APS of P. gingivalis was recently shown to be critical in the binding of μ-oxo-bisheme and is required for pigmentation (31). In addition, it is possible that PGN_0361/PG_0264 directly glycosylates Rgp, which affects its catalytic activity (32). Since gingipain activity is dramatically reduced in the PG_0264 deletion mutant and deletion of Rgp or Kgp alone leads to pigmentation defects, the effect of PG_0264 deletion on pigmentation may be through modulation of gingipain activity by glycosylation.
Glycan staining shows that the PG_0264 phenotype is WaaL (O-antigen ligase; PGN_1302/PG_1051) dependent since glycan staining is absent in the waaL mutant. This suggests that PG_0264 requires O-LPS or A-LPS to perform its function. Being O-antigen ligase dependent does potentially compartmentalize the region where the PGN_0361/PG_0264 modification takes place, given that O-LPS and A-LPS are synthesized prior to export to the cell surface. If the O-LPS and A-LPS are the sites of action for PGN_0361/PG_0264, then WbaP (PGN_1896), Wzx (PGN_1033), Wzy (PGN_1242), and WzzP (PGN_2005) as well as LptA, LptC, LptD, and LptO could be used to determine the localization of PGN_0361/PG_0264 action.
As with all screens, there are some important caveats to our results. First, some genes involved in colony pigmentation may be missed due to essentiality or insertion preferences of the transposon vector. Second, experimenter bias during selection or the timing of selection could have skewed the collection of clones. Lastly, although we screened over 144,000 colonies for pigmentation mutants, representing 4-fold coverage of the approximately 36,000 mutants in the ATCC 33277 library, we cannot guarantee that rare mutants were not missed.
In conclusion, we have utilized a Mariner transposon-based mutagenesis system with Porphyromonas gingivalis in order to isolate novel colony pigmentation-affecting loci in the species. When we screened en masse and coupled the screening with high-throughput sequencing, we identified 235 sites within the mutant library, 72 genes, and 17 intergenic regions, putatively involved in the colony pigmentation network. We believe that this is the most complete reporting of P. gingivalis genes involved in pigmentation to date. We have confirmed the validity of the screen through identification of known pigment-associated loci as well as through additional testing of a selected putative glycosyltransferase mutant identified through multiple insertions in different strains. Future testing of the remaining newly identified loci will lead to additional insight into the mechanisms and regulation of pigmentation by P. gingivalis.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Bacterial strains used in the study are listed in Table 2. P. gingivalis strains W83 and ATCC 33277 were grown on blood agar plates containing Trypticase soy agar supplemented with defibrinated sheep's blood (5%, vol/vol), hemin (5 μg/ml), and menadione (0.5 μg/ml) (BAPHK). Cultures were also grown in brain heart infusion broth containing yeast extract (1 mg/ml), hemin (5 μg/ml), menadione (0.5 μg/ml), sodium bicarbonate (1 μg/ml), sodium thioglycolate (0.25 μg/ml), and cysteine (0.5 μg/ml) (BHIHKSbcStgC). Both media were used for solid and liquid cultures of P. gingivalis. Gentamicin (25 to 50 μg/ml), erythromycin (2 to 10 μg/ml), and tetracycline (1 to 2 μg/ml) were used when appropriate for prevention of contamination as well as for isolation and maintenance of P. gingivalis mutants. Strains were grown at 37°C in GasPak EZ Anaerobe Pouch Systems (BD Biosciences) for 24 or 48 h for broth-based assays and for 7, 14, or 21 days for plate-based assays. Escherichia coli DH5α, TOP10, and S17-1 λpir were used for cloning, plasmid maintenance, and conjugation. Ampicillin (100 μg/ml) was used for isolation and maintenance of transformants containing plasmids. E. coli strains were grown and maintained on LB (Lennox) agar or in LB (Lennox) broth (Invitrogen).
TABLE 2.
Bacterial strains and plasmids
| Bacterial strain or plasmid | Description or purpose |
|---|---|
| Porphyromonas gingivalis | |
| ATCC 33277 | Wild type |
| W83 | Wild type |
| Tn-PGN_0361 mutant | ATCC 33277 background with glycosyltransferase transposon insertion |
| Tn-PG_0264 mutant | W83 background with glycosyltransferase transposon insertion |
| ΔPG_0361 mutant | ATCC 33277 background with glycosyltransferase deletion |
| ΔPG_0264 mutant | W83 background with glycosyltransferase deletion |
| Tn-PGN_1302 mutant | ATCC 33277 background waaL transposon mutant |
| Tn-PG_1051 mutant | W83 background waaL transposon mutant |
| Tn-1 Comp strain | Complemented W83 background PG_0264 transposon mutant |
| Δ-1 Comp strain | Complemented W83 background PG_0264 deletion mutant |
| Escherichia coli | |
| TOP10 | Cloning and plasmid maintenance |
| S17-1 λpir | Conjugation into P. gingivalis |
| Plasmids | |
| pT-COW | Complementation vector without insert |
| pT-COW::PGN_0361 | Complementation vector with ATCC 33277 PGN_0361 gene insert |
Growth curves.
P. gingivalis strains from 48-h cultures were inoculated into 1 ml BHIHKSbcStgC or 1 ml BHIHKSbcStgC–1 μg/ml tetracycline at an optical density at 600 nm (OD600) of 0.02. Growth was monitored using a Biotek Powerwave HT spectrophotometer placed within an anaerobic chamber (Coy Laboratory Products). OD600 measurements were taken every 15 min, following a 1-min shake, for 48 h.
Construction of P. gingivalis transposon libraries.
Transposon mutant libraries were constructed in P. gingivalis strains ATCC 33277 and W83 using a Mariner-based pSAM_Bt system as described previously (12, 13). Briefly, wild-type P. gingivalis was cultured anaerobically on BAPHK and used to inoculate BHIHKSbcStgC for overnight culture at 37°C anaerobically in a Gaspak (BD). Overnight cultures of P. gingivalis were back-diluted into BHIHKSbcStgC and incubated for 8 h to an OD600 of 0.5 to 1.0. E. coli S17-1 λpir carrying vector pSAM_Bt was inoculated into LB broth containing 100 μg/ml ampicillin to an OD600 of 0.5 to 1.0. P. gingivalis and E. coli were mixed in a conical tube and then centrifuged at low speed to pellet the bacteria; the pellets were resuspended in 1 ml of phosphate-buffered saline (PBS) and then “puddled” on BAPHK lacking antibiotics and allowed to conjugate for 5 h aerobically at 37°C. Areas of bacterial growth were scraped, and the cells were resuspended in PBS and then plated onto BAPHK containing gentamicin and erythromycin for selection. The resulting colonies were pooled into BHIHKSbcStgC containing 15% glycerol following 10 to 14 days of growth and then frozen at −80°C.
Construction of P. gingivalis mutants and complemented mutants.
Mutants with deletions of gene PGN_0361 in strain ATCC 33277 and gene PG_0264 in strain W83 were constructed using 3-way stitching and homologous recombination. PCR primers (containing complementary overlaps to ermG of vector pSAM_Bt) were designed immediately flanking the gene PGN_0361 in P. gingivalis strain ATCC 33277 to generate 500-bp products upstream and downstream of the coding sequence (CDS). The ErmG PCR product (containing complementary overlaps to PGN_0361) was designed using vector pSAM_Bt ermG. PCR products were generated using GoTaqLong master mix (Promega). An initial PCR for each of the three individual parts was performed, followed by a “stitching” PCR connecting all three pieces. Following PCR amplification and purification (Qiagen), the final stitched product was electroporated into electrocompetent wild-type ATCC 33277 and W83 cells. After overnight incubation at 37°C anaerobically in a Gaspak (BD), cells were plated onto BAPHK containing gentamicin and erythromycin and incubated at 37°C anaerobically in a Gaspak (BD). Transformants were confirmed by PCR and sequencing.
To generate complementation constructs for PGN_0361 and PG_0264 transposon and deletion mutants, the entire PGN_0361 gene was ligated into the replicating plasmid pT-COW at SphI and NheI restriction sites and transformed into E. coli TOP10. Use of these restriction sites in pT-COW removes 335 bp from the middle of the tet(R) (for E. coli) gene. Transformants were selected for ampicillin resistance generated by a recombination-insertion event. Clones were confirmed by restriction digestion, PCR, and sequencing. The complementation vector pT-COW::S0361 was then transferred to E. coli S17-1 λpir. Deletion and transposon mutant strains in the P. gingivalis W83 background were conjugated with E. coli S17-1 λpir/pT-COW::S0361 without antibiotic selection (as described above for transposon mutagenesis), and transconjugants were selected on BAPHK containing gentamicin and tetracycline and incubated at 37°C. Complementation was assessed by restoration of black colony pigmentation and sequencing.
Bioinformatic analyses.
Genome sequence FASTA and GenBank files were downloaded from the NCBI database. The Pfam and InterProScan databases and programs were used to determine the presence and characteristics of nucleic acid and protein motifs (33, 34). Query inputs were FASTA sequences from NCBI files. For Pfam, an E value of 1.0 and checking Pfam-B motifs were selected options prior to submission.
NCBI BLAST suites were utilized to identify homologues and determine locations within P. gingivalis strains and other species (35). Query inputs were FASTA sequences from NCBI genome sequencing projects. Default settings for BLASTn and BLASTp were used, as well as BLASTn of WGS data.
The PHYRE2 software platform was used to determine putative structure, structured domains, and structure changes due to single nucleotide polymorphisms (SNPs) or truncations (36).
The Geneious software platform (version R8) was used to generate sequence alignments (37).
Phenotypic analyses.
A colony pigmentation screen was performed using aliquots of the initial BAPHK-based pooled transposon mutant library (12). A single glycerol-stocked aliquot was thawed, diluted in BHIHKSbcStgC, and plated onto BAPHK to obtain singlefold coverage of the colonies contained in the library (∼35,000). The screen was repeated four times. Plates were incubated for 7 to 14 days, and clones were selected throughout the incubation on the basis of pigmentation defects including complete loss of pigmentation (white colonies) and partial and delayed pigmentation. All selected clones were repurified to confirm the pigmentation defect. Patches were made of each selected clone for Tn-seq preparation; each patch was divided into two and pooled in duplicate for genomic DNA (gDNA) preparation. Tn-seq was performed as described previously (12, 13).
Heme quantification assay.
W83, PG_0264 mutant, and complemented mutant strains were scraped from blood agar plates after 6 days of growth and resuspended in sterile PBS. The OD600 of each cell suspension was adjusted to 1.25. Total bound heme in 50 μl of cell suspension for each strain was determined using the QuantiChrom heme assay kit (BioAssay Systems) according to the manufacturer's instructions.
Hydrogen peroxide stress assay.
A hydrogen peroxide stress assay was done by measuring strain sensitivity to hydrogen peroxide compared to that of the wild-type (WT) W83 strain. All strains were grown on supplemented tryptic soy blood agar (TSBA) for 3 to 5 days to allow pigment to develop. Cells were then resuspended in liquid medium (BHI) to an OD of ∼0.2. Hydrogen peroxide (H2O2) was then added to liquid cultures to a concentration of 100 μM. Cultures were incubated aerobically with H2O2 for 20 min and then diluted for spot plating. Survival rates were determined based on a comparison of CFU present in a culture before and after exposure to H2O2. Statistical significance was determined with Dunnett's multiple-comparison test (P ≥ 0.05) and calculated by Graph Pad Prism 7 software (GraphPad Software, Inc., La Jolla, CA).
Gingipain proteinase activity.
Enzyme components of assays consisted of either whole, live cells suspended in BHIHKSbcStgC or cell-free supernatant medium taken from cultures. To obtain cells grown on solid medium, cells grown on BAPHK were resuspended in BHIHKSbcStgC medium. Cell suspensions were diluted to an OD600 of 0.02 and immediately assayed as “solid medium/cell” fractions. To obtain cells grown in liquid medium, cultures were grown to exponential phase in BHIHKSbcStgC and pelleted by centrifugation at 3,000 × g for 8 min. Cells were washed and resuspended in fresh BHIHKSbcStgC to an OD600 reading of 1.0. Part of each culture was removed and diluted to an OD600 reading of approximately 0.02 and immediately assayed as “liquid medium/cell” fractions. The remaining culture was incubated for 3 h at 37°C under anaerobic conditions. At the end of the incubation, cells were pelleted by centrifugation at 3,000 × g for 8 min. Supernatant medium was removed and clarified by centrifugation at 17,000 × g for 10 min. Clarified medium samples were assayed as “liquid medium/supernatant” fractions. All cell suspensions and cultures used for the enzyme component of assays were evaluated for CFU via spot plating.
Lysine gingipain activity was assessed with the fluorogenic substrate benzyloxycarbonyl-l-histidyl-l-glutamyl-l-lysine 4-methyl-coumaryl-7-amide (Z-His-Glu-Lys-MCA) obtained from Peptides International (Louisville, KY). Arginine gingipain activity was assessed with the fluorogenic substrate t-butyloxycarbonyl-l-phenylalanyl-l-seryl-l-arginine 4-methyl-coumaryl-7-amide (Boc-Phe-Ser-Arg-MCA), also obtained from Peptides International (Louisville, KY). Assay mixes contained 32 μl gingipain buffer (200 mM Tris, 100 mM NaCl, 10 mM l-cysteine, 5 mM CaCl2), 2 μl fluorogenic substrate (2 mM stock solution in dimethyl sulfoxide; final assay concentration, 100 μM), and 6 μl enzyme. After aerobic incubation for 1 h at 37°C, assays were read at excitation and emission settings of 360 and 480 nm, respectively. Data were analyzed by calculating fluorescence readout per 106 CFU and reported relative to the fluorescence of the wild-type control.
RNA isolation and reverse transcription-quantitative PCR (qRT-PCR).
Total RNA from P. gingivalis was isolated from W83 and the PG_0264 mutant and PG_0264 complemented strains. Mid-logarithmic-phase P. gingivalis cultures were centrifuged at 10,000 rpm for 5 min and resuspended in TRIzol reagent (Ambion). RNA was isolated as per the manufacturer's instructions. Total RNA was examined for integrity by gel electrophoresis. The total RNA was DNase treated using the Turbo DNA-free kit according to the manufacturer's instructions (Ambion). RNA was reverse transcribed using the ImProm-II reverse transcription system according to the manufacturer's instructions (Promega).
qRT-PCR was performed using iTaq Universal SYBR green Supermix (Bio-Rad). The 16S gene was used for normalization. Primer pairs amplifying an approximately 150-bp product from 16S, rgpA, kgp, and hagA cDNA were added to the reaction mixture to a final concentration of 400 nM. Thermal cycling conditions were 95°C for 3 min followed by 39 cycles of 95°C for 10 s and 60°C for 30 s. Melt curves for the products were checked to ensure that only a single amplicon was produced. All samples were run in triplicate, and reverse transcriptase-free controls were included to ensure the absence of contaminating genomic DNA. W83 genomic DNA was used to generate a standard curve for each primer pair. The CFX Connect Real-Time PCR detection system (Bio-Rad) was used for the analysis.
LPS preparations and testing.
Initially, cells were resuspended at a concentration corresponding to 3 OD600 units of an overnight culture of E. coli (as a control) or P. gingivalis, separately, in 150 μl lysis buffer (2% SDS, 4% β-mercaptoethanol, 10% glycerol, 1 M Tris-HCl [pH 6.8]). Preparations were heated at 100°C for 10 min, followed by incubation with 2 μl proteinase K (20 mg/ml; Roche Applied Science) at 60°C for 2 h. Phenol (150 μl, 95%) was added and incubated at 70°C for 15 min. Preparations were then cooled on ice for 10 min and centrifuged at 18,000 × g for 10 min. The aqueous phase was transferred to a sterile tube. Finally, LPS was precipitated by the addition of 2.5 volumes of ethanol and resuspended in 50 μl distilled water (dH2O). Subsequent confirmatory experiments using LPS isolations were prepared using the Intron LPS extraction kit (Intron Biotechnology, South Korea).
LPS silver and glycan and A-LPS staining and blotting assays were carried out on purified LPS from wild-type, transposon mutant, deletion, and complemented mutant P. gingivalis strains. SDS-PAGE was carried out in 4 to 15% mini-Protean gels using a Bio-Rad minigel system. Silver staining of gels was performed using the SilverQuest silver staining kit (Invitrogen/Life Technologies) according to the manufacturer's instructions. Glycoprotein identification staining was performed using the Pierce glycoprotein staining kit (Invitrogen/Life Technologies) according to the manufacturer's instructions. For A-LPS Western blotting, nitrocellulose membranes were probed with monoclonal antibody (MAb) 1B5 directed against a glycan epitope on APS and arginine gingipain (20, 38).
Supplementary Material
ACKNOWLEDGMENTS
We greatly thank the members of the Hu lab and of the Duncan Lab and Michael Malamy for insightful discussion during the course of these studies.
This work was funded by R01 DE024308 (to L.T.H. and M.J.D.), F31 DE022491 (to B.A.K.), F31 DE025523 (to L.P.C.), and T32 AI007329 (to M.C.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00832-16.
REFERENCES
- 1.Liu GY, Nizet V. 2009. Color me bad: microbial pigments as virulence factors. Trends Microbiol 17:406–413. doi: 10.1016/j.tim.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smalley JW, Silver J, Marsh PJ, Birss AJ. 1998. The periodontopathogen Porphyromonas gingivalis binds iron protoporphyrin IX in the μ-oxo dimeric form: an oxidative buffer and possible pathogenic mechanism. Biochem J 331:681–685. doi: 10.1042/bj3310681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holt SC, Kesavalu L, Walker S, Genco CA. 1999. Virulence factors of Porphyromonas gingivalis. Periodontol 2000 20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 4.Nakayama K. 2003. Molecular genetics of Porphyromonas gingivalis: gingipains and other virulence factors. Curr Protein Pept Sci 4:389–395. doi: 10.2174/1389203033486983. [DOI] [PubMed] [Google Scholar]
- 5.Chen T, Dong H, Yong R, Duncan MJ. 2000. Pleiotropic pigmentation mutants of Porphyromonas gingivalis. Microb Pathog 28:235–247. doi: 10.1006/mpat.1999.0338. [DOI] [PubMed] [Google Scholar]
- 6.Paramonov N, Bailey D, Rangarajan M, Hashim A, Kelly G, Curtis MA, Hounsell EF. 2001. Structural analysis of the polysaccharide from the lipopolysaccharide of Porphyromonas gingivalis strain W50. Eur J Biochem 268:4698–4707. doi: 10.1046/j.1432-1327.2001.02397.x. [DOI] [PubMed] [Google Scholar]
- 7.Herath TDK, Darveau RP, Seneviratne CJ, Wang C-Y, Wang Y, Jin L. 2013. Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One 8:e58496. doi: 10.1371/journal.pone.0058496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darveau RP, Pham T-TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. 2004. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both Toll-like receptors 2 and 4. Infect Immun 72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin J, Bi L, Yu X, Kawai T, Taubman MA, Shen B, Han X. 2014. Porphyromonas gingivalis exacerbates ligature-induced, RANKL-dependent alveolar bone resorption via differential regulation of Toll-like receptor 2 (TLR2) and TLR4. Infect Immun 82:4127–4134. doi: 10.1128/IAI.02084-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshimura F, Murakami Y, Nishikawa K, Hasegawa Y, Kawaminami S. 2009. Surface components of Porphyromonas gingivalis. J Periodont Res 44:1–12. doi: 10.1111/j.1600-0765.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 11.Lewis JP. 2010. Metal uptake in host–pathogen interactions: role of iron in Porphyromonas gingivalis interactions with host organisms. Periodontol 2000 52:94–116. doi: 10.1111/j.1600-0757.2009.00329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT. 2012. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13:578. doi: 10.1186/1471-2164-13-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein B, Duncan M, Hu L. 2015. Defining essential genes and identifying virulence factors of Porphyromonas gingivalis by massively parallel sequencing of transposon libraries (Tn-seq), p 25–43. In Lu LJ. (ed), Gene essentiality. Springer, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein BA, Chen T, Scott JC, Koenigsberg AL, Duncan MJ, Hu LT. 2015. Identification and characterization of a minisatellite contained within a novel miniature inverted-repeat transposable element (MITE) of Porphyromonas gingivalis. Mob DNA 6:18. doi: 10.1186/s13100-015-0049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hug I, Feldman MF. 2011. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology 21:138–151. doi: 10.1093/glycob/cwq148. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Ratnayake DB, Okamoto K, Abe N, Yamamoto K, Nakayama K. 1999. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J Biol Chem 274:17955–17960. doi: 10.1074/jbc.274.25.17955. [DOI] [PubMed] [Google Scholar]
- 17.Okamoto K, Kadowaki T, Nakayama K, Yamamoto K. 1996. Cloning and sequencing of the gene encoding a novel lysine-specific cysteine proteinase (Lys-gingipain) in Porphyromonas gingivalis: structural relationship with the arginine-specific cysteine proteinase (Arg-gingipain). J Biochem 120:398–406. doi: 10.1093/oxfordjournals.jbchem.a021426. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi M, Sato K, Yukitake H, Noiri Y, Ebisu S, Nakayama K. 2010. A Porphyromonas gingivalis mutant defective in a putative glycosyltransferase exhibits defective biosynthesis of the polysaccharide portions of lipopolysaccharide, decreased gingipain activities, strong autoaggregation, and increased biofilm formation. Infect Immun 78:3801–3812. doi: 10.1128/IAI.00071-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanterpool E, Roy F, Fletcher HM. 2005. Inactivation of vimF, a putative glycosyltransferase gene downstream of vimE, alters glycosylation and activation of the gingipains in Porphyromonas gingivalis W83. Infect Immun 73:3971–3982. doi: 10.1128/IAI.73.7.3971-3982.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paramonov NA, Aduse-Opoku J, Hashim A, Rangarajan M, Curtis MA. 2009. Structural analysis of the core region of O-lipopolysaccharide of Porphyromonas gingivalis from mutants defective in O-antigen ligase and O-antigen polymerase. J Bacteriol 191:5272–5282. doi: 10.1128/JB.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis JP, Dawson JA, Hannis JC, Muddiman D, Macrina FL. 1999. Hemoglobinase activity of the lysine gingipain protease (Kgp) of Porphyromonas gingivalis W83. J Bacteriol 181:4905–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Letain TE, Postle K. 1997. TonB protein appears to transduce energy by shuttling between the cytoplasmic membrane and the outer membrane in Escherichia coli. Mol Microbiol 24:271–283. doi: 10.1046/j.1365-2958.1997.3331703.x. [DOI] [PubMed] [Google Scholar]
- 23.Simpson W, Olczak T, Genco CA. 2000. Characterization and expression of HmuR, a TonB-dependent hemoglobin receptor of Porphyromonas gingivalis. J Bacteriol 182:5737–5748. doi: 10.1128/JB.182.20.5737-5748.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slakeski N, Dashper SG, Cook P, Poon C, Moore C, Reynolds EC. 2000. A Porphyromonas gingivalis genetic locus encoding a heme transport system. Oral Microbiol Immunol 15:388–392. doi: 10.1034/j.1399-302x.2000.150609.x. [DOI] [PubMed] [Google Scholar]
- 25.Roy F, Vanterpool E, Fletcher HM. 2006. HtrA in Porphyromonas gingivalis can regulate growth and gingipain activity under stressful environmental conditions. Microbiology 152:3391–3398. doi: 10.1099/mic.0.29147-0. [DOI] [PubMed] [Google Scholar]
- 26.Sato K. 2011. Por secretion system of Porphyromonas gingivalis. J Oral Biosci 53:187–196. doi: 10.1016/S1349-0079(11)80001-0. [DOI] [Google Scholar]
- 27.Beale SI. 1990. Biosynthesis of the tetrapyrrole pigment precursor, δ-aminolevulinic acid, from glutamate. Plant Physiol 93:1273–1279. doi: 10.1104/pp.93.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naito M, Hirakawa H, Yamashita A, Ohara N, Shoji M, Yukitake H, Nakayama K, Toh H, Yoshimura F, Kuhara S, Hattori M, Hayashi T, Nakayama K. 2008. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res 15:215–225. doi: 10.1093/dnares/dsn013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felek S, Muszyński A, Carlson RW, Tsang TM, Hinnebusch BJ, Krukonis ES. 2010. Phosphoglucomutase of Yersinia pestis is required for autoaggregation and polymyxin B resistance. Infect Immun 78:1163–1175. doi: 10.1128/IAI.00997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smalley JW, Birss AJ, Szmigielski B, Potempa J. 2007. Sequential action of R- and K-specific gingipains of Porphyromonas gingivalis in the generation of the haem-containing pigment from oxyhaemoglobin. Arch Biochem Biophys 465:44–49. doi: 10.1016/j.abb.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 31.Rangarajan M, Aduse-Opoku J, Paramonov NA, Hashim A, Curtis MA. 2017. Hemin binding by Porphyromonas gingivalis strains is dependent on the presence of A-LPS. Mol Oral Microbiol doi: 10.1111/omi.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallagher A, Aduse-Opoku J, Rangarajan M, Slaney JM, Curtis MA. 2003. Glycosylation of the Arg-gingipains of Porphyromonas gingivalis and comparison with glycoconjugate structure and synthesis in other bacteria. Curr Protein Pept Sci 4:427–441. doi: 10.2174/1389203033486974. [DOI] [PubMed] [Google Scholar]
- 33.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2013. Pfam: the protein families database. Nucleic Acids Res 42(Database issue):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong S-Y, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtis MA, Thickett A, Slaney JM, Rangarajan M, Aduse-Opoku J, Shepherd P, Paramonov N, Hounsell EF. 1999. Variable carbohydrate modifications to the catalytic chains of the RgpA and RgpB proteases of Porphyromonas gingivalis W50. Infect Immun 67:3816–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.