

Abstract
Many organisms control initiation of DNA replication by limiting supply or activity of initiator proteins. In plasmids, such as P1, initiators are limited primarily by transcription and dimerization. However, the relevance of initiator limitation to plasmid copy number control has appeared doubtful, because initiator oversupply increases the copy number only marginally. Copy number control instead has been attributed to initiator-mediated plasmid pairing (“handcuffing”), because initiator mutations to handcuffing deficiency elevates the copy number significantly. Here, we present genetic evidence of a role for initiator limitation in plasmid copy number control by showing that autorepression-defective initiator mutants also can elevate the plasmid copy number. We further show, by quantitative modeling, that initiator dimerization is a homeostatic mechanism that dampens active monomer increase when the protein is oversupplied. This finding implies that oversupplied initiator proteins are largely dimeric, partly accounting for their limited ability to increase copy number. A combination of autorepression, dimerization, and handcuffing appears to account fully for control of P1 plasmid copy number.
Keywords: autorepression, DNA replication control, homeostatic control, plasmid copy number control
From early studies of Escherichia coli and its F plasmid, Jacob et al. (1) proposed that initiation of DNA replication was under positive control of a factor, the “initiator,” that binds to a specific DNA site to set in train the process of replication. Pritchard (2) later argued that positive control alone would not provide dynamic stability and that an “inhibitor” is needed to prevent runaway initiation.
Subsequent studies have shown that inhibition generally works by limiting the activity or availability of initiators. For plasmid ColE1, one of the simplest and best understood replicons, binding of a plasmid-encoded inhibitor RNA to the RNA that primes replication inhibits priming (3, 4). For the E. coli chromosome, several negative regulatory steps prevent the initiator DnaA protein from reinitiating replication prematurely. These include sequestration of DnaA binding sites that are situated at the origin of replication and at the dnaA promoter, reduction of free DnaA by binding to new sites created by replication, and accelerated hydrolysis of ATP in the active form of the protein, ATP-DnaA (5). The two initiation factors in Schizosaccharomyces pombe, Cdc18 and Cdt1, are inactivated by phosphorylation and proteolysis at the onset of S phase so that reinitiation of replication cannot take place within the same cell cycle (6).
Although initiator limitation seems widely conserved in replication control, an apparent exception is found in a family of bacterial plasmids controlled by repeated initiator binding sites (iterons). Well studied members of this group include plasmids F, P1, R6K, RK2, pSC101, and pP50 (7, 8). In these plasmids, saturation of initiator binding to origin iterons allows initiation. Iteron–initiator interactions also underlie negative control of replication.
Many attempts have been made to explain iteron-based control. The first was the initiator–titration model (9). It proposes that after replication, daughter origins compete for the limited amount of initiators, preventing saturation of either origin. However, this proposal overlooked the fact that the number of initiator genes also increases upon replication, leading to an increase in initiator synthesis. Moreover, the initiator genes were found to be transcriptionally autoregulated (10). In plasmid P1, the initiator promoter maps within the origin iterons (Fig. 1A). Thus, initiator binding to the origin also results in promoter repression. Autoregulation normally counteracts protein reduction by titration: as titration reduces the free initiator concentration, autorepression is proportionally relieved and compensates for the reduction (11). To keep titration from counteracting autorepression, it was proposed that the titrated initiators pair with promoter-bound initiators and thereby help maintain the repression (12). This mechanism, now called handcuffing (13), is common among transcriptional repressors that loop DNA, where the titrated repressors, instead of relieving autorepression, actually increase it (14–17).
Fig. 1.

Map of P1 plasmid replicon and amino acid changes in RepA mutants. (A) At the top are P1 map coordinates (43). The open bar represents P1 DNA, and arrowheads represent the iterons. ori, repA, and incA are functional units of P1 replicon. The miniP1 plasmid pSP102 has ori and repA genes but not incA. The origin iterons are numbered 10–14 for easy reference, and –35 and –10 are the elements of repA promoter that flank iteron no. 11. The plasmid is linearized at a PstI (P) site for display purposes. (B) Amino acid changes in RepA mutants and their position on the RepA polypeptide, which is 286 aa long. The changes above the line are for mutants studied here, and those below the line are for those studied earlier. The phenotypes used to select the mutants are shown on the right. The three mutants studied here (F120L, K123G, and K143E) are highlighted in red. (C) A model of 3D structure of RepA–DNA complexes showing the position of amino acid changes in the mutants studied here (adapted from ref. 44).
Handcuffing thus could solve the problem facing initiator titration, but a second observation seemed fatal to the model: Vast increase of initiator supply from constitutive sources increased plasmid copy number by no more than 1.5-fold (18–21). This finding suggested that initiators could not be limiting. Instead it was argued that handcuffing, apart from contributing to autorepression, also causes steric hindrance to replication to prevent reinitiation (13, 18). This view was reinforced when the initiator mutants that increase plasmid number (copy-up mutants) were found to be handcuffing defective (22–25).
Some results on plasmid P1, however, still seem better explained by limited initiator supply. When initiators are supplied in trans at a level only 2-fold below the physiological level, the plasmid copy number declines drastically (10). The copy number also falls when extra iterons are supplied in trans, but this drop can be overcome by supplying extra initiators (18, 26–28). Iteron-mediated control thus can be sensitive to initiator concentration.
Here, we show that selection of copy-up mutants of the P1 initiator RepA, isolated under physiological conditions, yields RepA mutants defective in autorepression but only slightly altered in other properties, including the capacity for handcuffing. Limiting RepA by autorepression thus contributes to copy number control under physiological conditions. We also present a quantitative model to show that initiator dimerization, which autoinactivates the protein (29), is functionally equivalent to autorepression in that they both dampen increase of (active) initiator monomer, which results from increase of copy number. This means of dampening RepA monomer increase works just as well when the protein is supplied from constitutive sources. Thus, the experiments that oversupplied the initiator actually supplied more dimers than monomers, which explains in part the marginal increase in copy number seen previously (18–21). Our results also indicate that restraining plasmid overreplication demands reduction of initiator concentration below that which autorepression or autoinactivation can achieve. Therefore, initiator limitation by autorepression and dimerization is insufficient for replication control, making the additional requirement for a mechanism like handcuffing obligatory. It is the combination of these mechanisms that makes the control most efficient, where one can benefit from the presence of others.
Materials and Methods
MiniP1 Copy Number. Plasmid-carrying DH5Δlac cells were grown from a single colony to OD600 = 0.4, and copy numbers were measured as described in ref. 30.
RepA Binding in Vivo. Repression of the repA promoter by RepA supplied in trans was used as a measure of RepA–iteron interactions in vivo (12). The assay system was as described except that the RepA source plasmid, pMVG02, was deleted for the incA iterons 9 + 8′ present in the previously used plasmid pRJM362.
Topoisomer Distribution by 2D Gel Electrophoresis. Topoisomer distribution by 2D gel electrophoresis was performed exactly as described in ref. 25.
RepA Purification, EMSA, and Ligation Kinetics. The wild-type (WT) and mutant RepAs (all untagged) were purified identically from an overproducer strain as described for the WT RepA (31). Ref. 31 also describes details of EMSA and ligation kinetics.
Surface Plasmon Resonance. Kinetic constants of RepA–iteron interactions were determined by using the Biacore 2000 instrument (Biacore, Piscataway, NJ). A 31-bp oligonucleotide containing the consensus iteron or nonspecific sequences was biotinylated at one end and conjugated to streptavidin-coated Biacore Sensor Chip SA. The binding buffer contained 50 mM Tris (pH 7.5), 50 mM NaCl, 50 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 5 μg/ml poly(dI-dC), and 0.05% (vol/vol) Biacore Surfactant P20. RepA was diluted to 30 nM in the binding buffer and flowed through all of the cells. The reference cell was identical to experimental cells except that it contained the nonspecific oligonucleotide. RepA binding to the reference cell was subtracted to account for nonspecific binding. Data analysis was performed by using the biaevaluation 3.0 software.
Cross-Linking of RepA. WT and mutant RepAs were diluted to 10 ng/μl by using RepA dilution buffer [50 mM Tris, pH 7.5/250 mM NaCl/1 mM DTT/1 mM EDTA/10% glycerol (vol/vol)] and incubated overnight at 4°C. An equal volume of cross-linking buffer (10 mM K-phosphate, pH 7.8/150 mM NaCl/1 mM MgCl2) was added. To 20 μl of the above mixture, 2 μl of variously diluted sulfo-bis[2-(succinimido-oxycarbonyloxy)ethyl-]sulfone (Pierce) solution in cross-linking buffer was added. The reactions were incubated at room temperature for 10 min, stopped by adding 3 μl of 1 M Tris (pH7.5), and further incubated for 15 min. Subsequently, 13 μl of 3× SDS sample buffer (pH 6.8) was added. The solution was boiled, chilled, and centrifuged, and the supernatant was run on a 12% Tris·glycine SDS gel. Bands representing RepA were detected by immunoblotting.
Results
Copy-Up Mutants of RepA. The P1 plasmid replication initiator RepA participates in three specific interactions potentially relevant to the negative control of copy number: autorepression, dimerization, and handcuffing. The autorepression is rather efficient because the repA promoter is repressed ≈100-fold when RepA is supplied in trans from a WT P1 plasmid (10). Dimerization is also efficient because purified RepA is predominantly dimeric, although the monomers are required for replication and autorepression (29, 31). Monomerization requires remodeling primarily by chaperones DnaJ and DnaK (32, 33). Only the remodeled monomers bind iterons efficiently, allowing replication and autorepression. The dimers may participate in handcuffing because it happens more efficiently in the absence of chaperones in vitro (31).
To assess the importance of these initiator interactions on plasmid copy number control, new RepA copy-up mutants were isolated without biasing the selection for a RepA phenotype and with RepA expressed from its natural promoter. The repA gene was amplified by using error-prone PCR, and inserted in place of the WT gene in a miniP1 plasmid, pSP102 (Fig. 1 A) (34). E. coli transformants with mutagenized plasmids were then screened on L agar medium containing 400 μg/ml chloramphenicol, on which cells carrying the WT plasmid formed tiny colonies. Approximately 2% of the transformed colonies were distinctly larger and potentially carried copy-up mutant plasmids. Plasmids were isolated from 17 such colonies, and their repA gene was sequenced. In 12 of 17 cases, single base substitutions were found (Fig. 1B). The remaining five had more than one substitution and were not studied further. All 12 plasmids showed a moderate increase in copy number (1.2- to 2.1-fold; see Table 1, column 2 from the left). Despite the modest extent of the copy number increase, in contrast to the WT plasmid, all of the mutant plasmids could transform a bacterial strain containing extra iterons [provided by a miniF clone of P1incA, pALA323 (34)]. In the presence of pALA323, the copy number of the mutant plasmids was 0.3- to 2.2-fold that of the WT (Table 1, column 4). The mutant RepAs thus were considered to be defective in iteron-mediated negative control of replication.
Table 1. Properties of copy-up RepA mutants.
| miniP1ΔincA copy number*
|
PrepA repression,† % (RepA in trans)
|
[RepA] in cis (Western)
|
|||||
|---|---|---|---|---|---|---|---|
| RepA | WT/miniF | ΔdnaJ/miniF | WT/miniF + incA | WT | ΔdnaJ | WT | ΔdnaJ |
| WT | 1 | 0.7 | 0 | 97 | 76 | 1 | 3.9 |
| D55G | 1.9 | 3.0 | 2.2 | 98 | 97 | ||
| A56T | 2.1 | 1.8 | 1.5 | 96 | 92 | ||
| A117G | 1.3 | 1.3 | 0.7 | 99 | 92 | ||
| F120L | 1.2 | 1.1 | 0.3 | 54 | 44 | 2.3 | 11 |
| K123G | 1.3 | 1.0 | 1.2 | 30 | 19 | 4.3 | 23 |
| K143E | 1.5 | 1.6 | 1.1 | 49 | 36 | 3.1 | 12 |
| D152V | 1.2 | 1.3 | 0.4 | 94 | 82 | ||
| F167S | 1.6 | 3.0 | 1.3 | 97 | 83 | ||
| D172E | 1.2 | 1.3 | 0.4 | 85 | 69 | ||
| D180G | 1.5 | 1.2 | 0.8 | 98 | 83 | ||
| K181E | 1.3 | 1.1 | 0.4 | 92 | 80 | ||
The three mutants shown in bold were not only chaperone-dependent but also DNA-binding defective. WT also is shown in bold.
Copy numbers are relative to the WT plasmid pSP102 and are mean values from three cultures started from independent colonies. The average one SD was ≈20% of the mean.
The repA promoter was fused to lacZ and was present in one copy in the chromosome. RepA was supplied in trans from constitutive promoter roughly at physiological concentration. The average one SD was ≈12% of the mean.
The mutations were located in the same region of repA where previously isolated mutations with the copy-up phenotype had been mapped (Fig. 1B) (30, 35). The previous mutants were selected for proficiency in iteron-binding in the absence of the chaperone DnaJ, but they simultaneously gained the copy-up (and incA insensitive) phenotype. Because of the strong correlation of chaperone-independent DNA binding with copy-up phenotypes, the new mutants were tested for chaperone independence. DNA binding in vivo was tested by RepA's ability to repress its own promoter when the protein was supplied from a constitutive promoter in trans at concentrations close to those in a P1 lysogen (called 1X). The repression values were comparable with and without DnaJ at least for the first three mutants, similar to those characterized previously (Table 1, columns 5 and 6) (30). These mutants were judged chaperone-independent for iteron binding. Results were similar in Δ(dnaJ-dnaK) (30) and ΔdnaJΔcbpA cells [data not shown; CbpA is a functional analog of DnaJ (36)]. Thus, chaperone-mediated monomerization appears to be a rate-limiting step for replication.
Interestingly, the three mutants shown in bold in Table 1 (F120L, K123G and K143E; hereafter called 120, 123 and 143, respectively) were not only chaperone-dependent but also DNA binding defective. When the positions of the mutant amino acids were mapped into a 3D model of RepA–iteron complex, mutants 120 and 123 were found to be in one of the helix–turn–helix regions implicated in DNA binding (Fig. 1C). Mutant 143 was just outside this DNA binding domain. These results are consistent with the DNA binding deficiencies of the mutants. The copy-up phenotype of RepA proteins despite the DNA-binding defect encouraged us to study these three mutants further.
When the three mutant proteins were supplied at 1X in trans, they could not support replication of a minP1 plasmid deleted for its own repA gene. This result is to be expected because reducing the level of WT RepA to ≈0.5× abolishes miniP1 replication (10). When the mutant proteins were produced in cis from their natural promoter, they supported miniP1 replication, probably because of initiator overexpression due to the autorepression defect (Table 1, columns 7 and 8). The level of overexpression depended on the degree of binding deficiency, as would be expected from an autoregulated source. However, the compensation for the binding defect by overexpression is expected to let the copy number approach the WT level, but not to exceed it or to make the replication incA insensitive. The three mutants therefore were examined for other functions in which the initiators are known to participate.
Phenotypes of Autorepression-Defective Initiator Mutants in ΔdnaJ Cells. In the absence of DnaJ, the initiator proteins were overproduced even more than in WT cells, confirming that DNA binding of the mutant proteins is chaperone-dependent (Table 1, columns 7 and 8). The overexpression apparently allowed mutant plasmids to replicate in ΔdnaJ cells efficiently (Table 1, column 3). However, copy number measurements indicated that the WT and mutant RepAs are qualitatively different. In the promoter repression assay, the WT RepA showed only modest defect in ΔdnaJ cells: repression reduced from 97% to 76% (Table 1, columns 5 and 6). Although the protein level increased 3.9× from an autoregulatory source in ΔdnaJ cells, it was apparently not high enough for optimal replication because plasmid copy number fell from 1 to 0.7 (Table 1, columns 2 and 3). The fact that the copy number of the mutant miniP1 plasmids was relatively unchanged in ΔdnaJ cells (Table 1, columns 2 and 3) suggests that there could be weakening of iteron binding specific to autorepression, resulting in increased initiator synthesis. Preliminary evidence to this effect has been obtained (see Fig. 5, which is published as supporting information on the PNAS web site). Alternatively, the mutants could be defective in mechanisms that facilitate autorepression. As discussed earlier, handcuffing could be one such mechanism. The mutant proteins therefore were tested for handcuffing.
Handcuffing Efficiency of RepA Mutants in Vivo. We used three different assays to measure handcuffing. First, we compared copy numbers of isogenic plasmid monomers and dimers. Because pairing in cis is expected to be more efficient than that in trans, the origin copy number in dimer-carrying cells should be lower than in cells carrying monomers. We have shown previously that dimer plasmid copy number is >2-fold lower than that of the monomer (25). This finding was confirmed in the present study (Fig. 2A). However, within experimental error, the dimer/monomer plasmid copy number ratio was not significantly different when the miniP1 plasmid (pSP102) carried the mutant genes, indicating that the mutants are handcuffing proficient.
Fig. 2.

Handcuffing efficiency of RepA mutants in vivo. (A) Copy number ratio of isogenic dimeric and monomeric forms of miniP1 plasmids when the RepA protein is WT or mutant 120, 123, or 143. (B) A topological assay for interactions between two arrays of five consensus iterons of a homodimeric plasmid derived from pBR322. The semicircular arrows show the direction of transcription of the plasmid bla gene. Transcription-generated positive (+) and negative (–) supercoils (SC) are shown. The small arrow indicates that the ω protein targets the –SC. The topoisomers are separated on a 2D gel. In the first dimension, +SC and –SC run together. In the second dimension, when chloroquine was present, +SC run ahead of –SC. The boxed region of the distribution, where the SC are well separated, was used to calculate the ratio of +SC and –SC as described in ref. 25. (C) MiniP1 copy number with an extra iteron placed 31 bp downstream of the five origin iterons in two orientations. The WT and mutant RepAs were supplied from constitutive sources in different amounts to achieve similar levels of iteron binding. Copy number of miniP1 without any extra iteron in the presence of 1× WT RepA was taken as 8.0 (30).
The next assay for handcuffing is based on the principle that pairing of sites in a homodimeric plasmid can separate the DNA into two topologically closed domains, preventing diffusion of transcription-generated superhelical tension from one domain to the other (25). A pBR322 derivative carrying five tandem consensus iterons was used in this assay, and a constant level of RepA (40×) was supplied in trans in all of the cases. By this assay, considering the experimental error, only mutant 123 was judged defective in iteron pairing (Fig. 2B). In a previous study, the ratio of positive to negative supercoils decreased ≈2.6-fold in handcuffing-defective mutants compared with the WT (25). Although 123 was similarly defective, this result does not necessarily indicate a specific handcuffing defect because handcuffing is contingent upon DNA binding activity of RepA. Mutant 123 was the most defective in DNA binding, which could have caused the handcuffing defect. A third assay was used where the degree of binding was made nearly uniform.
By using the promoter repression assay for DNA binding (Table 1, column 5), we first varied RepA concentration in trans to achieve nearly identical repression in all cases (data not shown). Under these conditions, the ability of an extra iteron to reduce copy number of a miniP1 plasmid was determined. We have shown previously that an extra iteron reduces miniP1 copy number by ≈2-fold when properly oriented and phased with respect to the origin iterons, apparently due to cis-handcuffing between the extra iteron and the origin (31). The extra iteron was ineffective in reducing copy number when previously characterized handcuffing-defective mutants were used (31). In the present study, the WT and the three RepA mutants reduced the plasmid copy number similarly, and, therefore, they were judged handcuffing proficient (Fig. 2C). Taken together, the results indicate that handcuffing is unlikely to be the primary defect of the mutants studied here. Further characterization of the mutants was carried out in vitro where it was easier to adjust for binding differences.
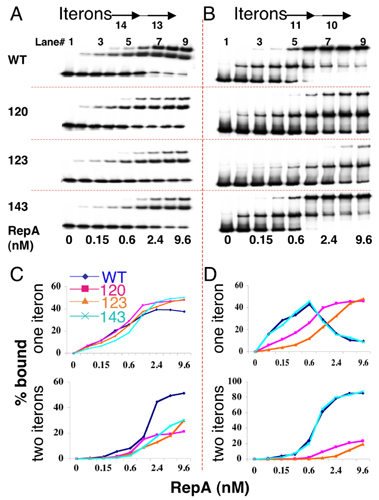
RepA Binding to Origin Iterons in Vitro. We first studied binding in vitro by using DNA fragments containing a single iteron. By using an excess of fragments and the DnaJ and DnaK chaperones, we determined the active fraction of the RepA proteins (37). Protein concentrations were adjusted to compensate for variations in the active fraction. Iteron binding of the mutants was significantly improved when both DnaJ and DnaK were present, but the improvement was less than for WT RepA (Fig. 3A). The results confirmed that the mutants are chaperone-dependent and DNA binding-defective. The KD of binding increased ≈2- to 3-fold, mostly due to increased dissociation rates (Fig. 3B).
Fig. 3.

DNA binding of RepA. (A) An EMSA showing dependence of binding of WT and mutant RepAs on chaperones DnaJ and DnaK. The proteins when present were 0.75 nM for RepA, 60 nM for DnaJ, and 70 nM for DnaK. The probe contained a single iteron at 0.15 nM. (B) Binding kinetics of RepA to a single iteron by surface plasmon resonance. (C) EMSA showing RepA binding to a fragment (0.15 nM) carrying all five origin iterons. The chaperones were present as in A.
The binding of the mutants to fragments carrying all five origin iterons was studied in the presence of chaperones (Fig. 3C). The pattern of binding for the mutants differed significantly from that of the WT. The mutants showed fewer retarded bands. Their maximum number (five) corresponded to the number of iterons present in the probe DNA. Moreover, saturation of binding was difficult to achieve in all three cases, particularly for the mutant 123. Increasing the 123 mutant protein concentration from 3.2 to 12 nM resulted in only a marginal increase in the fraction of DNA bound. RepA–iteron interactions seem to have changed both qualitatively and quantitatively in the mutants.
Handcuffing of Origin Iterons in Vitro. It is known that the kinetics of intermolecular DNA ligation is enhanced if the DNA molecules are brought into close proximity by interactions between their bound proteins, as in handcuffing (13, 38). Starting with short DNA fragments (a few hundred base pairs), a ladder of ligated multimers was generated (Fig. 4A). When the WT and the mutant RepAs were used at low protein concentration, they caused nearly identical ladder formation, indicating similarity in their handcuffing proficiency (Fig. 4B). At higher protein concentration, the ladder formation was more extensive, except for mutant 123. Increasing protein concentration from 3 to 15 nM did not significantly increase the extent of ladder formation, as was also the case for DNA binding (Fig. 3C). Because only minor differences were observed in the ligation assay, the mutants were considered largely handcuffing proficient.
Fig. 4.

Handcuffing and RepA dimerization in vitro.(A) A model of multimer formation by sequential pairing of RepA–DNA complexes, ligation at one of the DNA ends, reversal of either RepA–RepA, or RepA–iteron contacts, and pairing with a new RepA–DNA complex or DNA fragment. Black circles, RepA; black rectangles, iterons; gray circles, ligated ends. (B) A ladder of RepA catalyzed multimers in the presence of ligase resolved after deproteinization in an agarose gel. The probe was 251 bp long and was used at 0.15 nM. The fraction ligated represents the intensity of bands containing dimers and higher multimers as a fraction of total DNA. (C) RepA dimerization by cross-linking with increasing concentrations of sulfo-bis[2-(succinimido-oxycarbonyloxy)ethyl]sulfone (BSOCOES). The products were separated by SDS/PAGE and identified by immunoblotting. The fraction cross-linked represents the intensity of the dimer (D) band as a fraction of intensities of monomer (M) and D bands. The M and D bands were identified by using molecular weight markers shown in the first column.
Dimerization of RepA Mutants in Vitro. Because we have suggested that RepA dimers could be required for handcuffing, the mutants were characterized for their strength of dimerization (31). From the handcuffing proficiency of the mutants, our expectation was that they would be dimerization proficient. The proteins were diluted to the same final concentration and allowed to equilibrate overnight before exposing to a cross-linking agent (Fig. 4C). No significant differences in dimerization between the WT and the mutants were detected.
In summary, it appears that weakening of iteron binding such that autorepression is specifically compromised (Fig. 5) remains the best explanation for the mutant phenotypes. The present mutants are different from previously characterized copy-up mutants where apparent KD for iteron binding actually decreases (24).
Discussion
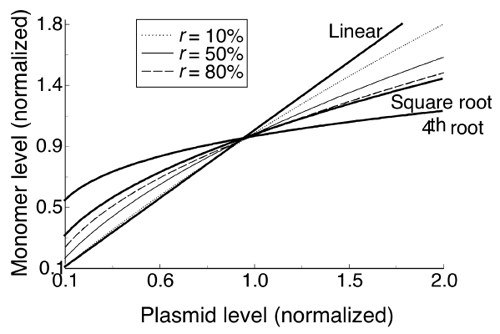
Transcriptional autorepression of the initiator gene and initiator inactivation by dimerization are the two well conserved features of iteron-based plasmid replication control systems. They strongly reduce initiator availability, but how they bear on plasmid copy number control has remained unclear. Here, we found that selection of copy-up initiator mutants of plasmid P1 under physiological conditions showed some to be autorepression defective. The mutant initiators were overproduced, which we believe caused the copy number increase. Why, then, does overproduction of WT initiators not cause a proportional increase of plasmid copy number? We argue that initiator inactivation by dimerization is one reason. By using kinetic theory, we show that dimerization dampens the increase of monomers when initiators are overproduced (see Fig. 6, which is published as supporting information on the PNAS web site). The theory also predicts that initiator limitation by autorepression and dimerization, although helpful in dampening plasmid overreplication, cannot be adequate for plasmid copy number control. Additionally, direct control by dimers or handcuffing is required. In fact, the copy number control is best explained if the mechanisms cooperate, as we discuss below.
The goal of replication control is to maintain plasmid copy number within narrow limits. For this process, the replication control mechanisms must be dynamic, meaning that they must respond to an increase in copy number by a decrease in the replication rate. Currently three mechanisms seem to contribute to dynamic responses as follows.
(i) Homeostatic handcuffing: An increase in plasmid copy number increases the plasmid–plasmid handcuffing probability, which sterically inhibits replication.
(ii) Homeostatic monomer–dimer competition: Initiator monomers and dimers promote and inhibit replication, respectively. An increase in plasmid concentration leads to an increase in total initiator concentration, which increases the ratio between dimers and monomers and thereby promotes inhibition over initiation.
(iii) Homeostatic initiator limitation: Autoregulatory mechanisms dampen the increase of total initiator concentration in response to higher plasmid copy numbers, and iteron-mediated titration of the initiator then reduces the free initiator concentration.
These mechanisms also seem to be interdependent where one contributes to mechanically execute the dynamics of the other, as explained below for the case of plasmid P1.
Homeostatic Handcuffing. Handcuffing can in principle provide dynamic control on its own, even if RepA is always present in saturating concentrations. The role of controls on RepA then could be to ensure that the initiator supply is always adequate but not wastefully high. Handcuffing possibly relies on RepA dimerization to link plasmid copies. Both RepA autoregulation and dimerization thus may be necessary to mechanically execute the homeostatic dynamics inherent to handcuffing.
Homeostatic Monomer–Dimer Competition. The binding of RepA monomer is required for replication initiation (29), and the binding of RepA dimer, although weaker, has the potential to inhibit replication (31, 39, 40). The dimers could also participate in replication inhibition without contacting DNA by serving as a bridge between bound monomers (41). Handcuffing then may stabilize dimer interactions as has been found in other systems (14–17). The role of handcuffing then can be to mechanically execute the homeostatic dynamics inherent to the RepA monomer–dimer competition. We note that although the two homeostatic principles may rely mechanically on each other for executing control, the basis of dynamics is different in the two cases. “Homeostatic handcuffing” relies on a quadratic (bimolecular) increase in the number of interplasmid encounters, whereas “homeostatic monomer–dimer competition” relies on a quadratic increase in the rate of turning RepA monomers to dimers.
Homeostatic Initiator Limitation. For initiator limitation to prevent runaway replication, the concentration of free initiator must be lower at higher plasmid copy numbers. If it were higher, each iteron would face a higher initiator association rate that would lead to a higher probability of replication. However, simple autoregulation can only dampen the increase in available initiator with higher plasmid copy number (Fig. 6), not produce the actual decrease that is necessary. An increase in plasmid copy number still produces an increase in the initiator concentration, just a smaller increase. As mentioned, initiator titration could produce an actual decrease, provided that handcuffing prevents initiator replenishment. Once again, handcuffing thus could help to mechanically execute the homeostatic dynamics inherent in initiator limitation by titration. Recently, replication dynamics of F plasmid was described quantitatively assuming initiators to be limiting, and the presence of handcuffing was obligatory in that model (42).
Transcriptional repression is not the only process that can autoregulate RepA monomer level. Dimerization can be as efficient as autorepression in obtaining initiator homeostasis (see Supporting Text, which is published as supporting information on the PNAS web site). Limiting monomers by dimerization does not rely on the dimers to be active in iteron binding as in the homeostatic monomer–dimer competition scenario. The dimers may be unable to bind DNA but still indirectly autoregulate the monomer concentration, mimicking feedback control without ever “feeding back.” The experiments that greatly oversupplied initiators failed to oversupply the monomer significantly because they only bypassed autorepression-based RepA autoregulation, not dimerization-based RepA autoregulation (18, 19, 21). However, the highly damped copy number increase seen in these experiments may still seem inconsistent with the limited ability of dimerization to dampen the increase in RepA monomer (Fig. 6). Most likely, the other two homeostatic mechanisms become dominant when RepA is supplied in unphysiological excess. The existence of multiple control mechanisms also explains the modest increase of copy number by the present mutants. Relaxing autorepression by mutation would start to increase the plasmid copy number, but the effect would be partly counteracted by preferential accumulation of dimers that would dampen the final increase in plasmid copy number by the other two mechanisms. Having multiple homeostasis mechanisms thus can make the system insensitive to changes in any one of them.
Supplementary Material
Acknowledgments
We thank Sue Wickner for providing Fig. 1C and David Lane, Paul Morrison, Michael Yarmolinsky, and the National Cancer Institute Center for Cancer Research Fellows Editorial Board for comments on the manuscript.
References
- 1.Jacob, F., Brenner, S. & Cuzin, F. (1964) Cold Spring Harbor Symp. Quant. Biol. 28, 329–348. [Google Scholar]
- 2.Pritchard, R. H. (1978) in DNA Synthesis: Present and Future, eds. Molineux, I & Kohiyama, M. (Plenum, New York), pp. 1–26.
- 3.Tomizawa, J., Itoh, T., Selzer, G. & Som, T. (1981) Proc. Natl. Acad. Sci. USA 78, 1421–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paulsson, J., Nordström, K. & Ehrenberg, M. (1998) Plasmid 39, 215–234. [DOI] [PubMed] [Google Scholar]
- 5.Dasgupta, S. & Lobner-Olesen, A. (2004) Plasmid 52, 151–168. [DOI] [PubMed] [Google Scholar]
- 6.Gopalakrishnan, V., Simancek, P., Houchens, C., Snaith, H. A., Frattini, M. G., Sazer, S. & Kelly, T. J. (2001) Proc. Natl. Acad. Sci. USA 98, 13114–13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.del Solar, G., Giraldo, R., Ruiz-Echevarria, M. J., Espinosa, M. & Diaz-Orejas, R. (1998) Microbiol. Mol. Biol. Rev. 62, 434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chattoraj, D. K. (2000) Mol. Microbiol. 37, 467–476. [DOI] [PubMed] [Google Scholar]
- 9.Tsutsui, H., Fujiyama, A., Murotsu, T. & Matsubara, K. (1983) J. Bacteriol. 155, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chattoraj, D. K., Snyder, K. M. & Abeles, A. L. (1985) Proc. Natl. Acad. Sci. USA 82, 2588–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becskei, A. & Serrano, L. (2000) Nature 405, 590–593. [DOI] [PubMed] [Google Scholar]
- 12.Chattoraj, D. K., Mason, R. J. & Wickner, S. H. (1988) Cell 52, 551–557. [DOI] [PubMed] [Google Scholar]
- 13.McEachern, M. J., Bott, M. A., Tooker, P. A. & Helinski, D. R. (1989) Proc. Natl. Acad. Sci. USA 86, 7942–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semsey, S., Tolstorukov, M. Y., Virnik, K., Zhurkin, V. B. & Adhya, S. (2004) Genes Dev. 18, 1898–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruusala, T. & Crothers, D. M. (1992) Proc. Natl. Acad. Sci. USA 89, 4903–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frappier, L., Goldsmith, K. & Bendell, L. (1994) J. Biol. Chem. 269, 1057–1062. [PubMed] [Google Scholar]
- 17.Hochschild, A. (2002) Curr. Biol. 12, R87–R89. [DOI] [PubMed] [Google Scholar]
- 18.Pal, S. K. & Chattoraj, D. K. (1988) J. Bacteriol. 170, 3554–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durland, R. H. & Helinski, D. R. (1990) J. Bacteriol. 172, 3849–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia de Viedma, D., Giraldo, R., Rivas, G., Fernandez-Tresguerres, E. & Diaz-Orejas, R. (1996) EMBO J. 15, 925–934. [PMC free article] [PubMed] [Google Scholar]
- 21.Uga, H., Matsunaga, F. & Wada, C. (1999) EMBO J. 18, 3856–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasina, A., Kittel, B., Toukdarian, A. E. & Helinski, D. R. (1996) Proc. Natl. Acad. Sci. USA 93, 3559–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miron, A., Patel, I. & Bastia, D. (1994) Proc. Natl. Acad. Sci. USA 91, 6438–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukhopadhyay, G., Sozhamannan, S. & Chattoraj, D. K. (1994) EMBO J. 13, 2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park, K., Han, E., Paulsson, J. & Chattoraj, D. K. (2001) EMBO J. 20, 7323–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chattoraj, D., Cordes, K. & Abeles, A. (1984) Proc. Natl. Acad. Sci. USA 81, 6456–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park, K., Mukhopadhyay, S. & Chattoraj, D. K. (1998) J. Biol. Chem. 273, 24906–24911. [DOI] [PubMed] [Google Scholar]
- 28.Mukhopadhyay, S. & Chattoraj, D. K. (2000) Proc. Natl. Acad. Sci. USA 97, 7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wickner, S., Hoskins, J. & McKenney, K. (1991) Proc. Natl. Acad. Sci. USA 88, 7903–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sozhamannan, S. & Chattoraj, D. K. (1993) J. Bacteriol. 175, 3546–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das, N. & Chattoraj, D. K. (2004) Mol. Microbiol. 54, 836–849. [DOI] [PubMed] [Google Scholar]
- 32.Dibbens, J. A., Muraiso, K. M. & Chattoraj, D. K. (1997) Mol. Microbiol. 25, 185–195. [DOI] [PubMed] [Google Scholar]
- 33.Pak, M. & Wickner, S. (1997) Proc. Natl. Acad. Sci. USA 94, 4901–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal, S. K., Mason, R. J. & Chattoraj, D. K. (1986) J. Mol. Biol. 192, 275–285. [DOI] [PubMed] [Google Scholar]
- 35.Baumstark, B. R., Lowery, K. & Scott, J. R. (1984) Mol. Gen. Genet. 194, 513–516. [DOI] [PubMed] [Google Scholar]
- 36.Ueguchi, C., Shiozawa, T., Kakeda, M., Yamada, H. & Mizuno, T. (1995) J. Bacteriol. 177, 3894–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DasGupta, S., Mukhopadhyay, G., Papp, P. P., Lewis, M. S. & Chattoraj, D. K. (1993) J. Mol. Biol. 232, 23–34. [DOI] [PubMed] [Google Scholar]
- 38.Mukherjee, S., Erickson, H. & Bastia, D. (1988) Proc. Natl. Acad. Sci. USA 85, 6287–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urh, M., Wu, J., Forest, K., Inman, R. B. & Filutowicz, M. (1998) J. Mol. Biol. 283, 619–631. [DOI] [PubMed] [Google Scholar]
- 40.Abhyankar, M. M., Reddy, J. M., Sharma, R., Bullesbach, E. & Bastia, D. (2004) J. Biol. Chem. 279, 6711–6719. [DOI] [PubMed] [Google Scholar]
- 41.Toukdarian, A. E. & Helinski, D. R. (1998) Gene 223, 205–211. [DOI] [PubMed] [Google Scholar]
- 42.Morrison, P. F. & Chattoraj, D. K. (2004) Plasmid 52, 13–30. [DOI] [PubMed] [Google Scholar]
- 43.Abeles, A. L., Snyder, K. M. & Chattoraj, D. K. (1984) J. Mol. Biol. 173, 307–324. [DOI] [PubMed] [Google Scholar]
- 44.Sharma, S., Sathyanarayana, B. K., Bird, J. G., Hoskins, J. R., Lee, B. & Wickner, S. (2004) J. Biol. Chem. 279, 6027–6034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}