

Abstract
In several species, GAF domains, which are widely expressed small-molecule-binding domains that regulate enzyme activity, are known to bind cyclic nucleotides. However, the molecular mechanism by which cyclic nucleotide binding affects enzyme activity is not known for any GAF domain. In the cyanobacterium, Anabaena, the cyaB1 and cyaB2 genes encode adenylyl cyclases that are stimulated by binding of cAMP to their N-terminal GAF domains. Replacement of the tandem GAF-A/B domains in cyaB1 with the mammalian phosphodiesterase 2A GAF-A/B tandem domains allows regulation of the chimeric protein by cGMP, suggesting a highly conserved mechanism of activation. Here, we describe the 1.9-Å crystal structure of the tandem GAF-A/B domains of cyaB2 with bound cAMP and compare it to the previously reported structure of the PDE2A GAF-A/B. Unexpectedly, the cyaB2 GAF-A/B dimer is antiparallel, unlike the parallel dimer of PDE2A. Moreover, there is clear electron density for cAMP in both GAF-A and -B, whereas in PDE2A, cGMP is found only in GAF-B. Phosphate and ribose group contacts are similar to those in PDE2A. However, the purine-binding pockets appear very different from that in PDE2A GAF-B. Differences in the β2–β3 loop suggest that this loop confers much of the ligand specificity in this and perhaps in many other GAF domains. Finally, a conserved asparagine appears to be a new addition to the signature NKFDE motif, and a mechanism for this motif to stabilize the cNMP-binding pocket is proposed.
Keywords: cAMP, phosphodiesterase
GAF domains (named for cyclic GMP, Adenylyl cyclase, FhlA) are small-molecule-binding domains expressed in many organisms that are important regulatory elements for a host of different enzymes (1, 2). In mammals, GAF domains are mostly found in cyclic nucleotide phosphodiesterases (PDEs), which are crucial cellular enzymes controlling cGMP and cAMP second-messenger levels. Five of the 11 mammalian PDE families contain two GAF domains (PDE2, -5, -6, -10, and -11), all in tandem and N-terminal to the catalytic domain (3–5). So far, two separate functions, namely cyclic nucleotide binding and dimerization, have been described for different PDE GAF domains. In the cGMP-stimulated PDE2A, a crystallographic study showed that it is the GAF-A domain that mediates protein dimerization, whereas it is the GAF-B domain that binds a cyclic nucleotide ligand (6). In contrast, in PDE5 and -6, GAF-A, but not GAF-B, appears to possess both the ligand-binding (7) and dimerization contacts (8, 9). The detailed mechanism by which these GAF domains regulate the phosphodiesterase activities of PDEs remains unclear.
The class III adenylyl cyclases (ACs) of the cyanobacterium Anabaena, which are at least 2 billion years separated in evolution from mammals, also contain tandem GAF domains in their N termini (10). The cyaB1 adenylyl cyclase has been shown to be stimulated by its product, cAMP. This regulation is due to cAMP binding and signaling to the catalytic domain via the GAF-B domain (11). Another very similar adenylyl cyclase, cyaB2, likewise is stimulated by cAMP [see the accompanying article (12)]. Remarkably, a chimeric AC consisting of the PDE2 GAF domains and the cyaB1 catalytic domain can be activated by cGMP (11), suggesting that the tandem GAF domains in the mammalian PDEs and the cyanobacterial AC share common mechanisms for regulation of the enzymatic activities of their neighboring catalytic domains.
Here, we report the 1.9-Å crystal structure of the tandem GAF-A/B domains from the Anabaena adenylyl cyclase cyaB2. Surprisingly, the structure shows an antiparallel dimer, in contrast to the parallel PDE2A GAF-A/B dimer, with both cyaB2 GAF domains involved in dimer formation. Moreover, cAMP is found in complex with both GAF-A and -B. These structural results have revealed previously uncharacterized modes of ligand binding and dimerization for the tandem GAF domains, suggesting that their regulatory mechanisms are likely to involve multiple states of structural configuration.
Materials and Methods
Crystallization. The cyaB2 GAF-A/B residues 58–445-GSRS-H6 crystallizes in space group P1 with one dimer in the asymmetric unit, Vm = 2.74 Å3/Da, and a solvent content of 55.1%. The same crystal form was grown with sulfomethionine (SuMet) and selenomethionine (SeMet) protein at room temperature, using sodium formate or ammonium sulfate as the precipitant. A fast desalting column or Centricon 50 was used to exchange protein into crystallization buffer (10 mM Tris·HCl, pH 7.5/10% glycerol/1 mM MgCl2/1 mM 2-mercaptoethanol; no glycerol was present for the ammonium sulfate crystals). Two millimolar Na-cAMP was then added. The protein concentration was 10–12 mg/ml. Sitting drops were set up in Cryschem plates from Hampton Research. Drops were 4 μl of protein plus 4 μl of well solution unless otherwise indicated. For the single-wavelength anomalous dispersion (SAD) SeMet data set, the well was 0.5 ml of 3.9 M Na formate and 100 mM bis-Tris propane (pH 9.0). For the single isomorphous replacement with anomalous scattering (SIRAS) data set, the SuMet crystal was grown from 3.8 M Na formate (pH 8.8); the SeMet crystal was grown from 3.6 M Na formate (pH 8.8). The crystal for a 1.9-Å ammonium sulfate data set was grown by using a well of 1.60 M ammonium sulfate, 100 mM bis-Tris propane (pH 8.8), and 12% glycerol. For data collection at 100 K, all crystals were frozen directly out of the drop into liquid nitrogen.
Data Collection and Structure Determination. The structure was solved by σA combination of two sets of phases from a 4.2-Å SAD peak SeMet data set and 3.0-Å SIRAS SeMet and SuMet data sets. After a fluorescent scan, the peak Se wavelength was determined to be 12,657.6 eV (0.9795 Å) (1 eV = 1.602 × 10-19 J). An ≈3.0-Å SAD data set was collected at Advanced Light Source (ALS) beamline 5.0.2. Also, at ALS beamline 5.0.1 (1.0000 Å), the Na formate SIRAS (SuMet and SeMet) data sets and an ammonium sulfate SuMet data set (for refinement) were collected. Both 4.2-Å SAD and 3.0-Å SIRAS phases [from solve (13)] were fed into the ccp4 sigmaa program (14). The combined, weighted phases were fed into resolve (15) with twofold noncrystallographic symmetry averaging and solvent flattening. The HA positions were supplied from the SIRAS solve run. The latter found 14 of the 18 Se sites (9 Se per monomer, and 1 dimer per asymmetric unit). A model was built manually with the program o (16). The model was refined with a 1.9-Å ammonium sulfate native Met data set and the program cns (17), using restrained individual B factors, then with refmac (14, 18), using TLS refinement. Continuous electron density is present from the first residue, V58, to residue 440, except for a few short loops. Residues 441–445 in chain A and 442–445 in chain B, followed by the histidine tag GSRSH6, were disordered and not included in the model. Cys-368 in chain A and Glu-213 in chain B were modeled as double conformers. The final free R factor is 21.5%, and Rwork is 18.2%. Of the residues in the final model, 93.7% are in the most favored area of the Ramachandran plot (Table 1, which is published as supporting information on the PNAS web site).
For details of protein expression and purification, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Results
Overall Structure of the cyaB2 Tandem GAF Dimer. The cyaB2 tandem GAF domains crystallized as a homodimer in one asymmetric unit (Fig. 1 a and b). This finding is consistent with the results of the light-scattering analysis in conjunction with size-exclusion chromatography showing that the protein also forms a dimer in solution (data not shown). In each cyaB2 monomer, the GAF-A and -B domains have very similar structures with identical topology, except for the β2–β3 loop, which is central to purine binding, and the conserved signature NKFDE motif [a conserved signature motif of the amino acids Asn(N), Lys(K), Phe(F), Asp(D), and Glu(E) found in all GAF-domain-containing enzymes that have cyclic-nucleotide-binding capacity], which appears to be important for stabilization of the cyclic-nucleotide-binding pocket (see below).
Fig. 1.

Ribbon-structure views of cyaB2 tandem GAF A/B domains compared with the PDE2A GAF A/B domains. (a) View of the quaternary structure of the regulatory segment of cyanobacterial Anabaena sp. PCC 7120 AC cyaB2. Each AC monomer (red or green) contains a GAF-A and a GAF-B domain, each with a cAMP-binding site. The cAMP-binding domains, connecting helices, and N- and C-terminal extensions (to GAF-A and -B, respectively) form the dimer interface. GAF-A dimerizes with GAF-B′. The four cAMP molecules are shown in CPK colors. The overall dimensions of the regulatory segment dimer are 111 × 102 × 55 Å. In the monomer, an N-terminal helical extension (residues 58–78) precedes the first GAF domain (residues 79–224, GAF-A). It is followed by a linker (residues 225–230), then a connecting helix (residues 231–263) and another linker (residues 264–270) to GAF-B (residues 271–430). A last helical extension (residues 431–440 or 441) ends in the last ordered density. (b) Same view as a but rotated 90° about the vertical axis. (c) Tandem GAF domains from PDE2A (dimer axis vertical). The figure was prepared with molscript (25) and raster3d (26).
In the crystal, the cyaB2 dimer is arranged in an antiparallel manner and adopts a highly compact overall shape. Like the previously described PDE2A tandem GAF domains, the two cyaB2 GAF domains are connected by a long helix (6). In each cyaB2 monomer, the two GAF domains are located on the same side of the connecting helix and likely contact each other through a single salt bridge. A single monomer is 86 Å in the longest dimension and thus about 15% shorter than the PDE2 GAF monomer. This difference is due to a sharp backward bend of the cyaB2 GAF-B domain toward the connecting helix (Fig. 1; see also Fig. 7, which is published as supporting information on the PNAS web site). Between the two monomers, dimerization occurs mainly through the connecting helices, two N-terminal helices of GAF-A, and one C-terminal helix of GAF-B, allowing the GAF-A domain of one monomer to closely pack against the GAF-B domain of the other. The total area buried in the dimer interface is ≈7,300 Å2. The maximum dimension of the dimer is 111 Å. Overall, the arrangement of the four GAF domains results in a “flat” rectangular shape, although the two pairs of GAF-A, B′ and GAF-B, A′ domains are in fact related by an ≈30° rotation around the connecting helix (Fig. 1b).
Dimerization of the cyaB2 Tandem GAF Domains Is Distinct from the PDE2A GAF Domains. The antiparallel dimerization of the cyaB2 tandem GAF domains is in complete contrast to the parallel dimer of the GAF-A/B from PDE2A (Fig. 1c). In the latter, GAF-A is a dimerizing domain, whereas the GAF-B domains are not in contact with each other (6). Within a monomer, the two PDE2A GAF-A and -B domains are also far away from each other. Although parts of the connecting helices in PDE2A play a role in mediating dimerization of the GAF-A domains, their C-terminal portions gradually bifurcate. In contrast, the cyaB2 connecting helices run almost in parallel over their entire length, creating an expanded interaction surface of 1,318 Å2 (Fig. 1a). The sequence identity between the connecting helices, which are of essentially identical length, is low (16%, 6 of 37 residues), suggesting that the specific dimerization modes observed in the two cases might be unique to each individual protein. This is particularly true for the intermolecular disulfide bond formed by PDE2 Cys-386, whose corresponding residue in cyaB2 is an alanine.
The cyaB2 GAF-A and -B Domains Share a Similar Cyclic-Nucleotide-Binding Fold. Each GAF-A and -B has a bound cAMP that is ≈97% buried. All four cAMPs in the asymmetric unit are bound in the anti-conformation, as is cGMP in PDE2A GAF-B (6). Because the cAMP-binding sites in GAF-A and -B are very similar (Figs. 2 and 3), only the GAF-B in chain A is chosen for discussion here. With the exception of the purine part of the binding pocket, cyaB2 GAF-B is generally similar to PDE2A GAF-B with bound cGMP. The most unexpected difference is the H-bond from Arg-291 to the N1 of cAMP (Fig. 2b). This would appear to be an important residue for cAMP binding because Arg-291 cannot H-bond to a protonated N1 in guanine. This H-bond is therefore likely to be a positive determinant for cAMP. The counterpart of Arg-291 in PDE2A GAF-B is Ile-422, which contacts the guanine ring. The orientation of Arg-291 appears to be constrained by the side chains of two other residues. From Cγ up through the guanidinium group, the side chain of Arg-291 is nearly flat against the side chain of Tyr-361, at a distance of 3.6 Å, whereas the carbonyl oxygen of the Gln-310 side chain makes a long 3.3-Å H-bond to NH1 of Arg-291. In GAF-A, Tyr-361 is conserved as Tyr-174. Gln-310 does not have a counterpart in GAF-A because it is in the β2–β3 loop that varies between GAF-A and -B.
Fig. 2.

Close-up view of the cAMP-binding sites in GAF-A (a) and -B (b) from AC cyaB2, chain A. There are four polar side chains directly contacting cAMP (GAF-A: R103, T105, T176, and Q196; GAF-B: R291, T293, T363, and Q383). In GAF-A, T172 hydroxyl group contacts cAMP through a water. In GAF-B, N359 and D356 side chains contact cAMP through a water molecule. Backbone amides or carbonyls of two or five residues contact cAMP through a water (GAF-A: A161 and A165; GAF-B: T309, K324, G328, D348, and A352). Depending on the GAF domain, there are three to four bound waters. Last are three to four hydrophobic contacts (GAF-A: Ile-131, Leu-198, and Tyr-174; GAF-B: Trp-295, Ile-308, I355, and Tyr-361). Helix α4 is partially (b) and strand β3 is entirely (a and b) depicted as coils for visibility of the cAMP. Water molecules are given arbitrary numbers for purposes of discussion. The figure was prepared with molscript (25) and raster3d (26).
Fig. 3.
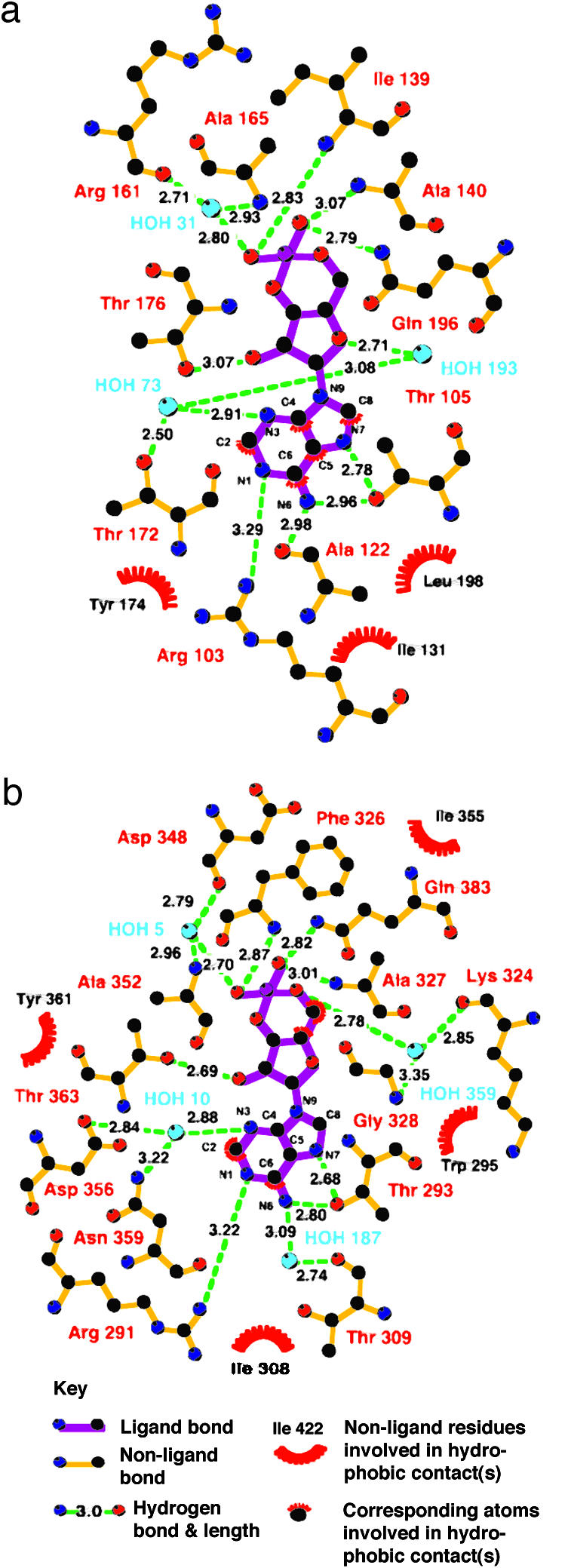
Diagram showing all of the close interactions between cAMP and AC cyaB2 GAF-A and -B, chain A. The figure was prepared with ligplot (27).
Two residues after Arg-291, Thr-293 forms direct 2.8- and 2.7-Å H-bonds to the cAMP N6 and N7. In PDE2A, the equivalent Ser-424 makes a 2.7-Å H-bond to N7 of cGMP. In either cyclase GAF domain, the conformation of this side chain is turned ≈130° to allow H-bonds of similar length to both N6 and N7.
There are two side chains that contact cAMP through a water. These include Asp-356 and Asn-359 that contact cAMP via HOH10 to the N3 position of cAMP. These residues are conserved in PDE2A GAF-B as Asp-485 and Thr-488 that coordinate to HOH1, which makes an H-bond to both the N2 amino group and N3 of cGMP. Therefore, a water molecule in this position is conserved in all three GAF domains. In the cyaB2 GAF-A, the equivalent residues are Glu-169, Thr-172, and HOH73. However, Glu-169, probably due to steric clash from its longer side chain, is turned away from the cAMP, exposing the carboxyl group to solvent.
In PDE2A, Asp-439 makes an H-bond with its side chain to cGMP N1, and also with its backbone amide to the O6 carbonyl (6). The preceding Phe-438 base stacks with the guanine ring. In cyaB2 GAF-B, the equivalent residues are Ile-308 and Thr-309. However, the backbone of these residues is shifted away from the cAMP (Fig. 5). Ile-308 cannot base stack but still makes a hydrophobic contact to the adenine ring, and Thr-309 is too far away for an H-bond. In cyaB2 GAF-A, the equivalent residues are Ala-121 and Ala-122. These residues overlap reasonably well in the structural alignment with their PDE2A counterparts. The amide of Ala-122 (chain A) is 3.4 Å from N6, at van der Waals distance. Instead, the carbonyl makes a 3.0-Å H-bond. The loop has a less well defined density, but the backbone density is continuous except for short gaps on either side of the Ala-122 carbonyl.
Fig. 5.

The β2–β3 loop of cNMP-binding GAF domain structures has a variable conformation and length. GAF-A (green) and -B (magenta) from the cyclase, and GAF-B (yellow) from PDE2A, were superimposed. Residues corresponding to PDE2A Phe-438–Asp-439 are shown. The figure was prepared with swiss-pdb viewer (28) and rendered with pov-ray.
In PDE2A, the 2′ hydroxyl makes an H-bond with Thr-492 (6). Although this is absolutely conserved as Thr-176 and Thr-363 in cyaB2 GAF-A and -B, 2′-deoxy-cAMP is as effective an activator as is cAMP itself (data not shown). Therefore, the functional significance of this conserved ensemble presently remains unresolved. In PDE2A, another contact of the 2′ hydroxyl is to HOH2, anchored by the Asp-485 backbone amide and Tyr-481 carbonyl. There is no equivalent water in either cyclase GAF-A or -B. The ribose has a C3′-endo puckering configuration in both PDE2A and the cyclase.
The 1.9-Å cyaB2 structure allows a high-resolution look at the binding of the cyclic phosphate. Overall, it is similar to that of cGMP in PDE2A GAF-B. The helix dipole α3 points to the phosphate group. The equatorial and axial oxygens form H-bonds to the sequential residues Ile-139 and Ala-140 in GAF-A and Phe-326 and Ala-327 in GAF-B. Both of these residues are at the N terminus of helix α3. The density shows clearly that cAMP is in the chair phosphoester conformation in both GAF-A and -B.
Conserved NKFDE Motif. Both cyaB2 GAF domains contain the conserved NKFDE motif found in all PDE tandem GAF domains (19). Consistent with the previously reported PDE2A GAF structures, the NKFDE motif in the cyaB2 GAF domains is not part of the binding pocket or dimer interface. However, superpositioning of the three cAMP or cGMP-bound GAF structures shows that these conserved sequences are accompanied by a conserved network of H-bonds, a conserved salt bridge, and a mutual proximity of the NKFDE residues (Fig. 4; also see Fig. 8 and Table 2, which are published as supporting information on the PNAS web site). The cyaB2 GAF-B structure will be used to illustrate. In this structure, Asn-386 is buried and forms H-bonds with the backbone of the residue preceding and the two residues following Thr-363 that in turn forms an H-bond to the ribose 2′ OH of the cyclic nucleotide. The salt bridge between Lys-387 and Asp-416 is not isolated, as the Lys amino group and Asp carboxyl group also make H-bonds to the backbone carbonyl of Met-287 and the backbone amide of Asp-413, respectively. The backbone carbonyl of Phe-412 itself forms an H-bond to the side chain amide of an Asn (Table 2 and Fig. 4; Asn-151 and Asn-338 in cyaB2 GAF-A and B; Asn-413 in PDE2 GAF-B). This Asn is very well conserved in PDE and cyclase GAF domains and perfectly conserved in all seven GAF domains that have been shown experimentally to bind cyclic nucleotide (Fig. 6). Therefore, the canonical NKFDE motif may be extended to an NNKFDE motif because the first Asn is conserved in all GAF domains known to bind cyclic nucleotide. All six residues form a continuous cluster in cyaB2 and PDE2 (Fig. 4). In this motif, the Glu shows the most variability in contacts. It interacts with a nearby Asn in PDE2A GAF-B and cyaB2 GAF-B but is solvent-exposed in cyaB2 GAF-A.
Fig. 4.

H-bonds of NKFDE motif in cyaB2 GAF-B. NKFDE residues are orange, cAMP is green, and T363 is magenta.
Fig. 6.

Sequence alignment of GAF domains known to bind cGMP or cAMP. From top to bottom, the sequences are cyaB2 AC (AC) GAF-B and -A in a structural alignment with mouse PDE2A GAF-B, cyaB1 AC GAF-B, human PDE5A GAF-A, chicken cone photoreceptor PDE6C GAF-A, and Trypanosoma brucei PDE2B GAF-A. For the homologous cyaB1, a mutation in the NKFDE motif implicates GAF-B in cAMP binding (11). For the PDE5A (7), PDE6C (9), and T. brucei PDE2B, individually expressed GAF-A domains have been studied with the filter-binding assay. The blue background denotes 50% or more identity. Lower conservation is denoted by the green background. A magenta bar indicates the β2–β3 loop that is the least conserved part of the sequence. The 17 residues that make direct and water-mediated contacts to cAMP in cyaB2 GAF-B are labeled and colored. Orange, polar side chains; purple, hydrophobic side chains; red, backbone amides and carbonyls. Yellow residues denote the NNKFDE motif.
Noticeably, the cyaB2 sequences containing the NKFDE motif are distinct from that of PDE2A by having a much longer loop between the Asn-Lys and the Phe (19 residues in cyaB2 GAF-A and 24 in GAF-B versus 5 residues in PDE2A GAF-B). In cyaB2 GAF-A, the loop points away from the cyclic-nucleotide-binding site, but in GAF-B, the loop lies on top of the α4-helix, which makes several contacts to cAMP.
Discussion
Dimerization. The antiparallel dimerization of GAF-A and -B is in complete contrast to the parallel dimer of the GAF-A/B from PDE2A (Fig. 1 a and c). In the latter, GAF-A is a dimerizing domain, and only GAF-B binds cGMP (6). Moreover, electron micrograph reconstructions of PDE5 and PDE6 also show parallel dimers, albeit at a much lower resolution (28 Å) (8, 20). Therefore, it appears that there can be more than one type of dimerization interface for GAF proteins.
A superposition along the connecting helices of cyaB2 GAF-A/B chain A (231–263) and the PDE2A GAF-A/B chain (367–398) (one chain in the asymmetric unit; see Fig. 7) shows these helices to be similar, but the GAF domains are disposed quite differently about them. The GAF-B domain of cyaB2 is tilted much closer to the connecting helix than in PDE2. The difference is due to the linker residues 399–402 in PDE2, which have a different conformation than the equivalent linker residues 268–271 of cyaB2. In PDE2A, the GAF-B domain does not dimerize and is ≈65 Å from the other GAF-B in the dimer.
Nucleotide Binding. The ability of each cyclase GAF domain to bind cAMP is also in contrast to what is seen with the PDE2A GAF domains. Nevertheless, the general topologies of the binding sites are similar between the GAF-B domain of PDE2A and either GAF domain of cyaB2. However, upon closer inspection, three differences are apparent. First, in PDE2A, the phosphoester ring was modeled in the boat conformation. Although, at a 2.9-Å resolution it was difficult to project the correct conformation, the chair conformation was rejected because the equatorial oxygen would have become axial, and within H-bonding distance of the carboxyl of Glu-512. Because presumably both of these oxygens are negatively charged, this would have caused an electrostatic repulsion. In the cyaB2 cyclase, Glu-512 is Gln-196 in GAF-A or Gln-383 in GAF-B, removing charge repulsion.
The second major structural difference between the PDE2A and the cyclase GAF domains is in the β2–β3 loop that may be important for ligand binding and possibly specificity (Fig. 5). In PDE2A, this loop contains Phe-438–Asp-439, which base stack with the guanine ring and form H-bonds to the N1 nitrogen, respectively (6). In cyaB2 GAF-A, Asp is Ala-122, whose carbonyl makes an H-bond to the N6 amino group. In GAF-B, the carbonyl of Thr-309 H-bonds to a water which itself H-bonds to N6. A superposition of these loops (Fig. 5) shows a variety of conformations. An alignment of GAF sequences known to bind cGMP or cAMP (Fig. 6) shows that this loop is the most variable part of the GAF domain in sequence and length and therefore may provide a major role in determining ligand specificity of the site.
The third major difference is in Arg-291 in GAF-B (or Arg-103 in GAF-A) that makes a 3.2- to 3.3-Å contact to N1 of cAMP. Of the seven to eight side-chain contacts to cAMP in cyaB2, only four are polar (GAF-B Arg-291, Thr-293, Thr-363, and Gln-383). The latter three H-bond to common contacts on either cAMP or cGMP. This leaves Arg-291 and its H-bond to the N1 of cAMP. Because in this conformation it would not form an H-bond with the protonated N1 of cGMP, it may well provide specificity for cAMP. However, the presence of Arg at this position is probably not a universal determinant of cAMP specificity because an Arg also occurs in the corresponding position of GAF-A of PDE5A, and of GAF-A of PDE6C, which are highly specific for cGMP binding (9, 21).
Activation Mechanism. So far, little biochemical data are available for the cyaB2 AC holoenyzme, because of difficulties in expression. However, the highly similar cyaB1 AC has been enzymatically characterized and shown to be activated by cAMP binding to its GAF-B domains (11). A chimera containing the tandem GAF-A/B domains from cyaB2 and the cyaB1 AC is activated almost exclusively by cAMP with an EC50 of ≈1 μM [see accompanying manuscript (12)]. Also, chimeras containing the tandem GAF-A/B domains from rat PDE2A or PDE5 could be activated by cGMP and not cAMP as in the native PDE holoenzymes (ref. 11 and unpublished data). These chimeras present a puzzle in light of the structure reported here. The mouse PDE2A GAF-A/B structure is a parallel dimer, with GAF-A dimerizing with itself, and the two cGMP-binding GAF-B domains spaced 65 Å apart. In the cyaB2 GAF-A/B antiparallel dimer reported here, GAF-A dimerizes with GAF-B of the opposite monomer. Furthermore, both GAF domains bind cAMP. At present it is difficult to see how such a radically different dimerization will be compatible with the activation mechanism in the cyaB1 AC chimeras. Although both holoenzymes are activated by cyclic nucleotide binding to one or more GAF domains, the different dimerization mechanisms would suggest different activation mechanisms, yet we know that the PDE2A and PDE5 GAF-A/B can activate the cyaB1 adenylyl cyclase. One possibility is that in both cases the catalytic domain directly contacts a GAF domain regardless of whether the dimer is parallel or antiparallel (or whether there is an intervening PAS domain as in the cyclase). Another possibility is that both parallel and antiparallel conformations provide a similar movement of their C termini upon binding of the cyclic nucleotide that is transduced to the respective catalytic domains. In this case, the orientation of the dimers would not be important for activation.
Conserved NKFDE Motif. The NKFDE motif is found in GAF domains of cyclases and phosphodiesterases, enzymes intrinsic to cyclic nucleotide signaling. A role of this motif in binding cyclic nucleotide is supported by several publications describing the effects of several mutations to the motif, nearly all to Ala. In a chimera containing PDE6 GAF-A/B and PDE5 catalytic domain, an Asn-to-Ala mutation in either GAF domain abolished noncatalytic cGMP binding (22). The Asp to Ala mutation in PDE5 in GAF-A or -B reduced affinity for cGMP (23). Asn, Lys, and Asp mutations to Ala in GAF-A did likewise, except for Glu to Ala, which had no effect (19). The Asp to Ala mutation in a GST fusion of PDE5 GAF-A modestly decreased binding (21). In PDE2A GAF-B, all five NKFDE residues were mutated to Ala (24). Each one abolished binding of cGMP and cAMP.
Inspection of the conserved hydrogen bonding, the placement of the salt bridge, and the proximity of the NKFDE motif residues (Fig. 4; also see Fig. 8 and Table 2) in all three GAF domain crystal structures with bound cyclic nucleotide (cyaB2 GAF-A and -B, and PDE2A GAF-B) show a number of conserved interactions. This conservation suggests that a general role for the NKFDE motif is to hold the α4-helix and the α4–β5 linker, which both close over the cAMP, in the proper orientation relative to the more rigid unit composed of the β-sheet, and the α2 and α5 helices on the opposite side of the β-sheet. These interactions are likely to be essential for closing the α4-helix over the bound cNMP. Overall, the function of the NKFDE motif would be to form the links that stabilize the closed conformation of the nucleotide binding pocket. It may also have additional roles in transmitting the conformation changes in the GAF domains that must occur upon nucleotide binding that in turn allow activation of the catalytic domains.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants DK-21723 and HL-44948 (to J.A.B.) and the Deutsche Forschungsgemeinschaft. N.Z. is a PEW Scholar.
Abbreviations: AC, adenylyl cyclase; PDE, phosphodiesterase; SeMet, selenomethionine; SuMet, sulfomethionine.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 1YKD).
References
- 1.Aravind, L. & Ponting, C. P. (1997) Trends Biochem. Sci. 22, 458-459. [DOI] [PubMed] [Google Scholar]
- 2.Anantharaman, V., Koonin, E. V. & Aravind, L. (2001) J. Mol. Biol. 307, 1271-1292. [DOI] [PubMed] [Google Scholar]
- 3.Charbonneau, H., Prusti, R. K., LeTrong, H., Sonnenburg, W. K., Mullaney, P. J., Walsh, K. A. & Beavo, J. A. (1990) Proc. Natl. Acad. Sci. USA 87, 288-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soderling, S. H., Bayuga, S. J. & Beavo, J. A. (1999) Proc. Natl. Acad. Sci. USA 96, 7071-7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fawcett, L., Baxendale, R., Stacey, P., McGrouther, C., Harrow, I., Soderling, S., Hetman, J., Beavo, J. A. & Phillips, S. C. (2000) Proc. Natl. Acad. Sci. USA 97, 3702-3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez, S., Wu, A., Glavas, N., Tang, X., Turley, S., Hol, W. & Beavo, J. (2002) Proc. Natl. Acad. Sci. USA 99, 13260-13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu, L., Underwood, T., Li, H., Pamukcu, R. & Thompson, W. J. (2002) Cell. Signalling 14, 45-51. [DOI] [PubMed] [Google Scholar]
- 8.Tcheudji, J. F., Lebeau, L., Virmaux, N., Maftei, C. G., Cote, R. H., Lugnier, C. & Schultz, P. (2001) J. Mol. Biol. 310, 781-791. [DOI] [PubMed] [Google Scholar]
- 9.Huang, D., Hinds, T. R., Martinez, S. E., Doneanu, C. & Beavo, J. A. (2004) J. Biol. Chem. 279, 48143-48151. [DOI] [PubMed] [Google Scholar]
- 10.Katayama, M. & Ohmori, M. (1997) J. Bacteriol. 179, 3588-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanacher, T., Schultz, A., Linder, J. U. & Schultz, J. E. (2002) EMBO J. 21, 3672-3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruder, S., Linder, J. U., Martinez, S. E., Zheng, N., Beavo, J. A. & Schultz, J. E. (2005) Proc. Natl. Acad. Sci. USA 102, 3088-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terwilliger, T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 15.Terwilliger, T. C. (2000) Acta Crystallogr. D 56, 965-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 17.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 5, 905-921. [DOI] [PubMed] [Google Scholar]
- 18.Murshudov, G. N. (1997) Acta Crystallogr. D 53, 240-255. [DOI] [PubMed] [Google Scholar]
- 19.Turko, I. V., Haik, T. L., McAllister-Lucas, L. M., Burns, F., Francis, S. H. & Corbin, J. D. (1996) J. Biol. Chem. 271, 22240-22244. [DOI] [PubMed] [Google Scholar]
- 20.Kajimura, N., Yamazaki, M., Morikawa, K., Yamazaki, A. & Mayanagi, K. (2002) J. Struct. Biol. 139, 27-38. [DOI] [PubMed] [Google Scholar]
- 21.Sopory, S., Balaji, S., Srinivasan, N. & Visweswariah, S. S. (2003) FEBS Lett. 539, 161-166. [DOI] [PubMed] [Google Scholar]
- 22.Granovsky, A. E., Natochin, M., McEntaffer, R. L., Haik, T. L., Francis, S. H., Corbin, J. D. & Artemyev, N. O. (1998) J. Biol. Chem. 273, 24485-24490. [DOI] [PubMed] [Google Scholar]
- 23.McAllister-Lucas, L. M., Haik, T. L., Colbran, J. L., Sonnenburg, W. K., Seger, D., Turko, I. V., Beavo, J. A., Francis, S. H. & Corbin, J. D. (1995) J. Biol. Chem. 270, 30671-30679. [DOI] [PubMed] [Google Scholar]
- 24.Wu, A. Y., Tang, X. B., Martinez, S. E., Ikeda, K. & Beavo, J. A. (2004) J. Biol. Chem. 279, 37928-37938. [DOI] [PubMed] [Google Scholar]
- 25.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 26.Merritt, E. A. (1994) Acta Crystallogr. 50, 869-873. [DOI] [PubMed] [Google Scholar]
- 27.Wallace, A. C., Laskowski, R. A. & Thornton, J. M. (1995) Protein Eng. 8, 127-134. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan, W. & Littlejohn, T. G. (2001) Brief Bioinform. 2, 195-197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.