

Abstract
Background
For single-cell or metagenomic sequencing projects, it is necessary to sequence with a very high mean coverage in order to make sure that all parts of the sample DNA get covered by the reads produced. This leads to huge datasets with lots of redundant data. A filtering of this data prior to assembly is advisable. Brown et al. (2012) presented the algorithm Diginorm for this purpose, which filters reads based on the abundance of their k-mers.
Methods
We present Bignorm, a faster and quality-conscious read filtering algorithm. An important new algorithmic feature is the use of phred quality scores together with a detailed analysis of the k-mer counts to decide which reads to keep.
Results
We qualify and recommend parameters for our new read filtering algorithm. Guided by these parameters, we remove in terms of median 97.15% of the reads while keeping the mean phred score of the filtered dataset high. Using the SDAdes assembler, we produce assemblies of high quality from these filtered datasets in a fraction of the time needed for an assembly from the datasets filtered with Diginorm.
Conclusions
We conclude that read filtering is a practical and efficient method for reducing read data and for speeding up the assembly process. This applies not only for single cell assembly, as shown in this paper, but also to other projects with high mean coverage datasets like metagenomic sequencing projects.
Our Bignorm algorithm allows assemblies of competitive quality in comparison to Diginorm, while being much faster. Bignorm is available for download at https://git.informatik.uni-kiel.de/axw/Bignorm.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1724-7) contains supplementary material, which is available to authorized users.
Keywords: Read filtering, Read normalization, Bignorm, Diginorm, Singe cell sequencing, Coverage
Background
Next generation sequencing systems (such as the Illumina platform) tend to produce an enormous amount of data — especially when used for single-cell or metagenomic protocols — of which only a small fraction is essential for the assembly of the genome. It is thus advisable to filter that data prior to assembly.
A coverage of about 20 for each position of the genome has been empirically determined as optimal for a successful assembly of the genome [1]. On the other hand, in many setups, the coverage for a large number of loci is much higher than 20, often rising up to tens or hundreds of thousands, especially for single-cell or metagenomic protocols (see Table 1, “max” column for the maximal coverage of the datasets that we use in our experiments). In order to speed up the assembly process — or in extreme cases to make it possible in the first place, given certain restrictions on available RAM and/or time — a sub-dataset of the sequencing dataset is to be determined such that an assembly based on this sub-dataset works as good as possible. For a formal description of the problem, see Additional file 1: Section S1.
Table 1.
Coverage statistics for Bignorm with Q 0=20, Diginorm, and the raw datasets
| Dataset | Algorithm | Mean | Max | ||
|---|---|---|---|---|---|
| Aceto | Bignorm | 6 | 132 | 216 | 6801 |
| Diginorm | 7 | 171 | 295 | 12,020 | |
| Raw | 15 | 9562 | 17,227 | 551,000 | |
| Alphaproteo | Bignorm | 10 | 43 | 92 | 884 |
| Diginorm | 7 | 173 | 481 | 6681 | |
| Raw | 25 | 5302 | 14,070 | 303,200 | |
| Arco | Bignorm | 1 | 98 | 54 | 2103 |
| Diginorm | 1 | 362 | 200 | 6114 | |
| Raw | 3 | 10,850 | 4091 | 220,600 | |
| Arma | Bignorm | 8 | 23 | 32 | 358 |
| Diginorm | 8 | 79 | 141 | 5000 | |
| Raw | 17 | 629 | 1118 | 31,260 | |
| ASZN2 | Bignorm | 40 | 70 | 83 | 2012 |
| Diginorm | 23 | 143 | 354 | 3437 | |
| Raw | 50 | 1738 | 4784 | 43,840 | |
| Bacteroides | Bignorm | 3 | 74 | 90 | 6768 |
| Diginorm | 3 | 123 | 205 | 7933 | |
| Raw | 7 | 6051 | 8127 | 570,900 | |
| Caldi | Bignorm | 25 | 63 | 110 | 786 |
| Diginorm | 15 | 67 | 135 | 3584 | |
| Raw | 27 | 1556 | 3643 | 33,530 | |
| Caulo | Bignorm | 7 | 228 | 216 | 10,400 |
| Diginorm | 8 | 362 | 491 | 35,520 | |
| Raw | 8 | 10,220 | 9737 | 464,300 | |
| Chloroflexi | Bignorm | 8 | 72 | 101 | 2822 |
| Diginorm | 9 | 412 | 878 | 20,850 | |
| Raw | 9 | 5612 | 7741 | 316,900 | |
| Crenarch | Bignorm | 8 | 104 | 159 | 3770 |
| Diginorm | 10 | 560 | 1285 | 29,720 | |
| Raw | 10 | 8086 | 14,987 | 316,700 | |
| Cyanobact | Bignorm | 9 | 144 | 153 | 5234 |
| Diginorm | 10 | 756 | 1450 | 26,980 | |
| Raw | 10 | 9478 | 11,076 | 356,600 | |
| E.coli | Bignorm | 37 | 45 | 56 | 234 |
| Diginorm | 50 | 382 | 922 | 7864 | |
| Raw | 112 | 2522 | 6378 | 56,520 | |
| SAR324 | Bignorm | 24 | 49 | 71 | 1410 |
| Diginorm | 18 | 53 | 107 | 2473 | |
| Raw | 26 | 1086 | 2761 | 106,000 |
Previous work
We briefly survey two prior approaches for read pre-processing, namely trimming and error correction. Read trimming programs (see [2] for a recent review) try to cut away the low quality parts of a read (or drop reads whose overall quality is low). These algorithms can be classified into two groups: running sum (Cutadapt, ERNE, SolexaQA with -bwa option [3–5]) and window based (ConDeTri, FASTX, PRINSEQ, Sickle, SolexaQA, and Trimmomatic [5–10]). The running sum algorithms take a quality threshold Q as input, which is subtracted from the phred score of each base of the read. The algorithms vary with respect to the functions applied to these differences to determine the quality of a read, the direction in which the read is processed, the function’s quality threshold upon which the cutoff point is determined, and the minimum length of a read after the cutoff to be accepted.
The window based algorithms, on the other hand, first cut away the reads’s 3’ or 5’ ends (depending on the algorithm) whose quality is below a specified minimum quality parameter and then determine a contiguous sequence of high quality using techniques similar to those used in the running sum algorithms.
All of these trimming algorithms generally work on a per-read basis, reading the input once and processing only a single read at a time. The drawback of this approach is that low quality sequences within a read are being dropped even when these sequences are not covered by any other reads whose quality is high. On the other hand, sequences whose quality and abundance are high are added over and over although their coverage is already high enough, which yields higher memory usage than necessary.
Most of the error correction programs (see [11] for a recent review) read the input twice: a first pass gathers statistics about the data (often k-mer counts) which in a second pass are used to identify and correct errors. Some programs trim reads which cannot be corrected. Again, coverage is not a concern: reads which seem to be correct or which can be corrected are always accepted. According to [11], currently the best known and most used error correction program is Quake [12]. Its algorithm is based on two assumptions:
“For sufficiently large k, almost all single-base errors alter k-mers overlapping the error to versions that do not exist in the genome. Therefore, k-mers with low coverage, particularly those occurring just once or twice, usually represent sequencing errors.”
Errors follow a Gamma distribution, whereas true k-mers are distributed as per a combination of the Normal and the Zeta distribution.
In the first pass of the program, a score based on the phred quality scores of the individual nucleotides is computed for each k-mer. After this, Quake computes a coverage cutoff value, that is, the local minimum of the k-mer spectrum between the Gamma and the Normal maxima. All k-mers having a score higher than the coverage cutoff are considered to be correct (trusted or solid in error correction terminology), the others are assumed to be erroneous. In a second pass, Quake reads the input again and tries to replace erroneous k-mers by trusted ones using a maximum likelihood approach. Reads which cannot be corrected are optionally trimmed or dumped.
But the main goal of error correctors is not the reduction of the data volume (in particular, they do not pay attention to excessive coverage), hence they cannot replace the following approaches.
Brown et al. invented an algorithm named Diginorm [1, 13] for read filtering that rejects or accepts reads based on the abundance of their k-mers. The name Diginorm is a short form for digital normalization: the goal is to normalize the coverage over all loci, using a computer algorithm after sequencing. The idea is to remove those reads from the input which mainly consist of k-mers that have already been observed many times in other reads. Diginorm processes reads one by one, splits them into k-mers, and counts these k-mers.
In order to save RAM, Diginorm does not keep track of those numbers exactly, but instead keeps appropriate estimates using the count-min sketch (CMS [14], see Additional file 1: Section S1.2 for a formal description). A read is accepted if the median of its k-mer counts is below a fixed threshold, usually 20. It was demonstrated that successful assemblies are still possible after Diginorm removed the majority of the data.
Our algorithm — Bignorm
Diginorm is a pioneering work. However, the following points, which are important from the biological or computational point of view, are not covered in Diginorm. We consider them as the algorithmic innovation in our work:
-
(i)
We incorporate the important phred quality score into the decision whether to accept or to reject a read, using a quality threshold. This allows a tuning of the filtering process towards high-quality assemblies by using different thresholds.
-
(ii)
When deciding whether to accept or to reject a read, we do a detailed analysis of the numbers in the count vectors. Diginorm merely considers their medians.
-
(iii)
We offer a better handling of the N case, that is, when the sequencing machine could not decide for a particular nucleotide. Diginorm simply converts all N to A, which can lead to false k-mer counts.
-
(iv)
We provide a substantially faster implementation. For example, we include fast hashing functions (see [15, 16]) for counting k-mers through the count-min sketch data structure (CMS), and we use the C programming language and OpenMP.
A technical description of our algorithm, called Bignorm, is given in Additional file 1: Section S1.3, which might be important for computer scientists and mathematicians working in this area.
Methods
Experimental setup
For the experimental evaluation, we collected the following datasets. We use two single cell datasets of the UC San Diego, one of the group of Ute Hentschel (now GEOMAR Kiel) and 10 datasets from the JGI Genome Portal. The datasets from JGI were selected as follows. On the JGI Genome Portal [17], we used “single cell” as search term. We narrowed the results down to datasets with all of the following characteristics:
status “complete”;
containing read data and an assembly in the download section;
aligning the reads to the assembly using Bowtie 2 [18] yields an “overall alignment rate” of more than 70%.
From those datasets, we arbitrarily selected one per species, until we had a collection of 10 datasets. We refer to each combination of species and selected dataset as a case in the following. In total, we have 13 cases; the details are given in Table 2.
Table 2.
Selected species and datasets (Cases)
| Short name | Species/Description | Source | URL |
|---|---|---|---|
| ASZN2 | Candidatus Poribacteria sp. WGA-4E_FD | Hentschel Group [27] | [28] |
| Aceto | Acetothermia bacterium JGI MDM2 LHC4sed-1-H19 | JGI Genome Portal | [29] |
| Alphaproteo | Alphaproteobacteria bacterium SCGC AC-312_D23v2 | JGI Genome Portal | [30] |
| Arco | Arcobacter sp. SCGC AAA036-D18 | JGI Genome Portal | [31] |
| Arma | Armatimonadetes bacterium JGI 0000077-K19 | JGI Genome Portal | [32] |
| Bacteroides | Bacteroidetes bacVI JGI MCM14ME016 | JGI Genome Portal | [33] |
| Caldi | Calescamantes bacterium JGI MDM2 SSWTFF-3-M19 | JGI Genome Portal | [34] |
| Caulo | Caulobacter bacterium JGI SC39-H11 | JGI Genome Portal | [35] |
| Chloroflexi | Chloroflexi bacterium SCGC AAA257-O03 | JGI Genome Portal | [36] |
| Crenarch | Crenarchaeota archaeon SCGC AAA261-F05 | JGI Genome Portal | [37] |
| Cyanobact | Cyanobacteria bacterium SCGC JGI 014-E08 | JGI Genome Portal | [38] |
| E.coli | E.coli K-12, strain MG1655, single cell MDA, Cell one | UC San Diego | [39] |
| SAR324 | SAR324 (Deltaproteobacteria) | UC San Diego | [39] |
For each case, we analyze the results obtained with Diginorm and with Bignorm using quality parameters Q 0∈{5,8,10,12,15,18,20,…,45}. Analysis is done on the one hand in terms of data reduction, quality, and coverage. On the other hand, we study actual assemblies that are computed with SPAdes [19] based on the raw and filtered datasets. For comparison, we also did assemblies using IDBA_UD [20] and Velvet-SC [21] (for Q 0=20 only). All the details are given in the next section.
The dimensions of the count-min sketch are fixed to m=1,024 and t=10, thus 10 GB of RAM were used.
Results
For our analysis, we mainly considered percentiles and quartiles of measured parameters. The ith quartile is denoted by , where we use for the minimum, for the median, and for the maximum. The ith percentile is denoted by ; we often use the 10th percentile .
Number of accepted reads
Statistics for the number of accepted reads are given as a box plot in Fig. 1 a. This plot is constructed as follows. Each of the blue boxes corresponds to Bignorm with a particular Q 0, while Diginorm is represented as the wide orange box in the background (recall that Diginorm does not consider quality values). Note that the “whiskers” of Diginorm’s box are shown as light-orange areas. For each box, for each case the raw dataset is filtered using the algorithm and algorithmic parameters corresponding to that box, and the percentage of the accepted reads is taken into consideration. For example, if the top of a box (which corresponds to the 3rd quartile, also denoted ) gives the value x %, then we know that for 75% of the cases, x % or less of the reads were accepted using the algorithm and algorithmic parameters corresponding to this box.
Fig. 1.
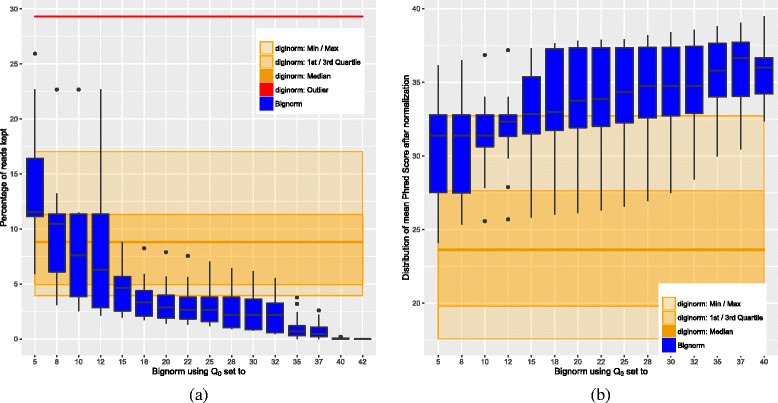
Box plots showing reduction and quality statistics. a Percentage of accepted reads (i.e. reads kept) over all datasets. b Mean quality values of the accepted reads over all datasets
There are two prominent outliers: one for Diginorm with value ≈29% (shown as the red line at the top) and one for Bignorm for Q 0=5 with value ≈26%. In both cases, the Arma dataset is responsible, which is the dataset with the worst mean phred score and the strongest decline of the phred score over the read length (see Additional file 1: Section S4 for more information and per base sequence quality plots). This suggest that the high rate of read kept is caused by a high error rate of the dataset. For 15≤Q 0, even Bignorm’s outliers fall below Diginorm’s median, and for 18≤Q 0 Bignorm keeps less than 5% of the reads for at least 75% of the datasets. In the range 20≤Q 0≤25, Bignorm delivers similar results for the different values of Q 0, and the gain in reduction for larger Q 0 is small up to Q 0=32. For even larger Q 0, there is another jump in reduction, but we will see that coverage and the quality of the assembly suffer too much in that range. We conjecture that in the range 18≤Q 0≤32, we remove most of the actual errors, whereas for larger Q 0, we also remove useful information.
Quality values
Statistics for phred quality scores in the filtered datasets are given in Fig. 1. The data was obtained using fastx_quality_stats from the FASTX Toolkit [7] on the filtered fastq files and calculating the mean phred quality scores over all read positions for each dataset. Looking at the statistics for these overall means, for 15≤Q 0, Bignorm’s median is better than Diginorm’s maximum. For 20≤Q 0, this effect becomes even stronger. For all values for Q 0, Bignorm’s minimum is clearly above Diginorm’s median. Note that an increase of 10 units means reducing error probability by factor 10.
In Table 3, we give quartiles of mean quality values for the raw datasets and Bignorm’s datasets produced with Q 0=20. Bignorm improves slightly on the raw dataset in all five quartiles.
Table 3.
Comparing quality values for the raw dataset and Bignorm with Q 0=20
| Quartile | Bignorm | Raw |
|---|---|---|
| (max) | 37.82 | 37.37 |
| 37.33 | 36.52 | |
| (median) | 33.77 | 32.52 |
| 31.91 | 30.50 | |
| (min) | 26.14 | 24.34 |
Of course, all this could be explained by Bignorm simply cutting away any low-quality reads. However, the data in the next section suggests that Bignorm may in fact be more careful than this.
Coverage
In Fig. 2, we see statistics for the coverage. The data was obtained by remapping the filtered reads onto the assembly from the JGI using Bowtie 2 and then using coverageBed from the bedtools [22] and R [23] for the statistics. In Fig. 2 a, the mean is considered. For 15≤Q 0, Bignorm reduces the coverage heavily. For 20≤Q 0, Bignorm’s is below Diginorm’s . This may raise the concern that Bignorm could create areas with insufficient coverage. However, in Fig. 2 b, we look at the 10th percentile () of the coverage instead of the mean. We consider this statistics as an indicator for the impact of the filtering on areas with low coverage. For Q 0≤25, Bignorm’s is at or above Diginorm’s maximum, and Bignorm’s minimum coincides with Diginorm’s (except for Q 0=10, where we are slightly below). In terms of the median, both algorithms are very similar for Q 0≤25. We consider all this as a strong indication that we cut away in the right places.
Fig. 2.
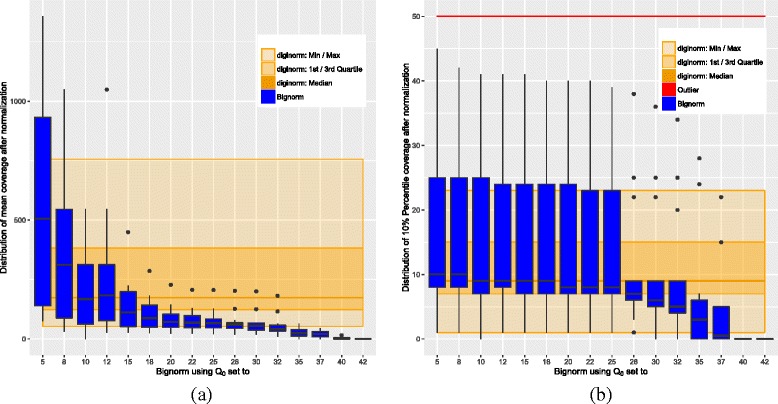
Box plots showing coverage statistics. a Mean coverage over all datasets. b 10th percentile of the coverage over all datasets
For 28≤Q 0, there is a clear drop in coverage, so we do not recommend such Q 0 values.
In Table 1, we give coverage statistics for each dataset. The reduction compared to the raw dataset in terms of mean, , and maximum is substantial. But also the improvement of Bignorm over Diginorm in mean, , and maximum is considerable for most datasets.
Assessment through assemblies
The quality and significance of read filtering is subject to complete assemblies, which is the final “road test” for these algorithms. For each case, we do an assembly with SPAdes using the raw dataset and those filtered with Diginorm and Bignorm for a selection of Q 0 values. The assemblies are then analyzed using quast [24] and the assembly from the JGI as reference. Statistics for four cases are shown in Fig. 3. We give the quality measures N50, genomic fraction, and largest contig, and in addition the overall running time (pre-processing plus assembler Wall time). Each measure is given in percentage relative to the raw dataset.
Fig. 3.
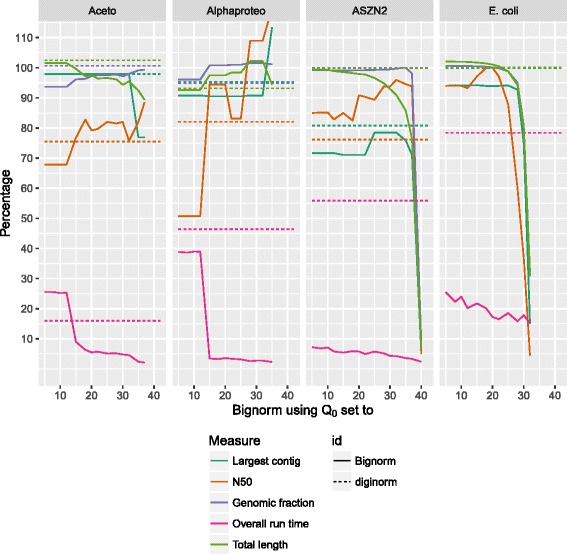
Assembly statistics for four selected datasets; measurements of assemblies performed on the datasets with prior filtering using Diginorm and Bignorm, relative to the results of assemblies performed on the unfiltered datasets
Generally, our biggest improvements are for N50 and running time. For 15≤Q 0, Bignorm is always faster than Diginorm, for three of the four cases by a large margin. In terms of N50, for 15≤Q 0, we observe improvements for three cases. For E.coli, Diginorm’s N50 is 100%, that we also attain for Q 0=20. In terms of genomic fraction and largest contig, we cannot always attain the same quality as Diginorm; the biggest deviation at Q 0=20 is 10 percentage points for the ASZN2 case. The N50 is generally accepted as one of the most important measures, as long as the assembly represents the genome well (as measured by the genomic fraction here) [25].
In Tables 4 and 5, we give statistics for Q 0=20 and each dataset. In terms of genomic fraction, Bignorm is generally not as good as Diginorm. However, excluding the Aceto and Arco cases, Bignorm’s genomic fraction is still always at least 95%. For Aceto and Arco, Bignorm misses 3.21% and 3.48%, respectively, of the genome in comparison to Diginorm. In 8 cases, Bignorm’s N50 is better or at least as good as Diginorm’s. The 4 cases where we achieved a smaller N50 are Arco, Caldi, Caulo, Crenarch, and Cyanobact.
Table 4.
Filter and assembly statistics for Bignorm with Q 0=20, Diginorm, and the raw datasets (Part I)
| Dataset | Algorithm | Reads kept | Mean phred | Contigs | Filter time | SPAdes time |
|---|---|---|---|---|---|---|
| in % | score | ≥10 000 | in sec | in sec | ||
| Aceto | Bignorm | 3.16 | 37.33 | 1 | 906 | 1708 |
| Diginorm | 3.95 | 27.28 | 1 | 3290 | 4363 | |
| Raw | 36.52 | 3 | 47,813 | |||
| Alphaproteo | Bignorm | 3.13 | 34.65 | 18 | 623 | 420 |
| Diginorm | 7.81 | 28.73 | 17 | 1629 | 11,844 | |
| Raw | 33.64 | 17 | 29,057 | |||
| Arco | Bignorm | 2.20 | 33.77 | 4 | 429 | 207 |
| Diginorm | 8.76 | 21.39 | 6 | 1410 | 1385 | |
| Raw | 32.27 | 6 | 15,776 | |||
| Arma | Bignorm | 7.90 | 28.21 | 44 | 240 | 135 |
| Diginorm | 29.30 | 21.19 | 50 | 588 | 1743 | |
| Raw | 26.96 | 44 | 5371 | |||
| ASZN2 | Bignorm | 5.66 | 37.66 | 118 | 1224 | 1537 |
| Diginorm | 12.62 | 32.73 | 130 | 5125 | 21,626 | |
| Raw | 36.85 | 112 | 47,859 | |||
| Bacteroides | Bignorm | 2.85 | 37.47 | 6 | 653 | 3217 |
| Diginorm | 4.94 | 27.64 | 5 | 2124 | 3668 | |
| Raw | 37.25 | 9 | 32,409 | |||
| Caldi | Bignorm | 3.97 | 37.82 | 41 | 842 | 455 |
| Diginorm | 5.61 | 30.67 | 36 | 1838 | 793 | |
| Raw | 37.37 | 38 | 7563 | |||
| Caulo | Bignorm | 2.40 | 36.95 | 10 | 679 | 712 |
| Diginorm | 4.70 | 25.16 | 9 | 2584 | 765 | |
| Raw | 36.01 | 13 | 18,497 | |||
| Chloroflexi | Bignorm | 1.40 | 31.91 | 32 | 694 | 134 |
| Diginorm | 9.70 | 18.91 | 33 | 2304 | 1852 | |
| Raw | 30.50 | 34 | 15,108 | |||
| Crenarch | Bignorm | 1.46 | 33.18 | 19 | 1107 | 790 |
| Diginorm | 9.72 | 19.80 | 18 | 2931 | 3754 | |
| Raw | 31.49 | 26 | 20,590 | |||
| Cyanobact | Bignorm | 1.65 | 30.45 | 12 | 679 | 450 |
| Diginorm | 11.30 | 17.58 | 13 | 1487 | 1343 | |
| Raw | 28.49 | 13 | 9417 | |||
| E. coli | Bignorm | 1.91 | 26.14 | 67 | 2279 | 598 |
| Diginorm | 17.03 | 19.34 | 63 | 9105 | 3995 | |
| Raw | 24.34 | 64 | 16,706 | |||
| SAR324 | Bignorm | 4.34 | 33.05 | 55 | 1222 | 708 |
| Diginorm | 4.69 | 23.58 | 52 | 3706 | 3085 | |
| Raw | 32.52 | 51 | 26,237 |
Table 5.
Filter and assembly statistics for Bignorm with Q 0=20, Diginorm, and the raw datasets (Part II)
| Dataset | Algorithm | N50 | Longest contig length | Genomic fraction | Misassembled contig length | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| abs | % of raw | % of Diginorm | abs | % of raw | % of Diginorm | abs | % of raw | % of Diginorm | abs | % of raw | % of Diginorm | ||
| Aceto | Bignorm | 2324 | 79 | 105 | 11,525 | 98 | 100 | 91 | 97 | 97 | 52,487 | 148 | 178 |
| Diginorm | 2216 | 76 | 11,525 | 98 | 94 | 100 | 29,539 | 84 | |||||
| Raw | 2935 | 11,772 | 94 | 35,351 | |||||||||
| Alphaproteo | Bignorm | 11,750 | 94 | 115 | 43,977 | 91 | 95 | 98 | 101 | 105 | 52,001 | 120 | 89 |
| Diginorm | 10,213 | 82 | 46,295 | 95 | 93 | 95 | 58,184 | 134 | |||||
| Raw | 12,446 | 48,586 | 98 | 43,388 | |||||||||
| Arco | Bignorm | 3320 | 81 | 97 | 12,808 | 57 | 57 | 85 | 100 | 97 | 76,797 | 99 | 91 |
| Diginorm | 3434 | 84 | 22,463 | 100 | 88 | 103 | 84,613 | 109 | |||||
| Raw | 4092 | 22,439 | 85 | 77,888 | |||||||||
| Arma | Bignorm | 18,432 | 102 | 107 | 108,140 | 100 | 100 | 98 | 100 | 100 | 774,291 | 91 | 103 |
| Diginorm | 17,288 | 96 | 108,498 | 100 | 98 | 100 | 748,560 | 88 | |||||
| Raw | 18,039 | 108,498 | 98 | 849,085 | |||||||||
| ASZN2 | Bignorm | 19,788 | 91 | 88 | 72,685 | 71 | 88 | 97 | 99 | 99 | 2,753,167 | 94 | 105 |
| Diginorm | 16,591 | 76 | 82687 | 81 | 97 | 100 | 2,617,095 | 89 | |||||
| Raw | 21,784 | 102,287 | 97 | 2,941,524 | |||||||||
| Bacteroides | Bignorm | 3356 | 68 | 100 | 25,300 | 100 | 100 | 95 | 98 | 99 | 70,206 | 105 | 112 |
| Diginorm | 3356 | 68 | 25,300 | 100 | 96 | 99 | 62,882 | 94 | |||||
| Raw | 4930 | 25,299 | 98 | 66,626 | |||||||||
| Caldi | Bignorm | 50,973 | 82 | 83 | 143,346 | 89 | 91 | 100 | 100 | 100 | 573,836 | 94 | 68 |
| Diginorm | 61,108 | 98 | 157,479 | 98 | 100 | 100 | 839,126 | 138 | |||||
| Raw | 62,429 | 160,851 | 100 | 609,604 | |||||||||
| Caulo | Bignorm | 4515 | 69 | 95 | 20,255 | 100 | 107 | 96 | 98 | 98 | 60,362 | 86 | 113 |
| Diginorm | 4729 | 72 | 18,907 | 93 | 98 | 101 | 53,456 | 76 | |||||
| Raw | 6562 | 20,255 | 97 | 70,161 | |||||||||
| Chloroflexi | Bignorm | 13,418 | 102 | 109 | 79,605 | 102 | 102 | 99 | 100 | 100 | 666,519 | 95 | 93 |
| Diginorm | 12,305 | 93 | 78,276 | 100 | 100 | 100 | 716,473 | 102 | |||||
| Raw | 13,218 | 78,276 | 99 | 703,171 | |||||||||
| Crenarch | Bignorm | 6538 | 77 | 91 | 31,401 | 81 | 66 | 97 | 99 | 99 | 484,354 | 89 | 95 |
| Diginorm | 7148 | 84 | 47,803 | 124 | 98 | 100 | 510,256 | 94 | |||||
| Raw | 8501 | 38,582 | 98 | 544,763 | |||||||||
| Cyanobact | Bignorm | 5833 | 95 | 99 | 33,462 | 98 | 100 | 99 | 101 | 100 | 236,391 | 113 | 110 |
| Diginorm | 5907 | 96 | 33,516 | 98 | 99 | 101 | 214,574 | 103 | |||||
| Raw | 6130 | 34,300 | 98 | 209,269 | |||||||||
| E. coli | Bignorm | 112,393 | 100 | 100 | 268,306 | 94 | 94 | 96 | 100 | 100 | 28,966 | 65 | 65 |
| Diginorm | 112,393 | 100 | 285,311 | 100 | 96 | 100 | 44,465 | 100 | |||||
| Raw | 112,393 | 285,528 | 96 | 44,366 | |||||||||
| SAR324 | Bignorm | 135,669 | 100 | 114 | 302,443 | 100 | 100 | 99 | 100 | 100 | 4,259,479 | 98 | 100 |
| Diginorm | 119,529 | 88 | 302,443 | 100 | 99 | 100 | 4,264,234 | 98 | |||||
| Raw | 136,176 | 302,442 | 99 | 4,342,602 | |||||||||
In Table 6, we show the total length of the assemblies for Q 0=20 absolute and relative to the length of the reference. In most cases, all assemblies are clearly longer than the reference, with Diginorm by trend causing slightly larger and Bignorm causing slightly shorter assemblies compared to the unfiltered dataset (see Additional file 1: Figure S6 for a box plot).
Table 6.
Reference length and total length of assemblies for Bignorm with Q 0=20, Diginorm, and the raw datasets
| Dataset | Reference | Raw | Diginorm | Bignorm | |||
|---|---|---|---|---|---|---|---|
| Ref length | Total length | % of ref | Total length | % of ref | Total length | % of ref | |
| Aceto | 426,710 | 750,316 | 175.80 | 769,090 | 180.20 | 731,850 | 171.50 |
| Alphaproteo | 463,456 | 405,020 | 87.40 | 377,293 | 81.40 | 394,979 | 85.20 |
| Arco | 231,937 | 408,571 | 176.20 | 419,403 | 180.80 | 380,191 | 163.90 |
| Arma | 1,364,272 | 2,123,588 | 155.70 | 2,131,958 | 156.30 | 2,077,037 | 152.20 |
| ASZN2 | 3,669,182 | 4,938,079 | 134.60 | 4,930,677 | 134.40 | 4,836,216 | 131.80 |
| Bacteroides | 560,676 | 826,566 | 147.40 | 818,799 | 146.00 | 792,384 | 141.30 |
| Caldi | 1,961,164 | 2,044,270 | 104.20 | 2,041,841 | 104.10 | 2,037,901 | 103.90 |
| Caulo | 423,390 | 601,709 | 142.10 | 616,942 | 145.70 | 590,319 | 139.40 |
| Chloroflexi | 863,677 | 1,317,768 | 152.60 | 1,326,848 | 153.60 | 1,186,531 | 137.40 |
| Crenarch | 716,004 | 1,009,122 | 140.90 | 1,016,485 | 142.00 | 946,606 | 132.20 |
| Cyanobact | 343,353 | 635,368 | 185.00 | 636,876 | 185.50 | 591,367 | 172.20 |
| E. coli | 4,639,675 | 4,896,992 | 105.50 | 4,898,422 | 105.60 | 4,948,739 | 106.70 |
| SAR324 | 4,255,983 | 4,676,938 | 109.90 | 4,674,540 | 109.80 | 4,669,774 | 109.70 |
Bignorm’s mean phred score is always slightly larger than that of the raw dataset, whereas Diginorm’s is always smaller. For some cases, the difference is substantial; the quartiles for the ratio of Diginorm’s mean phred score to that of the raw dataset are given in Table 7 in the first row.
Table 7.
Quartiles for comparison of mean phred score, filter and assembler Wall time in %
| Min | Median | Mean | Max | |||
|---|---|---|---|---|---|---|
| Diginorm mean phred score | 62 | 66 | 74 | 74 | 79 | 89 |
| raw mean phred score | ||||||
| Bignorm filter time | 24 | 28 | 31 | 33 | 38 | 46 |
| Diginorm filter time | ||||||
| Bignorm SPAdes time | 4 | 08 | 18 | 26 | 35 | 88 |
| Diginorm SPAdes time |
Clearly, our biggest gain is in running time, for the filtering as well for the assembly. Quartiles of the corresponding improvements are given in rows two and three of Table 7.
IDBA_UD and Velvet-SC
For a detailed presentation of the results gained with IDBA_UD and Velvet-SC, please see “Comparison of different assemblers” section in the Additional file 1. We briefly summarize the results:
IDBA_UD does not considerably benefit from read filtering, while Velvet-SC clearly does.
Velvet-SC is clearly inferior to both SPAdes and IDBA_UD, though in some regards the combination of read filtering and Velvet-SC is as good as IDBA_UD.
SPAdes nearly always produced better results than IDBA_UD, but in median (on unfiltered datasets) IDBA_UD is about 7 times faster than SPAdes.
SPAdes running on a dataset filtered using Diginorm is approximately as fast as IDBA_UD on the unfiltered dataset while SPAdes on a dataset filtered using Bignorm is roughly 4 times faster.
Discussion
The quality parameter Q 0 that Bignorm introduces as an innovation to Diginorm has shown to have a strong impact on the number of reads kept, coverage, and quality of the assembly. A reasonable upper bound of Q 0≤25 was obtained by considering the 10th percentile of the coverage (Fig. 2 b). With this constraint in mind, in order to keep a small number of reads, Fig. 1 a suggests 18≤Q 0≤25. Given that N50 for E.coli starts to decline at Q 0=20 (Fig. 3), we decided for Q 0=20 as the recommended value. As presented in detail in Table 4, Q 0=20 gives good assemblies for all 13 cases. The gain in speed is considerable: in terms of the median, we only require 31% and 18% of Diginorm’s time for filtering and assembly, respectively. This speedup generally comes at the price of a smaller genomic fraction and shorter largest contig, although those differences are relatively slight.
We believe that the increase of the N50 and largest contig for high values of Q 0, which we observe for some datasets just before the breakdown of the assembly (compare for example the results for the Alphaproteo dataset in Fig. 3), is due to the reduced number of branches in the assembly graph: SPAdes, as every assembler, ends a contig when it reaches an unresolvable branch in its assembly graph. As the number of reads in the input decreases more and more with increasing Q 0, the number of these branches also decreases and the resulting contigs get longer.
Conclusions
For 13 bacteria single cell datasets, we have shown that good and fast assemblies are possible based on only 5% of the reads in most of the cases (and on less than 10% of the reads in all of the cases). The filtering process, using our new algorithm Bignorm, also works fast and much faster than Diginorm. Like Diginorm, we use a count-min sketch for counting k-mers, so the memory requirements are relatively small and known in advance. Our algorithm Bignorm yields filtered datasets and subsequent assemblies of competative quality in much shorter time. In particular, the combination of Bignorm and SPAdes gives superior results to IDBA_UD while being faster. Furthermore, the mean phred score of our filtered dataset is much higher than that of Diginorm.
Acknowledgements
Not applicable.
Funding
This work was funded by DFG Priority Programme 1736 Algorithms for Big Data, Grant SR7/15-1.
Availability of data and materials
The datasets analyzed in the current study can be found in the references in Table 2. The source code for Bignorm is available at [26].
Author’s contributions
All authors planned and designed the study. AW implemented the software and performed the experiments. AW, LK, and CS wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
See file ’supplement.pdf’ for formal definitions and details on results from different assemblers. (PDF 259 kb)
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1724-7) contains supplementary material, which is available to authorized users.
Contributor Information
Axel Wedemeyer, Email: axw@informatik.uni-kiel.de.
Lasse Kliemann, Email: lki@informatik.uni-kiel.de.
Anand Srivastav, Email: asr@informatik.uni-kiel.de.
Christian Schielke, Email: csch@informatik.uni-kiel.de.
Thorsten B. Reusch, Email: treusch@geomar.de
Philip Rosenstiel, Email: admin1@ikmb.uni-kiel.de.
References
- 1.Brown CT, Howe A, Zhang Q, Pyrkosz AB, Brom TH. A Reference-Free Algorithm for Computational Normalization of Shotgun Sequencing Data. ArXiv e-prints. 2012:1–18. http://arxiv.org/abs/1203.4802.
- 2.Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. An Extensive Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS ONE. 2013;8(12):1–13. doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–2. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 4.Prezza N, Del Fabbro C, Vezzi F, De Paoli E, Policriti A. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine. BCB ’12. New York: ACM; 2012. ERNE-BS5: Aligning BS-treated Sequences by Multiple Hits on a 5-letters Alphabet. [Google Scholar]
- 5.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinforma. 2010;11(1):1–6. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smeds L, Künstner A. ConDeTri - A Content Dependent Read Trimmer for Illumina Data. PLoS ONE. 2011;6(10):1–6. doi: 10.1371/journal.pone.0026314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FASTX-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit/. Accessed 18 July 2016http://hannonlab.cshl.edu/fastx_toolkit/.
- 8.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–4. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi N, Fass J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33). 2011. https://github.com/najoshi/sickle.
- 10.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alic AS, Ruzafa D, Dopazo J, Blanquer I. Objective review of de novo stand-alone error correction methods for NGS data. Wiley Interdiscip Rev Comput Mol Sci. 2016;6(2):111–46. doi: 10.1002/wcms.1239. [DOI] [Google Scholar]
- 12.Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11(11):1–13. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Pell J, Canino-Koning R, Howe AC, Brown CT. These Are Not the K-mers You Are Looking For: Efficient Online K-mer Counting Using a Probabilistic Data Structure. PLoS ONE. 2014;9(7):1–13. doi: 10.1371/journal.pone.0101271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormode G, Muthukrishnan S. An improved data stream summary: the count-min sketch and its applications. J Algoritm. 2005;55(1):58–75. doi: 10.1016/j.jalgor.2003.12.001. [DOI] [Google Scholar]
- 15.Dietzfelbinger M, Hagerup T, Katajainen J, Penttonen M. A Reliable Randomized Algorithm for the Closest-Pair Problem. J Algoritm. 1997;25(1):19–51. doi: 10.1006/jagm.1997.0873. [DOI] [Google Scholar]
- 16.Wölfel P. Über die Komplexität der Multiplikation in eingeschränkten Branchingprogrammmodellen. PhD thesis, Universität Dortmund, Fachbereich Informatik. 2003.
- 17.JGI Genome Portal - Home. http://genome.jgi.doe.gov. Accessed 18 July 2016http://genome.jgi.doe.gov.
- 18.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012;19(5):455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng Y, Leung HCM, Yiu SM, Chin FYL. Idba-ud: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 21.Chitsaz H, Yee-Greenbaum Joyclyn L, Tesler G, Lombardo M-J. Dupont CL, Badger JH, Novotny M, Rusch DB, Fraser LJ, Gormley NA, Schulz-Trieglaff O, Smith GP, Evers DJ, Pevzner PA, Lasken RS. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotech. 2011;29(10):915–21. doi: 10.1038/nbt.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R Core Team . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2016. [Google Scholar]
- 24.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Earl D, Bradnam K, John JS, Darling A, Lin D, Fass J, Yu HOK, Buffalo V, Zerbino DR, Diekhans M, et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011;21(12):2224–241. doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wedemeyer A. Bignorm. https://git.informatik.uni-kiel.de/axw/Bignorm. Accessed 10 Oct 2016https://git.informatik.uni-kiel.de/axw/Bignorm.
- 27.Kamke J, Sczyrba A, Ivanova N, Schwientek P, Rinke C, Mavromatis K, Woyke T, Hentschel U. Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 2013;7(12):2287–300. doi: 10.1038/ismej.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Candidatus Poribacteria Sp. WGA-4E. http://genome.jgi.doe.gov/CanPorspWGA4E_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/CanPorspWGA4E_FD.
- 29.Acetothermia Bacterium JGI MDM2 LHC4sed-1-H19. http://genome.jgi.doe.gov/AcebacLHC4se1H19_FD/AcebacLHC4se1H19_FD.info.html. Accessed 18 July 2016http://genome.jgi.doe.gov/AcebacLHC4se1H19_FD/AcebacLHC4se1H19_FD.info.html.
- 30.Alphaproteobacteria Bacterium SCGC AC-312_D23v2. http://genome.jgi.doe.gov/AlpbacA312_D23v2_FD/AlpbacA312_D23v2_FD.info.html. Accessed 18 July 2016http://genome.jgi.doe.gov/AlpbacA312_D23v2_FD/AlpbacA312_D23v2_FD.info.html.
- 31.Arcobacter Sp. SCGC AAA036-D18. http://genome.jgi.doe.gov/ArcspSAAA036D18_FD/ArcspSAAA036D18_FD.info.html. Accessed 18 July 2016http://genome.jgi.doe.gov/ArcspSAAA036D18_FD/ArcspSAAA036D18_FD.info.html.
- 32.Armatimonadetes Bacterium JGI 0000077-K19. http://genome.jgi.doe.gov/Armbac0000077K19_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/Armbac0000077K19_FD..
- 33.Bacteroidetes bacVI JGI MCM14ME016. http://genome.jgi.doe.gov/BacbacMCM14ME016_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/BacbacMCM14ME016_FD.
- 34.Calescamantes Bacterium JGI MDM2 SSWTFF-3-M19. http://genome.jgi.doe.gov/CalbacSSWTFF3M19_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/CalbacSSWTFF3M19_FD.
- 35.Caulobacter Bacterium JGI SC39-H11. http://genome.jgi.doe.gov/CaubacJGISC39H11_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/CaubacJGISC39H11_FD..
- 36.Chloroflexi Bacterium SCGC AAA257-O03. http://genome.jgi.doe.gov/ChlbacSAAA257O03_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/ChlbacSAAA257O03_FD.
- 37.Crenarchaeota Archaeon SCGC AAA261-F05. http://genome.jgi.doe.gov/CrearcSAAA261F05_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/CrearcSAAA261F05_FD..
- 38.Cyanobacteria Bacterium SCGC JGI 014-E08. http://genome.jgi.doe.gov/CyabacSJGI014E08_FD. Accessed 18 July 2016http://genome.jgi.doe.gov/CyabacSJGI014E08_FD.
- 39.Single Cell Data Sets. http://bix.ucsd.edu/projects/singlecell/nbt_data.html. Accessed 18 July 2016http://bix.ucsd.edu/projects/singlecell/nbt_data.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed in the current study can be found in the references in Table 2. The source code for Bignorm is available at [26].